

Abstract
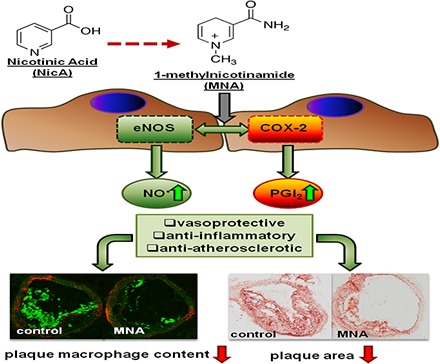
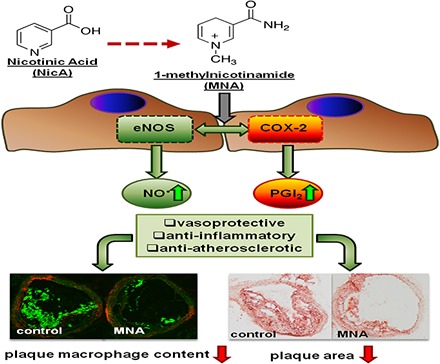
1-Methylnicotinamide (MNA), the major endogenous metabolite of nicotinic acid (NicA), may partially contribute to the vasoprotective properties of NicA. Here we compared the antiatherosclerotic effects of MNA and NicA in apolipoprotein E (ApoE)/low-density lipoprotein receptor (LDLR)–deficient mice. ApoE/LDLR−/− mice were treated with MNA or NicA (100 mg/kg). Plaque size, macrophages, and cholesterol content in the brachiocephalic artery, endothelial function in the aorta, systemic inflammation, platelet activation, as well as the concentration of MNA and its metabolites in plasma and urine were measured. MNA and NicA reduced atherosclerotic plaque area, plaque inflammation, and cholesterol content in the brachiocephalic artery. The antiatherosclerotic actions of MNA and NicA were associated with improved endothelial function, as evidenced by a higher concentration of 6-keto-prostaglandin F1α and nitrite/nitrate in the aortic ring effluent, inhibition of platelets (blunted thromboxane B2 generation), and inhibition of systemic inflammation (lower plasma concentration of serum amyloid P, haptoglobin). NicA treatment resulted in an approximately 2-fold higher concentration of MNA and its metabolites in urine and a 4-fold higher nicotinamide/MNA ratio in plasma, compared with MNA treatment. In summary; MNA displays pronounced antiatherosclerotic action in ApoE/LDLR−/− mice, an effect associated with an improvement in prostacyclin– and nitric oxide–dependent endothelial function, inhibition of platelet activation, inhibition of inflammatory burden in plaques, and diminished systemic inflammation. Despite substantially higher MNA availability after NicA treatment, compared with an equivalent dose of MNA, the antiatherosclerotic effect of NicA was not stronger. We suggest that detrimental effects of NicA or its metabolites other than MNA may limit beneficial effects of NicA-derived MNA.
Introduction
Nicotinic acid (NicA; niacin), first introduced as early as 1955 as a hypolipidemic drug (Carlson, 2005), lowers non–high-density lipoprotein (HDL) cholesterol, lipoprotein A, and triglycerides and increases HDL cholesterol. NicA also displays vasoprotective and anti-inflammatory activities, which are either linked to the activation of specific G protein–coupled receptors (GPR) such as GPR109A (Lukasova et al., 2011a,b; Chai et al., 2013) or mediated by receptor-independent mechanisms. Recent clinical trials [Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health and the HPS2-THRIVE (Treatment of HDL to Reduce the Incidence of Vascular Events)] did not confirm any clinical benefit of NicA treatment given in addition to statins or combined with laropiprant (Ginsberg and Reyes-Soffer, 2013), despite earlier clinical studies suggesting clear antiatherosclerotic effects for NicA. It has been suggested that the beneficial effect of NicA could be linked to the lowering of non-HDL cholesterol, the lowering of lipoprotein (a), or the elevation of HDL cholesterol (Kühnast et al., 2013). On the other hand, some authors have claimed that the vasoprotective action of NicA is an important factor in the pharmacological activity of this agent. In clinical studies, NicA reduced carotid atherosclerosis (Lee et al., 2009) and improved nitric oxide (NO)–dependent vasodilation in the brachial artery (Thoenes et al., 2007; Warnholtz et al., 2009). Improvement of endothelial function by NicA cannot be explained by the activation of GPRs, because they are not present on the surface of endothelial cells and specific GPR agonists such as MK-6892 (2-[[3-[3-(5-hydroxy-2-pyridinyl)-1,2,4-oxadiazol-5-yl]-2,2-dimethyl-1-oxopropyl]amino]-1-cyclohexene-1-carboxylic acid) do not display direct vasoprotective properties (Shen et al., 2010). Moreover, in vitro studies on endothelial cell cultures have demonstrated the ability of NicA to reduce the expression of adhesion molecules (Ganji et al., 2009, 2014; Stach et al., 2012) and to diminish the release of tumor necrosis factor-α (TNFα) (Ganji et al., 2009). This may suggest the involvement of a metabolite of NicA in the compound’s vasoprotective and anti-inflammatory properties.
The main metabolic route of NicA leads to the activation of nicotinamide-N-methyltransferase (NNMT) via nicotinamide (NA) and the subsequent formation of 1-methylnicotinamide (MNA) (Aksoy et al., 1994). Approximately one-half of an administered NicA dose is transformed into MNA and its metabolites (Menon et al., 2007). MNA has long been considered to be an inactive biomarker of NicA/NA catabolism. However, a number of studies have demonstrated pharmacological activity for MNA, including antithrombotic (Chlopicki et al., 2007), anti-inflammatory (Gebicki et al., 2003; Bryniarski et al., 2008), and gastroprotective (Brzozowski et al., 2008a,b) effects, mediated by the activation of cyclooxygenase (COX)-2 and the production of prostacyclin (PGI2). In addition, MNA was found to restore endothelial function in hypertriglyceridemic rats (Bartuś et al., 2008) and to improve endothelial function in humans (Domagala et al., 2012).
Accordingly, the NicA-MNA pathway seems to play a pivotal role in the GPR-independent anti-inflammatory, vasoprotective, and antiatherosclerotic effects of NicA. In fact, MNA could mediate many of the beneficial effects of NicA. However, one important consideration is that during prolonged NicA treatment, the robust metabolism of NicA to MNA may alter the NAD metabolome and induce a detrimental effect due to the high endogenous NA concentration and enhanced NA methylation resulting in the depletion of methyl group donors (Guyton and Bays, 2007; Sun et al., 2012; Li et al., 2013a,b). In this study, we compare the antiatherosclerotic effects of exogenous NicA and MNA in female apolipoprotein E (ApoE)/low-density lipoprotein receptor (LDLR)–deficient mice with advanced atherosclerosis, to examine the relative potency of NicA and MNA to act as vasoprotective agents and to inhibit atherosclerosis.
Materials and Methods
Animals and Experimental Groups.
To compare the antiatherosclerotic and anti-inflammatory actions of MNA and NicA, 36 female ApoE/LDLR−/− mice on C57Bl/6J background (Jackson Laboratories, Bar Harbor, ME), aged 16 weeks, were randomly divided into control (n = 12, untreated), MNA-treated (n = 12), and NicA-treated (n = 12) experimental groups. After an acclimatization period, the experimental groups received MNA or NicA (Sigma-Aldrich, St. Louis, MO) administered with drinking water at a mean daily dose of 100 mg/kg from week 16 to week 24. All groups were fed a standard rodent diet (Altromin Maintenance Diet; Altromin GmbH, Lage, Germany). Water intake was measured daily and body weight increase was monitored once a week. After 7 weeks, the mice were individually housed for 24 hours in specially designed metabolic cages (Tecniplast, West Chester, PA) to collect urine specimens. Urine samples after 15 minutes of centrifugation (1500g) were stored at −80°C for further analysis.
