

Abstract

The metalloproteinase ADAM10 has been reported as an important target for drug discovery in several human diseases. In this vein, (6S,7S)-N-hydroxy-5-methyl-6-(4-(5-(trifluoromethyl)pyridin-2-yl)piperazine-1-carbonyl)-5-azaspiro[2.5]octane-7-carboxamide (compound 1) has been reported as a selective ADAM10 inhibitor. We synthesized this compound and document that it lacks both potency and selectivity in inhibition of ADAM10. This finding necessitated a structure-based computational analysis to investigate potency and selectivity of ADAM10 inhibition. The model that emerged indeed excluded compound 1 as an inhibitor for ADAM10, while suggesting another reported compound, (1R,3S,4S)-3-(hydroxycarbamoyl)-4-(4-phenylpiperidine-1-carbonyl)cyclohexyl pyrrolidine-1-carboxylate (compound 2), as an ADAM10 selective inhibitor. Compound 2 was synthesized and its potency, and selectivity in inhibition of ADAM10 were documented with a panel of several related enzymes. Pharmacokinetic studies of compound 2 in mice documented that the compound crosses the blood–brain barrier and may be useful as a pharmacological agent or mechanistic tool to delineate the role of ADAM10 in neurological diseases.
Keywords: ADAM10, inhibitor, selectivity, animal studies
A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) is a membrane-bound zinc-dependent metalloproteinase that has been proposed as molecular target for diseases ranging from cancer to neurodegenerative disorders in humans.1−4 In light of the fact that the active sites of ADAMs—of which 22 are known3—and those of the related matrix metalloproteinases (MMPs; 24 human variants are known5) are highly similar, discovery of selective inhibitors of this enzyme is critical to delineate pathophysiological roles of this enzyme. Selective inhibitors could potentially serve as pharmaceutical agents or as mechanistic tools in delineating the function of the enzyme in physiology and pathology. Although several inhibitor classes have been reported for ADAM10,6−16 selectivity of these compounds is generally poor, with documentation of cross-inhibitory effect in related enzymes. Identification of features that contribute to the potency and selectivity of these known inhibitors will pave the way for the design of the next generation inhibitors. We were particularly interested in a compound that has been reported in the literature as a selective inhibitor of ADAM10 (1, Figure 1).6,9 Here, we have undertaken computational, synthetic, and in vitro experimental efforts to understand features of selectivity of this inhibitor class. Contrary to the literature report, we document that compound 1 lacks both potency and selectivity for ADAM10 inhibition. Rather another compound of the same chemical class (2,Figure 1)6 exhibits both potency and the desirable selective inhibition of ADAM10 (Table 1). Utilizing structural and biochemical data, we report insights on the origins of selectivity for ADAM10 inhibition and provide pharmacokinetic attributes for this compound.
Figure 1.
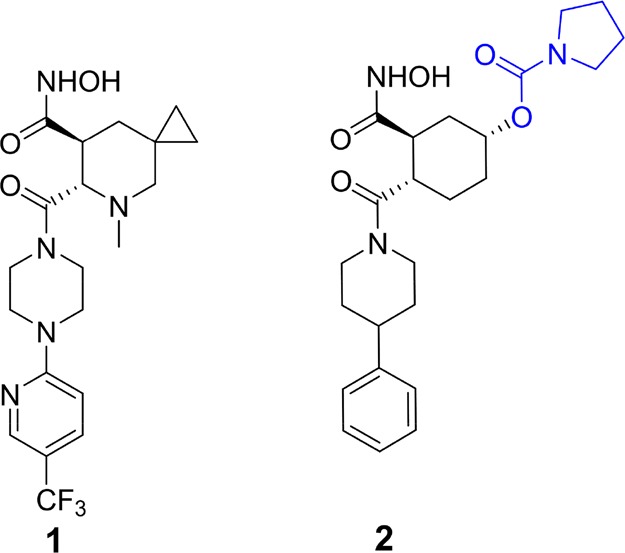
Chemical structures of compounds 1 and 2.
