

Abstract

The emergence and evolution of new immunological cancer therapies has sparked a rapidly growing interest in discovering novel pathways to treat cancer. Toward this aim, a novel series of pyrrolidine derivatives (compound 5) were identified as potent inhibitors of ERK1/2 with excellent kinase selectivity and dual mechanism of action but suffered from poor pharmacokinetics (PK). The challenge of PK was overcome by the discovery of a novel 3(S)-thiomethyl pyrrolidine analog 7. Lead optimization through focused structure–activity relationship led to the discovery of a clinical candidate MK-8353 suitable for twice daily oral dosing as a potential new cancer therapeutic.
Keywords: MK-8353, MAPK pathway, ERK inhibitor, kinase selectivity, lead optimization, oncology
The MAPK pathway plays a central role in regulating mammalian cell growth, differentiation, survival, and migration by relaying extracellular signals. Activation of the MAPK pathway occurs via a cascade of protein phosphorylation events. One of the critical components in this pathway is the small GTPase RAS, which recruits and activates the serine/threonine kinase RAF. Activated RAF phosphorylates and activates MEK1/2, which in turn phosphorylates and activates ERK1/2. Once activated, ERK1/2 promotes the transcription of genes that are involved in cell cycle regulation, differentiation, and survival.1 Aberrant activation of the MAPK pathway has been demonstrated to be an important feature common to several human tumor types, and several components of the pathway have become attractive drug targets. Gain of function mutations of the RAS oncogene are found in several human cancers including pancreatic (90%), colorectal (50%), and lung (30%). Gain of function B-RAF mutations, which lead to constitutive activation of the MEK-ERK pathway, are found in melanoma (∼70%), colorectal (15%), and thyroid tumors (50%).2 In addition, nonsmall cell lung cancer and pancreatic tumors have been shown to have high incidence of RAS mutation. Tumors with mutations in BRAF or RAS are usually nonoverlapping in occurrence. The transforming activity of the RAS and BRAF mutations suggests that inhibition of the downstream targets such as ERK would be a good strategy for the development of therapeutic agents. Inhibition of ERK should effectively inhibit all signal transduction downstream from MEK since ERK is the only known substrate for MEK. To date, several signal transduction inhibitors, e.g., Vemurafenib (BRAF), Dabrafenib (BRAF), and Trametinib (MEK), targeting this pathway have been approved and various RAS-ERK inhibitors are being studied at various phases of development.3 Reactivation of the ERK1/2 phosphorylation and signaling has been frequently associated with secondary resistance to these upstream inhibitors.4 Recently several highly optimized ERK inhibitors such as the pyrrole 1 by Vertex/Biomed Valley Discoveries,5 the pyridone 2 by Genentech,6 fused pyrrolo-diazepanone 3 by Novartis,7 and irreversible acrylate inhibitor 4 by Astrazeneca8 have been reported (Figure 1) highlighting the importance of exploring targeted inhibition of more downstream components of MAPK signaling.
Figure 1.

Representative structures of ERK inhibitors 1–5.
We previously reported the discovery of a pyrrolidine based ERK inhibitor 5 (SCH-772984),9 which was derived from an initial high throughput screening hit 6 utilizing an automated ligand identification system (ALIS)10 and validation11 (Figure 2). Compound 5 showed excellent kinase selectivity against a panel of kinases while displaying nanomolar ERK1/2 potency in both biochemical and cellular assays. Interestingly, compound 5 displayed dual mechanism of action effecting both the inhibition of MEK phosphorylation of ERK1/2 and inhibition of its intrinsic kinase function with nanomolar potency. This pyrrolidine compound 5 served as a proof-of-concept (PoC) lead that demonstrated single agent efficacy in several xenograft mouse models with either KRAS or BRAF mutant cell lines.
Figure 2.
