

Abstract
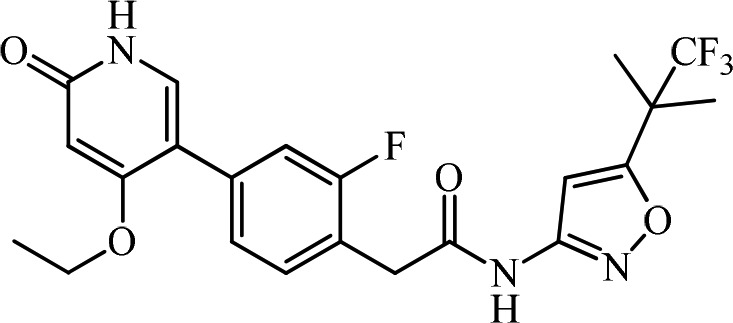
Abdominal pain and abnormal bowel habits represent major symptoms for irritable bowel syndrome (IBS) patients that are not adequately managed. Although the etiology of IBS is not completely understood, many of the functions of the gastrointestinal (GI) tract are regulated by the enteric nervous system (ENS). Inflammation or stress-induced expression of growth factors or cytokines may lead to hyperinnervation of visceral afferent neurons in GI tract and contribute to the pathophysiology of IBS. Rearranged during transfection (RET) is a neuronal growth factor receptor tyrosine kinase critical for the development of the ENS as exemplified by Hirschsprung patients who carry RET loss-of-function mutations and lack normal colonic innervation leading to colonic obstruction. Similarly, RET signaling in the adult ENS maintains neuronal function by contributing to synaptic formation, signal transmission, and neuronal plasticity. Inhibition of RET in the ENS represents a novel therapeutic strategy for the normalization of neuronal function and the symptoms of IBS patients. Herein, we describe our screening effort and subsequent structure–activity relationships (SARs) in optimizing potency, selectivity, and mutagenicity of the series, which led to the discovery of a first-in-class, gut-restricted RET kinase inhibitor, 2-(4-(4-ethoxy-6-oxo-1,6-dihydropyridin-3-yl)-2-fluorophenyl)-N-(5-(1,1,1-trifluoro-2-methylpropan-2-yl)isoxazol-3-yl)acetamide (15, GSK3179106), as a clinical candidate for the treatment of IBS. GSK3179106 is a potent, selective, and gut-restricted pyridone hinge binder small molecule RET kinase inhibitor with a RET IC50 of 0.3 nM and is efficacious in vivo.
Keywords: RET kinase, IBS, gut restricted, genotoxicity, AMES
Irritable bowel syndrome (IBS) is a relatively common gastrointestinal illness characterized by a constellation of clinical symptoms including abdominal pain and discomfort, abnormal bowel habits, and bloating.1−3 The pathophysiology of IBS is thought to result from alterations in sensory inputs/outputs in the peripheral and central nervous system such that a patient with IBS has a heightened and disproportionate sensory experience for a given stimulus, i.e., visceral hypersensitivity.4,5 While it is unclear whether an enhancement of central or peripheral mechanisms underlies visceral hypersensitivity, sensitization of visceral afferent nociceptors has been demonstrated to induce visceral hypersensitivity.1 Furthermore, gastrointestinal (GI) insults, such as infection, can lead to sensitization of the enteric nervous system (ENS)4 and have been associated with neurotrophic factors and/or inflammatory cytokine expression that may mediate neuronal outgrowth or plasticity in IBS.6−8
Rearranged during transfection (RET) kinase is a neuronal growth factor receptor tyrosine kinase that is activated upon binding to one of four neurotrophic factors glial cell line-derived neurotrophic factor (GDNF), neurturin, artemin, and persephin in combination with a GPI-anchored coreceptor GFRα-1, 2, 3, and 4, respectively.9 RET activity is critical for the development of the ENS, the kidney, and spermatogenesis.10 The role of RET kinase in the development of the ENS has been aided by the study of patients with Hirschsprung disease who frequently suffer from colonic obstruction due to variable lengths of bowel aganglionosis, often in association with loss-of-function RET mutations.11−13 Mechanistically, RET signaling is associated with the maturation of presynaptic axon terminals through regulating the expression of presynaptic proteins.14−16 Systemic administration of GDNF in adult rodents significantly increases submucosal neuron density in both the small intestine and colon,17 while a conditional knockout of the RET kinase coreceptor, GFRα3, attenuates visceral hypersensitivity suggesting that RET kinase plays a role in the signaling of visceral nociception.18 These observations led us to hypothesize that antagonizing RET signaling in the gut may offer a novel therapeutic approach to treat visceral pain in conditions such as IBS.
