

Abstract
After hindlimb ischemia (HI), increased catecholamine levels within the ischemic muscle can cause dysregulation of β2-adrenergic receptor (β2AR) signaling, leading to reduced revascularization. Indeed, in vivo β2AR overexpression via gene therapy enhances angiogenesis in a rat model of HI. G protein-coupled receptor kinase 2 (GRK2) is a key regulator of βAR signaling, and β adrenergic receptor kinase C-terminal peptide (βARKct), a peptide inhibitor of GRK2, has been shown to prevent βAR down-regulation and to protect cardiac myocytes and stem cells from ischemic injury through restoration of β2AR protective signaling (i.e., protein kinase B/endothelial nitric oxide synthase). Herein, we tested the potential therapeutic effects of adenoviral-mediated βARKct gene transfer in an experimental model of HI and its effects on βAR signaling and on endothelial cell (EC) function in vitro. Accordingly, in this study, we surgically induced HI in rats by femoral artery resection (FAR). Fifteen days of ischemia resulted in significant βAR down-regulation that was paralleled by an approximately 2-fold increase in GRK2 levels in the ischemic muscle. Importantly, in vivo gene transfer of the βARKct in the hindlimb of rats at the time of FAR resulted in a marked improvement of hindlimb perfusion, with increased capillary and βAR density in the ischemic muscle, compared with control groups. The effect of βARKct expression was also assessed in vitro in cultured ECs. Interestingly, ECs expressing the βARKct fenoterol, a β2AR-agonist, induced enhanced β2AR proangiogenic signaling and increased EC function. Our results suggest that βARKct gene therapy and subsequent GRK2 inhibition promotes angiogenesis in a model of HI by preventing ischemia-induced β2AR down-regulation.
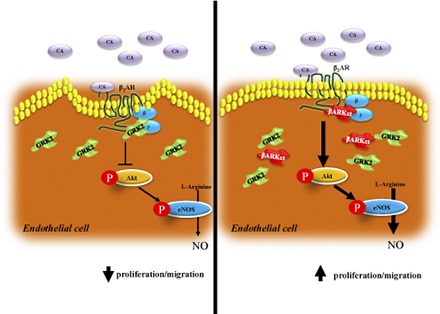
Introduction
Peripheral arterial occlusive disease and critical limb ischemia, its most advanced form, represent a major clinical problem that affects 10%–15% of the aged adult population (Norgren et al., 2007). Despite advances in endovascular revascularization and drug therapies, its prognosis remains poor, with about 40% of patients requiring limb amputation. Therefore, new therapeutic options are urgently needed. In this regard, therapeutic angiogenesis has emerged as a promising investigational strategy for the treatment of patients with limb ischemia, and gene therapy has been established as a potential method to manipulate levels/activity of key molecules to induce revascularization in patients with ischemic cardiovascular diseases (Lederman et al., 2002; Pugh and Ratcliffe, 2003; Rajagopalan et al., 2003; Kusumanto et al., 2006; Nikol et al., 2008; Belch et al., 2011).
Angiogenesis is a biologic process that generates new blood vessels from existing vasculature (Carmeliet 2005). In physiologic conditions, this process occurs during embryonic development, pregnancy, and through the ovarian cycle, but angiogenesis is also reactivated in a variety of pathologic conditions, including ischemia, inflammation, wound healing, tumor growth, and diabetic retinopathy (Melillo et al., 1997; Rafii and Lyden, 2003). Importantly, in all these processes, angiogenesis is primarily regulated via endothelial cell (EC) proliferation and migration (D’Amore and Thompson, 1987). At the molecular level, it is known that the β2-adrenergic receptor (β2AR), the most abundant β adrenergic receptor (βAR) isoform in ECs, is involved in the control of these functions (Howell et al., 1988). However, ischemia can result in increased sympathetic catecholamine levels that can cause β2AR signaling dysfunction in ECs, resulting in an inadequate angiogenic response and loss of tissue integrity and/or function (Iaccarino et al., 2005; Rengo et al., 2012).
Mechanistically, G protein-coupled receptor kinases phosphorylate and desensitize activated βARs, thus preventing deleterious receptor overstimulation, but chronically this process continues through receptor internalization and degradation (Rengo et al., 2011). G protein-coupled receptor kinase 2 (GRK2) is the isoform that appears to be the most important for regulated βARs in muscle (Rengo et al., 2011). The β adrenergic receptor kinase C-terminal peptide (βARKct), a peptide derived from the carboxyl terminal portion of GRK2 that blocks Gβγ recruitment of this kinase to the activated membrane-embedded receptor, can inhibit βAR desensitization and improve signaling down-regulation/desensitization (Rengo et al., 2009b; Cannavo et al., 2013a; Salazar et al., 2013). Of note, different reports have proposed βARKct as a new therapeutic molecule for various model of cardiovascular disease, mainly through the potentiation of β2AR signaling (Cannavo et al., 2013a; Salazar et al., 2013; Khan et al., 2014). Accordingly, because the effects of βARKct and GRK2 manipulation on EC function and angiogenesis are not well understood, we posited that this peptide could have the potential to improve postischemic revascularization through restoration of β2AR signaling/function both in vivo and in vitro.
Materials and Methods
Rat Hindlimb Ischemia Model and In Vivo Gene Therapy.
Hindlimb ischemia was induced in adult Sprague-Dawley male rats (300 g) by excision of the right common femoral artery, as previously reported elsewhere (Leosco et al., 2007). Briefly, rats were anesthetized with isoflurane (2%, v/v). A surgical incision was made in the skin overlying the middle portion of the right hindlimb. After ligation of the proximal end of the femoral artery, the distal portion of the saphenous artery was legated, and the artery and all side branches were dissected free and excised. Then the skin was closed with a 2.0 silk suture. Sham-operated animals underwent the same procedure without ligation and excision of the right common femoral artery.
In a group of rats receiving in vivo gene therapy, a silastic catheter was placed into the femoral artery distal to the resection through which a solution containing 1012 total viral particles of an adenovirus (Ad) encoding for βARKct or green fluorescent protein (GFP), or vehicle (saline), was infused into the hindlimb and allowed to remain there for 30 minutes while the saphenous vein was temporarily occluded. Afterward, the virus was removed through the catheter, the common femoral artery was removed, and the wound was closed in layers. All animal care and experimental protocols were approved by the ethics committee for the use of animals in research of our institution.
β-Adrenergic Receptor Radioligand Binding.
Plasma membrane fractions from excised skeletal muscles (gastrocnemius) were prepared and used for βAR radioligand binding studies using the nonselective βAR antagonist ligand (125I)-cyanopindolol (125I-CYP), as described previously elsewhere (Rengo et al., 2010). The concentration of 125I-CYP used in each series of reactions (assay) was 68.9 pmol/ml CYP and total activity was 5 µCi (125I-CYP specific activity: 2.2 Ci/µmol).