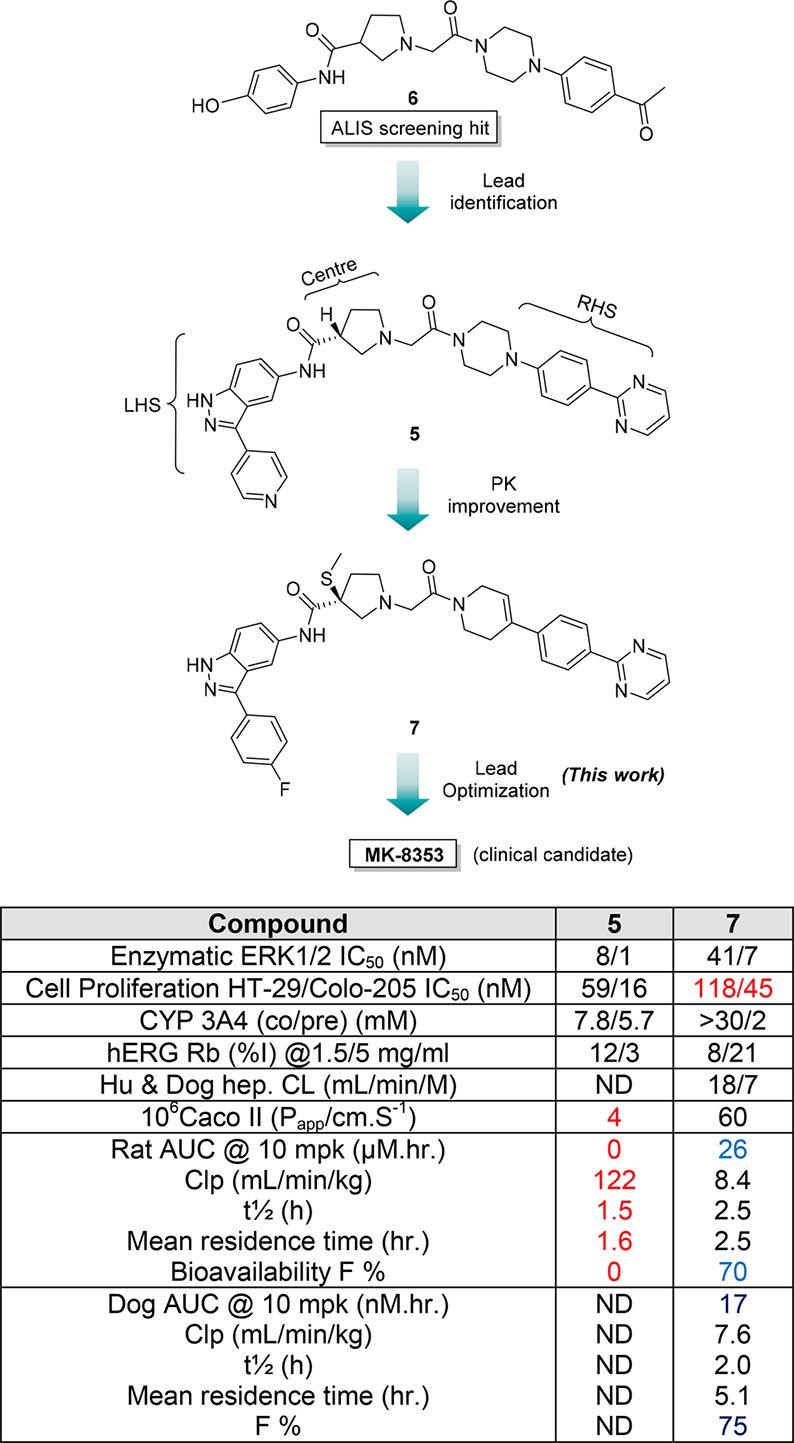
Progression of discovery of ERK inhibitor MK-8353. ND, not determined.
Compound 5 suffered from poor pharmacokinetics (PK), which precluded further development due to high clearance and low permeability leading to poor absorption and bioavailability in rat, as seen in the plasma drug concentration measured by area under the concentration–time curve (AUC) (rat AUC @ 10 mpk = 0 μM·h; F % = 0; Figure 2). The conventional approaches of lowering the clearance by reducing hydrophobicity and lowering logD did not improve oral exposure as lowering logD further deteriorated the permeability. Athough compound 5 provided an excellent starting point for our medicinal chemistry program, our goal was to develop a potent, selective, and orally bioavailable ERK1/2 inhibitor for oncology. Cold metabolite identification studies revealed pyrrolidine amide hydrolysis between center core and left-hand side (LHS) due to proteolytic enzymes. We suspected that this hydrolysis was responsible for poor PK. Systematic introduction of steric hindrance groups at the 3-position of the pyrrolidine led to the discovery of 3(S)-thiomethyl pyrrolidine analog 7 (Figure 2). This group vastly improved the PK (rat/dog AUC PK @10 mpk = 26/17 μM·h; rat/dog F% = 70/75, see ref (12) for detailed structure–activity relationship (SAR) analysis) but with the loss of 2–3-fold cell potency when compared to PoC compound 5. Herein we report further optimization of the lead 7 to which led to the discovery of a clinical candidate MK-8353, a highly potent and selective ERK1/2 inhibitor with good oral bioavailability across multiple preclinical species. MK-8353 displayed anti-tumor efficacy in several BRAF mutant models.