After 8 weeks of treatment, the mice were euthanized under anesthesia by ketamine/xylazine injection (intraperitoneally). Blood was taken from the right ventricle using an 18G intravenous cannula (Van Oostveen Medical B.V, Wilnis, Netherlands) to avoid platelet activation. The blood was then divided into two samples, one of which was centrifuged for plasma isolation (1000g for 10 minutes) and biochemical measurements and the other was used for thromboxane TXB2 (a stable metabolite of TXA2) measurement using a validated full blood ex vivo assay. For histochemical, immunohistochemical (IHC), and spectroscopic evaluation of atherosclerotic plaques, the brachiocephalic artery (BCA) was collected, cleaved from the perivascular adventitia, placed into OCT (Optimal Cutting Temperature) compound, and then either snap-frozen at −80°C or preserved in 4% buffered formalin solution and embedded in paraffin. The thoracic aorta was also isolated and cut into several 2-mm–long rings for short-term incubation in Krebs’ buffer for nitrate/nitrite and 6-keto-prostaglandin F1α (PGF1α) measurements. All animal procedures conformed to European Union Directive 2010/63/EU for animal experiments and all experimental procedures were approved by the Jagiellonian University First Local Ethical Committee on Animal Testing (955/2012).
Lipoprotein and Acute Phase Protein Analysis in Plasma.
Low-density lipoprotein (LDL) cholesterol, very low density lipoprotein cholesterol, HDL cholesterol, total cholesterol, and triglyceride plasma concentration were analyzed in plasma samples using the field flow fractionation method, with quantitative determination of cholesterol in lipoprotein fractions performed by enzymatic colorimetric assay (Kostogrys et al., 2015). High-performance liquid chromatography (HPLC) analysis was performed using an Agilent 1100 system and Agilent OpenLAB (C.01.05) software in combination with the appropriate Eclipse plugin (version 4; Agilent Technologies, Santa Clara, CA). The separation was performed by an Eclipse DUALTEC apparatus, using a polyethersulfone hollow fiber field flow fractionation channel with an inner radius of 400 µm and a molecular weight cut-off of 10 kDa. Separations were performed at a detector flow rate of 0.3 ml/min, while a cross-flow gradient, beginning at 0.5 ml/min and linearly reduced to zero over 30 minutes, was applied. The enzyme buffer flow for the postseparation colorimetric reaction was 0.3 ml/min; thus, the mixing ratio was 1:1 and the total flow entering the UV-Vis detector (Agilent G1314A) was 0.6 ml/min.
The acute phase protein panel (haptoglobin, α2-macroglobulin, serum amyloid P) was estimated in plasma samples using a designed multiplex assay (Millipore, Billerica, MA) and a Bioplex 200 (Bio-Rad, Hercules, CA) multiplex array system.
Analysis of Atherosclerotic Plaques in the BCA.
OCT-frozen BCAs were cut into 10-μm–thick cross-sections using a Leica CM1920 automatic cryostat (Leica, Wetzlar, Germany) and placed on poly(l-lysine)–covered microscopic slides (Menzel-Glaser SuperFrost; Thermo Scientific, Waltham, MA), followed by fixation with 4% buffered formalin or acetone for 10 minutes. Formalin-fixed slides were stained with Oil Red O (ORO) dye (Sigma-Aldrich, St. Louis, MO) to visualize the general plaque area. Acetone-fixed slides were immunostained using rat anti-mouse CD68 (AbD Serotec, Raleigh, NC) primary antibody, followed by Alexa Fluor 488–conjugated goat anti-rat secondary antibody (Jackson ImmunoResearch, West Grove, PA).
Paraffin-embedded BCAs were cut into 5-μm–thick cross-sections using a histologic microtome, placed on microscopic slides, and deparaffinized prior to staining using a standard protocol. Sections were stained according to a modified Orcein Martius Scarlet Blue (OMSB) protocol to differentiate between collagen, elastic lamina, and smooth muscle cells (stained blue, violet, and pink, respectively). Images of sections were acquired using a DotSlide automatic scanning station (Olympus, Tokyo, Japan), stored as TIFF files, and analyzed by Image Browser software (Carl Zeiss, Jena, Germany). For IHC staining, after deparaffinization, sections were pretreated according to the citrate-based heat-induced epitope retrieval protocol and then incubated with rat anti-mouse MAC3 (lysosomal-associated membrane protein 2; LAMP-2) (Chemicon, Billerica, MA) primary antibody, followed by biotinylated goat anti-rat secondary antibody and Alexa Fluor 488–conjugated streptavidin (Jackson ImmunoResearch).
Images were acquired using an AxioCam MRc5 digital camera and an AxioObserver.D1 inverted fluorescent microscope (Carl Zeiss), stored as TIFF files, and analyzed automatically using Columbus software (PerkinElmer, Waltham, MA). The areas of atherosclerotic plaques (µm2 × 105) were assessed in nine (for ORO staining), seven (for OMSB staining), or six (for IHC staining) cross-sections per mouse, representing approximately three-quarters of the length of the whole vessel, starting from the aortic arch to the bifurcation. Data were normalized using the plaque area/vessel area ratio for each section. The mean percentage of lesion-specific area was calculated for each BCA height.
Infrared Imaging of Atherosclerotic Plaque Components in the BCA.
Spectroscopic analysis was performed on OCT (Optimal Cutting Temperature)-embedded tissues. The infrared (IR) spectra of BCA cross-sections were obtained using a Varian 620-IR microscope (Varian Medical Systems, Palo Alto, CA) coupled to a 670 spectrometer with liquid nitrogen–cooled mercury-cadmium-telluride and a 128 × 128-pixel focal plane array detector. Dimensions of acquired maps were 128 × 128 pixels, with a field of view of 700 × 700 μm2. For background and sample measurements, 128 and 32 scans were collected, respectively. Spectra were recorded in transmission mode with a spectral resolution of 8 cm−1 in the range of 3600 to 850 cm−1. Protein allocation throughout the tissue was visualized by integrating the amide I (1700 to 1600 cm−1) range of the IR spectra (Fig. 4, A and B). The other band used for the integration was a CH2/CH3 stretching mode observed at 2925 cm−1, assigned to vibrations of organic matter; in addition, the stretching mode for the carbonyl ester groups in the 1770 to 1710 cm−1 range was used since it has been found to be far more selective for lipids (Wrobel et al., 2011). This band is assigned to cholesteryl esters, triglycerides, and free fatty acids. Estimation of cholesterol and its ester content in aortic tissue was based on the intensity of bands at 1058 cm−1 (C-O bending vibration) and 3005 cm−1, respectively. The latter is generally characteristic of unsaturated aliphatic compounds (Kodali et al., 1991; Wrobel et al., 2011; Marzec et al., 2014).
Fig. 4.

Results of IR imaging of plaque biochemical content in the BCA. (A) Representative microphotographs of the cross-sections of BCAs taken from control (top), MNA-treated (middle), and NicA-treated (bottom) ApoE/LDLR−/− mice, with spectroscopic images in the ranges of 1700 to 1600 cm−1 (amide I, proteins), 1770 to 1710 cm−1 (esters and triglycerides), and 3000 to 2800 cm−1 (lipids and proteins). (B) Representative FT-IR spectrum of a murine aortic tissue. Arrows indicate bands used to map the distribution of major plaque components. (C) Plaque lipid content of MNA- and NicA-treated mice shown as a percentage of plaque lipid content from the control group. Values are means ± S.E.M. (n = 12). *P ≤ 0.05.
