

Abstract
Of the twenty classical cadherin subtypes identified in mammals, the functions of the two initially identified family members E- (epithelial) and N- (neural) cadherin have been most extensively studied. E- and N-cadherin have mostly mutually exclusive expression patterns, with E-cadherin expressed primarily in epithelial cells whereas N-cadherin is found in a variety of cells, including neural, muscle and mesenchymal cells. N-cadherin function in particular appears to be cell-context-dependent, as it can mediate strong cell–cell adhesion in the heart, but induce changes in cell behavior in favor of a migratory phenotype in the context of epithelial–mesenchymal transition (EMT). The ability of tumor cells to alter their cadherin expression profile, e.g. E- to N-cadherin, is critical for malignant progression. Recent advances in mouse molecular genetics, and specifically tissue-specific knockout and knock-in alleles of N-cadherin, have provided some unexpected results. This chapter highlights some of the genetic studies that explored the complex role of N-cadherin in embryonic development and disease.
Keywords: adherens junction, gap junction, cardiac intercalated disc, cardiomyopathy, arrhythmia, connexin43, cancer
I. Introduction
In the early 1980s, several groups identified a 135-KD cell surface protein responsible for calcium-dependent cell–cell adhesion in neural retina [1], heart muscle [2], and brain [3]. The subsequent cloning of the gene encoding this cell adhesion molecule and the realization that it belongs to a family of related molecules eventually led to the name N (neural) cadherin [4]. The function of N-cadherin depends much on the cellular context. N-cadherin plays a critical role in maintaining the structural integrity of the heart by mediating strong adhesion between cardiomyocytes. On the other hand, N-cadherin is expressed in migratory fibroblastic or mesenchymal cells. Tumor cells take advantage of this latter characteristic of N-cadherin to promote their invasive and metastatic behavior (see also chapter by Gheldof and Berx). These different adhesive characteristics are likely controlled by modification of protein interactions with its cytoplasmic domain, thus affecting linkage with the actin cytoskeleton and overall adhesion strength of the cadherin/catenin complex. Thirty years after the identification of N-cadherin, researchers continue to actively investigate the complex relationship between N-cadherin-mediated adhesion and signaling in development and disease.
In most cell types, N-cadherin is concentrated at cell–cell contact sites called adherens junctions. Cadherins form cis-dimers in the plasma membrane that interact in an anti-parallel fashion with like cadherins on a neighboring cell, creating an adhesion zipper between the cells [5]. Intracellularly, cadherins interact with the armadillo proteins β-catenin or γ-catenin (plakoglobin), and with p120 catenin. Structurally unrelated to these direct binding partners, α-catenin indirectly links the cadherin/catenin complex to the actin cytoskeleton. Besides mediating intercellular adhesion, N-cadherin might influence signaling networks involved in various cellular processes, including cell proliferation, differentiation and apoptosis (Fig. 1). In addition to its cell adhesive role, β-catenin is also involved in signal transduction and developmental patterning as part of the canonical Wnt signaling pathway (see also chapter by Miller et al.). By sequestering β-catenin at the plasma membrane, N-cadherin can regulate the cytosolic pool of β-catenin and thereby modulate the Wnt pathway. As discussed later in this chapter, N-cadherin can stabilize the fibroblast growth factor receptor (FGFR) on the cell surface, thus facilitating intracellular signaling pathways.
Figure 1.