Although thiomethyl compound 7 improved PK properties from the PoC lead compound 5, it required further optimization to improve potency, reduce hERG, and improve the overall profile in selecting a single clinical candidate for progression. Crystal structure of compound 5 and other related molecules in this series revealed that the pyrimidine group in the right-hand side (RHS) forms a key π–π interaction with Tyr-62 of the ERK protein.12,13 The nature of the linker connecting the pyrimidine to the center core is critical for optimal binding as it orients the pyrimidine to pick up the π–π interaction with Tyr-62. We began SAR scrutinizing the piperidine-ene (double bond oxidation, a potential liability) on the RHS of the molecule 7 while holding the p-fluorophenyl indazole on the LHS constant, and the results are summarized in Table 1. Replacing piperidine-ene in 7 with piperazine to obtain compound 8 maintained ERK1/2 potency but showed a decreased rat AUC by 7-fold. Substitution of fluorine on the o-position of phenyl ring to obtain compound 9 resulted in 16-fold loss in ERK2 potency, potentially changing the trajectory angle of pyrimidine to a less optimal position. Reducing the double bond of piperidine-ene 7 to a tetrahydropyridine group to obtain compound 10 and substitution of fluorine on the o-position of phenyl ring to obtain compound 11 both resulted in 2–3-fold loss in ERK2 potency.
Table 1. Targeted Modifications on the Right Hand Side Portion of Compound 7.

Rat AUC (μM.hr) @ 10 mpk.
Attempted replacement of the phenyl group between the piperidine-ene ring and the pyrimidine ring in 7 with a five-membered thiazole ring resulted in two regioisomers 12 and 13 with much decreased ERK1/2 potency. This limited SAR illustrated that the piperidine-ene phenyl pyrimidine construction of the RHS of the molecule 7 was optimal in maintaining the ERK potency with improved PK.
Concurrently, we rapidly investigated 3(S)-thiomethoxy pyrrolidine compound 7 that provided the balanced enzymatic ERK potency and PK properties, while we continued medicinal chemistry efforts to further improve physicochemical properties such as cell potency (HT-29 and colo-205) and other off-targets viz., hERG by combining various LHS and RHS modifications, and the results are summarized in Table 2(14,15). Change of the p-fluoro phenyl group attached to the indazole on the LHS of compound 7 to a 4,5-methylenedioxy phenyl compound 14 gave an excellent 4-fold boost in rat AUC (106 μM at 10 mpk) and 1.5-fold improvement in cellular potency but suffered from strong CYP 3A4 inhibition (preincubation) with an in vitro IC50 = 0.3 μM. In general, this series of ERK compounds are not potent inhibitors of human CYPs 1A2, 2C9, 2C19, or 2D6 but are time-dependent inhibitors of CYP3A4 and thus potentially raise the concerns for drug–drug interactions when coadministered with drugs that are primarily metabolized by CYP 3A4. The 5-methoxy pyridyl, compound 15, showed a similar profile but with only marginal improvement in hERG activity and rat AUC. Various heterocyclic rings were explored on the RHS and LHS. For example, 2-ethylamino-1,3,4-oxadiazole compound 16 (with p-fluoro phenyl indazole in LHS) and 2-ethylamino-1,3-thiazole compound 17 (with 2-methylpyridine indazole in LHS) showed only a modest rat AUC 4.9 and 4.8 μM at 10 mpk, respectively. However, a more electron deficient 1-methyl-1,2,4-triazole compound 18 improved both ERK1/2 enzymatic and cellular (HT-29) potency by 3- to 4-fold and at the same time restored the rat oral AUC exposure. To remove the potential metabolic liability of the methoxy group in 5-methoxy pyridyl compound 18, introduction of a 5-isopropoxy pyridyl group to obtain compound 19 led to a 10-fold loss of ERK1/2 potency potentially due to steric hindrance and nonalignment of favorable Lys112 interaction of ERK. Enabled by structure-based drug design (SBDD), an effort to reorient the 5-isopropoxy O atom, which forms a key H-bond with Lys112 of ERK when changed to 4-isopropoxy pyridyl indazole on the LHS (see X-ray, Figure 4) and in combination with the potent, optimal 1-methyl-1,2,4-triazole RHS resulted in the discovery of compound 20. Compound 20 displayed overall improved ERK1/2 enzymatic, cell potency, hERG selectivity, and good rat oral AUC. Further replacement of 1-methyl-1,2,4-triazole with pyrimidine, compound 21, led to a loss of ERK1/2 and cell potency (2-fold) with no advantage in hERG selectivity. At this point, compound 20 was selected as a preclinical candidate (MK-8353) for further development and evaluated in vivo studies based on its in vitro potency (ERK1/2 IC50 = 20/7 nM), cellular activity (HT29/Colo-205 IC50 = 51/23 nM), and in conjunction with its favorable off-target hERG activity and pharmacokinetic profile. Interestingly, the sulfone methyl pyrrolidine analog of compound 20 gave compound 22, and the opposite enantiomer 3(R)-thiomethoxy of compound 20 gave compound 23. Both compounds were shown to be weaker in ERK1/2 potency, illustrating the importance of a stereospecific structural requirement for this class of inhibitors for optimal potency.12
Table 2. Combination of LHS and RHS Modifications of the 3(S)-Thiomethoxy Pyrrolidine Core.