There are many reported orally active RET inhibitors; however, highly selective gut-restricted RET inhibitors have not yet been identified.19−21 Yang and colleagues recently published a report on N-phenyl-7,8-dihydro-6H-pyrimido[5,4-b][1,4]oxazin-4-amine derivatives and detailed a list of available RET inhibitors in the publication and references within.22 In addition, since chronic dosing is required for the treatment for IBS, therefore, our initial focus was on identifying a highly selective and gut-restricted RET inhibitor to improve tolerability and the safety margin. A high throughput screen (HTS) campaign was undertaken and resulted in several potent but kinase nonselective RET inhibitors. The most attractive of those came from a biphenyl urea series as shown in Table 1.23 While triaging the HTS hits, it became evident that kinase insert domain receptor (KDR/VEGFR2) selectivity could be used as a surrogate for overall off-target kinase selectivity for many of the RET inhibitors, including published RET inhibitors. Therefore, we used KDR as the off-target selectivity assay during primary screening. We targeted >100-fold selectivity over other kinases. One of the most promising leads (compound 1, Table 1) was a directly linked A and B ring biaryl urea that was a potent RET inhibitor, with 45-fold selectivity over KDR and was structurally similar to the known inhibitor FLT3 inhibitor AST-487 with the ethyl piperazine.24 Both RET and KDR potency were determined in in vitro enzyme assays. Movement of the methyl-amine (1) to the para-position of a meta-pyridine (2) showed an improvement in RET biochemical potency from 22 to 0.7 nM (compound 1 to compound 2, Table 1). While trying to improve physiochemical properties of the compounds, we explored pyridones as hinge binders due to their decreased lipophilicity such as clogP. While we noted similar RET potency as the methyl-amine pyridines, we did note an improvement in KDR selectivity (compound 2, 8-fold vs compound 3, 79-fold) and lipophilicity as measured by clogP (compound 2, 6.18 vs compound 3, 6.01) while maintaining RET potency. In addition, pyridone hinge binders (compounds 3 and 4) removed the potential genotoxicity associated with the amino-pyridine scaffold present in compounds 1 and 2 in the A ring.
Table 1. A-Ring (Hinge Binder) Optimization.

IC50 is a mean of least two experiments.
Value in parentheses for KDR represent fold selectivity over RET.
To improve KDR selectivity, we sought an understanding of the structural differences in the RET and KDR kinase domains. DFG-out cocrystal structures for RET kinase were not available, which led the team to build homology models based on KDR and KIT kinase for RET kinase. Based on docking of compounds into the homology model, it suggested substitution on the B ring (compound 4, Table 1) could reduce KDR activity thereby increasing overall kinase selectivity. A figure of compound 4 docked in the model is provided in the Supporting Information (Figure S1). The homology model showed key hinge interactions at Glu805 and Ala807, as well as Glu775 and Asp892 for the central linker. The model also showed a small pocket in the hinge, which was optimized with an ethyl ether on the A ring. Compound 4 with an optimized hinge and F-substitution on the B ring showed a RET IC50 of 0.1 nM and KDR IC50 of 20.8 nM, a 208-fold selectivity window.