Table 1. Kinetic Parameters for Inhibition (Ki in nM).
| Enzyme | Compound 1 | Compound 2 |
|---|---|---|
| MMP-1 | 348 ± 0.5 | NIa |
| MMP-2 | 1.37 ± 0.10 | 13000 ± 6000 |
| MMP-3 | 484 ± 52 | 2%b |
| MMP-7 | 75 ± 13 | NIa |
| MMP-8 | 3.01 ± 0.70 | 8700 ± 600 |
| MMP-9 | 10.4 ± 1.4 | 25%b |
| MMP-12 | 29.5 ± 0.9 | 270 ± 40 |
| MMP-14 | 67.6 ± 1.6 | 19%b |
| ADAM9 | 2200 ± 1100 | 15%b |
| ADAM10 | 3%b | 73 ± 4 |
| ADAM17 | 61 ± 10 | 900 ± 40 |
No inhibition at 10 μM.
The level of inhibition at 10 μM.
We were drawn to compound 1 as a reportedly selective inhibitor of ADAM10.6 This compound was synthesized in our lab according to a reported synthesis in 11 steps and tested with a panel of metalloproteinases (Table 1).17 To our dismay, the compound did not show inhibitory activity for ADAM10 (merely 3% inhibition at 10 μM). Subsequently, we noted from our calculations (which we describe below) that another compound reported in the same literature (compound 2; Figure 1),6 should exhibit favorable structural features for potential ADAM10 selectivity. We sought to explain the unexpected outcome and attempted to investigate the basis for selectivity of ADAM10 inhibition. At the time, a crystal structure for the ADAM10 catalytic domain had not been reported. This absence of structural information hindered structure-based design of inhibitors and investigations of molecular recognition. Hence, we generated a computational model of the catalytic domain of ADAM10. A sequence search of the Protein Data Bank (PDB) identified TNF-alpha-converting enzyme (TACE/ADAM17) as a closely related protein with sufficient homology to ADAM10 catalytic site (35% identity and 52% similarity, Figure S1) to serve as template for protein modeling. Comparative modeling was performed with Modeler v 9.12.18 The model revealed that the overall catalytic domain organization of ADAM10 is similar to those of MMP-2 and MMP-9, consistent with frequent cross-inhibition of these enzymes by inhibitors conceived for ADAM10. Notable differences for the ADAM10 model, compared to the structures of MMP-2 and MMP-9, are at the S1′ and S1 subsites (Figure 2). The S1′ subsite in ADAM10 is defined on one side by a loop, which creates a shallow cavity, in contrast to deeper cavities for MMP-2 and MMP-9.19 The model was further studied by molecular-dynamics simulation with the AMBER12 program,20 which demonstrated that the active-site region is quite stable and should be useful for the purpose of structure-based inhibitor design (Figure S2). Molecular dynamism was seen mainly for two regions, located away from the active site.
Figure 2.

(A) Stereoview of the binding mode of compound 2 (stick representation) with the ADAM10 model (green ribbon and carbon atoms), as calculated by molecular docking. MMP-2 (gray ribbon) and MMP-9 (red ribbon) X-ray structures (PDB IDs, ICK7 and 2OVX, respectively) are superimposed to the ADAM10 model to show the variable residues at the S1 subsite. The hydroxamate group chelates with the zinc ion (sphere representation), while the phenyl group fits in the shallow S1′ pocket (6 o’clock position). The pyrrolidine-1-carboxylate moiety of compound 2 (colored in blue in Figure 1) binds at the S1 pocket (1 o’clock position), which is different for MMP-2 and MMP-9. The conserved structural calcium ion present in MMPs is pointed to with the black arrow (sphere representation, colored off-white). The zinc-chelating amino-acid residues are represented as a thin line with green for carbon atoms (B) The ADAM10 model is rendered in green surface representation (for protein, except for a few residues of the S1′ loop and zinc-coordinating residues for clarity) to show the S1 and S1′ pockets.
To investigate potency and selectivity, a library of 120 potent ADAM10 inhibitors (defined by Ki or IC50 of ≤1 μM) and 9 less active (or inactive) compounds6−8,10−14 (defined by Ki or IC50 of >5 μM) was docked to the active site of the model of ADAM10 with the program Glide (XP, v.5.6, Schrödinger LLC, NY, USA).21 The docked poses of the inhibitors affirmed interactions of their zinc-binding groups with the catalytic zinc ion of ADAM10, while providing insights on the moieties that would bind to the adjacent subsites of the enzyme (Figure S3). As a second validation of the model, a molecular-docking enrichment study was carried out. In this study, we docked a library of 1000 random decoy compounds from Schrödinger LLC, mixed together with the aforementioned library of 129 compounds for ADAM10 inhibitors. The randomly pooled druglike compounds were from the chemical collections of pharmaceutical corporate databases (Schrödinger decoys) with an average molecular mass of 400 Da.22 The analysis was able to differentiate among known inhibitors and the decoy compounds (Figure S4), which bolstered confidence in the model and in the analysis.