ND, not determined.
Rat AUC (μM.hr) @ 10 mpk.
hERG (IW) %I@ 1/10 μM.
Figure 4.
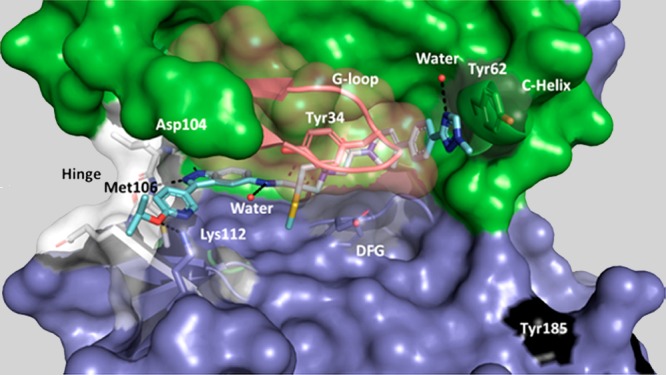
X-ray crystal structure of MK-8353 (PDB: 6DCG) in the active site of ERK2 with hydrogen bond interactions to the key residues highlighted in dashed lines.
The full profile of the clinical compound MK-8353 is depicted in Figure 3. MK-8353 is a potent and selective inhibitor of both active and inactive ERK1 and ERK2 kinases (IC50 = 20 and 7 nM, respectively). The overall kinase selectivity profile of MK-8353 was characterized against a panel of 233 mammalian serine/threonine and tyrosine kinases. Only three kinases showed greater than 50% inhibition at the 1 μM test concentration, and none of the 231 off-target kinases were inhibited at 100 nM.
Figure 3.

Detailed profile of MK-8353.
The excellent selectivity of MK-8353 for ERK1/2 can be attributed to a large compound induced conformational change in the poly glycine loop of ERK (see crystal structure, Figure 4). A2058 melanoma cells were used to characterize the in-cell target engagement profile of MK-8353. A typical Western analysis profile of MK-8353 (see Supporting Information) caused a dose proportional decrease in pERK1/2 and pRSK levels with complete suppression of pERK1/2 at 30 nM in A2058 cells, illustrating the dual mechanism of action. MK-8353 was not a potent inhibitor of human CYPs 1A2, 2C9, 2C19, or 2D6 but inhibits CYP 3A4 (preincubation) in vitro and showed inhibition of CYP 3A4 and 2C8 (IC50 = 1.7 and 3.5 μM), which can cause drug–drug interactions when coadministered with drugs that are primarily metabolized by CYP 2C8 or 3A4. MK-8353 is a weak inhibitor of hERG current, producing 16% inhibition at 0.6 μM. There were no test article-related changes in PR, QRS, and QT/QTc intervals in telemetered guinea pigs exposed at 30 and 100 mg/kg with exposure multiples of 2–3-fold (based on total Cmax). Based on the metabolite characterization in rat bile, urine, and plasma, the primary metabolic pathways appeared to be oxidative dealkylation of the parent compound leading to a loss of the isopropyl group as well as the amide cleavage of the parent with subsequent oxidative and conjugation reactions. No glutathione conjugate metabolites were found.18
The in vivo pharmacokinetics and metabolism of MK-8353 were evaluated in male CD1 mice, Sprague–Dawley (SD) rats, guinea pigs, beagle dogs, and cynomologus monkeys. With the exception of monkeys, MK-8353 showed moderate clearance after IV administration in all species, with a half-life range of 1.3–2.8 h and a mean residence time range of 1.5–4.0 h (Tables 3 and 4). Acceptable oral bioavailability was seen in mice, rats, and dogs (23–80%) but low oral bioavailability in monkeys (2%). The permeability observed in Caco-2 cells was high (135 nm/s), suggesting that intestinal absorption and permeability in humans should also be high. The steady-state volume of distribution in mice, dogs, and monkeys was in the range of 0.9–3.3 L/kg, while in rats it was 0.1 L/kg.