Cell Culture.
Bovine aortic endothelial cells (BAECs) were purchased from Lonza (Basel, Switzerland) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 200 mg/ml l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in 95% air and 5% CO2. For all experiments, BAECs were used at passage 14 or less.
Primary ECs were isolated from wild-type (WT) and global endothelial nitric oxide synthase (eNOS) knockout (KO) mice. Thoracic aorta was dissected, placed rapidly in ice-cold phosphate-buffered saline (PBS), and gently flushed using a 1-ml syringe fitted with a 23-G needle to remove blood clots. After removing the fibroadipose tissue and small lateral blood vessels, we cut the aorta into 1-mm rings. The aortic rings were further washed using sterile ice-cold PBS and then transferred in six-well plates coated with Matrigel (Corning Life Sciences, Tewksbury, MA). The aortic rings were then covered with few drops of Matrigel and endothelial cell growth medium (DMEM with 25 mM HEPES,10% FBS, 90 μg/ml heparin sulfate, 90 μg/ml endothelial cell growth factor, 10,000 U/ml penicillin, 10 mg/ml streptomycin). The EC sprouts were observed after 2 days. On day 4, the aortic rings were carefully removed, and the ECs were allowed to proliferate in Matrigel until reaching confluence.
To collect the ECs, the Matrigel was digested using Dispase, then the cells were cultured to 80% confluence and sorted for CD-31 using CD-31 Endothelial Cell Dynabeads (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. The CD31+ cells were then plated and used for experimental procedures.
BAECs and primary ECs were stimulated with fenoterol (1 µM; Sigma-Aldrich, St. Louis, MO) for 5, 15, and 30 minutes and for 6 hours for the immunoblot assay. We used 6 and 24 hours of fenoterol stimulation to assess the EC function. Before fenoterol stimulation, some cells were pretreated for 30 minutes with the highly selective β2AR-antagonist ICI 118551 [(2R,3S)-1-[(7-methyl-2,3-dihydro-1H-inden-4-yl)oxy]-3-(propan-2-ylamino)butan-2-ol] (IC50 value: 1.2 nM (Ki); 10 µM), as previously reported elsewhere (Cannavo et al., 2013c).
Adenoviral Constructs.
For in vivo and in vitro procedures we used recombinant Ad vectors encoding for the bovine WT GRK2 gene (Ad-GRK2) or one encoding for the C-terminal region containing the last 194 amino acids of GRK2, which makes up βARKct (Ad-βARKct). An Ad encoding for GFP (Ad-GFP) was used as the control (Lymperopoulos et al., 2008).
In Vitro Adenoviral Infection.
Infections were accomplished on ECs at 50%–60% confluence. Cells were infected with Ad expressing GRK2, βARKct, or GFP as the control at a multiplicity of infection of 100 per cell for 24 hours at 37°C, as previously described elsewhere (Lymperopoulos et al., 2008; Homan et al., 2014). The cells were then incubated in fresh medium for an additional 24 hours before experimentation.
Immunoblot.
Cells and skeletal muscle samples were lysed in a radioimmunoprecipitation assay buffer with protease and phosphatase inhibitors cocktail (Basel, Switzerland) as previously described elsewhere (Cannavo et al., 2013b). Protein concentrations in all lysates were measured using a dye-binding protein assay kit (Bio-Rad Laboratories, Hercules, CA) and a spectrophotometer reader (Bio-Rad Laboratories) at a wavelength of 750 nm. Protein levels of GRK2 (1:1000; Santa Cruz Biotechnology, Dallas, TX), phospho-protein kinase B (pAkt, 1:1000; Sigma-Aldrich), tAkt (1:1000; Santa Cruz Biotechnology), eNOS (1:1000; Cell Signaling Technology, Beverly, MA), p-eNOS (1:1000; Cell Signaling Technology), β2AR (1:1000; Santa Cruz Biotechnology), p-β2AR (1:1000; EMD Millipore Corporation, Billerica, MA) and GAPDH (1:1000; EMD Millipore Corporation) were assessed. Secondary antibodies were purchased from Amersham Life Sciences (Buckinghamshire, United Kingdom). Bands were visualized by enhanced chemiluminescence (EMD Millipore) according to the manufacturer’s instructions, and were quantified using densitometry (Chemidoc; Bio-Rad Laboratories). Each experiment and densitometric analysis was separately repeated 3 times.
Immunohistochemistry.
Immunohistochemistry was performed as previously described elsewhere (Cannavo et al., 2013b). Briefly, deparaffinized slides were treated with the following polyclonal antibodies: GRK2 (1:200; Santa Cruz Biotechnology), GFP (1:200; Santa Cruz Biotechnology). The visualization was performed using an ABC kit (Thermo Scientific, Rockford, IL) and diaminobenzidine (Pierce Biotechnology, Rockford, IL) chromogen.
Confocal Microscopy.
Confocal microscopy studies were performed as previously described elsewhere (Cannavo et al., 2013c). After fixation with 3% paraformaldehyde, cells were incubated with an anti-β2AR antibody (1:200 anti-rabbit IgG; Santa Cruz Biotechnology) in 1% bovine serum albumin. Next, the cells were incubated with the secondary rabbit polyclonal antibody (1:200, Texas Red conjugated; Sigma-Aldrich). Visualization with confocal laser scanning microscopy was performed at 568 nm (Cy3) with a Zeiss 510 confocal laser scanning microscope (Carl Zeiss Light Microscopy, Göttingen, Germany). The fluorescent data sets were analyzed by LSM 510 software (Carl Zeiss).
EC Function In Vitro Assays.
For EC proliferation and migration assays, the stimulation was performed in the presence of DMEM supplemented with 2% FBS. Cell migration was assessed by a wound-healing scratch assay performed in 12-well tissue culture plates. Twenty-four hours after Ad infection, scratches were made using 100-µl pipette tips, and the wells were washed twice with PBS. After 6 hours of fenoterol (1 µM) stimulation, the cells were fixed in 3.7% paraformaldehyde and stained with 0.1% crystal violet staining solution as previously described elsewhere (Liu et al., 2013). Photographs were taken on Nikon TE2000-U inverted microscope connected to a Nikon camera (Nikon, Tokyo, Japan). Quantification of cell migration was done by measuring the distance between 10 random points within the wound edge. The gap distance of the wound was measured using ImageJ software (http://imagej.nih.gov/ij/), and the data were normalized to the average of the wound of control cells fixed at the time of scratches.