The inhibition data for many of the ADAM10 inhibitors of the same class as compounds 1 and 2 showed cross-inhibition of the closely related MMP-2 and MMP-9 (Table S1). To understand this promiscuity in inhibition, we performed a comparative analysis of the binding-site residues of ADAM10 with respect to MMP-2 and MMP-9 (Table S2). Whereas high conservation of structures was seen within the three active sites, especially near the zinc-chelation locus, differences in the pockets and the features of potential selectivity emerged from the analysis. Notable differences, as described below, are observed at the S1 subsite (Figure 2A and Table S2). The spatial position of Val297 in ADAM10 is substituted by Phe184 and Pro180 in MMP-2 and MMP-9, respectively. Additionally, Phe323, Asp325, and Val327 in ADAM10 are also substituted by other residues in the S1 pocket for MMP-2 and MMP-9 enzymes (see Table S2), suggesting a change in steric and electrostatic properties at the subsite among the closely related metalloproteinases. Moreover, a structural calcium ion is present in the vicinity of the S1 subsite of MMPs but absent in ADAM10 (Figure 2A). This could impact a differential behavior in the dynamics of the S1 subsite of these enzymes.
As mentioned earlier, compound 2 demonstrated structural features that could suit for selectivity toward ADAM10. The molecular-docking calculations suggested that the phenyl piperidine group of compound 2 would occupy the S1′ pocket, which is a shallow cavity in the case of ADAM10, as indicated earlier (Figure 2A and 2B). More importantly, the pyrrolidine-1-carboxylate functionality of compound 2, which is attached to the cyclohexyl group, binds to the S1 subsite (Figures 2A and 2B). Based on the anticipation of selectivity predicted by our computational analysis, we synthesized compound 2 in our laboratory in 14 steps, following a previously reported method,23 and tested it for inhibition in the same panel of metalloproteinases (Table 1). Indeed, compound 2 showed selective inhibition of ADAM10 (Ki of 73 ± 4 nM for ADAM10; poor inhibition for MMP-2 and MMP-9), nicely fitting to the pattern that emerged from our computational analysis.
In order to assess the utility of the compound for in vivo animal efficacy studies, the pharmacokinetic and brain distribution properties of compound 2 were evaluated in mice (n = 3 mice per time point per route of administration, total 33 mice) after intraperitoneal (ip) and subcutaneous (sc) dose administration (Figure 3 and Table 2). After a 10 mg/kg ip dose, plasma concentrations of compound 2 were above Ki for about 6 h, while those in brain remain above Ki for 20 min. Concentrations in brain after a 25 mg/kg sc dose were sustained, remaining above Ki for 100 min. The ratio of systemic exposure, as measured by the area-under-the-curve (AUC), of brain to plasma ranged from 0.0693 to 0.0761, indicating that compound 2 crossed the blood–brain barrier. As such, compound 2 may find utility as a pharmacological agent or mechanistic tool to delineate the role of ADAM10 in neurological diseases.
Figure 3.