Table 3. Mean Pharmacokinetic Parameters of MK-8353 Following Intravenous Administrationa.
| species | dose (mg/kg) | n | AUC (0-∞) (μM·h) | half-life (h) | MRT (h) | clearance (mL/min/kg) | Vd,ss (L/kg) |
|---|---|---|---|---|---|---|---|
| mouse | 10 | 30 | 19.4 | 1.8 | 1.5 | 38 | 3.3 |
| SD rat | 3 | 3 | 85 | 1.6 | 2.3 | 1.0 | 0.1 |
| dog | 3 | 3 | 20 | 2.8 | 4.0 | 3.9 | 0.9 |
| monkey | 1 | 3 | 1.3 | 1.3 | 1.9 | 17.9 | 2.0 |
Aqueous solution of 20% hydroxypropyl-β-cyclodextrin (HPβCD).
Table 4. Mean Pharmacokinetic Parameters of MK-8353 Following Oral Administrationa.
| species | dose (mg/kg) | n | Cmax (μM) | Tmax (h) | AUC (0–∞) (μM.h) | bioavailability (%) |
|---|---|---|---|---|---|---|
| mouse | 30 | 27 | 4.6 | 1.0 | 15.3 | 80 |
| SD rat | 10 | 3 | 24 | 1.7 | 143 | 50 |
| dog | 30 | 3 | 6.5 | 4.0 | 45 | 23 |
| monkey | 10 | 3 | 0.06 | 1.4 | 0.2 | 2 |
0.4% hydroxypropyl methylcellulose (0.4% MC).
The crystal structure of ERK with the ATP competitive inhibitor MK-8353 has been determined at 1.45 Å resolutions. An overall view of this structure is shown below (Figure 4). The indazole ring of the inhibitor inserts into the ATP binding cavity and mimics the hydrogen bonding of adenine to the hinge region of ERK and H-bonds to the backbone Asp104 CO and Met106 NH.
Another strong interaction is the H-bond from the catalytic Lys52 to the pyrrolidine nitrogen, which requires the deprotonated form of the amino nitrogen; a pKa near neutral pH is essential. Tyr34 folds under the Gly-rich loop (conformational shift) and stacks with the pyrrolidine ring. This rearrangement opens a cavity that is occupied by the pendant methyl-triazole where it stacks with Tyr62. The 3(S)-thiomethoxy group that allows to retain strong binding to ERK point toward the binding region defined by residues Asn152 and Cys164. The numerous interactions that MK-8353 makes with ERK contribute to its potency and selectivity versus other kinases.
In addition to inhibiting the kinase activity of ERK, MK-8353 prevents the phosphorylation of ERK by MEK.18 By contrast, a published Vertex ERK inhibitor,4 which maintains the original apo-ERK conformation of Tyr34, inhibits the enzymatic activity of ERK, but not its phosphorylation by MEK. Apparently, phosphorylation of ERK requires not only the full length ERK protein but also a precise conformation of ERK. Successful ERK phosphorylation by MEK appears to critically depend on retaining the native conformation within Tyr34 and the Gly-rich loop. Efficacy of anti-tumor activity of MK-8353 was established in the Colo-205 human colon xenograft model. Female nude mice bearing Colo-205 human colon tumor xenografts were randomized for treatment with MK-8353 at the doses shown Figure 5. After 28 days of treatment, MK-8353 at all the doses tested significantly inhibited Colo-205 tumor growth compared to vehicle control (P < 0.05 or P < 0.0001). At the 20 and 40 mg/kg, p.o., bid dose, MK-8353 caused 37% and 88% TGI, respectively. At the 60 mg/kg, p.o. bid doses, MK-8353 caused mean tumor regressions of 40%. Projection of the human dose regimen was carried out based on an effective average plasma exposure of 31 μM·h, corresponding to a 72% tumor growth inhibition in the Colo-205 mouse model. The projected human dose regimen is bid with a dose range of 400 mg (based on dogs) to 1800 mg (based on monkeys).18
Figure 5.