An optimal compound profile for an IBS indication necessitated a preclinical candidate devoid of genotoxicity. Metabolite identification (MET ID) studies and in silico metabolism predictions showed aniline formation from urea hydrolysis as a predominant route of elimination. In addition, there was a risk of aniline synthetic intermediates appearing as impurities (or degradants), warranting an early genotoxicity risk assessment of the embedded anilines in the structure. At this point in the early lead optimization process, owing to the risk of genotoxicity from anilines from both synthetic intermediates and potential urea hydrolysis metabolism, we evaluated these anilines in the standard Ames test for genotoxic liability. Initial Ames25 testing of both the left-hand and right-hand side anilines of compound 4 showed positive in the TA98 bacterial strain, which is typically the most sensitive strain. Additionally, we tested several structurally similar anilines (see Supporting Information), and they were also found to be Ames positive. Due to the lack of Ames negative A/B-rings, we explored urea isosteres and noted that amides retained biochemical potency with a pyridone hinge binder. However, compounds were still not progressible due to the presence of the Ames positive C-ring aniline; specifically, the anilines with the ethyl piperazine attached that helps drive RET potency. Therefore, a full exploration of the C ring was undertaken to find Ames negative C-ring anilines, while maintaining RET potency and KDR selectivity as seen in Table 2.
Table 2. C-Ring Optimization.

IC50 is a mean of least two experiments.
IC50 is a mean of one experiment.
Given the relatively low throughput of the Ames assay and the need for rapid lead optimization, we employed computational modeling to prioritize anilines for synthesis and testing in the Ames assay. Multiple computational approaches were investigated, but ultimately nitrenium ion formation energy26 was found to be most predictive for the RET potent aniline fragments. The nitrenium ion formation model was validated using GSK Ames data on a series of structurally similar anilines. The model was critical in rapidly identifying readily available aromatic amines that fitted established RET SAR. Using the internal data set to validate the model, a nitrenium ion formation energy of <153 kcal/mol was predictive of a positive Ames result and an energy of >164 kcal/mol was predictive of a negative result. Energies between 154 and 163 kcal/mol could not lead to accurate predictions. Examples listed in the Supporting Information showed both A/B-ring and C-ring anilines and their nitrenium ion formation energies alongside their experimental Ames result.
Table 2 showcases an array of C-ring phenyl substitutions, as well as heteroaryl replacements optimized for RET potency. The ortho-fluoro was maintained in the B-ring, as well as an ethyl ether in the ortho position of the A-ring. Both ortho- and meta-ethyl ethers displayed similar RET potency. While a wide range of substitutions with various steric and electronic effects were tolerated for RET potency, compound triage was now focused on removing the genotoxic liability, as well as acceptable PK parameters and low nanomolar cellular potency. In addition to the RET and KDR biochemical assays, compounds were evaluated in a cellular assay (TT cell) for the inhibition of phosphorylation of a constitutively active RET kinase. Figures S2–S5 show optimized amino acid interactions of RET kinase with compound 15 in the homology model.
The highly convergent synthesis of representative amide-linked RET inhibitors is described in Scheme 1 and has been previously published.27 The A-ring is constructed in four synthetic steps starting from the commercially available 2-chloro-4-nitropyridine. Nucleophilic aromatic substitution utilizing sodium ethoxide provides the desired aryl ether in excellent yield. Subsequent directed bromination followed by displacement of the 2-chloro functionality with p-methoxybenzyl alcohol furnishes the fully elaborated, PMB-protected A-ring. Synthesis of the B-ring begins with the commercially available benzyl bromide, which is converted to the corresponding phenylacetic acid in two synthetic steps and excellent yield. Esterification of the carboxylic acid followed by Suzuki coupling with bis(pinacolato) diborane affords the completed B-ring, which is then coupled to the A-ring under standard Suzuki conditions. Saponification of the elaborated methyl ester provides the carboxylic acid necessary for amide formation utilizing T3P in pyridine. Finally, PMB deprotection affords the desired RET inhibitor. Experimentals of biaryl urea RET inhibitors are provided (Supporting Information) and have previously been published.28
Scheme 1. General Synthesis of Pyridone Hinge Binder Amides,

Reagents and conditions: (a) EtONa, THF, 92% yield; (b) NBS, H2SO4; (c) chromatography, 40% yield; (d) PMBOH, KOH, toluene, 70% yield; (e) NaCN, EtOH, 99% yield; (f) NaOH, MeOH, 92% yield; (g) H2SO4, H2O, MeOH, 94% yield; (h) Pd(dppf)Cl2, KOAc, dioxane, 90% yield; (i) Pd(dppf)Cl2, Cs2CO3, 90% yield; (j) LiOH·H2O, THF, 93% yield; (k) T3P, pyridine, 74% yield; (l) TFA; (m) THF, H2O, 77% yield.