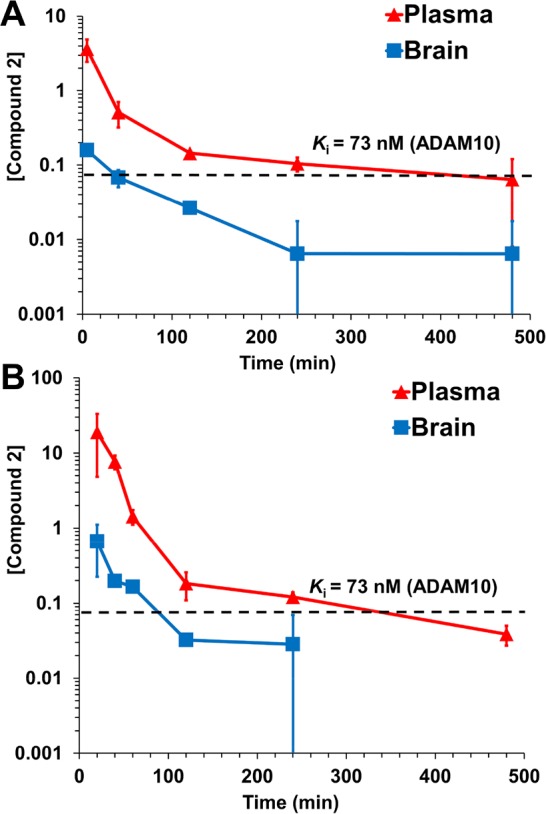
Concentration–time curve of compound 2 in plasma (red) and brain (blue) after (A) a single ip dose at 10 mg/kg and (B) a single sc dose at 25 mg/kg to mice (n = 3 per time point per route of administration, total 33 mice). Concentrations for plasma are in μM and for brain in pmol/mg tissue (equivalent to μM assuming a density of 1 g/mL).
Table 2. Pharmacokinetic Parameters of Compound 2.
|
Intraperitoneal 10 mg/kg |
Subcutaneous 25 mg/kg |
|||
|---|---|---|---|---|
| Parameter | Braina | Plasmaa | Braina | Plasmaa |
| AUC0-lasta | 12.1 | 155 | 51.2 | 1103 |
| AUC0-∞a | 14.0 | 184 | 77.0 | 1112 |
| t1/2α (min) | 28.8 | 12.4 | 11.4 | 15.4 |
| t1/2β (min) | 204 | 315 | 630 | 158 |
| AUCbrain/AUCplasma | 0.0761 | 0.0693 | ||
AUC in pmol·min/mg tissue for brain and in μM·min for plasma.
While this manuscript was in preparation, we noticed a recent report on the X-ray structure of ADAM10.24 A comparison of our computational model to the experimental X-ray structure is shown in Figure 4, where superimposition of the two catalytic domains is depicted. The computational model showed high similarity to the X-ray structure with root-mean-square deviation of 0.75 Å for Cα atoms of the active site residues (within 6 Å of the docked compound 2). Notably, the spatial positions of the residues in the S1 subsite in our model were validated by the new X-ray (Figure 4). For comparison, we have conducted additional molecular-docking calculation of the two compounds to the active site of the ADAM10 X-ray structure (PDB code 6BE6). The result reaffirmed the binding interactions of compound 2 at the S1 and S1′ sites (Figure S5A) that we have disclosed above. As a parenthetic observation, we add here that consistent with our in vitro inhibition experiments showing a lack of ADAM10 inhibition, compound 1 did not dock to the ADAM10 active site. This is likely because of the steric repulsion that hindered accommodating the trifluoromethyl group (Figure 1) in the shallow S1′ pocket of ADAM10 (Figure 2B).
Figure 4.
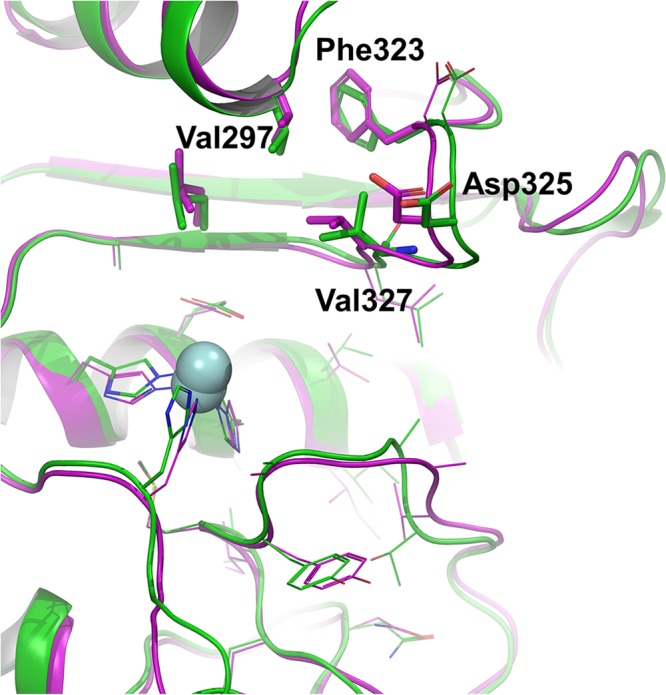
Comparison of our computational ADAM10 model (green for ribbon and carbon atoms), with the X-ray structure (PDB code 6BE6,24 purple for ribbon and carbon atoms). Residues of the S1 site, which play key roles in imparting ADAM10 inhibition and selectivity are labeled. The zinc ions are represented as spheres.