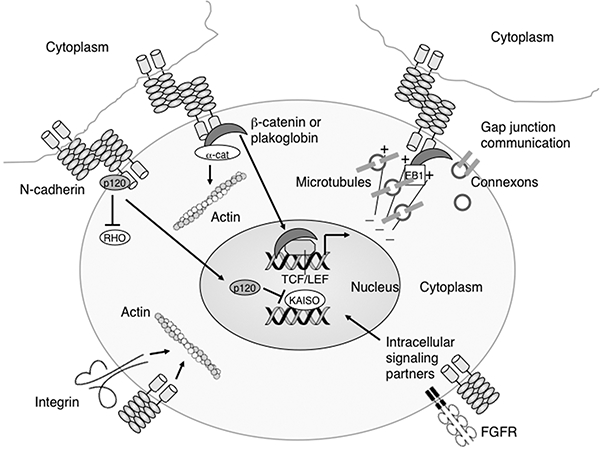
N-cadherin affects a range of biological activities. Schematic representation of the multiple functions of N-cadherin. Besides mediating cell-cell interactions between neighboring cells, N-cadherin may influence cell signaling by binding β-catenin, plakoglobin and p120 catenin thus regulating their ability to enter the nucleus and modulate gene expression. Additionally, unbound p120 can also regulate cell motility by inhibiting the activity of RHO GTPases. The forward trafficking of connexon-containing vesicles to the membrane is dependent on N-cadherin/β-catenin interactions with microtubule plus-end-tracking proteins, including EB1. N-cadherin stabilizes fibroblast growth factor receptor (FGFR) at the membrane thus enhancing its downstream signaling pathways. N-cadherin and integrins cooperate to regulate actin dynamics important for cell motility.
In mammals, the classical cadherin family is composed of 20 members (see also chapter by Hulpiau et al.), which exhibit specific spatio-temporal expression patterns associated with embryonic morphogenesis. Interestingly, only three cadherin subtypes are essential for embryonic development of for instance mouse: E (epithelial), N, and VE (vascular endothelial) cadherin. In contrast, germline deletion of other classical cadherins such as P (placental), R (retinal), and M (muscle) is compatible with embryonic development and these mutant mice are viable with only subtle phenotypic changes [6–8]. On the other hand, N-cadherin’s importance is illustrated by the various tissue-specific knockout experiments that affect embryonic morphogenesis and tissue homeostasis (Table I).
Table I:
N-cadherin mutant phenotypes in mice
| Mutation | Tissue | Phenotypes | Reference |
|---|---|---|---|
| Ncad−/− | global | Embryonic lethal, multiple abnormalities, severe cardiovascular defect | [14, 15] |
| Ncad−/−;Cad11−/− | global | Embryonic lethal, more severe somite defects compared to Ncad−/− | [43] |
| Ncad+/– | global | Altered gap junction organization in the heart, susceptibility to arrhythmia; bone loss in ovarectomized females |
[39, 49] |
| Ncad+/–;Cx43+/– | global | Severe gap junction remodeling in the heart, increase susceptibility to arrhythmias compared to Ncad+/− | [39] |
| Ncad+/–;Cad11−/− | global | Viable, small bones, osteopenia, and reduced bone strength | [50] |
| Ncadfl/fl;αMHC-MerCre | adult myocardium | Cardiomyopathy, sudden arrhythmic death, gap junction and ion channel remodeling | [29, 36, 38] |
| Ncadfl/fl;αMHC-Cre | embryonic myocardium | Embryonic lethal, cardiomyocyte cell adhesion defect | [92] |
| Ncadfl/fl;Tie2-Cre | endothelium | Embryonic lethal, disruption of EC-EC interactions, vascular defects | [23] |
| Ncadfl/fl;Wnt1-Cre | neural crest cells/epicardium | Embryonic lethal, malformed outflow tract, hypoplastic myocardium | [21] |
| Ncadfl/fl;D6-Cre | cerebral cortex | Disorganized cerebral cortical structures, loss of laminar organization | [93] |
| Ncadfl/fl;Col2.3-Cre | osteoblast | Viable, modest trabecular osteopenia | [50] |
| Ncadfl/fl;Pdx1-Cre | pancreas | Viable, pancreas develops normally, defect in beta-cell granule turnover | [94] |
| Ncadfl/fl;Ht-PA-Cre | enteric neural crest cells | Directionality of migration perturbed, delay in colonization of the developing gut | [90] |
| Ncadfl/fl;β1-integrinfl/fl; Ht-PA-Cre |
enteric neural crest cells | Failure to colonize the distal part of the gut, severe aganglionosis in the proximal hindgut compared to single mutants | [90] |
| Ncadfl/fl;Lens-Cre | embryonic lens | Microphthalmia, defects Microphthalmia, defects in lens epithelia and fiber cell integrity | [95] |
| Ncadfl/fl;Ecadfl/fl; Lens-Cre |
embryonic lens | Failure in lens vesicle separation | [95] |
| Ncadfl/fl;POMC-Cre | pituitary | Viable, aberrant corticotrope cell clustering | [96] |
fl, floxed (loxP-flanked) allele; EC, endothelial cell.