Reported yields are for compound 15.
Compound 15 was found to have the best overall profile with respect to potency and selectivity and was progressed to additional DMPK studies and in vivo evaluation. Compound 15 (GSK3179106) was a potent RET inhibitor with IC50s of 0.4 and 11 nM in the biochemical assay and cellular assay, respectively. In addition, it had a clean genotoxic profile with no embedded genotoxicity liabilities. Compound 15 possessed good kinase selectivity; only 26 out of a set of >300 recombinant kinases were found to be inhibited at a 1 μM test concentration (Supporting Information, Table S4). To further determine the physiologically relevant kinase target(s) of GSK3179106 in colon tissues isolated from rats, a chemoproteomics technique identified RET kinase as the most potent target that was competed for binding to the kinobeads by GSK3179106 (pKdapp = 7.9 or Kd = 0.02 μM; Supporting Information Table S5).29,30 However, two other kinases, DDR1 (pKdapp = 7.6 or Kd = 0.04 μM) and DDR2 (pKdapp = 7.3 or Kd = 0.09 μM) were also competed by GSK3179106 with comparable potency and may represent additional targets of the compound. Compound 15 has relatively poor solubility with the kinetic chemiluminescent nitrogen detection (CLND) solubility of 21 μM and 4 h fasted state simulated intestinal fluid (FaSSIF) solubility of 0.4 μg/mL.
Single dose IV (bolus, 0.06 mg/kg) PK in male Sprague–Dawley rats of compound 15 formulated as 0.04 mg/mL in DMSO/6% HP-beta-CD = 5:95 with a pH of 7 as a clear solution showed low exposure with an AUC of 102 ng·h/mL (Figure 1, full details in the Supporting Information). Oral PK was evaluated (Supporting Information) with the same dosing regimen as the in vivo colonic hypersensitivity model, seven doses of 10 mg/kg given over 3.5 days. Full gut PK measurements were also taken to help understand the PK/PD relationship.
Figure 1.
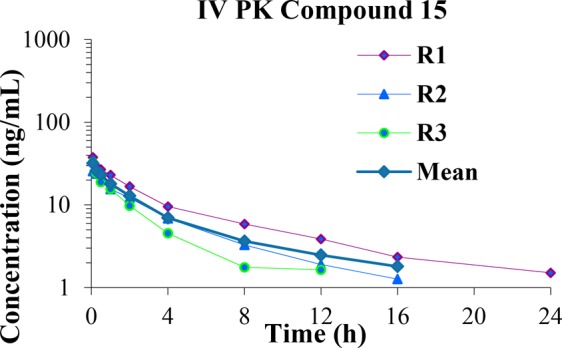
Rat IV PK (0.06 mg/kg) of compound 15.
A full gut pharmacokinetics analysis revealed high concentrations of compound 15 in the colon contents, jejunum, duodenum, and ileum, over that in plasma (Table 3). Upon further investigation, pharmacokinetic exposures of most compounds in the chemical series were predominantly gut restricted when administered orally. Specifically, following 10 mg/kg given seven doses in 3.5 days, plasma concentrations were only 40 ng/mL at Cmax (Table 3), while colon tissue homogenate concentrations were 3358 ng/mL at Cmax. The full PK curve data is available in the Supporting Information (Figure S5).