Measurement of PGI2 Metabolite, Nitrite, and Nitrate Production in Aortic Rings.
For 6-keto-PGF1α measurement, the aorta was washed with cold phosphate-buffered saline solution, cleansed of perivascular fatty tissue, and divided into three 2-mm–long rings. Each ring was placed separately in Krebs’ solution (37°C) under constant CO2 flow and kept for 15 minutes under stabilizing conditions. The rings were then placed in buffer with added dimethylsulfoxide (control sample) or a COX inhibitor: indomethacin (5 µmol) or DUP-697 [5-bromo-2-(4-fluorophenyl)-3-(4-(methylsulfonyl)phenyl)-thiophene] (1 µmol). After 10 minutes, the rings were moved into fresh Krebs’ buffer, and 100-µl samples were taken after 3 and 60 minutes of incubation. 6-Keto-PGF1α was measured using an enzyme-linked immunosorbent assay (ELISA) 96-well plate kit (Enzo Life Sciences, Farmingdale, NY) and the Synergy4 multiplate reader (BioTek, Winooski, VT). The mean concentration of 6-keto-PGF1α was calculated by Prism 5.0 (GraphPad Software, La Jolla, CA), using the Mann–Whitney test and Kruskal–Wallis one-way analysis of variance (P ≤ 0.05). Afterward, the aortic rings were dried at 60°C and weighed.
For the measurement of nitrite and nitrate, the thoracic aorta was isolated and cleaned at room temperature using ultrapure Krebs’ buffer made with low-resistance (≥18.2 mΩ) distilled water (purified by Millipore UltraQ3; Millipore, Billerica, MA). The aortas were divided into three 2-mm–long rings and put into a 96-well plate containing ultrapure Krebs’ buffer with nitric oxide synthase (nos) inhibitor NG-nitro-l-arginine methyl ester (500 µmol; Sigma-Aldrich). The plate with the aortic rings was placed into a BIO-V gas treatment chamber (Noxygen Science, Elzach, Germany), where it remained for 15 minutes under CO2 flow at 37°C. After preincubation, the rings were removed to wells containing fresh ultrapure Krebs’ buffer and incubated for 1 hour in the presence of 1 µM calcium ionophore A23187 [5-(methylamino)-2-({(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec-2-yl}methyl)-1,3-benzoxazole-4-carboxylic acid] (Sigma-Aldrich). Ultrapure Krebs’ buffer samples, with and without the addition of ionophore, were used as a negative control for the wells containing the aortic rings. The samples were used for HPLC measurement of nitrate and nitrite concentrations by an ENO-20 NOx Analyzer (EiCom, Kyoto, Japan).
Measurement of PGI2/TXA2 Metabolites, Nitrate, and Nitrite in Urine.
Quantitative analysis of the PGI2 and TXB2 metabolites (2,3-dinor-6-keto-PGF1α and 2,3-dinor-TXB2) was accomplished using a UFLC Nexera system (Shimadzu, Kyoto, Japan) coupled to a triple quadrupole mass spectrometer (QTRAP 5500; ABSciex, Framingham, MA) equipped with a Turbo V ion source. The best separation of analytes was achieved using an Acquity BEH C18 analytical column (3.0 × 100 mm, 1.7 μm; Waters, Milford, MA). The mobile phases, consisting of pure acetonitrile and 0.1% ammonium hydroxide in water, were delivered in gradient elution at a flow rate of 0.25 ml/min.
The electrospray ionization process was performed in negative ion mode. Data acquisition was carried out in multiple reaction monitoring mode, with mass-to-charge ion transitions of 367.2→305.0 and 341.6→134.6 chosen for 2,3-dinor-TXB2 and 2,3-dinor-6-keto-PGF1α, respectively. Purification of urine samples (100 µl) was performed via a liquid–liquid extraction technique using 1 ml acidified ethyl acetate as an extrahent. Nine-hundred microliters of supernatant was completely evaporated under nitrogen stream and the dry residue was reconstituted by adding a mixture of EtOH and water. The limit of quantification was 0.25 ng/ml for both 2,3-dinor-TXB2 and 2,3-dinor-6-keto-PGF1α.
The following reagents were used as measurement standards: 11-dehydro-TXB2, 2,3-dinor-6-keto- PGF1α, and deuterated internal standards (11-dehydro-TXB2-d4, 2,3-dinor-6-keto-PGF1α-d9) purchased from Cayman Chemicals Co. (Ann Arbor, MI). HPLC-grade solvents such as acetonitrile, ethanol, ethyl acetate, and acetic acid (≥99.9%) were obtained from Sigma-Aldrich. Ammonium hydroxide was supplied by J.T. Baker (Deventer, The Netherlands). Ultrapure water was delivered by a MilliQ Water Purification System from Merck (Darmstadt, Germany).
For nitrate and nitrite measurement in urine, samples were centrifuged after collection (1500g for 5 minutes) and frozen at −80°C. Undiluted samples were measured using an ENO-20 analyzer; results were normalized for urinary creatinine concentrations as assessed by an ABX Pentra 400 workstation (Horiba, Kyoto, Japan).
Measurement of TXB2 and TNFα Release Using Ex Vivo Full Blood Assay.
Immediately after sampling, the blood was diluted 5× with 0.9% NaCl solution and incubated at 37°C for 1 hour with constant stirring by microdipol to activate platelets (1500 rpm; rotation direction changed every 3 seconds). The entire procedure was performed using a specially designed Xyzyk apparatus (XYZYK Ltd., Krakow, Poland), which combines 16 magnetic centrifuges and enables centrifugation of multiple blood samples simultaneously and independently at a defined speed, in stable conditions and at a stable temperature. Each centrifuge is powered by an individual engine, and the whole system is cooled by a Peltier module. At specific time points of the ex vivo assay in the Xyzyk apparatus (0, 30, and 60 minutes), blood samples were placed into centrifuge tubes containing acetylsalicylic acid solution (500 µmol), incubated for 2 minutes, and then centrifuged (3000 rpm, 4°C, 12 minutes). TXB2 concentration was measured in the supernatant using an ELISA 96-well plate kit (Enzo Life Sciences). In addition, TNFα concentrations were estimated in samples collected after 0, 30, and 60 minutes of ex vivo full blood assay, using an ELISA 96-well plate kit (R&D Scientific, Flanders, NJ). All ELISA measurements were performed by a Synergy4 multiplate reader (BioTek).
Measurement of MNA Metabolites in Plasma and Urine.
Chromatographic analysis of plasma samples was performed on an UltiMate 3000 LC system (Thermo Scientific Dionex, Sunnyvale, CA) consisting of a pump (DGP 3600RS), a column compartment (TCC 3000RS), an autosampler (WPS-3000TRS), and an SRD-3600 solvent rack (degasser). Chromatographic separation was carried out on an Aquasil C18 analytical column (4.6 mm × 150 mm, 5 μm; Thermo Scientific). The mobile phase consisted of acetonitrile (A) and water with 0.1% formic acid (B). The flow rate was set at 0.8 ml/min with the following linear elution steps: 0.0 minutes (A:B, 90/10), 1.5 minutes (A:B, 90/10), 4.5 minutes (A:B, 20/80), 5.5 minutes (A:B, 20/80), 5.7 minutes (A:B, 90/10), and 10.0 minutes (A:B, 90/10). Plasma samples were prepared by deproteinization with acidified acetonitrile.