Many studies over the years have employed function-blocking antibodies or dominant-negative cadherin molecules to perturb N-cadherin function both in vitro and in vivo. Such antibody-mediated blocking experiments have been reported to interfere with cellular processes whereas in some cases the genetic loss-of-function (LOF) approach had no apparent effect on the same processes [9]. All studies have their limitations and it is important not to misinterpret LOF studies. When a gene is eliminated and a cellular process continues in its absence, it proves that the gene is not essential for that process. However, it remains possible that the gene (and its product) is normally involved in that process but that in its absence, its function is taken over by other genes, what is called biological redundancy. One explanation for this phenomenon is that two (or more) genes have overlapping functions. Both genes might be simultaneously expressed and their products active in the process. However, in the absence of one of the genes (G1), the other one (G2) is sufficient for the process to occur normally. Alternatively, in the case of compensation, gene G2 might not be normally involved in a particular process but it is upregulated in response to elimination of G1, thus substituting for the function of G1. In either scenario, overlapping or compensatory function, the generation of double (or triple) mutants might be necessary to define the genes involved in a particular biological process.
This chapter will focus on the role of N-cadherin in regulating cell adhesion, cellular communication and signaling in different cellular contexts. The most dramatic cell adhesion defects observed in vivo are associated with tissues undergoing active cellular rearrangements and/or increased mechanical stress (e.g. heart muscle). Genetic manipulation of N-cadherin expression in various murine models has provided new insight into the complex role of N-cadherin in disease pathogenesis. In this regard, knock-in of the N-cadherin gene into the E-cadherin locus has generated some unexpected phenotypes that may help us understand the significance of ectopic expression of this mesenchymal cadherin with respect to different signaling pathways.
II. N-cadherin function in cardiovascular development and disease
II.1. N-cadherin function in embryonic myocardium
Most cells express multiple cadherin subtypes. For example, skeletal muscle expresses R-cadherin, M-cadherin, and N-cadherin. In contrast, heart muscle depends on one classical cadherin, N-cadherin. The heart is the first organ to form in the embryo with the establishment of the primitive circulatory system. N-cadherin has been implicated in various aspects of cardiac development, including sorting out of the precardiac mesoderm [10], establishment of left-right asymmetry [11], cardiac looping morphogenesis [12], and trabeculation of the myocardial wall [13]. N-cadherin is strongly expressed in precardiac mesoderm and its expression continues throughout cardiomyocyte development and in the adult differentiated myocardium. Despite this expression in precardiac mesoderm, cardiomyocytes develop in N-cadherin-null embryos and the primitive heart tube forms normally, indicating that N-cadherin is not required for specification of the cardiac lineage [14]. However, N-cadherin-deficient myocytes are unable to maintain their cell–cell contacts when the heart begins to pump, resulting in disassociated rounded myocytes surrounding the endocardial cell layer. Obstruction of the cardiac outflow tract is likely the primary cause of the dramatic pericardial swelling seen in the N-cadherin-null embryos [15]. The cell adhesion defect in N-cadherin-null embryos is rescued by cardiac-specific expression of either N-cadherin or E-cadherin, demonstrating that these two classical cadherins are functionally interchangeable in the primitive heart [15]. In another study, chimeric mouse embryos derived from N-cadherin-null embryonic stem (ES) cells showed that N-cadherin-deficient cardiomyocytes initially interact with N-cadherin-positive cells, but as the primitive heart tube develops, they are excluded from the myocardial wall. The timing of the segregation and exclusion of the N-cadherin-null cells from the wild-type cells corresponds to the transition from simple cuboidal cells to a flattened, tightly associated myocardial cell layer [16]. Myofibrillogenesis occurs in the absence of N-cadherin, but alignment of the myofibrils through regions of cell–cell contact is lost, resulting in their random orientation [17]. As earlier studies demonstrated the importance of N-cadherin for myofibril formation and maintenance, this result was not expected. These former studies employed function-blocking antibodies [18, 19] or dominant-negative cadherin molecules [20] to perturb N-cadherin function in cultured cardiomyocytes. The discrepancy in these results may be explained by the different experimental approaches. In the case of cultured N-cadherin-null cardiomyocytes, integrins might have compensated for the loss of N-cadherin by providing sufficient cytoskeletal integrity to support myofibril formation [17].