Proliferation was assessed by quantitative measurement of DNA synthesis using a 5-bromo-2ʹ-deoxyuridine (BrdU) enzyme-linked immunosorbent assay kit (Roche), according to manufacturer instructions. Briefly, after 24 hours from Ad infection, BrdU was added to the medium at a final concentration of 10 μM, and the cells were incubated for 6 or 24 hours in presence or absence of fenoterol (1 µM). The BrdU incorporation was then assayed using a colorimetric detection system. The number of proliferating cells was represented by the level of BrdU incorporation, which directly correlates to the color intensity and the absorbance values.
Nitric Oxide Measurement.
BAECs or murine aortic endothelial cells infected with Ad-βARKct and Ad-GFP, were stimulated or not with fenoterol (1 µM) for 6 hours. After stimulation, the concentrations of nitric oxide (NO) in the culture medium were measured using the NO colorimetric assay kit (BioVision, Mountain View, CA) according to the manufacturer’s protocol. Briefly, the nitrate was converted to nitrite using nitrate reductase, then the nitrite was converted to a deep purple azo compound using Griess reagent. The amount of the azochromophore was detected by the colorimetric determination at 540 nm using a microplate reader.
Blood Flow Determination.
Blood flow in the posterior tibial artery of ischemic and nonischemic hindlimb was evaluated by ultrasound Doppler using a VisualSonics Vevo 770 imaging system (Fujifilm VisualSonics, Amsterdam, the Netherlands) with a 708 MHz scan head in isofluorane-anesthetized rats (2% v/v) immediately before and at 12 hours and 3, 7, 10, and 15 days after surgery, as previously described elsewhere (Ciccarelli et al., 2011). Data are expressed as ischemic to nonischemic ratio. Fifteen days after surgery, blood flow was also measured by dyed beads assay, as previously described elsewhere (Ciccarelli et al., 2011).
Histology.
Tibialis anterior muscle specimens were fixed in 4% paraformaldehyde and embedded in paraffin. After deparaffinization and rehydration, 5-μm-thick sections were prepared and mounted on glass slides. The capillary density measurement was performed as previously described elsewhere (Ciccarelli et al., 2011; Rengo et al., 2013).
Statistical Analysis.
Data are summarized as mean ± S.E.M. Comparisons were made with the use of t tests or analysis of variance, as appropriate. A Bonferroni correction was applied to the probability values whenever multiple comparisons arose. P < 0.05 was considered significant.
Results
βARKct Gene-Therapy Improves Angiogenesis and Restores βAR Density in the Ischemic Hindlimb.
After hindlimb ischemia (HI), increased levels of catecholamines are responsible for βAR-signaling dysfunction, leading to reduced angiogenesis in the ischemic tissue (Iaccarino et al., 2005; Sorriento et al., 2012). Importantly, a similar mechanism has been observed also in the heart, where βARKct gene-therapy has been proposed as a potential strategy to prevent βAR dysregulation and to ameliorate cardiac response to ischemia (White et al., 2000). However, the effects of βARKct gene-therapy in a model of HI have not been specifically investigated. Thus, we surgically induced HI in adult male Sprague-Dawley rats by resection of the right common femoral artery (FAR). A group of sham-operated rats served as control. At 15 days after surgery, we measured the blood flow with a dyed beads perfusion assay, which showed a significant reduction in hindlimb perfusion in the ischemic muscle of the group undergoing FAR compared with sham-operated rats, as expected (Fig. 1A).
Fig. 1.

βAR down-regulation is associated with increased GRK2 protein levels in the ischemic muscle. (A) Bar graph showing blood perfusion in the ischemic hindlimb in rats at 15 days after femoral artery resection (HI; n = 6) and in sham-operated rats (sham; n = 6) as assessed by dyed beads dilution method. Data are expressed as percentage of ischemic to nonischemic hindlimb. (B) Bar graphs showing total βAR density on skeletal muscle plasma membrane preparations from rats 15 days after femoral artery resection (HI; n = 6) and sham-operated rats (n = 6). (C) Representative immunoblots (upper panel) and densitometric analysis (lower panel) of multiple (n = 3, including two samples per group each) independent experiments showing GRK2/GAPDH protein levels in skeletal muscle lysates from the ischemic hindlimb of rats undergoing femoral artery resection (HI, n = 6) and from sham-operated rats (n = 6). GAPDH was used as a loading control. Data are expressed as mean ± S.E.M. *P < 0.05 versus sham.
In line with previous reports (Iaccarino et al., 2005), 2 weeks after surgically induced ischemia, we observed a significant reduction in βAR density within the ischemic skeletal muscle compared with the skeletal muscle of sham-operated rats (Fig. 1B). The reduction in βAR plasma membrane density was paralleled by a robust up-regulation of GRK2 protein levels in the ischemic skeletal muscle compared with the limbs of sham-operated rats (Fig. 1C).
Next, to test the effects of βARKct expression, a group of rats was injected into the femoral artery with an Ad encoding for βARKct (Ad-βARKct) or Ad-GFP as control at the time of FAR. A separate group of animals was treated with a saline injection to provide an additional control to assess any potential effect of Ad infection or GFP expression (Fig. 2A). At the end of the study period, transgene expression was successfully detected by immunohistochemistry and resulted in predominant perivascular localization (Fig. 2B). However, consistent with previous reports (Iaccarino et al., 2005), transgene expression was not limited to the endothelium but was also observed in the skeletal muscle. Moreover, we assessed βARKct expression in the ischemic skeletal muscle of rats at the end of the study period by immunoblot analysis (Fig. 2C). As expected, βARKct was clearly detectable in the muscle of rats treated with Ad-βARKct (Fig. 2D).
Fig. 2.

In vivo gene delivery study design. (A) Overall study design. Rats underwent femoral resection and were randomized to receive intravascular injection of Ad-βARKct or Ad-GFP or saline (vehicle). Hindlimb perfusion was evaluated by Doppler ultrasound before, immediately after, and 3, 7, 10, and 15 days after surgery. (B) Representative immunohistochemistry images showing βARKct/endogenous GRK2 expression in skeletal muscle of rats treated with Ad-βARKct or saline. βARKct was detected using an anti-GRK2 antibody. (Scale bar: 100 µm, C–D.) Representative immunoblot (C) and densitometric analysis (D) of multiple independent experiments (n = 4) to evaluate GRK2 and βARKct expression in the skeletal muscle lysates of rats treated with Ad-βARKct or saline. GRK2 expression was normalized with GAPDH, and βARKct expression is expressed as a fold of the endogenous GRK2 level.