HPLC analysis of urine samples was performed on a Transcend TLX-2 system with an HTS PAL System autosampler (Thermo Scientific). Instrument control was done using Aria 1.6 software. Compounds were separated from the matrix using a TurboFlow Cyclone-P polymer column (0.5 × 50 mm; Thermo Scientific). From the TurboFlow column, the analytes were eluted with acidified acetonitrile onto an Aquasil C18 analytical column (4.6 × 150 mm, 5 μm). The mobile phase consisted of acetonitrile (A) and acidified water (0.1% formic acid) (B) with the following linear eluting steps: 0.0 minutes (A:B, 80/20), 1.5 minutes (A:B, 80/20), 5.5 minutes (A:B, 50/50), 6.5 minutes (A:B, 50/50), 7.0 minutes (A:B, 80/20), and 10.0 minutes (A:B, 80/20). The flow rate was set at 0.8 ml/min. Urine samples were diluted 1:10 prior to analysis.
The following reagents were used as measurement standards: NicA, NA, and MNA obtained from Sigma-Aldrich and N-methyl-4-pyridone-3-carboxamide (Met-2Pyr) and N-methyl-2-pyridone-5-carboxamide (Met-4Pyr) purchased from TLC PharmaChem (Vaughan, ON, Canada). Deuterated analytes were used as internal standards. NA-d4 and NAM-d4 were purchased from Sigma-Aldrich and Dr. Ehrenstorfer GmbH (Augsburg, Germany), respectively. Met-2Pyr-d3 and Met-4Pyr-d3 were obtained from TLC PharmaChem and MNA-d3 was synthesized by Dr. Adamus (Technical University, Lodz, Poland). HPLC gradient–grade acetonitrile and formic acid were purchased from Sigma-Aldrich. Ultrapure water was obtained from a Millipore system (Direct-Q 3UV).
Statistical Analysis.
The mean concentrations of NicA metabolites and TXB2/PGI2 metabolites in urine, plasma, and aortic effluent, as well as plasma lipid profile, acute phase proteins, TNFα concentration (if full blood assay), atherosclerotic plaque area, and macrophage-specific plaque area were analyzed by GraphPad Prism 5.0 software, using a nonparametric Mann–Whitney test and Kruskal–Wallis one-way analysis of variance (P ≤ 0.05). IR data were analyzed using CytoSpec (version 2.00.01; CytoSpec Inc., Berlin, Germany) and MatLab (MathWorks, Natick, MA) software.
Results
Effects of MNA and NicA Treatment on Lipid Profile and Acute Phase Proteins in Plasma.
There were no significant effects of MNA and NicA treatment on total cholesterol, triglycerides, and very low density lipoprotein cholesterol; however, a tendency for elevated HDL cholesterol and reduced LDL cholesterol in both MNA- and NicA-treated groups was noted (P ≥ 0.05), as compared with the control (Fig. 1).
Fig. 1.

Effects of MNA and NicA treatment on plasma lipid profile in ApoE/LDLR−/− mice. (A and B) In MNA- and NicA-treated groups, HDL tended to increase (A), whereas LDL tended to decrease (B); however, the differences were statistically insignificant (P ≥ 0.05). (C–E) Very low density lipoprotein (C), total cholesterol (D), and total triglyceride (E) concentrations were unaffected by MNA and NicA treatment. Values are means ± S.E.M. (n = 12). TG, triglyceride; totC, total cholesterol; VLDL, very low density lipoprotein.
Treatment with MNA and NicA was associated with lower concentrations of serum amyloid P (408.56 and 356.83 versus 564 µg/ml) and haptoglobin (79.35 and 76.93 versus 153.67 µg/ml) as well as slightly lower concentrations of α2-macroglobulin (1.40 and 1.34 versus 1.57 µg/ml, P ≤ 0.05; data not shown).
Effects of MNA and NicA on BCA Atherosclerotic Plaque Size and Plaque Inflammation.
In MNA- and NicA-treated groups of mice, the mean area ratios between atherosclerotic plaque area and vessel area, as assessed by ORO staining, were significantly lower. Statistically significant (P ≤ 0.05) differences in the treated versus control groups were present in the cross-sections collected from the middle areas of the BCA, becoming less distinct closer to the aortic arch and more prominent toward the distal bifurcation. Reduction in plaque area was comparable between MNA- and NicA-treated groups (23% and 25%, respectively, versus 45% in control group), as shown in Fig. 2B. In the BCA analyzed by OMSB staining (Fig. 2D), the mean percentage of vessel area occupied by atherosclerotic plaques was significantly lower in the MNA-treated mice, as compared with control mice (35% versus 55% in the control group). Plaque progression was also reduced in the BCA of the NicA-treated group, but only in cross-sectional areas taken from the distal parts of the BCA.
Fig. 2.

Effects of MNA and NicA treatment on atherosclerotic plaque size in the BCA of ApoE/LDLR−/− mice. (A and C) Representative images show ORO (A) and OMSB (C) staining of plaques in the BCA at various distance from the aortic arch. (B and D) Summarized results show atherosclerotic plaque area estimated on the basis of ORO (B) and OMSB (D) staining, normalized to vessel area for the entire serial sections of the BCA. Values are means ± S.E.M. (n = 12). *P ≤ 0.05 versus untreated control.
Reduction in plaque area was accompanied by diminished macrophage infiltration (Fig. 3). The mean CD68-specific plaque area was reduced in both the MNA- and NicA-treated groups versus the untreated control (15% and 17% versus 30%). In MAC3-immunostained plaques, statistically significant differences between MNA-treated mice and the control group were observed (15% versus 38%, respectively), whereas only a slight reduction in MAC3-positive area was seen in the NicA-treated group.
Fig. 3.

Effects of MNA and NicA treatment on macrophage content in plaques of the BCA in ApoE/LDLR−/− mice. (A and C) Representative images show anti-CD68 (A) and anti-MAC3 (C) immunostainings of plaque area in BCA at various distances from the aortic arch. (B and D) Results of macrophage staining demonstrate a reduction in mean CD68-specific plaque area in MNA- and NicA-treated mice versus untreated control (B) and reduction in mean MAC-specific plaque area, but only in MNA- treated mice (D). Values are means ± S.E.M. (n = 12). *P ≤ 0.05 versus untreated control.
Effects of MNA and NicA on Plaque Chemical Content, as Evaluated by IR Imaging.
MNA and NicA treatment showed a remarkable effect on the cholesterol content of BCA plaques (Fig. 4C). Compared with the untreated control group, significantly lowered cholesterol content was observed (reduction by 44% for MNA and by 39% for NicA versus control groups, P ≤ 0.05). Moreover, cholesteryl ester storage was reduced by approximately 50% in the MNA-treated group (P ≤ 0.05 versus control). Treatment with MNA and NicA had no significant effect on the saturation profile of lipids (P ≥ 0.05 versus control).
Effect of MNA and NicA on PGI2 Metabolite, Nitrite, and Nitrate Production in the Thoracic Aorta.