II.2. N-cadherin is required in multiple cell lineages during cardiovascular development
In addition to mediating adhesion between cardiomyocytes, N-cadherin plays an essential role in other cell lineages required for mouse cardiovascular development. Using a Wnt1-Cre transgene, it was shown that N-cadherin-deficient cardiac neural crest cells are unable to undergo the normal morphological changes associated with outflow tract remodeling, and this resulted in persistent truncus arteriosus in the majority of mutant embryos [21]. Some mutant embryos did undergo septation, but rotation of the outflow tract was incomplete, suggesting that alignment of the aortic and pulmonary channels is dependent on N-cadherin-generated cytoskeletal forces. Furthermore, N-cadherin is required for heterotypic epicardial-myocardial cell–cell interactions in the developing heart [21]. Disruption of these cellular interactions leads to myocardial cell hypoplasia that is likely caused by the loss of proliferative signaling from the epicardium.
In endothelial cells (EC), N-cadherin exhibits an unusual nonjunctional distribution thought to be important for interactions of the EC with mural cells (i.e. pericyte and smooth muscle cells) [22]. However, more recently it was shown that N-cadherin localizes to EC–EC junctions in addition to its well-known diffuse membrane expression [23]. Unexpectedly, deletion of N-cadherin in EC using Tie2-Cre caused embryonic death at mid-gestation due to severe vascular defects [23]. This phenotype manifested before mural cell investment, a finding that supports a previously unappreciated role for N-cadherin in EC–EC interactions. The Tie2-Cre;N-cad phenotype is remarkably similar to the VE-cadherin-null phenotype [24, 25]. Interestingly, loss of N-cadherin caused a significant decrease in VE-cadherin and p120ctn levels. These studies demonstrate that N-cadherin function is required for angiogenesis and redefine the hierarchical relationship of the structural components of the interendothelial cell junction.
II.3. N-cadherin is essential for the structural integrity of the adult myocardium
Efficient cardiac contractile function is highly dependent on the coordinated mechanical and electrical activation of myocardial tissue. In this regard, it is essential that the structural integrity of the myocardium is tightly maintained. This is largely achieved by the end-to-end connections between myocytes, called intercalated discs (ICD). The ICD is classically defined as containing three distinct junctional complexes: adherens junctions and desmosomes provide strong cell–cell adhesion, and gap junctions electrically couple myocytes. Recently, a novel, exclusive type of ‘hybrid adhering junction’ or ‘area composita’ was identified in the mammalian heart [26, 27]. In this mixed-type junctional complex, classical adherens junction components, including N-cadherin, β-catenin and αE-catenin are co-localized with desmosomal proteins. Interestingly, the area composita is not found in lower vertebrates [28], which suggests that it might have evolved to support the increased mechanical load of the mammalian four-chamber heart by anchoring both actin and intermediate filaments over an extended junctional area of the ICD.
A cardiac-specific, inducible Cre transgene (i.e. tamoxifen administration required for gene deletion) was used to ablate N-cadherin in the working myocardium to generate what is known as the N-cadherin conditional knockout (N-cad CKO) model [29]. Loss of N-cadherin in the adult heart led to disassembly of the ICD, including adherens junctions and desmosomes (Fig. 2). N-cad CKO mice exhibit modest dilated cardiomyopathy and impaired cardiac function, with most animals dying suddenly within two months after deletion of the N-cadherin gene. Telemetric monitoring of conscious free moving N-cad CKO animals captured the abrupt onset of ventricular tachyarrhythmia coincident with sudden death (Fig. 3). It is quite remarkable that the heart can function at all without the ICD structure and its adhesive junctions connecting the individual cardiomyocytes. In this regard, it is worth noting that β1-integrin is upregulated in the N-cadherin-depleted myocardium suggesting that the structural integrity of the heart might be partially maintained by enhanced cell-extracellular matrix interactions. This animal model provided the first demonstration of the hierarchical relationship of the structural components of the intercalated disc in the working myocardium, thus establishing N-cadherin’s paramount importance in maintaining the structural integrity of the heart.