Next, to evaluate the effects of βARKct gene-therapy on postischemic angiogenesis, we measured the blood flow in all groups by Doppler ultrasound over the course of 15-day study period. As shown in Fig. 3A, immediately after FAR, blood flow was not detectable in the ischemic tibial posterior artery of all study groups. As expected, blood flow was partially and progressively restored in rats treated with saline or GFP over the 15-day study period (Fig. 3A). However, βARKct gene therapy resulted in a significant increase in blood flow compared with the control groups (Fig. 3A). Data from the dyed beads perfusion assay confirmed these results (Fig. 3B). In line with blood flow and perfusion data, histologic analysis of the tibial anterior muscle revealed that 15 days of HI induced a robust capillary rarefaction in saline- and Ad-GFP-treated rats (Fig. 3C). Of note, we found a complete restoration of the capillary density in βARKct-treated rats that was not statistically different from that measured in sham-operated animals (Fig. 3C).
Fig. 3.

βARKct gene delivery in skeletal muscle improves postischemic angiogenesis and restores β2AR density. (A) Blood flow measured by Doppler ultrasound in the tibial posterior artery of rats (n = 6 to 8 rats for each group) over the course of 15 days after surgery. Data are expressed as a percentage of the ischemic to nonischemic limb. (B) Blood perfusion (n = 6 to 8 rats for each group) in the ischemic hindlimb of all four study groups as evaluated by dyed beads dilution assay performed at the end of the study period (15 days after gene transfer). Perfusion data are expressed as the ischemic-to-nonischemic ratio percentage of dyed beads content per milligram of hindlimb muscle tissue. (C) Representative images of Lectin Bandeiraea simplicifolia I staining of capillaries in the ischemic hindlimb (scale bar: 100 µm, left panels); and bar graph (right panel) showing capillary-to-myocyte ratio in ischemic muscles (n = 8 each group) of all four groups and in sham-operated rats as control. Arrows indicate capillaries. (D) βAR Plasma membrane density in skeletal muscle homogenates, purified from the hindlimb (n = 6 rats for each group) from the sham and ischemic groups (saline, GFP, and βARKct) at 15 days after surgery and gene delivery. *P < 0.05 versus sham; #P < 0.05 versus saline and GFP.
Finally, we assessed βAR plasma membrane density in the ischemic gastrocnemius from all study groups (Fig. 3D). In line with previous reports (Iaccarino et al., 2005), a significant down-regulation of total βAR density was observed in saline-treated and GFP-treated groups compared with the sham-operated animals. More importantly, βARKct gene therapy resulted in the restoration of βAR density in the ischemic muscles, at levels that were almost similar to the sham groups.
βARKct Improves In Vitro β2AR-Signaling in Endothelial Cells.
Our in vivo data showing that βARKct-dependent revascularization is associated with a restoration of βAR density in the ischemic skeletal muscle prompted us to investigate the effects of this peptide on EC function. BAECs were stimulated in vitro with the selective β2AR agonist fenoterol (1 μM) (January et al., 1997) to test the activation of Akt and eNOS because these proteins are known to be nodal regulators of EC function (Howell et al., 1988). Indeed, we found that fenoterol was able to increase both Akt and eNOS activation after 15 minutes of stimulation (Fig. 4, A and B). Notably, the pretreatment of cells for 30 minutes with the selective β2AR antagonist ICI-118,551 (10 μM) completely prevented the ability of fenoterol to increase in Akt and eNOS phosphorylation (Fig. 4, A and B), indicating that these effects are completely dependent on β2AR activation.
Fig. 4.

Fenoterol selectively stimulates endothelial β2AR proangiogenic signaling in BAECs. (A, B) Representative immunoblots (upper panels) and densitometric quantitative analysis (lower panel) of multiple (n = 3) independent experiments to evaluate in BAECs unstimulated (Ns) or stimulated with fenoterol (Fen, 1 µM) for 15 minutes. (A) Akt phosphorylation levels on serine 473 (ser473p-Akt) and (B) eNOS phosphorylation levels on serine 1177 (ser1177p-eNOS) and total protein levels. Total Akt and total e-NOS served as loading controls, respectively. Before Fen stimulation, a group of cells was pretreated with selective β2AR antagonist ICI-118,551 (ICI, 10 µM). *P < 0.05 versus Ns.
Next, ECs were infected in vitro with Ad-GRK2 or Ad-βARKct or Ad-GFP (as control), and we assessed the effects of fenoterol on β2AR phosphorylation/desensitization (p-β2AR) by measuring the phospho-threonine (pThr) 384 levels of the bovine β2AR (i.e., the homologous residue to the threonine targeted by GRK2 in the human β2AR, triggering rapid receptor desensitization) (Fredericks et al., 1996). In absence of agonist stimulation, there were no differences in terms of β2AR phosphorylation between GFP-infected, GRK2-infected, and βARKct-infected cells (Fig. 5A). Five minutes of fenoterol stimulation resulted in a significant increase in p-β2AR levels in all EC groups compared with unstimulated ECs. However, GRK2 overexpression induced a further significant increase in p-β2AR levels compared with both GFP and βARKct cells, suggesting that increased GRK2 activity is responsible for augmented β2AR phosphorylation and consequent desensitization also in ECs (Fig. 5A).
Fig. 5.

Effects of GRK2 levels on β2AR phosphorylation and Akt and eNOS activation after fenoterol (Fen) stimulation in BAECs. (A–D) Representative immunoblots (upper) and densitometric analysis (bottom) of multiple independent experiments (n = 3) to evaluate: (A) β2AR phosphorylation levels on threonine 384 (thr384p-β2AR), (B) ser473p-Akt, and (C) ser1177p-eNOS, and (D) total protein levels of GRK2 in total lysates of BAECs infected with adenoviruses (Ad) encoding for GFP, GRK2, or βARKct. Cells were not stimulated Ns or stimulated with Fen (1 μM), respectively, for (A) 5 minutes or (B–D) 15 minutes. Total β2AR, total Akt, total eNOS and GAPDH served as loading controls. Data are expressed as mean ± S.E.M. *P < 0.05 versus GFP Ns; #P < 0.05 versus GFP Fen.
Next, we tested the direct effects of GRK2 and βARKct on fenoterol-dependent Akt and eNOS activation (Queen et al., 2006; Figueroa et al., 2009). Indeed, in GFP-treated and βARKct-treated cells, 15 minutes of fenoterol administration induced a significant enhancement in both Akt and eNOS phosphorylation (Fig. 5, B and C), while GRK2 overexpression resulted in a blunted activation of these factors. Notably, after 15 minutes of fenoterol stimulation no significant changes in GRK2 protein levels were observed between GFP-infected and βARKct-infected cells (Fig. 5D).