Further, we sought to understand the structural basis of selectivity/inhibition of compounds 1 and 2 with respect to MMP-2, MMP-9, ADAM10, and ADAM17. We docked the two compounds to the catalytic site of the X-ray structures of these enzymes. Compound 2 demonstrated significantly poorer in vitro dissociation constant for MMP-2 and MMP-9 (>10,000-folds) compared to compound 1 (Table 1). The docked poses suggested binding of the pyrrolidine-1-carboxylate moiety of 2 to the S1 site of these enzymes, where the larger leucine residues in MMPs (Leu190 in MMP-2 and Leu187 in MMP-9) in place of Val327 in ADAM10 likely disfavored the binding (Figures S5B and S5C). Moreover, a stretch of residues in MMP-2 and MMP-9 (residues 185–190 and 183–187, respectively) near the S1 sites interact with a structural calcium ion (Figures 2, S5B, and S5C)—absent in ADAM10—altering the steric and electrostatic features of the S1 site. The basis for the 12-fold selectivity of compound 2 to ADAM10 over ADAM17 (Ki of 73 ± 4 nM vs 900 ± 40 nM) is less obvious. However, variation of amino acids at the S1 sites of ADAM10 and ADAM17 (especially Met345 and Thr347 in ADAM17 compared to Asp325 and Val327, respectively, in ADAM10; Figure S5D) appears to affect the electrostatic properties of the site (Figure S5, lower panel). On the other hand, in vitro selectivity was observed for compound 1 toward ADAM17 in comparison to ADAM10 (Ki of 61 ± 10 nM and 3% inhibition at 10 μM, respectively). These data suggested that the trifluoromethyl group of compound 1 is tolerated at the S1′ pocket of ADAM17 but not in ADAM10.
In conclusion, we have investigated the selectivity of ADAM10 inhibition by small-molecule compounds. Selective inhibition of ADAM10 is achievable by compounds that bind to the S1 subsite, whereas the S1′ subsite does not provide opportunity to exploit for selective inhibitor design. Compound 2 is identified as a molecule with potency, selectivity, and appropriate pharmacokinetic properties for use as a tool in the investigation of diseases in which ADAM10 plays a role. Moreover, the data reported herein serve as foundation for the design of new classes of selective inhibitors targeting ADAM10.
Experimental Section
Protein Modeling
Homology modeling of the ADAM10 catalytic domain was performed with Modeler v 9.12 program18 using two ADAM17 X-ray structures (PDB codes 1BKC and 3G42) as template (See Supporting Information for details).
Molecular-Dynamics Simulations
Molecular-dynamics simulation of the model was performed with the AMBER12 suite20 following a protocol described previously.25 The Amberff12SB force field provided the necessary molecular-mechanics parameters. The protein model was inserted in a truncated octahedron box of TIP3P water molecules, energy-minimized and equilibrated for 400 ps. The system was subjected to a 50 ns simulation with the PMEMD module. Postsimulation analysis was performed with ptraj module, implemented in the AMBER12 package.
Compounds Data Set
Coordinates for compounds 1 and 2 were built using Maestro program (v. 9.5, Schrödinger LLC, NY, USA). Coordinates and activities of the additional compounds were downloaded from BindingDB (www.bindingdb.org).26 All compounds were verified for structural issues such as stereochemistry.
Molecular Docking
Compounds were prepared and energy-minimized with the LigPrep program (Schrödinger LLC, NY, USA). Molecular docking was conducted with the Glide program (v. 5.5, Schrödinger LLC, NY, USA).21 Docking grids for the model and X-ray structures were calculated for a 15-Å cube box with the catalytic site as the center. The X-ray structures were downloaded from the Protein Data Bank (rcsb.org) with codes 6BE6 (ADAM10), 1CK7 (MMP-2), 2OVX (MMP-9), and 3G42 (ADAM17) and prepared for docking with the Maestro program (Schrödinger LLC, NY, USA).
Syntheses
Compounds 1 and 2 were synthesized according to previously reported procedures.17,23 The purity of all final products was ≥95% (See Supporting Information for details).