After 60 minutes of incubation, the mean increase in 6-keto-PGF1α was remarkably higher in aortic rings taken from MNA-treated mice (1.69 versus 0.47 µg/mg in the control group, P ≤ 0.05) and was less pronounced, but still statistically significant, in the NicA-treated group (1.05 µg/mg, P ≤ 0.05) (Fig. 5A). In the presence of COX-2 inhibitor DUP-697, there was a marked decrease in 6-keto-PGF1α release by aortic rings isolated from the control (0.11 µg/mg), NicA-treated (0.13 µg/mg), and MNA-treated groups (0.38 µg/mg, P ≤ 0.05 versus control). Indomethacin, a nonselective COX inhibitor, also had profound inhibitory effects (0.04 µg/mg in the control group, 0.20 µg/mg in MNA-treated mice, and 0.02 µg/mg in NicA-treated mice).
Fig. 5.

Effects of MNA and NicA on PGI2 and nitrate/nitrite production by the thoracic aorta (A) Concentration of 6-keto-PGF1α in effluent samples taken from incubated ex vivo aortic rings, with or without the presence of COX inhibitors DUP-697 and indomethacin. (B and C) Concentration of nitrite (B) and nitrate (C) released by aortic rings taken from MNA- and NicA-treated mice and incubated with or without calcium ionophore. Values are means ± S.E.M. (n = 6). *P ≤ 0.05 and **P ≤ 0.01 versus untreated control,
After 60 minutes of incubation in the presence of ionophore, aortic rings taken from MNA-treated mice released a substantially higher (P ≤ 0.01 versus control) concentration of nitrate (0.97 versus 0.01 µmol/mg) and nitrite (3.9 versus 0.54 µmol/mg). Less distinct but still significant (P ≤ 0.05 versus control) elevations of nitrate (0.86 versus 0.01 µmol/mg) and nitrite (2.44 versus 0.54 µmol/mg) were detected in samples containing aortic rings taken from NicA-treated mice. Nitrite release was significantly diminished in the presence of NOS inhibitor NG-nitro-l-arginine methyl ester (to 0.005 and 0.01 µmol/mg in control and NicA- or MNA-treated mice, respectively; data not shown).
Effect of MNA and NicA on Concentrations of PGI2 and TXB2 Metabolites, Nitrate, and Nitrite in Urine.
Liquid chromatography with tandem mass spectrometry (LC-MS/MS)-based analysis of TXB2 and PGI2 metabolites in urine showed a significantly elevated concentration of 2,3-dinor-6-keto-PGF1α (a urine metabolite of PGI2) in urine samples collected from MNA-treated mice, but not from the NicA-treated group (Fig. 6A), compared with untreated controls (3.76 and 1.79 versus 2.17 ng/mg, respectively, P ≤ 0.05). Concentrations of 2,3-dinor-TXB2 were similar in all groups (1.43 ng/mg in the control group, 1.84 ng/mg in MNA-treated mice, and 1.68 ng/mg in NicA-treated mice).
Fig. 6.

Effects of MNA and NicA on PGI2/TXB2 metabolite and nitrate/nitrite concentrations in urine. (A) Results of LC-MS/MS analysis of 2,3-dinor-6-keto-PGF1α and 2,3-dinor-TXB2 in urine samples. (B) Results of HPLC-based measurement of urinary nitrite and nitrate. Values are means ± S.E.M. (n = 6). *P ≥ 0.05.
Measurement of nitrite and nitrate in urine samples (Fig. 6B) showed no statistically significant differences in nitrite concentrations between MNA- or NicA-treated mice and untreated controls (0.04 and 0.026 versus 0.036 mmol/mg, respectively), whereas significantly increased nitrate concentrations were observed in the urine samples collected from both the MNA- and NicA-treated mice (0.28 and 0.41, respectively, versus 0.11 mmol/mg in the control group, P ≤ 0.05).
Effect of MNA and NicA on TXB2 and TNFα Concentrations in Ex Vivo Full Blood Assay.
A significant difference in TXB2 concentration between the control and MNA-treated groups (P ≤ 0.05) was apparent after 60 minutes of blood activation in the ex vivo full blood assay (7.04 versus 3.66 ng/105 platelets, respectively). The mean production of TXB2 was similar at the beginning of blood activation and after 30 minutes, decreasing after 1 hour of stirring in blood samples taken from MNA-treated mice (P ≤ 0.05 versus control) (Fig. 7A). Reduced TXB2 release in samples taken from NicA-treated mice was also seen after 60 minutes of activation (5.04 ng/105 platelets), but the observed result was not statistically significant. Moreover, significantly lower concentrations of TNFα were detected after 60 minutes of activation in samples taken from the MNA- and NicA-treated groups versus the control group (2.67 and 2.53 versus 6.71 pg/ml, respectively; Fig. 7B).
Fig. 7.

Effects of MNA and NicA on TXB2 and TNFα release, assessed in ex vivo full blood assay. (A) Results of TXB2 measurement after 30 and 60 minutes of activation. Data were normalized to 105 platelet counts. (B) Concentration of TNFα in blood samples after 60 minutes of activation. Values are means ± S.E.M. (n = 6). *P ≤ 0.05; **P ≤ 0.01 versus untreated control.
Effects of MNA and NicA Treatment on MNA Metabolite Concentrations in Plasma and Urine.
LC-MS/MS measurement of NicA concentration in urine showed the highest NicA concentration in the NicA-treated group (0.048 versus 0.015 µmol/mg in controls). NA concentration was also significantly increased in the NicA-treated group (0.29 versus 0.052 µmol/mg in controls). As expected, the concentration of MNA pooled with Met-2Pyr and Met-4Pyr was higher in the MNA-treated group than in the control group (0.69 versus 0.47 µmol/mg, respectively). Surprisingly, an even higher concentration of MNA and its metabolites was detected in the NicA-treated mice (1.13 µmol/mg) compared with the MNA-treated group (0.69 µmol/mg; Fig. 8A).
Fig. 8.
Effects of MNA and NicA on the concentration of NicA metabolites in urine and plasma. (A) Results of LC-MS/MS measurement of NicA and NA and MNA plus Met-4Pyr plus Met-2Pyr concentration in urine. Values are means ± S.E.M. (n = 6). *P ≤ 0.05; **P ≤ 0.01 versus untreated control. (B) Diagram showing the NA/MNA ratio in plasma samples based on LC-MS/MS measurement of NA, MNA, Met-2Pyr, and Met-4Pyr in plasma samples taken from untreated control, MNA-, and NicA-treated mice. Values are means ± S.E.M. (n = 6). **P ≤ 0.01 versus MNA-treated group.
LC-MS/MS analysis of plasma samples showed a significant elevation (P ≤ 0.01) in endogenous NA in the NicA-treated group, compared with the MNA treatment and control groups (84 versus 6 and 4 nmol/mg, respectively). The NA/MNA plasma concentration ratio was 4-fold higher in the NicA-treated group compared with the MNA treatment and control groups (Fig. 8B). Plasma concentrations of MNA and its metabolites Met-2Pyr and Met-4Pyr were significantly higher in MNA- and NicA-treated mice compared with the control group (12 and 13 nmol/mg versus 4 nmol/mg, respectively).
Discussion
We have demonstrated for the first time that MNA, the major metabolite of nicotinamide and NicA, displays pronounced vasoprotective, anti-inflammatory, and anti-atherosclerotic in ApoE/LDLR−/− mice, associated with an improvement in PGI2- and NO-dependent endothelial function, inhibition of platelet activation, inhibition of inflammatory burden in plaques, and diminished systemic inflammation. Despite substantially higher MNA availability after NicA treatment compared with an equivalent dose of MNA, the antiatherosclerotic effect of NicA was not stronger. We suggest that detrimental effects of NicA itself or its metabolites other than MNA may limit beneficial effects of NicA-derived MNA.
