

Abstract
Fabry disease is an X-linked metabolic disorder resulting in widespread deposition of Globotriaosylceramide within a variety of human tissues. The classical Fabry phenotype is one of early onset disease, with extensive tissue involvement resulting in acroparaesthesia, gastrointestinal disturbances, angiokeratoma, cornea verticillata renal failure, and cardiovascular disease.
We describe two brothers exhibiting the GLA p.N215S mutation, a variant most often conferring a late-onset disease confined to the myocardium.
The proband was diagnosed aged 34, following investigation into proteinuria.
Despite Enzyme Replacement Therapy, he progressed to end-stage renal failure, and subsequently received a renal transplant. He also developed hypertrophic cardiomyopathy.
His sibling however, whose disease was detected aged 32 following screening, exhibits mild left ventricular hypertrophy, and no evidence of renal disease. He remains clinically asymptomatic.
This case report details a discordant phenotype in brothers with Fabry disease and p.N215S mutation. Despite the fact that in the majority of patients this mutation is associated with a late onset presentation with hypertrophic cardiomyopathy, we have clearly demonstrated that patients with GLA p.N215S mutation can present with the classical phenotype. Further studies are required to elucidate the underlying modifying factors that influence clinical presentation with a more severe phenotype.
Keywords: Fabry disease, p.N215S, Genetics, Metabolic disease
1. Background
Fabry disease is an X-linked disorder resulting in widespread deposition of glycosphingolipids within human tissues [1]. The disease itself is the result of a number of distinct, well documented mutations in the GLA gene, located at Xq22.1, which codes for the α-galactosidase A enzyme (α-Gal A) [2]. This enzyme is responsible for the hydrolysis of globotriaosylceramide (Gb3); a glycosphingolipid present in cell membranes [3]. Mutations lead to excess lysosomal accumulation of Gb3 within a variety of tissues, with disease severity determined by the amount of residual enzyme activity [4].
The classical phenotype of a patient with Fabry disease is one of widespread tissue involvement leading to manifestations including gastrointestinal disturbances and acroparaesthesia typically first appearing in early childhood [5]. Manifestations will then increase in number and severity with age and patients develop renal, cardiac and cerebrovascular disease [6]. However, this classical Fabry Disease phenotype is not seen in all affected patients and many will present with a less severe, later onset form of the disease. It is generally accepted that patients with p.N215S mutation, a missense mutation in the GLA gene, present later in life with manifestations primarily related to cardiac Gb3 deposition [7].
2. Case presentation
The proband was 34 years old at diagnosis. He had trained as a stonemason, drank little alcohol and had given up smoking 15 years prior. As a teenager he had struggled with intermittent loose stools and abdominal pain.
Examination revealed a Caucasian male of regular body habitus. He was well at rest with no pain or dyspnoea. Clinical signs of Fabry Disease such as angiokeratoma and hypohydrosis, were absent, though he had complained of neuropathic pain in his extremities, exacerbated by cold weather, since his teenage years. He complained of dizziness on exertion, and was experiencing arthralgic symptoms in both hands. Further investigations revelealed a blood pressure of 126/70 mmHg. Urinalysis revealed significant proteinuria and haematuria, which was attributed to Glomerulonephritis, an assumed complication of his Crohn's. No dysmorphic cells were present. However, a renal biopsy was arranged to investigate further.
The biopsy revealed tubular interstitial fibrosis and atrophy with no vascular involvement. pANCA was negative, indicating that the renal damage was likely to be of a separate aetiology to Inflammatory Bowel Disease. Of the 7 glomeruli visualised, 2 were globally sclerosed, with the remainder appearing normal on light microscopy. There was no evidence of mesangial proliferation or thickening of the basement membrane. Patchy moderate tubular atrophy was noted. No arteries were seen but all arterioles appeared free of any deposition. Electron Microscopy (EM) demonstrated Zebra bodies within podocytes and the renal tubular epithelium; suggestive of Fabry Disease (Fig. 1, Fig. 2). The histological stains used for the Light Microscopy samples were Haematoxylin & Eosin to assess overall morphology, Periodic acid Shiff stain (PAS) to assess the interstitium, Methanamine Silver (MethAg) to assess the glomerular basement membranes, and Elastic Van Gieson stain (EVG) to assess arterial blood vessels.
Fig. 1.