Kinetics
The enzymes and their corresponding substrates were obtained as previously reported.27 The enzyme inhibition studies were performed according to a previously reported protocol28 (See Supporting Information for details). Ki values were measured in triplicate.
Pharmacokinetics
Male CD-1 mice (6–7 weeks old, average body weight 30 g) were purchased from Charles River Laboratories, Inc. (Wilmington, MA). Mice were given Teklad 2019 Extruded Diet (Harlan, Madison, WI) and water ad libitum. Mice were housed in polycarbonate shoeboxes containing 1/4” Bed-o’Cobs (The Andersons Inc., Maumee, OH) and Alpha-dri (Sheperd Specialty Papers, Inc., Richardland, MI) as bedding. A 12-h light and 12-h dark cycle was provided at 72 ± 2 °F. Animal studies were conducted with approval by the Institutional Animal Care and Use Committee.
Mice (n = 3 per time point per route of administration) were given 100 μL of dosing formulation, equivalent to a dose of 10 mg/kg ip or 25 mg/kg sc. At specific time points, mice were sacrificed and blood was collected by cardiac puncture using heparinized syringes. Brain samples were harvested after transcardiac perfusion with saline, weighed, and immediately flash-frozen in liquid nitrogen and stored at −80 °C until analysis. Blood was centrifuged to obtain plasma and was stored at −80 °C until analysis.
Acknowledgments
We acknowledge computing resources and assistance provided by Center for Research Computing of the University of Notre Dame.
Glossary
Abbreviations
- ADAM
a disintegrin and metalloproteinase domain-containing protein
- ip
intraperitoneal
- MMP
matrix metalloproteinase
- MRM
multiple-reaction monitoring
- sc
subcutaneous
- TACE
tumor necrosis factor-alpha-converting enzyme
- UPLC
ultraperformance liquid chromatography
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00163.
Sequence alignment, activity of additional compounds, enzyme binding-site comparison, method of protein modeling, spectral details of synthesized compounds, purity determination of compounds, additional details of pharmacokinetic measurements, and images. (PDF)
This work was supported by a grant from the Craig H. Neilsen Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Marcello E.; Borroni B.; Pelucchi S.; Gardoni F.; Di Luca M. ADAM10 as a therapeutic target for brain diseases: from developmental disorders to Alzheimer’s disease. Expert Opin. Ther. Targets 2017, 21, 1017–1026. 10.1080/14728222.2017.1386176. [DOI] [PubMed] [Google Scholar]
- Venkatesh H. S.; Tam L. T.; Woo P. J.; Lennon J.; Nagaraja S.; Gillespie S. M.; Ni J.; Duveau D. Y.; Morris P. J.; Zhao J. J.; Thomas C. J.; Monje M. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. 10.1038/nature24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S.; Saftig P. Ectodomain shedding and ADAMs in development. Development 2012, 139, 3693–3709. 10.1242/dev.076398. [DOI] [PubMed] [Google Scholar]
- Wetzel S.; Seipold L.; Saftig P. The metalloproteinase ADAM10: A useful therapeutic target?. Biochim. Biophys. Acta, Mol. Cell Res. 2017, 1864, 2071–2081. 10.1016/j.bbamcr.2017.06.005. [DOI] [PubMed] [Google Scholar]
- Klein T.; Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. 10.1007/s00726-010-0689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. C.; Liu X.; Li Y.; Covington M.; Wynn R.; Huber R.; Hillman M.; Yang G.; Ellis D.; Marando C.; Katiyar K.; Bradley J.; Abremski K.; Stow M.; Rupar M.; Zhuo J.; Li Y. L.; Lin Q.; Burns D.; Xu M.; Zhang C.; Qian D. Q.; He C.; Sharief V.; Weng L.; Agrios C.; Shi E.; Metcalf B.; Newton R.; Friedman S.; Yao W.; Scherle P.; Hollis G.; Burn T. C. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. 10.4161/cbt.5.6.2708. [DOI] [PubMed] [Google Scholar]