Figure 2.
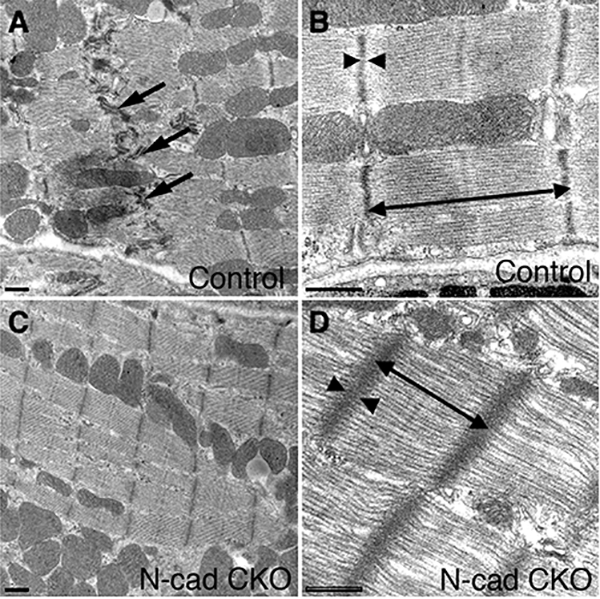
Transmission electron microscopy of N-cadherin CKO hearts. Electron micrographs of ventricular myocardium from control (N-cadfl/–, αMHC/MerCreMer mouse minus tamoxifen) and N-cad CKO (N-cadfl/–, αMHC/MerCreMer mouse plus tamoxifen). Intercalated discs (arrows) were readily visible in the control (A). In contrast, these structures were absent in the N-cad CKO heart (C). The myofibrils appear distorted in the N-cad CKO (D) compared to control (B), with increased sarcomere length (double headed arrow) and wider, less dense Z-lines (arrowheads). Bars, 500 nm. Reprinted with permission from Ref.[29].
Figure 3.
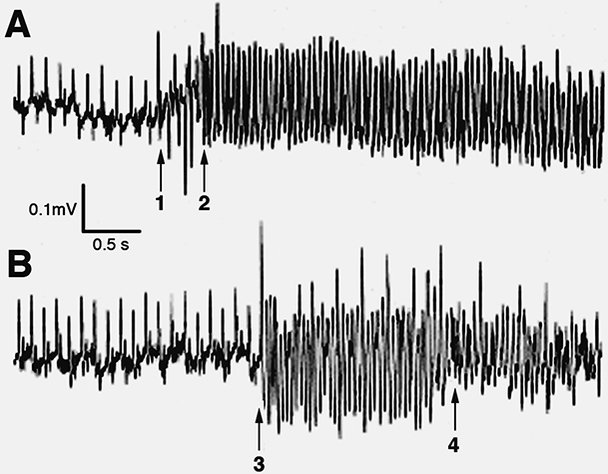
Spontaneous ventricular arrhythmias recorded from N-cadherin CKO mice that died suddenly. Continuous electrocardiographic recordings from a miniaturized transmitter implanted in conscious, freely mobile animals. (A) The left-hand side of the panel shows normal sinus activity followed by the onset of several premature ventricular beats (arrow #1), which then suddenly develops into ventricular tachycardia (arrow #2). (B) Again, the left-hand side of the panel shows normal sinus activity followed by the sudden onset of ventricular tachycardia (arrow #3), which then quickly degenerates into ventricular fibrillation (arrow #4). Reprinted with permission from Ref. [29].