Table 3. Satellite Full Gut PK for 10 mg/kg 3.5 days BID p.o. Dosing of Compound 15 in Rats.
| Cmax (ng/mL) | Tmax (h) | |
|---|---|---|
| colon contents | 287500 | 7 |
| duodenum | 15713 | 0 |
| jejunum | 12800 | 1 |
| ileum | 5520 | 2 |
| colon | 3358 | 7 |
| plasma | 40 | 4 |
A noninflammatory irritation model of colonic hypersensitivity in rats utilizes a low concentration of acetic acid to induce a transient sensitization of colonic afferents, which, upon colorectal distension, induces a visceromotor response that can be monitored visually as abdominal contractions and used to assess the severity of induced visceral pain.31,32 GSK3179106 dosed orally at 10 mg/kg (n = 7, all groups) for 3.5 days BID reduced the visceromotor response to colorectal distension in comparison to rats given an acetic acid enema and dosed with vehicle (Figure 2, *P < 0.05, ****P < 0.0001 vehicle compared to GSK3179106 treatment, repeated-measure two-way ANOVA, Bonferroni post-test). Treatment with tegaserod, a 5-HT4 agonist used as a positive control for the model, exhibited a decrease in observed abdominal contractions as expected (Supporting Information, Figure S6).33,34
Figure 2.

Colonic hypersensitivity induced by acute irritation stress is ameliorated by RET kinase inhibition with compound 15.
Compound 15 (GSK3179106) was further progressed to rodent safety studies and found to have a favorable safety profile to support clinical testing.35
In summary, a selective, gut-restricted RET kinase inhibitor has been developed for the treatment of IBS in a clinical setting. The calculation of nitrenium ion formation energies greatly aided the deprioritization of anilines with a genotoxic risk. In due course, the clinical study of GSK3179106 will be presented.35
Acknowledgments
The authors thank Karl Tyler (University of Oklahoma), as well as Jane Wang and Yuhong Shen (WuXi AppTec) for their contributions to data generation. All studies involving the use of animals were conducted after review by the GlaxoSmithKline (GSK) Institutional Animal Care and Use Committee and in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals.
Glossary
ABBREVIATIONS
- CLND
chemiluminescent nitrogen detection
- FaSSIF
fasted state simulated intestinal fluid
- IBS
irritable bowel syndrome
- KDR
kinase insert domain receptor
- RET
rearranged during transfection
- VEGFR2
vascular endothelial growth factor receptor type 2
- THF
tetrahydrofuran
- NBS
N-bromosuccinimide
- PMBOH
4-methoxybenzel alcohol
- Pd(dppf)Cl2
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)
- T3P
propylphosphonic anhydride
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00035.
Compound synthesis and spectroscopic characterization of compound 15, along with nitrenium ion formation energy calculations and Ames data, as well as homology model details and images. Enzyme and cell-based assay methods, kinase selectivity, chemoproteomics, pharmacokinetics, and in vivo models methods and standards (PDF)
The authors declare the following competing financial interest(s): H.S.E., J.R., K.R., M.D., D.Q., G.J., N.Z., S.L., S.R., A.W., A.G., A.O., S.K., and M.C. are current or past employees of GlaxoSmithKline and/or stockholders of GlaxoSmithKline.
Supplementary Material
References
- Camilleri M. Peripheral mechanisms in irritable bowel syndrome. N. Engl. J. Med. 2012, 367, 1626–1635. 10.1056/NEJMra1207068. [DOI] [PubMed] [Google Scholar]
- Halland M.; Talley N. J. New treatments for IBS. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 13–23. 10.1038/nrgastro.2012.207. [DOI] [PubMed] [Google Scholar]