Next, we tested the impact of longer fenoterol stimulation on β2AR signaling in ECs. Interestingly, after 6 hours of fenoterol administration, GRK2 expression was increased in both GFP and βARKct cells at levels almost comparable but statistically different from those observed in Ad-GRK2 treated cells (Fig. 6A). However, a robust β2AR phosphorylation/desensitization was evident only in GFP and GRK2 cells. Further, p-β2AR levels were significantly reduced in presence of βARKct (Fig. 6B). Consistent with increased β2AR desensitization, long-term fenoterol stimulation resulted in decreased Akt (Fig. 6C) and eNOS (Fig. 6D) activation in both GFP and GRK2 overexpressing cells. In contrast, the presence of βARKct, which prevented β2AR phosphorylation and subsequent desensitization (Fig. 6B), resulted in an increased activation of Akt and eNOS, at levels comparable to basal unstimulated conditions (Fig. 6, C and D).
Fig. 6.

Long-term effects of fenoterol (Fen) stimulation on β2AR function in BAECs. (A–D) Densitometric analysis of multiple independent experiments to evaluate (A) GRK2 protein levels and (B) thr384p-β2AR, (C) ser473p-Akt, or (D) ser1177p-eNOS in total lysates of BAECs infected with adenoviruses (Ad) encoding for GFP, GRK2, or βARKct. Cells were not stimulated Ns or stimulated with Fen (1 μM), respectively, for 6 hours (A–D). GAPDH, total β2AR, total Akt, and total eNOS served as loading controls, respectively. *P < 0.05 versus GFP Ns. #P < 0.05 versus GRK2.
These results strongly suggest a potential role of βARKct in preventing GRK2-dependent β2AR phosphorylation and consequent receptor down-regulation. To confirm this hypothesis, we performed plasma membrane purifications in extracts obtained from BAECs infected with GFP and βARKct, and either left unstimulated or stimulated with both with fenoterol for 6 hours. We found a significant reduction in β2AR plasma membranes levels in GFP cells (Fig. 7, A and B). Further, internalized β2AR levels were increased compared with unstimulated cells. In contrast, βARKct expression prevented the effects of fenoterol on β2AR-plasma membrane down-regulation (Fig. 7, A and B).
Fig. 7.

βARKct prevents fenoterol-induced β2AR-internalization in BAECs. (A, B) Representative immunoblots (A) and densitometric quantitative analysis (B) of multiple (n = 3) independent experiments to evaluate β2AR levels in crude plasma membrane preparations and in cytosolic fraction obtained from BAECs infected with Ad-GFP and Ad-βARKct. Cells were not stimulated (Ns) or were stimulated with fenoterol (Fen, 1 μM) for 6 hours. *P < 0.05 versus GFP Ns. #P < 0.05 versus GFP Fen. (C) Representative immunofluorescence images (scale bar: 10 µm) of β2AR in BAECs infected with Ad-GFP and Ad-βARKct. Cells were not stimulated (Ns) or were treated for 6 hours with Fen (1 μM). Arrows indicate receptors that are internalized.
In line with these results, confocal microscopy experiments confirmed that β2AR was mainly localized at cytosolic level after fenoterol stimulation in GFP ECs (Fig. 7C). In contrast, βARKct expression resulted in predominant β2AR localization at the plasma membrane similar to that observed in unstimulated cells (Fig. 7C).
βARKct Enhances In Vitro EC Function in Response to Selective β2AR Stimulation.
Because our data above showed that βARKct prevented the fenoterol-induced β2AR desensitization/down-regulation and rescued associated proangiogenic signaling, we evaluated the effects of β2AR-agonism on EC function. We first assessed NO release because it has been demonstrated to be a potential modulator of EC function. As showed in Fig. 8A, after 6 hours of fenoterol stimulation, we observed a significant increase in NO release in GFP cells compared with unstimulated ones. Of note, after fenoterol stimulation in the presence of βARKct, we observed a more pronounced NO release compared with all cell groups.
Fig. 8.

The presence of βARKct enhances BAEC function. (A) Bar graph showing NO release in the media obtained from BAECs infected with Ad-GFP or Ad-βARKct, not stimulated (Ns) or stimulated with fenoterol (Fen, 1 μM) for 6 hours. *P < 0.05 versus GFP Ns. #P < 0.05 versus GFP Fen. (B) Representative images (upper; scale bar: 25 µm) and bar graphs (lower) showing percentage of cell migration in response to 6 hours of Fen (1 µM) stimulation, evaluated by wound healing scratch assay. Confluent monolayers of BAECs infected with Ad-GFP and Ad-βARKct were wounded at time 0 (T = 0). The average rate of wound closure during the first 6 hours of wound healing was calculated from three independent experiments. *P < 0.05 versus GFP Ns. #P < 0.05 versus GFP Fen. (C) BrdU proliferation assay in BAECs infected with Ad-GFP and Ad-βARKct at different time points (0, 6, and 24 hours) of stimulation with Fen (1 µM). Results are expressed as the percentage of proliferation over nonstimulated (Ns) cells. *P < 0.05 versus GFP Ns. #P < 0.05 versus GFP Fen.
Next, we assessed the effects of β2AR-agonism on EC function. We evaluated EC migration by performing a wound healing scratch assay under basal conditions and after challenge with β2AR agonist. As shown in Fig. 8B, under basal conditions, the expression of βARKct did not induce any significant effect on cell migration compared with GFP cells. Importantly, fenoterol stimulation resulted in a ≈2-fold increase in migration in GFP cells, and the presence of βARKct induced significantly enhanced EC migration in response to β2AR stimulation that was almost double to that observed in control GFP cells. Similar results were obtained in an EC proliferation assay by 5-bromo-2ʹ-deoxyuridine (BrdU) incorporation (Fig. 8C). Fenoterol induced a significant increase in EC proliferation in GFP cells compared with nonstimulated cells at both 6 and 24 hours, and βARKct expression induced a robust proliferative response to β2AR-agonism that was significantly higher than that observed in GFP-treated cells at both time points (Fig. 8C).
Finally, to study the role of βARKct on β2AR-dependent eNOS activation, we isolated primary ECs from the aorta of WT and eNOS KO mice. The ECs were then infected with Ad-GFP and Ad-βARKct. Importantly, the lack of eNOS almost completely prevented the effects of βARKct on fenoterol-dependent increase in EC migration (Fig. 9A) and proliferation (Fig. 9B) compared with WT cells. In line with these results, at the molecular level we observed that the lack of eNOS abolished the ability of βARKct to activate/rescue β2AR-dependent Akt activation (Supplemental Fig. 1, A and B) and to increase NO release after fenoterol stimulation (Fig. 9C).
Fig. 9.