- Yao W.; Zhuo J.; Burns D. M.; Xu M.; Zhang C.; Li Y. L.; Qian D. Q.; He C.; Weng L.; Shi E.; Lin Q.; Agrios C.; Burn T. C.; Caulder E.; Covington M. B.; Fridman J. S.; Friedman S.; Katiyar K.; Hollis G.; Li Y.; Liu C.; Liu X.; Marando C. A.; Newton R.; Pan M.; Scherle P.; Taylor N.; Vaddi K.; Wasserman Z. R.; Wynn R.; Yeleswaram S.; Jalluri R.; Bower M.; Zhou B. B.; Metcalf B. Discovery of a potent, selective, and orally active human epidermal growth factor receptor-2 sheddase inhibitor for the treatment of cancer. J. Med. Chem. 2007, 50, 603–606. 10.1021/jm061344o. [DOI] [PubMed] [Google Scholar]
- Burns D. M.; He C.; Li Y.; Scherle P.; Liu X.; Marando C. A.; Covington M. B.; Yang G.; Pan M.; Turner S.; Fridman J. S.; Hollis G.; Vaddi K.; Yeleswaram S.; Newton R.; Friedman S.; Metcalf B.; Yao W. Conversion of an MMP-potent scaffold to an MMP-selective HER-2 sheddase inhibitor via scaffold hybridization and subtle P1′ permutations. Bioorg. Med. Chem. Lett. 2008, 18, 560–564. 10.1016/j.bmcl.2007.11.086. [DOI] [PubMed] [Google Scholar]
- Georgiadis D.; Yiotakis A. Specific targeting of metzincin family members with small-molecule inhibitors: progress toward a multifarious challenge. Bioorg. Med. Chem. 2008, 16, 8781–8794. 10.1016/j.bmc.2008.08.058. [DOI] [PubMed] [Google Scholar]
- Yao W.; Zhuo J.; Burns D. M.; Li Y. L.; Qian D. Q.; Zhang C.; He C.; Xu M.; Shi E.; Li Y.; Marando C. A.; Covington M. B.; Yang G.; Liu X.; Pan M.; Fridman J. S.; Scherle P.; Wasserman Z. R.; Hollis G.; Vaddi K.; Yeleswaram S.; Newton R.; Friedman S.; Metcalf B. Design and identification of selective HER-2 sheddase inhibitors via P1′ manipulation and unconventional P2′ perturbations to induce a molecular metamorphosis. Bioorg. Med. Chem. Lett. 2008, 18, 159–163. 10.1016/j.bmcl.2007.10.108. [DOI] [PubMed] [Google Scholar]
- Burns D. M.; Li Y. L.; Shi E.; He C.; Xu M.; Zhuo J.; Zhang C.; Qian D. Q.; Li Y.; Wynn R.; Covington M. B.; Katiyar K.; Marando C. A.; Fridman J. S.; Scherle P.; Friedman S.; Metcalf B.; Yao W. Compelling P1 substituent affect on metalloprotease binding profile enables the design of a novel cyclohexyl core scaffold with excellent MMP selectivity and HER-2 sheddase inhibition. Bioorg. Med. Chem. Lett. 2009, 19, 3525–3530. 10.1016/j.bmcl.2009.04.143. [DOI] [PubMed] [Google Scholar]
- Li Y. L.; Shi E.; Burns D.; Li Y.; Covington M. B.; Pan M.; Scherle P.; Friedman S.; Metcalf B.; Yao W. Discovery of novel selective HER-2 sheddase inhibitors through optimization of P1 moiety. Bioorg. Med. Chem. Lett. 2009, 19, 5037–5042. 10.1016/j.bmcl.2009.07.052. [DOI] [PubMed] [Google Scholar]
- Girijavallabhan V. M.; Chen L.; Dai C.; Feltz R. J.; Firmansjah L.; Li D.; Kim S. H.; Kozlowski J. A.; Lavey B. J.; Kosinski A.; Piwinski J. J.; Popovici-Muller J.; Rizvi R.; Rosner K. E.; Shankar B. B.; Shih N. Y.; Siddiqui M. A.; Tong L.; Wong M. K.; Yang D. Y.; Yang L.; Yu W.; Zhou G.; Guo Z.; Orth P.; Madison V.; Bian H.; Lundell D.; Niu X.; Shah H.; Sun J.; Umland S. Novel TNF-alpha converting enzyme (TACE) inhibitors as potential treatment for inflammatory diseases. Bioorg. Med. Chem. Lett. 2010, 20, 7283–7287. 10.1016/j.bmcl.2010.10.081. [DOI] [PubMed] [Google Scholar]