Anti-tumor activity of MK-8353 on the Colo-205 human colon tumor xenograft model.
The discovery synthesis of ERK inhibitor MK-8353 relied on a convergent approach utilizing three fragments viz., LHS-substituted iso-propoxy indazole (LHS 29), RHS piperidine-ene phenyl triazole (RHS 37), 3(S)-thiomethyl pyrrolidine center core 41, and final coupling as depicted in Scheme 1. Chemo-selective alkylation of 5-bromopyridin-2(1H)-one 24 with isopropyl iodide using K2CO3 followed by borylation gave 2-isopropoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine 26. Suzuki reaction of the commercially available 3-bromo-5-nitro-1-trityl-1H-indazole 27(17) with 26 gave the nitro indazole 28 in good yield after chromatography. Reduction of 28 gave LHS 29 as an oil in good yield without chromatography, with an overall yield of 20%. RHS 37 was prepared by reaction of the commercially available bromo-4-cyanobenzene 30 with ethanol under acidic conditions to give 31 followed by methyl hydrazine to form the hydrazinoimidate 32 in modest yield. After reaction with formic acid the bromophenyl-N-methyl triazole intermediate 33 was obtained. The tetrahydropyridine ring was introduced by a Suzuki reaction of the commercially available Boc-protected tetrahydropyridine-boronate 35 to obtain the tricyclic ring system 36. RHS fragment chloroacetamide 37 was obtained in excellent yield by reaction of the deprotected 36 with chloroacetyl chloride. The center core 3(S)-thiomethyl pyrrolidine 41 was obtained in good yield in three steps starting from commercially available pyrrolidine intermediate 38. Deprotonation with LDA at −78 °C and quenching with dimethyl disulfide gave the center core 3-thiomethyl pyrrolidine 39 in moderate yield.16 Subsequent Boc deprotection gave 40 and chiral resolution by crystallization with l-tartaric acid, and filtration from methanol gave the pure enantiomer 3(S)-thiomethyl pyrrolidine 41 in good yield with >99% purity. Boc protection followed by hydrolysis of the methyl ester gave 43 in good yield. The final coupling of the intermediates is proceeded by coupling LHS 29 with center core derivative 43 using HATU to give 44 in good yield. After deprotection of both trityl and Boc groups with TFA gave 44 in 73% after purification. Final coupling between 45 and RHS 37 using Hünig’s base at 50 °C resulted in MK-8353 in good yield after chromatography. The convergent discovery synthesis of MK-8353 is 19 steps with the longest linear synthesis of nine steps proceeding in an unoptimized 3% overall yield (Scheme 1). Optimization of this synthesis to provide kilogram quantities will be discussed in future publications.
Scheme 1. Synthesis of MK-8353.

Reagents and conditions: (a) iPrI, K2CO3 DMF, 59%; (b) B2Pin2, Pd(dppf)2Cl2, KOAc, DMSO; (c) 27, Pd(Ph3P)4, Na2CO3 Tol-EtOH-H2O; Pd/C, 100 °C, 80%; (d) Pd/C, H2iPrOH-toluene, 100%; (e) HCl, EtOH, 56%; (f) MeNHNH2, pyridine, 85%; (g) formic acid, 84%; (h) 34, Pd(dppf)2Cl2, K2CO3, DME/water, 73%; (i) HCl, dioxane, 100%; (j) chloro acetyl chloride, Et3N, CH2Cl2, 67%; (k) LDA, (SMe)2, THF, −78 °C, 67%; (l) TFA, CH2Cl2, 90%; (m) l-tartaric acid, MeOH, 78%; (n) (Boc)2O, Et3N, DMF, 100%; (o) aq. LiOH (2.0 M), THF/MeOH, 92%; (p) LHS 29, HATU, iPr2EtN, DMF, 60%; (q) TFA, CH2Cl2, 73%; (r) RHS 37, iPr2EtN, DMF, 50 °C, 70%.
In conclusion, structure-based drug design afforded a novel conformationally restricted potent pyrrolidine series of ERK inhibitors. 3(S)-Thiomethyl substitution of the pyrrolidine core retarded amide metabolism and ameliorated the challenge of PK in this chemotype. A systematic SAR exploration of LHS and RHS led to the discovery of orally bioavailable ERK inhibitor MK-8353. MK-8353 displayed excellent potency, high kinase selectivity, and dual mechanism of action inhibition. MK-8353 also demonstrated significant in vivo efficacy, shown by tumor growth inhibition and regressions in BRAF/KRAS tumor models. The detailed pharmacological and clinical evaluation of MK-8353 as a potential treatment of cancer showed that it was well tolerated up to 400 mg twice daily and exhibited antitumor activity in patients with BRAFV600 mutant melanoma.19
Acknowledgments
We thank Drs. John Piwinski, Cathy Strader, Cecil Pickett, and Malcolm MacCoss at Merck & Co., Inc., Kenilworth, NJ USA, for their support and encouragement, and Drs. William Greenlee and Robert Aslanian for support of the program.