- Longstreth G. F.; Thompson W. G.; Chey W. D.; Houghton L. A.; Mearin F.; Spiller R. C. Functional bowel disorders. Gastroenterology 2006, 130, 1480–1491. 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Mayer E. A. Clinical practice. Irritable bowel syndrome. N. Engl. J. Med. 2008, 358, 1692–1699. 10.1056/NEJMcp0801447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azpiroz F.; Bouin M.; Camilleri M.; Mayer E. A.; Poitras P.; Serra J.; Spiller R. C. Mechanisms of hypersensitivity in IBS and functional disorders. Neurogastroenterol. Motil. 2007, 19, 62–88. 10.1111/j.1365-2982.2006.00875.x. [DOI] [PubMed] [Google Scholar]
- Brierley S. M.; Linden D. R. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 611–627. 10.1038/nrgastro.2014.103. [DOI] [PubMed] [Google Scholar]
- Buhner S.; Li Q.; Berger T.; Vignali S.; Barbara G.; De Giorgio R.; Stanghellini V.; Schemann M. Submucous rather than myenteric neurons are activated by mucosal biopsy supernatants from irritable bowel syndrome patients. Neurogastroenterol. Motil. 2012, 24, 1134–e1572. 10.1111/nmo.12011. [DOI] [PubMed] [Google Scholar]
- Dothel G.; Barbaro M. R.; Boudin H.; Vasina V.; Cremon C.; Gargano L.; Bellacosa L.; De Giorgio R.; Le Berre-Scoul C.; Aubert P.; Neunlist M.; De Ponti F.; Stanghellini V.; Barbara G. Nerve fiber outgrowth is increased in the intestinal mucosa of patients with irritable bowel syndrome. Gastroenterology 2015, 148, 1002–1011. e1004 10.1053/j.gastro.2015.01.042. [DOI] [PubMed] [Google Scholar]
- Plaza-Menacho I.; et al. Current concepts in RET-related genetics, signaling and therapeutics. Trends Genet. 2006, 22, 627–636. 10.1016/j.tig.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Airaksinen M. S.; Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Butler Tjaden N. E.; Trainor P. E. The developmental etiology and pathogenesis of Hirschsprung disease. Transl. Res. 2013, 162, 1–15. 10.1016/j.trsl.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace A. S.; Anderson R. B. Genetic interactions and modifier genes in Hirschsprung’s disease. World J. Gastroenterol. 2011, 17, 4937–4944. 10.3748/wjg.v17.i45.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery P.; Lyonnet S.; Mulligan L. M.; Pelet A.; Dow E.; Abel L.; Holder S.; Nihoul-Fekete C.; Ponder B. A.; Munnich A. Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature 1994, 367, 378–380. 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- Barrenschee M.; Bottner M.; Harde J.; Lange C.; Cossais F.; Ebsen M.; Vogel I.; Wedel T. SNAP-25 is abundantly expressed in enteric neuronal networks and upregulated by the neurotrophic factor GDNF. Histochem. Cell Biol. 2015, 143, 611–623. 10.1007/s00418-015-1310-x. [DOI] [PubMed] [Google Scholar]
- Baudet C.; Pozas E.; Adameyko I.; Andersson E.; Ericson J.; Ernfors P. Retrograde signaling onto Ret during motor nerve terminal maturation. J. Neurosci. 2008, 28, 963–975. 10.1523/JNEUROSCI.4489-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottner M.; Harde J.; Barrenschee M.; Hellwig I.; Vogel I.; Ebsen M.; Wedel T. GDNF induces synaptic vesicle markers in enteric neurons. Neurosci. Res. 2013, 77, 128–136. 10.1016/j.neures.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Wang H.; Hughes I.; Planer W.; Parsadanian A.; Grider J. R.; Vohra B. P.; Keller-Peck C.; Heuckeroth R. O. The timing and location of glial cell line-derived neurotrophic factor expression determine enteric nervous system structure and function. J. Neurosci. 2010, 30, 1523–1538. 10.1523/JNEUROSCI.3861-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T.; Shinoda M.; Feng B.; Albers K. M.; Gebhart G. F. Modulation of visceral hypersensitivity by glial cell line-derived neurotrophic factor family receptor alpha-3 in colorectal afferents. American journal of physiology. Gastrointestinal and liver physiology 2011, 300, G418–424. 10.1152/ajpgi.00456.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mologni L.; et al. RET kinase inhibitors: a review of recent patents (2012–2015). Expert Opin. Ther. Pat. 2017, 27, 91–99. 10.1080/13543776.2017.1238073. [DOI] [PubMed] [Google Scholar]