In this work, 4-month-old mice with advanced atherosclerosis were used to study the effects of MNA and NicA. In this model, endothelial dysfunction is present at the age of 2 months, in the absence of visible atherosclerotic plaques. The first lesions, composed mostly of fatty streaks and infiltrating macrophages, appear at the age of 3 months and subsequently develop into advanced atherosclerotic plaques at the age of 4 months (Csányi et al., 2012). Accordingly, the antiatherosclerotic effects of MNA and NicA demonstrated here have clinical relevance and indicate the inhibition of progression of atherosclerosis, not prevention of atherosclerosis as if the treatment had been initiated before atherosclerotic plaque development.
NicA and MNA treatment did not significantly affect plasma cholesterol, however; ApoE/LDLR−/− mice display a lipid profile that is not similar to humans, as opposed to ApoE*3-Leiden (E3L) or ApoE*3-Leiden cholesteryl ester transfer protein (E3L.CETP) transgenic mice (de Haan et al., 2008). Thus, the lack of significant effects of NicA and MNA on the lipid profile shown here point to possible differences from humans, in which NicA is known to affect total cholesterol, LDL cholesterol, and HDL cholesterol. The effects of MNA on lipid profile, triglycerides, and HDL cholesterol, in particular, are currently being evaluated in a clinical trial (ClinicalTrials.gov identifier NCT02008084). The ongoing clinical study aims to demonstrate the pharmacological effects of MNA on the lipid profile in humans, and results are expected in 2016.
The lack of significant effects on the lipid profile in ApoE/LDLR−/− mice in this study suggests that the antiatherosclerotic effects of MNA and NicA are not linked to the hypolipidemic effects of either compound, but mainly to their vasoprotective properties and improvement in endothelial function. Indeed, despite the lack of significant effects on lipid profile, we clearly demonstrated, using the unique approach of Fourier transform infrared spectroscopy analysis, that cholesterol and cholesteryl ester content in atherosclerotic plaques was substantially reduced, suggesting that the major mechanism by which NicA and MNA decrease cholesterol content in plaques is linked to the improvement in vascular endothelial function. This should result in decreased permeability to lipoproteins and increased activity of anti-inflammatory endothelial mechanisms.
In fact, MNA and NicA treatment resulted in an improvement in PGI2-dependent and NO-dependent vascular function. Higher concentrations of 6-keto-PGF1α were detected in the effluent from ex vivo aortic rings taken from MNA- and NicA-treated mice. 6-Keto-PGF1α generation was blunted by COX-2 inhibitor DUP-697 as well as by nonselective COX inhibitor indomethacin, which may suggest a major role for COX-2 in vascular PGI2 production; this effect was augmented by MNA and NicA treatment. Moreover, a higher concentration of 2,3-dinor-6-keto-PGF1α was detected in urine samples collected from MNA-treated mice but not NicA-treated mice, suggesting a weaker stimulation of the PGI2 pathway by NicA. It has been previously reported that MNA enhances COX-2–dependent PGI2 release and affords an antithrombotic effect (Chlopicki et al., 2007). Other reports have supported a PGI2-dependent mechanism of MNA action (Bryniarski et al., 2008; Brzozowski et al., 2008a). Interestingly, NicA has also been shown to release PGI2, which we believe may occur through generation of MNA (Luria, 1990). Since PGI2 is the major antiplatelet agent produced by the vascular wall, our study also looked at whether MNA-dependent activation of COX-2/PGI2–dependent mechanisms affects platelets. Using our original ex vivo full blood assay, we demonstrated a significant antiplatelet effect in MNA-treated animals, whereas this effect was weaker in NicA-treated mice, compatible with a weaker effect of NicA on PGI2 metabolites in urine. Interestingly, MNA and NicA treatment was accompanied by intensified production of nitrate/nitrite by ex vivo aortic rings and led to increased nitrate concentration in urine, suggesting increased bioavailability of NO, probably linked to improved endothelial nitric oxide synthase function in the vascular wall. Although we did not look at the mechanisms involved, improvement of NOS-dependent function may be connected with PGI2 and cAMP-dependent phosphorylation of endothelial nitric oxide synthase (Niwano et al., 2006) and was also demonstrated previously in humans after MNA treatment (Domagala et al., 2012).
Altogether, our results point to a major role of the vascular mechanisms of action of MNA and its precursor NicA, linked to an improvement in COX-2/PGI2–dependent function, which could result in inhibition of platelet activation and, subsequently, an anti-inflammatory effect. Indeed, in the ex vivo model, the TNFα concentration was reduced in MNA- and NicA-treated mice, along with several acute phase proteins in plasma (haptoglobin, serum amyloid P, and α2-macroglobulin).
It should be also considered that the antiatherosclerotic action of NicA may be due to activation of the GPR109A receptor and its effect on the macrophage content of atherosclerotic plaque (Lukasova et al., 2011a,b). In our work, the effect of NicA on plaque macrophage content indicated by MAC-3 immunostaining was not robust, which would exclude the importance of this mechanism in the antiatherosclerotic action of NicA during advanced atherosclerosis. It is also unlikely that MNA has an affinity to GPR109A receptors. Perhaps a GPR109A-dependent action of NicA is important in the early development of atherosclerosis, when monocytes are infiltrating the vascular wall, but not in the late stage of plaque development, when significant macrophage content is seen in the plaques of 4-month old ApoE/LDLR−/− mice (Csányi et al., 2012). Indeed, unlike in our experiment (reversal of atherosclerosis), Lukasova et al. (2011a,b) used a preventive treatment of atherosclerosis to investigate the GPR109A-dependent effect of NicA, in which LDLR−/− mice were treated with NicA starting at the age of 2 months, prior to the appearance of visible lesions.
It is worth noting that NicA-derived MNA or exogenous MNA may inhibit macrophage activation and subsequent cholesterol phagocytosis by a PGI2-releasing effect, thus preventing foam cell formation and atherosclerotic plaque development. This hypothesis could explain the lower concentration of plaque free cholesterol and cholesteryl esters detected by Fourier transform infrared spectroscopy in NicA- and MNA-treated animals.
Considering the metabolism and pharmacokinetics of MNA and NicA, we compared the antiatherosclerotic efficacy of equivalent doses of the compounds and found approximately 2-fold higher MNA, Met-2Pyr, and Met-4Pyr concentrations in the urine of NicA-treated animals. These results are not surprising, considering the higher bioavailability of NicA (approximately 80%), which is metabolized into MNA by approximately 50% (Menon et al., 2007) and the much lower MNA bioavailability (approximately 10%, unpublished data). Obviously, NicA was detected only in urine taken from NicA-treated mice. The most striking difference in the metabolite profiles of NicA- and MNA-treated animals was the approximately 4-fold higher plasma concentration of the NA/MNA ratio in the NicA-treated group compared with MNA-treated group. We believe that high NA concentration after NicA treatment leads to the concomitant activation of NNMT and depletion of SAM, a common methyl group donor and various detrimental changes in the NAD+ metabolome. By contrast, MNA, the product of NA methylation does not affect methylation status nor influence NAD+ metabolome (Aksoy et al.,1994; Mullangi & Srinivas, 2011).
Among NAD+ consumers, sirtuins play a pivotal role as transcriptional regulators, acting as lysine deacetylases on H3 and H4 histones (Belenky et al., 2007; Kirkland, 2009). Since NA concentration and the NA/MNA ratio was significantly higher in the plasma of NicA-treated mice, the excess NA could have an inhibitory effect on the sirtuins, consequently disrupting the regulation of gene expression important for maintaining vascular homeostasis. Sirtuin inhibition by NA after NicA administration, along with S-adenosyl methionine depletion, may thus at least partially explain the weaker anti-inflammatory effect of NicA in ApoE/LDLR−/− mice, compared with MNA, despite higher MNA bioavailability; however, this hypothesis warrants further studies.