The lack of eNOS prevents the effects of βARKct in ECs. Murine aortic endothelial cells (ECs), isolated from WT and eNOS KO mice, infected with Ad-GFP or Ad-βARKct, not stimulated (Ns) or stimulated with fenoterol (Fen, 1 μM). (A) Bar graphs (lower) showing the percentage of cell migration, evaluated by wound healing scratch assay. Confluent monolayers of ECs were wounded at time 0 (T = 0). The average rate of wound closure during the first 6 hours of wound healing was calculated from three independent experiments. (B) BrdU proliferation assay in ECs at different time points (0, 6, and 24 hours). Results are expressed as the percentage of proliferation over nonstimulated cells. *P < 0.05 versus GFP Ns. # P < 0.05 versus GFP Fen. ^P < 0.05 versus WT GFP. (C) Bar graph showing NO release in the medium from ECs stimulated with Fen (1 μM) for 6 hours. *P < 0.05 versus WT GFP. #P < 0.05 versus WT βARKct.
Discussion
There is extensive literature supporting the therapeutic value of βARKct gene therapy to ameliorate cardiac function and remodeling after myocardial infarction (Cannavo et al., 2013a), especially for its ability to block GRK2-dependent β2AR dysfunctional signaling (Cannavo et al., 2013a; Salazar et al., 2013; Khan et al., 2014). Herein, we report for the first time the effects of βARKct gene therapy in a rat model of HI. In particular, we show that βARKct is a new potential therapeutic tool to improve postischemic angiogenesis through the preservation of β2AR signaling in the endothelium. We observed that ischemia induces the up-regulation of GRK2 protein levels in skeletal muscle, and this event appears to be critical in the processes of revascularization of the ischemic hindlimb as it is associated with βAR-desensitization/down-regulation.
Our present study confirms the relevant role of endothelial β2AR in the control of revascularization in vivo and EC function in vitro. In this context, previous studies have reported a crucial role of β2AR at promoting in vivo postischemic revascularization; in particular, its functional relevance in the regulation of EC function in vitro has emerged as a key mechanism (Iaccarino et al., 2005). β2AR KO mice exhibited impaired revascularization after ischemia with a high rate of tissue necrosis and subsequent autoamputation of ischemic limbs (Iaccarino et al., 2005). Moreover, in a rat model of HI, when β2AR was down-regulated the gene therapy using the receptor resulted in improved revascularization of the ischemic limb (Iaccarino et al., 2005).
Importantly, no mechanism has been proposed for triggering ischemic-mediated β2AR dysfunction. Accordingly, the ability of GRK2 to regulate βAR signaling and function in the heart has been well established (Cannavo et al., 2013a; Huang et al., 2011) and therapies that inhibit the activity of this kinase on receptor phosphorylation/down-regulation at the plasma membrane have been shown to be strongly protective toward cardiac injury and stress, in part by restoring myocardial βAR signaling abnormalities (Rengo et al., 2009a; Rengo et al., 2011; Cannavo et al., 2013a).
Herein, we show that the resection of the common femoral artery in rats, a well-recognized model of peripheral artery disease (Iaccarino et al., 2005; Leosco et al., 2007), results in ≈2-fold increase in GRK2 levels in the ischemic gastrocnemius compared with sham-operated rats. Moreover, we confirmed that GRK2 up-regulation is paralleled by significant βAR down-regulation in the ischemic skeletal muscle. Thus, we tested the in vivo properties of Ad-mediated intra-arterial gene transfer of the GRK2 inhibitor βARKct on HI.
Our data show that βARKct expression in the ischemic tissue induces a significant increase in blood flow recovery and perfusion of the ischemic hindlimb compared with control groups (saline and GFP treated) at 15 days after FAR. Of note, this result is associated with a significant increase in capillary density, suggesting improved postischemic angiogenesis. As reported by others (Iaccarino et al., 2005), ischemia is associated with significant βAR down-regulation and dysfunction in the affected muscle. Consistently, in our control groups undergoing HI, βAR density is reduced in the ischemic muscle. However, 15 days of βARKct expression significantly improved βAR density in the ischemic hindlimb, almost to levels observed in sham-operated rats, thus suggesting a possible explanation for the beneficial effects of this therapeutic strategy on in vivo revascularization.
Because a pivotal role of β2ARs exists for EC function (Howell et al., 1988; Iaccarino et al., 2005; Ciccarelli et al., 2011), particularly in respect to the regulation of postnatal ischemic angiogenesis, our mechanistic focus targeting GRK2 activity was to investigate the impact of βARKct on EC function in vitro. In BAECs, we have observed that GRK2 overexpression, obtained via Ad-mediated gene transfer, significantly impairs both cell migration and proliferation in response to selective β2AR stimulation. This shows that indeed GRK2 has influence at the level of the β2AR and can be a mediator of dysregulation of the system. At the molecular level, these deleterious effects exerted by GRK2 on EC function are paralleled by β2AR dysfunctional signaling, as suggested by levels of receptor phosphorylation and reduced Akt and eNOS activation after acute fenoterol stimulation (5 and 15 minutes, respectively). Interestingly, after 6 hours of fenoterol stimulation, these negative effects are also observed in GFP-infected cells, but it is important to underline that at this time point fenoterol induces a strong up-regulation of endogenous GRK2 levels (Fig. 6). However, after 15 minutes of fenoterol stimulation, a time point where GRK2 is not up-regulated, Ad-GFP treated ECs show reduced levels of p-β2AR and an increased level of Akt and eNOS activation when compared with Ad-GRK2 treated cells, thus indicating higher responsiveness to β2AR activation (Fig. 5).
Of further importance, βARKct expression improves EC migration and proliferation and preserves β2AR signaling and function after fenoterol stimulation (Fig. 7). These data suggest that βARKct is able to enhance β2AR signaling and to improve EC function after fenoterol administration.
Study Limitations.
GRK2 has been shown to directly interact with and inhibit both Akt and eNOS (Brinks at al., 2010; Huang at al., 2013), so it is quite likely that some of the observed effects of βARKct are due to direct inhibition of GRK2 acting on Akt/eNOS and independently of the β2AR. However, our results strongly suggest that acute β2AR stimulation by fenoterol can directly activate Akt and eNOS in ECs in vitro and that although βARKct expression does not affect the acute action of fenoterol it ameliorates later effects of fenoterol on β2AR phosphorylation, enhancing NO release in these cells. Moreover, our data obtained in eNOS KO ECs support the hypothesis that the preservation of β2AR/eNOS axis is relevant for the positive effects of βARKct in ECs.