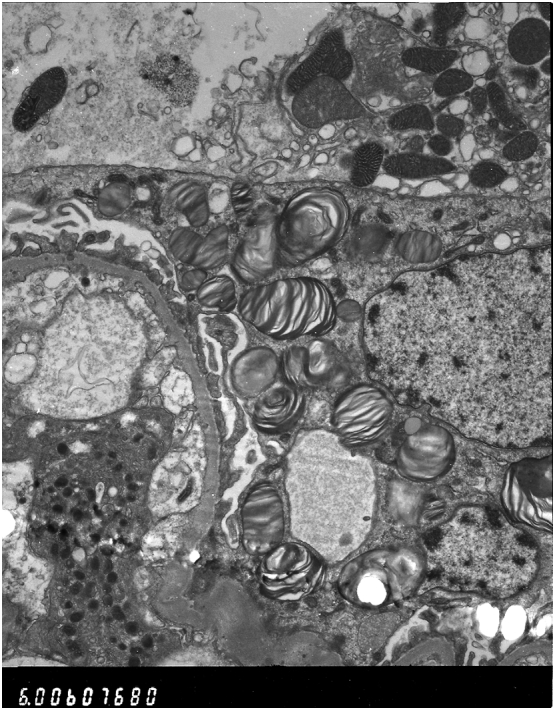
Proband first biopsy images under Electron Microscopy revealing numerous lamellated inclusion Zebra bodies in the glomerular visceral and tubular epithelial cells confirming Fabry Disease.
Fig. 2.
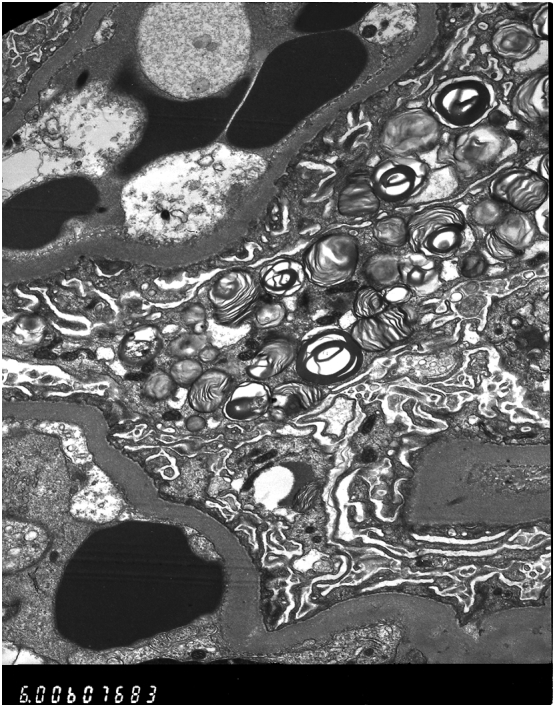
Proband first biopsy images under Electron Microscopy revealing numerous lamellated inclusion Zebra bodies in the glomerular visceral and tubular epithelial cells confirming Fabry Disease.
Plasma α-Gal A activity was measured at 1.4 μmol/l/h and genetic analysis of the GLA gene also revealed the p.N215S mutation, thus confirming the diagnosis.
A complete baseline Fabry assessment was performed. Exercise tolerance testing revealed a poor blood pressure response and the patient only achieved 75% of predicted heart rate. A resting ECG showed sinus rhythm and characteristic changes of Left Ventricular Hypertrophy. 24 h monitoring revealed no evidence of any arrhythmias. An echocardiogram demonstrated moderate symmetrical LVH with normal systolic function, typical findings related to cardiac involvement in Fabry Disease. MRI brain was normal. Renal function testing revealed significant proteinuria at 1.4 g/24 h, an eGFR of 41 ml/min/1.73 m2 and a Creatinine of 235 μmol/l with a UPCR of 133 mg/g. Enzyme Replacement Therapy (ERT) in the form of Agalsidase beta was initiated.
Two years later, his Creatinine had risen to 456 μmol/l (UPCR 172 mg/g) and eGFR declined to 10 ml/min/1.73 m2. A second biopsy (Fig. 3) revealed further glomerular sclerosis. There was no evidence of any basement membrane thickening or mesangial proliferation. Marked striped fibrosis of the tubular interstitium was accompanied by renal tubular atrophy. Arterioles again appeared unremarkable however reduplication of the internal elastic lamina was noted in a few arteries. EM exposed a reduction in Gb3 deposition although Zebra bodies were still present within podocytes and in the tubular epithelium No electron dense deposits were seen.
Fig. 3.