Interestingly, a number of reports have suggested that depletion of S-adenosyl methionine after treatment with high doses of NicA and NA or by the prolonged use of sustained-release NicA may lead to hyperhomocysteinemia, oxidative stress, and insulin resistance (Guyton and Bays, 2007; Sun et al., 2012; Li et al., 2013a,b) and may also increase the risk of drug-induced hepatotoxicity (Bhardwaj and Chalasani, 2007; Demyen et al., 2013).
In conclusion, we have demonstrated for the first time that MNA displays pronounced antiatherosclerotic and anti-inflammatory actions in ApoE/LDLR−/− mice. Interestingly, despite substantially higher availability of MNA after NicA treatment, compared with an equivalent dose of MNA, the antiatherosclerotic effect of NicA was not superior to that of MNA. This was most likely due to side effects of NicA linked to overproduction of endogenous NA that limited the beneficial effects mediated by NicA-derived MNA. MNA seems to lack most of the side effects of NicA and may possess important therapeutic potential as a vasoprotective and antiatherosclerotic agent. Yet its low bioavailability and relatively short half-live may limit its usefulness.
Abbreviations
- A23187
5-(methylamino)-2-({(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec-2-yl}methyl)-1,3-benzoxazole-4-carboxylic acid
- ApoE
apolipoprotein E
- BCA
brachiocephalic artery
- COX
cyclooxygenase
- DUP-697
5-bromo-2-(4-fluorophenyl)-3-(4-(methylsulfonyl)phenyl)-thiophene
- ELISA
enzyme-linked immunosorbent assay
- GPR
G protein–coupled receptor
- HDL
high-density lipoprotein
- HPLC
high-performance liquid chromatography
- IHC
immunohistochemical
- IR
infrared
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- LDLR
low-density lipoprotein receptor
- MAC3
lysosomal-associated membrane protein 2
- Met-2Pyr
1-methyl-2-pyridone-5-carboxamide
- Met-4Pyr
1-methyl-4-pyridone-5-carboxamide
- MK-6892
2-[[3-[3-(5-hydroxy-2-pyridinyl)-1,2,4-oxadiazol-5-yl]-2,2-dimethyl-1-oxopropyl]amino]-1-cyclohexene-1-carboxylic acid
- MNA
1-methylnicotinamide
- NA
nicotinamide
- NicA
nicotinic acid
- NNMT
nicotinamide-N-methyltransferase
- NO
nitric oxide
- NOS
nitric oxide synthase
- OMSB
Orcein Martius Scarlet Blue
- ORO
Oil Red O
- PGF1α
prostaglandin F1α
- PGI2
prostacyclin (prostaglandin I2)
- TNFα
tumor necrosis factor-α
- TX A2/B2
thromboxane A2/B2
Authorship Contributions
Participated in research design: Mateuszuk, Chlopicki.
Conducted experiments: Mateuszuk, Jasztal, Maslak, Gasior-Glogowska, Sitek, Zakrzewska, Kij, Walczak.
Contributed new reagents or analytic tools: Baranska, Kostogrys.
Performed data analysis: Mateuszuk, Gasior-Glogowska, Sitek, Zakrzewska, Kij.
Wrote or contributed to the writing of the manuscript: Mateuszuk, Gasior-Glogowska, Chlopicki.
Footnotes
This research was supported by the European Union from the resources of the European Regional Development Fund under the Innovative Economy Programme [Grant POIG.01.01.02-00-069/09]. S.C. is a coinventor of patent no. CA2547234 A1 (“The use of quaternary pyridinium salts as vasoprotective agents”).
We would like to thank Dr. Jan Adamus from Lodz University of Technology for kindly providing us with MNA, and Dr. Christoph Johann from Wyatt Tech. for FFF method-based measurements of lipid profile.
References
- Aksoy S, Szumlanski CL, Weinshilboum RM. (1994) Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J Biol Chem 269:14835–14840. [PubMed] [Google Scholar]
- Bartuś M, Łomnicka M, Kostogrys RB, Kaźmierczak P, Watała C, Słominska EM, Smoleński RT, Pisulewski PM, Adamus J, Gebicki J, et al. (2008) 1-Methylnicotinamide (MNA) prevents endothelial dysfunction in hypertriglyceridemic and diabetic rats. Pharmacol Rep 60:127–138. [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C. (2007) NAD+ metabolism in health and disease. Trends Biochem Sci 32:12–19. [DOI] [PubMed] [Google Scholar]
- Bhardwaj SS, Chalasani N. (2007) Lipid-lowering agents that cause drug-induced hepatotoxicity. Clin Liver Dis 11:597–613, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryniarski K, Biedron R, Jakubowski A, Chlopicki S, Marcinkiewicz J. (2008) Anti-inflammatory effect of 1-methylnicotinamide in contact hypersensitivity to oxazolone in mice; involvement of prostacyclin. Eur J Pharmacol 578:332–338. [DOI] [PubMed] [Google Scholar]
- Brzozowski T, Konturek PC, Chlopicki S, Sliwowski Z, Pawlik M, Ptak-Belowska A, Kwiecien S, Drozdowicz D, Pajdo R, Slonimska E, et al. (2008a) Therapeutic potential of 1-methylnicotinamide against acute gastric lesions induced by stress: role of endogenous prostacyclin and sensory nerves. J Pharmacol Exp Ther 326:105–116. [DOI] [PubMed] [Google Scholar]
- Brzozowski T, Konturek PC, Sliwowski Z, Drozdowicz D, Burnat G, Pajdo R, Pawlik M, Bielanski W, Kato I, Kuwahara A, et al. (2008b) Gastroprotective action of orexin-A against stress-induced gastric damage is mediated by endogenous prostaglandins, sensory afferent neuropeptides and nitric oxide. Regul Pept 148:6–20. [DOI] [PubMed] [Google Scholar]
- Carlson LA. (2005) Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. J Intern Med 258:94–114. [DOI] [PubMed] [Google Scholar]
- Chai JT, Digby JE, Choudhury RP. (2013) GPR109A and vascular inflammation. Curr Atheroscler Rep 15:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlopicki S, Swies J, Mogielnicki A, Buczko W, Bartus M, Lomnicka M, Adamus J, Gebicki J. (2007) 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br J Pharmacol 152:230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csányi G, Gajda M, Franczyk-Zarow M, Kostogrys R, Gwoźdź P, Mateuszuk L, Sternak M, Wojcik L, Zalewska T, Walski M, et al. (2012) Functional alterations in endothelial NO, PGI₂ and EDHF pathways in aorta in ApoE/LDLR-/- mice. Prostaglandins Other Lipid Mediat 98:107–115. [DOI] [PubMed] [Google Scholar]
- de Haan W, van der Hoogt CC, Westerterp M, Hoekstra M, Dallinga-Thie GM, Princen HMG, Romijn JA, Jukema JW, Havekes LM, Rensen PCN. (2008) Atorvastatin increases HDL cholesterol by reducing CETP expression in cholesterol-fed APOE*3-Leiden.CETP mice. Atherosclerosis 197:57–63. [DOI] [PubMed] [Google Scholar]