It is important to underline that we cannot exclude any potential involvement of other cell types (i.e., skeletal muscle cells or smooth muscle cells) other than ECs and endothelial progenitor cells, for which a relevant role has been proposed in postischemic angiogenesis in vivo (Galasso et al., 2013). In this regard, it has been recently demonstrated that in C2C12 myoblasts GRK2 overexpression led to a significant impairment in cell differentiation, thus suggesting for this kinase a relevant role in skeletal muscle myogenesis (Garcia-Guerra et al., 2014). Thus, we cannot exclude additional effects of βARKct in skeletal muscle cells in our in vivo model.
As an addition limitation, as reported by others previously (Iaccarino et al., 2005) and as shown in our immunohistochemical analysis (Supplemental Fig. 1), the gene delivery technique we used results in a predominant localization of the transgenes to the existing vascular structures within the ischemic tissue.
Finally, in our in vivo study we measured total βAR density in the ischemic muscle of rats rather than β2AR density. However, the β2AR is known to be the main isoform expressed in EC and in the skeletal muscles (Osswald and Guimarães, 1983; Guimarães and Moura, 2001; Lynch and Ryall, 2008), so we can assume that part of the effects of HI and βARKct gene therapy on total βAR density might be ascribed to changes in β2AR density. Moreover, our in vitro data in ECs have specifically investigated the effects of βARKct on β2AR signaling and function, clearly demonstrating a protective effect of βARKct on β2AR down-regulation.
In conclusion, in the present study we provide novel evidence that βARKct expression and subsequent GRK2 inhibition positively regulate β2AR signaling and function in ECs, with relevant implications for postischemic angiogenesis. For this reason, we propose βARKct gene therapy as a new therapeutic approach to reestablish blood flow to the limbs affected by critical ischemia.
Abbreviations
- Ad
adenovirus
- Akt
protein kinase B
- βAR
β adrenergic receptor
- β2AR
β2-adrenergic receptor
- βARKct
β adrenergic receptor kinase C-terminal peptide
- BAEC
bovine aortic endothelial cell
- BrdU
bromodeoxyuridine (5-bromo-2ʹ-deoxyuridine)
- 125I-CYP
(125I)-cyanopindolol
- DMEM
Dulbecco’s modified Eagle’s medium
- EC
endothelial cell
- eNOS
endothelial nitric oxide synthase
- FAR
femoral artery resection
- FBS
fetal bovine serum
- GRK2
G protein-coupled receptor kinase 2
- GFP
green fluorescent protein
- HI
hindlimb ischemia
- ICI 118551
(2R,3S)-1-[(7-methyl-2,3-dihydro-1H-inden-4-yl)oxy]-3-(propan-2-ylamino)butan-2-ol
- KO
knockout
- NO
nitric oxide
- PBS
phosphate-buffered saline
- WT
wild type
Authorship Contributions
Participated in research design: Cannavo, Liccardo, Rengo, Koch, Leosco.
Conducted experiments: Cannavo, Liccardo, Lymperopoulos, Gambino, D’Amico, Rengo.
Contributed new reagents or analytic tools: Cannavo, Liccardo, Rengo, Koch, Leosco.
Performed data analysis: Liccardo, Cannavo, Rengo.
Wrote or contributed to the writing of the manuscript: Cannavo, Liccardo, Rengo, Rengo, Koch, Ferrara, Leosco.
Footnotes
This work was supported in part by the Italian Ministry of University and Scientific Research-Year 2009 [prot. 2009L4X28T].
 This article has supplemental material available at (jpet.aspetjournals.org).
This article has supplemental material available at (jpet.aspetjournals.org).
References
- Belch J, Hiatt WR, Baumgartner I, Driver IV, Nikol S, Norgren L, Van Belle E, TAMARIS Committees and Investigators (2011) Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet 377:1929–1937. [DOI] [PubMed] [Google Scholar]
- Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, Huang ZM, Wang X, Qiu G, Gumpert A, et al. (2010) Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ Res 107:1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo A, Liccardo D, Koch WJ. (2013a) Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front Physiol 4:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo A, Rengo G, Liccardo D, Pironti G, Scimia MC, Scudiero L, De Lucia C, Ferrone M, Leosco D, Zambrano N, et al. (2013b) Prothymosin alpha protects cardiomyocytes against ischemia-induced apoptosis via preservation of Akt activation. Apoptosis 18:1252–1261. [DOI] [PubMed] [Google Scholar]
- Cannavo A, Rengo G, Liccardo D, Pagano G, Zincarelli C, De Angelis MC, Puglia R, Di Pietro E, Rabinowitz JE, Barone MV, et al. (2013c) β1-adrenergic receptor and sphingosine-1-phosphate receptor 1 (S1PR1) reciprocal downregulation influences cardiac hypertrophic response and progression to heart failure: protective role of S1PR1 cardiac gene therapy. Circulation 128:1612–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936. [DOI] [PubMed] [Google Scholar]
- Ciccarelli M, Sorriento D, Cipolletta E, Santulli G, Fusco A, Zhou RH, Eckhart AD, Peppel K, Koch WJ, Trimarco B, et al. (2011) Impaired neoangiogenesis in β₂-adrenoceptor gene-deficient mice: restoration by intravascular human β₂-adrenoceptor gene transfer and role of NFκB and CREB transcription factors. Br J Pharmacol 162:712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amore PA, Thompson RW. (1987) Mechanisms of angiogenesis. Annu Rev Physiol 49:453–464. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Poblete I, Fernández R, Pedemonte C, Cortés V, Huidobro-Toro JP. (2009) NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol 297:H134–H143. [DOI] [PubMed] [Google Scholar]
- Fredericks ZL, Pitcher JA, Lefkowitz RJ. (1996) Identification of the G protein-coupled receptor kinase phosphorylation sites in the human β2-adrenergic receptor. J Biol Chem 271:13796–13803. [DOI] [PubMed] [Google Scholar]
- Galasso G, De Rosa R, Ciccarelli M, Sorriento D, Del Giudice C, Strisciuglio T, De Biase C, Luciano R, Piccolo R, Pierri A, et al. (2013) β2-Adrenergic receptor stimulation improves endothelial progenitor cell-mediated ischemic neoangiogenesis. Circ Res 112:1026–1034. [DOI] [PubMed] [Google Scholar]
- Garcia-Guerra L, Vila-Bedmar R, Carrasco-Rando M, Cruces-Sande M, Martín M, Ruiz-Gómez A, Ruiz-Gómez M, Lorenzo M, Fernández-Veledo S, Mayor F, Jr, et al. (2014) Skeletal muscle myogenesis is regulated by G protein-coupled receptor kinase 2. J Mol Cell Biol 6:299–311. [DOI] [PubMed] [Google Scholar]
- Guimarães S, Moura D. (2001) Vascular adrenoceptors: an update. Pharmacol Rev 53:319–356. [PubMed] [Google Scholar]
- Homan KT, Wu E, Cannavo A, Koch WJ, Tesmer JJ. (2014) Identification and characterization of amlexanox as a G protein-coupled receptor kinase 5 inhibitor. Molecules 19:16937–16949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell RE, Albelda SM, Daise ML, Levine EM. (1988) Characterization of beta-adrenergic receptors in cultured human and bovine endothelial cells. J Appl Physiol (1985) 65:1251–1257. [DOI] [PubMed] [Google Scholar]