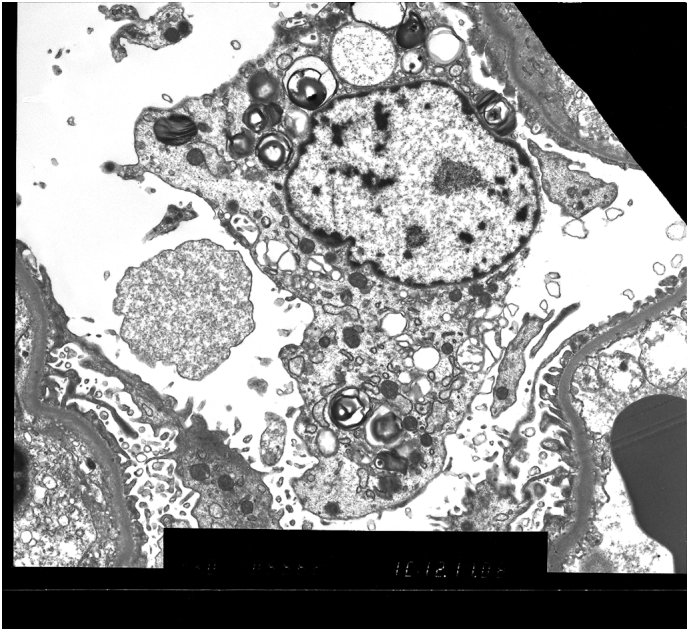
Renal Electron Microscopy images taken from the probands second biopsy. Zebra bodies are still present.
Despite ERT he had progressed to end stage renal failure and haemodialysis was initiated. The patient was subsequently accepted onto the renal transplant waiting list and, after three years, received a successful transplant. Three years later, a Myocardial Infarction following obstruction of the proximal LAD resulted in severe left ventricular systolic impairment. He remains short of breath on exertion but following a cardiac rehabilitation program, his cardiac symptoms are improving.
3. Proband's brother
Following the diagnosis of his brother, the next member of the family we present underwent screening. Gene analysis revealed the p.N215S mutation and a plasma α-galactosidase measurement of 0.83 μmol/l/h [reference range = 3.0–20.0] his diagnosis of Fabry Disease was confirmed.
He first presented at the age of 32. A computer-aided Designer living with his wife. He had no health issues, weighed 98 kg and regularly undertook exercise in the form of playing Squash. He admitted to smoking 20 cigarettes per day, with little intake of alcohol. He took no regular medications and had no known allergies.
Examination was unremarkable. Angiokeratoma, neuropathic pain, and gastrointestinal symptoms were absent. There was no clinical evidence of renal, cardiac or neurological damage. A full baseline Fabry Disease Assessment was performed and all investigations were within acceptable limits. Renal Function was satisfactory with a Creatinine of 99 μmol/L and an eGFR of 78 ml/min/1.73 m2. Renal biopsy revealed no glomerular sclerosis. One glomerulus showed scanty podocyte inclusions (Fig. 4). There were no adhesions or ischaemic features seen and no periglomerular fibrosis. The tubular interstitium, all arteries and arterioles, and the mesangium appeared normal.
Fig. 4.
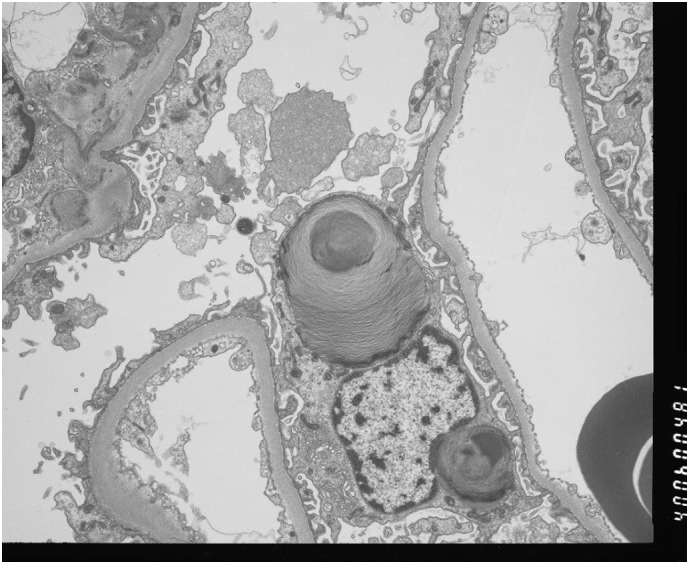
Renal Electron Microscopy image from the proband's brother revealing scanty glomerular podocyte Gb3 inclusions.
EM was arranged to further investigate the possible diagnosis. Again the biopsy appeared almost normal but for sparse inclusions in a few glomerular cells. These were only convincingly seen ultra-structurally and on semi-thin Toluidine blue stained sections. It was decided that annual monitoring was the most appropriate option.