- Rosner K. E.; Guo Z.; Orth P.; Shipps G. W. Jr.; Belanger D. B.; Chan T. Y.; Curran P. J.; Dai C.; Deng Y.; Girijavallabhan V. M.; Hong L.; Lavey B. J.; Lee J. F.; Li D.; Liu Z.; Popovici-Muller J.; Ting P. C.; Vaccaro H.; Wang L.; Wang T.; Yu W.; Zhou G.; Niu X.; Sun J.; Kozlowski J. A.; Lundell D. J.; Madison V.; McKittrick B.; Piwinski J. J.; Shih N. Y.; Arshad Siddiqui M.; Strickland C. O. The discovery of novel tartrate-based TNF-alpha converting enzyme (TACE) inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1189–1193. 10.1016/j.bmcl.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Camodeca C.; Nuti E.; Tepshi L.; Boero S.; Tuccinardi T.; Stura E. A.; Poggi A.; Zocchi M. R.; Rossello A. Discovery of a new selective inhibitor of A Disintegrin And Metalloprotease 10 (ADAM-10) able to reduce the shedding of NKG2D ligands in Hodgkin’s lymphoma cell models. Eur. J. Med. Chem. 2016, 111, 193–201. 10.1016/j.ejmech.2016.01.053. [DOI] [PubMed] [Google Scholar]
- Madoux F.; Dreymuller D.; Pettiloud J.-P.; Santos R.; Becker-Pauly C.; Ludwig A.; Fields G. B.; Bannister T.; Spicer T. P.; Cudic M.; Scampavia L. D.; Minond D. Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 2016, 6, 11. 10.1038/s41598-016-0013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W.; Zhuo J.; Xu M.; Zhang F.; Metcalf B. W.. AZA spiro alkane derivatives as inhibitors of metalloproteases. WO Patent 2004,096,139, November 11, 2004.
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M. Y.; Pieper U.; Sali A.. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007, Chapter 2, Unit 2 9, 50, 2.9.1. 10.1002/0471140864.ps0209s50 [DOI] [PubMed] [Google Scholar]
- Gooyit M.; Song W.; Mahasenan K. V.; Lichtenwalter K.; Suckow M. A.; Schroeder V. A.; Wolter W. R.; Mobashery S.; Chang M. O-phenyl carbamate and phenyl urea thiiranes as selective matrix metalloproteinase-2 inhibitors that cross the blood-brain barrier. J. Med. Chem. 2013, 56, 8139–8150. 10.1021/jm401217d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, e. a.AMBER 12; University of California, San Francisco, 2012.
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Li Y.-l.; Zhuo J.; Burns D.; Yao W.; Jalluri R. K.. Substituted cyclic hydroxamates as inhibitors of matrix metalloproteinases. WO Patent 2005,037,826, April 28, 2005.
- Seegar T. C. M.; Killingsworth L. B.; Saha N.; Meyer P. A.; Patra D.; Zimmerman B.; Janes P. W.; Rubinstein E.; Nikolov D. B.; Skiniotis G.; Kruse A. C.; Blacklow S. C. Structural Basis for Regulated Proteolysis by the alpha-Secretase ADAM10. Cell 2017, 171, 1638–1648. (e1637). 10.1016/j.cell.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahasenan K. V.; Bastian M.; Gao M.; Frost E.; Ding D.; Zorina-Lichtenwalter K.; Jacobs J.; Suckow M. A.; Schroeder V. A.; Wolter W. R.; Chang M.; Mobashery S. Exploitation of Conformational Dynamics in Imparting Selective Inhibition for Related Matrix Metalloproteinases. ACS Med. Chem. Lett. 2017, 8, 654–659. 10.1021/acsmedchemlett.7b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Zhang H.; Trivedi A.; Mahasenan K. V.; Schroeder V. A.; Wolter W. R.; Suckow M. A.; Mobashery S.; Noble-Haeusslein L. J.; Chang M. Selective Inhibition of MMP-2 Does Not Alter Neurological Recovery after Spinal Cord Injury. ACS Chem. Neurosci. 2016, 7, 1482–1487. 10.1021/acschemneuro.6b00217. [DOI] [PubMed] [Google Scholar]
- Page-McCaw A.; Ewald A. J.; Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.