Glossary
ABBREVIATIONS
- ERK
extracellular signal-regulated kinases
- MAPK
mitogen-activated protein kinases
- SBDD
structure-based drug design
- SAR
structure–activity relationship
- PK
pharmacokinetics
- AUC
area under curve
- TGI
tumor growth inhibiition
- PoC
proof of concept
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00220.
Kinase selectivity, PK profiles, X-ray data collection, experimental procedures, NMR, and MS data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Keshet Y.; Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- Samatar A. A.; Poulikakos P. I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discovery 2014, 13, 928.and references cited therein 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Ahronian L. G.; Sennott E. M.; Van Allen E. M.; Wagle N.; Kwak E. L.; Faris J. E.; Godfrey J. T.; Nishimura K.; Lynch K. D.; Mermel C. H.; Lockerman E. L.; Kalsy A.; Gurski J. M. Jr.; Bahl S.; Anderka K.; Green L. M.; Lennon N. J.; Huynh T. G.; Mino-Kenudson M.; Getz G.; Dias-Santagata D.; Iafrate A. J.; Engelman J. A.; Garraway L. A.; Corcoran R. B. Clinical acquired resistance to raf inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discovery 2015, 5 (4), 358–367. 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan R. J.; Infante J. R.; Janku F.; Wong D. J. L.; Sosman J. A.; Keedy V.; Patel M. R.; Shapiro G. I.; Mier J. W.; Tolcher A. W.; Wang-Gillam A.; Sznol M.; Flaherty K.; Buchbinder E.; Carvajal R. D.; Varghese A. M.; Lacouture M. E.; Ribas A.; Patel S. P.; De Crescenzo G. A.; Emery C. M.; Groover A. L.; Saha S.; Varterasian M.; Welsch D. J.; Hyman D. M.; Li B. T. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discovery 2018, 8, 184–195. 10.1158/2159-8290.CD-17-1119. [DOI] [PubMed] [Google Scholar]
- Blake J. F.; Burkard M.; Chan J.; Chen H.; Chou K. J.; Diaz D.; Dudley D. A.; Gaudino J. J.; Gould S. E.; Grina J.; Hunsaker T.; Liu L.; Martinson M.; Moreno D.; Mueller L.; Orr C.; Pacheco P.; Qin A.; Rasor K.; Ren L.; Robarge K.; Shahidi-Latham S.; Stults J.; Sullivan F.; Wang W.; Yin J.; Zhou A.; Belvin M.; Merchant M.; Moffat J.; Schwarz J. B. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J. Med. Chem. 2016, 59, 5650–5660. 10.1021/acs.jmedchem.6b00389. [DOI] [PubMed] [Google Scholar]
- Bagdanoff J. T.; Jain R.; Han W.; Zhu S.; Madiera A.-M.; Lee P. S.; Ma X.; Poon D. Tetrahydropyrrolo-diazepenones as inhibitors of ERK2 kinase. Bioorg. Med. Chem. Lett. 2015, 25, 3788–3792. 10.1016/j.bmcl.2015.07.091. [DOI] [PubMed] [Google Scholar]
- Ward R. A.; Colclough N.; Challinor M.; Debreczeni J. E.; Eckersley K.; Fairley G.; Feron L.; Flemington V.; Graham C. R.; Greenwood R.; Hopcroft P.; Howard T. D.; James M.; Jones C. D.; Jones C. R.; Renshaw J.; Roberts K.; Snow L.; Tonge M.; Yeung K. Structure-Guided Design of Highly Selective and Potent Covalent Inhibitors of ERK1/2. J. Med. Chem. 2015, 58, 4790. 10.1021/acs.jmedchem.5b00466. [DOI] [PubMed] [Google Scholar]