- Huang ZM, Gao E, Fonseca FV, Hayashi H, Shang X, Hoffman NE, Chuprun JK, Tian X, Tilley DG, Madesh M, et al. (2013) Convergence of G protein-coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Sci Signal 6:ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZM, Gold JI, Koch WJ. (2011) G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci (Landmark Ed) 16:3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, et al. (2005) Ischemic neoangiogenesis enhanced by β2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res 97:1182–1189. [DOI] [PubMed] [Google Scholar]
- January B, Seibold A, Whaley B, Hipkin RW, Lin D, Schonbrunn A, Barber R, Clark RB. (1997) β2-adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J Biol Chem 272:23871–23879. [DOI] [PubMed] [Google Scholar]
- Khan M, Mohsin S, Toko H, Alkatib M, Nguyen J, Truffa S, Gude N, Chuprun K, Tilley DG, Koch WJ, et al. (2014) Cardiac progenitor cells engineered with βARKct have enhanced β-adrenergic tolerance. Mol Ther 22:178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumanto YH, van Weel V, Mulder NH, Smit AJ, van den Dungen JJ, Hooymans JM, Sluiter WJ, Tio RA, Quax PH, Gans RO, et al. (2006) Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: a double-blind randomized trial. Hum Gene Ther 17:683–691. [DOI] [PubMed] [Google Scholar]
- Lederman RJ, Mendelsohn FO, Anderson RD, Saucedo JF, Tenaglia AN, Hermiller JB, Hillegass WB, Rocha-Singh K, Moon TE, Whitehouse MJ, et al. TRAFFIC Investigators (2002) Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): a randomised trial. Lancet 359:2053–2058. [DOI] [PubMed] [Google Scholar]
- Leosco D, Rengo G, Iaccarino G, Sanzari E, Golino L, De Lisa G, Zincarelli C, Fortunato F, Ciccarelli M, Cimini V, et al. (2007) Prior exercise improves age-dependent vascular endothelial growth factor downregulation and angiogenesis responses to hind-limb ischemia in old rats. J Gerontol A Biol Sci Med Sci 62:471–480. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ. (2008) Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther 16:302–307. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Ryall JG. (2008) Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev 88:729–767. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cao W, Zhang B, Liu YQ, Wang ZY, Wu YP, Yu XJ, Zhang XD, Ming PH, Zhou GB, et al. (2013) The natural compound magnolol inhibits invasion and exhibits potential in human breast cancer therapy. Sci Rep 3:3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo G, Scoccianti M, Kovesdi I, Safi J, Jr, Riccioni T, Capogrossi MC. (1997) Gene therapy for collateral vessel development. Cardiovasc Res 35:480–489. [DOI] [PubMed] [Google Scholar]
- Nikol S, Baumgartner I, Van Belle E, Diehm C, Visoná A, Capogrossi MC, Ferreira-Maldent N, Gallino A, Wyatt MG, Wijesinghe LD, et al. TALISMAN 201 investigators (2008) Therapeutic angiogenesis with intramuscular NV1FGF improves amputation-free survival in patients with critical limb ischemia. Mol Ther 16:972–978. [DOI] [PubMed] [Google Scholar]
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG, Rutherford RB, TASC II Working Group (2007) Inter-society consensus for the management of peripheral arterial disease. Int Angiol 26:81–157. [PubMed] [Google Scholar]
- Osswald W, Guimarães S. (1983) Adrenergic mechanisms in blood vessels: morphological and pharmacological aspects. Rev Physiol Biochem Pharmacol 96:53–122. [DOI] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ. (2003) Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9:677–684. [DOI] [PubMed] [Google Scholar]
- Queen LR, Ji Y, Xu B, Young L, Yao K, Wyatt AW, Rowlands DJ, Siow RC, Mann GE, Ferro A. (2006) Mechanisms underlying beta2-adrenoceptor-mediated nitric oxide generation by human umbilical vein endothelial cells. J Physiol 576:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii S, Lyden D. (2003) Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med 9:702–712. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Mohler ER, 3rd, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, Blebea J, Macko J, Kessler PD, Rasmussen HS, et al. (2003) Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 108:1933–1938. [DOI] [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Koch WJ. (2009a) Future g protein-coupled receptor targets for treatment of heart failure. Curr Treat Options Cardiovasc Med 11:328–338. [DOI] [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. (2009b) Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation 119:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G, Leosco D, Zincarelli C, Marchese M, Corbi G, Liccardo D, Filippelli A, Ferrara N, Lisanti MP, Koch WJ, et al. (2010) Adrenal GRK2 lowering is an underlying mechanism for the beneficial sympathetic effects of exercise training in heart failure. Am J Physiol Heart Circ Physiol 298:H2032–H2038. [DOI] [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Leosco D, Koch WJ. (2011) GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol 50:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G, Zincarelli C, Femminella GD, Liccardo D, Pagano G, de Lucia C, Altobelli GG, Cimini V, Ruggiero D, Perrone-Filardi P, et al. (2012) Myocardial β(2)-adrenoceptor gene delivery promotes coordinated cardiac adaptive remodelling and angiogenesis in heart failure. Br J Pharmacol 166:2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G, Cannavo A, Liccardo D, Zincarelli C, de Lucia C, Pagano G, Komici K, Parisi V, Scala O, Agresta A, et al. (2013) Vascular endothelial growth factor blockade prevents the beneficial effects of β-blocker therapy on cardiac function, angiogenesis, and remodeling in heart failure. Circ Heart Fail 6:1259–1267. [DOI] [PubMed] [Google Scholar]
- Salazar NC, Vallejos X, Siryk A, Rengo G, Cannavo A, Liccardo D, De Lucia C, Gao E, Leosco D, Koch WJ, et al. (2013) GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility. Cell Commun Signal 11:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorriento D, Santulli G, Del Giudice C, Anastasio A, Trimarco B, Iaccarino G. (2012) Endothelial cells are able to synthesize and release catecholamines both in vitro and in vivo. Hypertension 60:129–136. [DOI] [PubMed] [Google Scholar]
- White DC, Hata JA, Shah AS, Glower DD, Lefkowitz RJ, Koch WJ. (2000) Preservation of myocardial beta-adrenergic receptor signaling delays the development of heart failure after myocardial infarction. Proc Natl Acad Sci USA 97:5428–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]