- Moccia M.; Liu Q.; Guida T.; Federico G.; Brescia A.; Zhao Z.; Choi H.; Deng X.; Tan L.; Wang J.; Billaud M.; Gray N.; Carlomagno F.; Santoro M. Identification of Novel Small Molecule Inhibitors of Oncogenic RET kinase. PLoS One 2015, 10 (6), e0128364. 10.1371/journal.pone.0128364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H.; Shin I.; Nam Y.; Kim N.; Lee K.; Sim T. Identification of a novel 5-amino-3-(5-cyclopropylisoxazol-3-yl)-1-isopropyl-1H-pyrazole-4-carboxamide as a specific RET kinase inhibitor. Eur. J. Med. Chem. 2017, 125, 1145–1155. 10.1016/j.ejmech.2016.10.050. [DOI] [PubMed] [Google Scholar]
- Yang J.; Chen K.; Zhang G.; Yang Q.; Li Y.; Huang S.; Wang Y.; Yang W.; Jiang X.; Yan H.; Zhu J.; Xiang R.; Luo Y.; Li W.; Wei Y.; Li L.; Yang S. Structural optimization and structure-activity relationship studies of N-phenyl-7,8-dihydro-6H-pyrimido[5,4-b][1,4]oxazin-4-amine derivatives as a new class of inhibitors of RET and its drug resistance mutants. Eur. J. Med. Chem. 2018, 143, 1148–1164. 10.1016/j.ejmech.2017.09.018. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; et al. Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery?. ACS Chem. Biol. 2014, 9, 1230–1241. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeno-Stuart N.; et al. The RET kinase inhibitor NVP-AST487 blocks growth and calcitonin gene expression through distinct mechanisms in medullary thyroid cancer cells. Cancer Res. 2007, 67, 6956–6964. 10.1158/0008-5472.CAN-06-4605. [DOI] [PubMed] [Google Scholar]
- ICH S2(R1). “Guidance on Genotoxicity and data interpretation for pharmaceuticals intended for human use.” Adopted EMA/CHMP/ICH/126642/2008. December 2011.
- Mattioni B. E.; et al. Predicting the Genotoxicity of Secondary and Aromatic Amines Using Data Subsetting To Generate a Model Ensemble. J. Chem. Inf. Comput. Sci. 2003, 43, 949–963. 10.1021/ci034013i. [DOI] [PubMed] [Google Scholar]
- Eidam H.; DeMartino M.; Gong Z.; Guan A.; Raha K.; Wu C.; Yang H.; Yu H.; Zhang Z.; Cheung M.. Preparation of pyridinone and pyrimidinone derivatives as RET kinase inhibitors. PCT Int. Appl. WO2016037578, 2016.
- Cheung M.; DeMartino M.; Eidam H.; Guan H.; Qin D.; Wu C.; Gong Z.; Yang H.; Yu H.; Zhang Z.. Preparation of pyridine derivatives as rearranged during transfection (RET) kinase inhibitors. PCT Int. Appl. WO2014141187, 2014.
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Werner T.; Becher I.; Sweetman G.; Doce C.; Savitski M. M.; Bantscheff M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. 10.1021/ac301553x. [DOI] [PubMed] [Google Scholar]
- Werner T.; Becher I.; Sweetman G.; Doce C.; Savitski M. M.; Bantscheff M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. 10.1021/ac301553x. [DOI] [PubMed] [Google Scholar]
- Langlois A.; Diop L.; Riviere P. J.; Pascaud X.; Junien J. L. Effect of fedotozine on the cardiovascular pain reflex induced by distension of the irritated colon in the anesthetized rat. Eur. J. Pharmacol. 1994, 271, 245–251. 10.1016/0014-2999(94)90780-3. [DOI] [PubMed] [Google Scholar]
- Greenwood-Van Meerveld B.; Venkova K.; Hicks G.; Dennis E.; Crowell M. D. Activation of peripheral 5-HT receptors attenuates colonic sensitivity to intraluminal distension. Neurogastroenterol. Motil. 2006, 18, 76–86. 10.1111/j.1365-2982.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- Polverino A.; et al. AMG 706, an Oral, Multikinase Inhibitor that Selectively Targets Vascular Endothelial Growth Factor, Platelet-Derived Growth Factor, and Kit Receptors, Potently Inhibits Angiogenesis and Induces Regression in Tumor Xenografts. Cancer Res. 2006, 66 (17), 8715–21. 10.1158/0008-5472.CAN-05-4665. [DOI] [PubMed] [Google Scholar]
- Clinicaltrials.gov Identifiers: NCT02727283 and NCT02798991.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.