- Morris E. J.; Jha S.; Restaino C. R.; Dayananth P.; Zhu H.; Cooper A.; Carr D.; Deng Y.; Jin W.; Black S.; Long B.; Liu J.; Dinunzio E.; Windsor W.; Zhang R.; Zhao S.; Angagaw M. H.; Pinheiro E. M.; Desai J.; Xiao L.; Shipps G.; Hruza A.; Wang J.; Kelly J.; Paliwal S.; Gao X.; Babu B. S.; Zhu L.; Daublain P.; Zhang L.; Lutterbach B. A.; Pelletier M. R.; Philippar U.; Siliphaivanh P.; Witter D.; Kirschmeier P.; Bishop W. R.; Hicklin D.; Gilliland D. G.; Jayaraman L.; Zawel L.; Fawell S.; Samatar A. A. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discovery 2013, 3, 742–750. 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Shipps G. W. Jr; Cooper A. B.; English J. M.; Annis D. A.; Carr D.; Nan Y.; Wang T.; Zhu H. Y.; Chuang C.-C.; Dayananth P.; Hruza A. W.; Xiao Li.; Jin W.; Kirschmeier P.; Windsor W. T.; Samatar A. A. Discovery of Novel, Dual Mechanism ERK Inhibitors by Affinity Selection Screening of an Inactive Kinase. J. Med. Chem. 2014, 57, 8817–8826. 10.1021/jm500847m. [DOI] [PubMed] [Google Scholar]
- Zhu H. Y.; Desai J.; Deng Y.; Cooper A. B.; Wang J.; Shipps J.; Samatar A.; Carr D.; Windsor W. Discovery of hydroxyaniline amides as selective Extracellular Regulated Kinase (Erk) inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1627–1629. 10.1016/j.bmcl.2015.01.049. [DOI] [PubMed] [Google Scholar]
- Boga S. B.; Alhassan A.-B.; Cooper A. B.; Doll R.; Shih N.-Y.; Shipps G. Jr.; Deng Y.; Zhu H.; Nan Y.; Sun R.; Zhu L.; Desai J.; Patel M.; Muppalla K.; Gao X.; Wang J.; Yao X.; Kelly J.; Gudipati S.; Paliwal S.; Tsui H.-C.; Wang T.; Sherborne B.; Xiao L.; Hruza A.; Buevich A.; Zhang L.-K.; Hesk D.; Samatar A. A.; Carr D.; Long B.; Black S.; Dayananth P.; Windsor W.; Kirschmeier P.; Bishop R. Discovery of 3(S)-thiomethyl pyrrolidine ERK inhibitors for oncology. Bioorg. Med. Chem. Lett. 2018, 28, 2029–2034. 10.1016/j.bmcl.2018.04.063. [DOI] [PubMed] [Google Scholar]
- Chaikuad A.; Tacconi E. M.; Zimmer J.; Liang Y.; Gray N. S.; Tarsounas M.; Knapp S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxidation of 1-(tert-butyl) 3-methyl (S)-3-(methylthio)pyrrolidine-1,3-dicarboxylate 42 with mCPBA afforded the sulfone 1-(tert-butyl) 3-methyl (S)-3-(methylsulfonyl)pyrrolidine-1,3-dicarboxylate center core for the synthesis of compound 22.
- Crystallization of methyl 3-(methylthio)pyrrolidine-3-carboxylate 40 with d-tartaric acid gave the 3(R)-thiomethyl pyrrolidine centre core for the synthesis of compound 23.
- Boga S. B.; Alhassan A. B.; Cooper A. B.; Shih N.-Y.; Doll R. J. [3 + 2] Cycloaddition-mediated synthesis of 3-methylsulfanyl-pyrrolidine-3-carboxylic acid methyl ester. Tetrahedron Lett. 2009, 50, 5315–5316. 10.1016/j.tetlet.2009.06.141. [DOI] [Google Scholar]
- Cooper A. B.; et al. Polycyclic indazole derivatives that are ERK inhibitors and their preparation, pharmaceutical compositions and use in the treatment of cancer. U.S. Pat. Appl. Publ., 20090118284, 2009.
- See Supporting Information for more details.
- Moschos S. J.; Sullivan R. J.; Flaherty K. T.; Hwu W.-J.; Ramanathan R. K.; Adjei A. A.; Fong P. C.; Shapira-Frommer R.; Tawbi H. A.; Rubino J.; Rush T. S. 3rd; Zhang D.; Miselis N. R.; Samatar A. A.; Chun P.; Rubin E. H.; Schiller J.; Long B. J.; Dayananth P.; Carr D.; Kirschmeier P.; Bishop W. R.; Deng Y.; Cooper A.; Shipps G. W.; Moreno B. H.; Robert L.; Ribas A. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, 2379–3708. 10.1172/jci.insight.92352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.