The next assessment revealed no signs of disease progression and annual assessment continued in this manner until 9 years post-diagnosis. Our patient was now aged 41 and had remained asymptomatic. However, the evening before his assessment he presented to the Emergency Department following 2 episodes of loss of consciousness.
Cardiac MRI was performed and revealed: mild LVH with the basal anteroseptum measuring 13 mm, basal anterolateral segment measured 14 mm and mid-inferoseptum was 13 mm. 24 h ECG was within acceptable limits and renal function remained satisfactory.
These findings precipitated a change in the management plan for our patient and ERT was initiated. He remains clinically asymptomatic.
4. Discussion
In summary, we present two brothers with starkly contrasting phenotypes of Fabry Disease.
One brother is asymptomatic, demonstrating the typical p.N215S phenotype of a late-onset cardiac phenotype, with manifestations limited primarily to the myocardium and sparing of renal tissue. On the other hand, the other exhibits a distinct p.N215S phenotype, with history of neuropathic pains since his adolescence and later development of severe renal disease as well as the hypertrophic cardiomyopathy generally seen in Fabry patients with GLA p.N215S mutation.
The evidence of renal involvement is clear on from the EM images. Fig. 1, Fig. 2, taken from the symptomatic brother's renal biopsy in 2006, show numerous lamellated inclusion Zebra bodies in the glomerular visceral epithelial cells and tubular epithelial cells confirming Fabry Disease. His later biopsy (Fig. 3) shows Zebra bodies are still present within podocytes and in the tubular epithelium. Interestingly, and worthy of note, baseline enzyme activity was lower in the brother without renal involvement (0.83 μmol/l/h compared to 1.4 μmol/l/h). The differences in distribution of renal Gb3 deposition seem to correlate with the severity of renal disease. This concurs with the findings of Meehan et al. [8], who found that variation in distribution of Gb3 deposition may account for development of end stage renal failure, as cardiac variants generally experience sparing of Gb3 deposition in the renal endothelium and usually present in the latter decades of life with proteinuria. This is in stark comparison to the early renal failure that classical Fabry patients experience; generally associated with podocytic and endothelial accumulation. It is not clear why there were such differences in the case we present, but such polar outcomes resulting from these differences were clear.
In 1996, Eng and Desnick [9], described three patients with the GLA p.N215S mutation, all of whom exhibited cardiac involvement and sparing of any renal disease, much the same as our first brother. The second brother therefore presents a very interesting case. His development of renal disease due to Gb3 accumulation is not typical of p.N215S patients. Despite the fact that in the majority of patients this mutation is associated with late onset presentation with hypertrophic cardiomyopathy, we have clearly demonstrated that patients with GLA p.N215S mutation can present with the classical phenotype. Further studies are required to elucidate the underlying modifying factors that influence clinical presentation with a more severe phenotype.
References
- 1.Schiffmann R. Fabry disease. Handb. Clin. Neurol. 2015;132:231–248. doi: 10.1016/B978-0-444-62702-5.00017-2. [DOI] [PubMed] [Google Scholar]
- 2.National Institute of Health Genetics Home Reference 2017. https://ghr.nlm.nih.gov/gene/GLA#location Available from.
- 3.Germain D.P. Fabry disease. Orphanet J. Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brady R.O., Schiffmann R. Clinical features of and recent advances in therapy for Fabry disease. JAMA. 2000;284(21):2771–2775. doi: 10.1001/jama.284.21.2771. [DOI] [PubMed] [Google Scholar]
- 5.Eng C.M., Guffon N., Wilcox W.R., Germain D.P., Lee P., Waldek S. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N. Engl. J. Med. 2001;345(1):9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 6.Eng C.M., Resnick-Silverman L.A., Niehaus D.J., Astrin K.H., Desnick R.J. Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am. J. Hum. Genet. 1993;53(6):1186–1197. [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas A., Baker R., Mehta A., Hughes D. The N215S mutation results in a distinct subtype of Fabry disease. Mol. Genet. Metab. 2015;114(2):S113. [Google Scholar]
- 8.Meehan S.M., Junsanto T., Rydel J.J., Desnick R.J. Fabry disease: renal involvement limited to podocyte pathology and proteinuria in a septuagenarian cardiac variant. Pathologic and therapeutic implications. Am. J. Kidney Dis. 2004;43(1):164–171. doi: 10.1053/j.ajkd.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 9.Eng C.M., Desnick R.J. Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha-galactosidase A gene. Hum. Mutat. 1994;3(2):103–111. doi: 10.1002/humu.1380030204. [DOI] [PubMed] [Google Scholar]