- Demyen M, Alkhalloufi K, Pyrsopoulos NT. (2013) Lipid-lowering agents and hepatotoxicity. Clin Liver Dis 17:699–714, x. [DOI] [PubMed] [Google Scholar]
- Domagala TB, Szeffler A, Dobrucki LW, Dropinski J, Polanski S, Leszczynska-Wiloch M, Kotula-Horowitz K, Wojciechowski J, Wojnowski L, Szczeklik A, et al. (2012) Nitric oxide production and endothelium-dependent vasorelaxation ameliorated by N1-methylnicotinamide in human blood vessels. Hypertension 59:825–832. [DOI] [PubMed] [Google Scholar]
- Ganji SH, Kamanna VS, Kashyap ML. (2014) Niacin decreases leukocyte myeloperoxidase: mechanistic role of redox agents and Src/p38MAP kinase. Atherosclerosis 235:554–561. [DOI] [PubMed] [Google Scholar]
- Ganji SH, Qin S, Zhang L, Kamanna VS, Kashyap ML. (2009) Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis 202:68–75. [DOI] [PubMed] [Google Scholar]
- Gebicki J, Sysa-Jedrzejowska A, Adamus J, Woźniacka A, Rybak M, Zielonka J. (2003) 1-Methylnicotinamide: a potent anti-inflammatory agent of vitamin origin. Pol J Pharmacol 55:109–112. [PubMed] [Google Scholar]
- Ginsberg HN, Reyes-Soffer G. (2013) Niacin: a long history, but a questionable future. Curr Opin Lipidol 24:475–479. [DOI] [PubMed] [Google Scholar]
- Guyton JR, Bays HE. (2007) Safety considerations with niacin therapy. Am J Cardiol 99:22C–231C. [DOI] [PubMed] [Google Scholar]
- Kirkland JB. (2009) Niacin status impacts chromatin structure. J Nutr 139:2397–2401. [DOI] [PubMed] [Google Scholar]
- Kodali DR, Small DM, Powell J, Krishnan K. (1991) Infrared micro-imaging of atherosclerotic arteries. Appl Spectrosc 45:1310–1317. [Google Scholar]
- Kostogrys RB, Johann C, Czyżyńska I, Franczyk-Żarów M, Drahun A, Maślak E, Jasztal A, Gajda M, Mateuszuk Ł, Wrobel TP, Baranska M, Wybrańska I, Jezkova K, Nachtigal P, Chlopicki S. (2015) Characterisation of Atherogenic Effects of Low Carbohydrate, High Protein Diet (LCHP) in ApoE/LDLR-/- Mice. J Nutr Health Aging 19:710–718. [DOI] [PubMed] [Google Scholar]
- Kühnast S, Louwe MC, Heemskerk MM, Pieterman EJ, van Klinken JB, van den Berg SAA, Smit JWA, Havekes LM, Rensen PCN, van der Hoorn JWA, Princen HMG, Jukema JW. (2013) Niacin Reduces Atherosclerosis Development in APOE*3Leiden.CETP Mice Mainly by Reducing NonHDL-Cholesterol. PLoS One 19:(8)6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I, Digby JE, Bannister T, Handa A, Wiesmann F, et al. (2009) Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function. A randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol 54:1787–1794. [DOI] [PubMed] [Google Scholar]
- Li D, Luo N, Ma Q, Li SZ, Shi Q, Cao Y, Zhou SS. (2013a) Excessive nicotinic acid increases methyl consumption and hydrogen peroxide generation in rats. Pharm Biol 51:8–12. [DOI] [PubMed] [Google Scholar]
- Li D, Tian YJ, Guo J, Sun WP, Lun YZ, Guo M, Luo N, Cao Y, Cao JM, Gong XJ, et al. (2013b) Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br J Nutr 110:2156–2164. [DOI] [PubMed] [Google Scholar]
- Lukasova M, Hanson J, Tunaru S, Offermanns S. (2011a) Nicotinic acid (niacin): new lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol Sci 32:700–707. [DOI] [PubMed] [Google Scholar]
- Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. (2011b) Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest 121:1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria MH. (1990) Atherosclerosis: the importance of HDL cholesterol and prostacyclin: a role for niacin therapy. Med Hypotheses 32:21–28. [DOI] [PubMed] [Google Scholar]
- Marzec KM, Wrobel TP, Rygula A, Maslak E, Jasztal A, Fedorowicz A, Chlopicki S, Baranska M. (2014) Visualization of the biochemical markers of atherosclerotic plaque with the use of Raman, IR and AFM. J Biophotonics 7:744–756. [DOI] [PubMed] [Google Scholar]
- Menon RM, Adams MH, González MA, Tolbert DS, Leu JH, Cefali EA. (2007) Plasma and urine pharmacokinetics of niacin and its metabolites from an extended-release niacin formulation. Int J Clin Pharmacol Ther 45:448–454. [DOI] [PubMed] [Google Scholar]
- Mullangi R, Srinivas NR. (2011) Niacin and its metabolites: role of LC-MS/MS bioanalytical methods and update on clinical pharmacology. An overview. Biomed Chromatogr 25:218–237. [DOI] [PubMed] [Google Scholar]
- Niwano K, Arai M, Koitabashi N, Hara S, Watanabe A, Sekiguchi K, Tanaka T, Iso T, Kurabayashi M. (2006) Competitive binding of CREB and ATF2 to cAMP/ATF responsive element regulates eNOS gene expression in endothelial cells. Arterioscler Thromb Vasc Biol 26:1036–1042. [DOI] [PubMed] [Google Scholar]
- Shen HC, Ding FX, Raghavan S, Deng Q, Luell S, Forrest MJ, Carballo-Jane E, Wilsie LC, Krsmanovic ML, Taggart AK, et al. (2010) Discovery of a biaryl cyclohexene carboxylic acid (MK-6892): a potent and selective high affinity niacin receptor full agonist with reduced flushing profiles in animals as a preclinical candidate. J Med Chem 53:2666–2670. [DOI] [PubMed] [Google Scholar]
- Stach K, Zaddach F, Nguyen XD, Elmas E, Kralev S, Weiss C, Borggrefe M, Kälsch T. (2012) Effects of nicotinic acid on endothelial cells and platelets. Cardiovasc Pathol 21:89–95. [DOI] [PubMed] [Google Scholar]
- Sun WP, Li D, Lun YZ, Gong XJ, Sun SX, Guo M, Jing LX, Zhang LB, Xiao FC, Zhou SS. (2012) Excess nicotinamide inhibits methylation-mediated degradation of catecholamines in normotensives and hypertensives. Hypertens Res 35:180–185. [DOI] [PubMed] [Google Scholar]
- Thoenes M, Oguchi A, Nagamia S, Vaccari CS, Hammoud R, Umpierrez GE, Khan BV. (2007) The effects of extended-release niacin on carotid intimal media thickness, endothelial function and inflammatory markers in patients with the metabolic syndrome. Int J Clin Pract 61:1942–1948. [DOI] [PubMed] [Google Scholar]
- Warnholtz A, Wild P, Ostad MA, Elsner V, Stieber F, Schinzel R, Walter U, Peetz D, Lackner K, Blankenberg S, et al. (2009) Effects of oral niacin on endothelial dysfunction in patients with coronary artery disease: results of the randomized, double-blind, placebo-controlled INEF study. Atherosclerosis 204:216–221. [DOI] [PubMed] [Google Scholar]
- Wrobel TP, Mateuszuk L, Chlopicki S, Malek K, Baranska M. (2011) Imaging of lipids in atherosclerotic lesion in aorta from ApoE/LDLR−/− mice by FT-IR spectroscopy and Hierarchical Cluster Analysis. Analyst 136:5247–5255. [DOI] [PubMed] [Google Scholar]