II.4. Relationship between mechanical and electrical junctions in the heart
The loss of large gap junction plaques from the termini of cardiomyocytes, referred to as gap junction remodeling, is commonly observed in many forms of heart disease, including hypertrophy, ischemia, and dilated cardiomyopathies [30]. A major role of gap junctions in the myocardium is to enable rapid and coordinated electrical excitation, a prerequisite for normal rhythmic cardiac function. There is compelling evidence from human disease and animal models linking gap junction remodeling with a highly pro-arrhythmic substrate [31, 32]. Gap junctions are plaques of multiple intercellular channels that connect the cytoplasm of adjacent cells. An individual channel is created by stable, noncovalent interactions of two hemichannels, referred to as connexons. Each connexon is composed of six transmembrane connexin proteins. Many connexins, including those expressed in the heart, have been found to turn over quite rapidly. The principal connexin expressed in the working ventricular myocardium, Cx43, is surprisingly short-lived in the intact adult heart (half-life = 1.5 hours) [33]. Cx43 oligomerizes in the post-ER/Golgi compartment and vesicles containing connexons are transported to the plasma membrane. The forward trafficking of these Cx43-containing vesicles to the junctional membrane is dependent on N-cadherin/β-catenin interactions with microtubule plus-end-tracking proteins, including EB1, p150 (Glued), and the dynein/dynactin complex [34].
Thecarboxy terminus of Cx43 contains numerous sites that are subject to post-translational phosphorylation, and these modifications can affect gap junction activity [35]. Cx43 is phosphorylated when it forms an active gap junctional complex at the cell surface. In contrast, it is generally dephosphorylated during trafficking/endocytosis in the cytoplasm. The time course of N-cadherin depletion directly correlates with a reduction in Cx43-containing gap junctions in the N-cad CKO heart [36]. Moreover, a significant increase in dephosphorylated Cx43 is observed immediately after deleting N-cadherin, consistent with an increased turnover of Cx43 coincident with disassembly of the N-cadherin/catenin complex. Like the Cx43 CKO mice [37], N-cad CKO mice have decreased epicardial conduction velocity associated with sudden arrhythmic death. Loss of N-cadherin contributes to the incidence of arrhythmias not only by slowing conduction but also through significant remodeling of the major outward potassium currents [38]. The actin-binding scaffold protein, cortactin, is required for N-cadherin regulation of Kv1.5 channel function.
Figure 4.
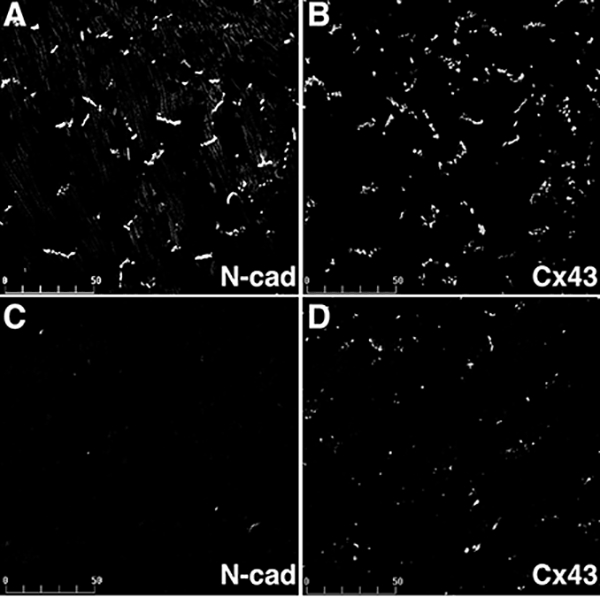
Expression of Cx43 in N-cadherin CKO hearts. Heart ventricles from wild-type (A, B) and N-cadherin CKO (C, D) animals seven weeks post-tamoxifen treatment were co-immunostained for N-cadherin (A, C) and Cx43 (B, D). N-cadherin was lost from the intercalated discs in the CKO heart, while Cx43 (D) was significantly decreased in the ventricular myocardium. Bar, 50 μm. Reprinted with permission from Ref. [36].
The severity of gap junction remodeling in N-cad CKO hearts suggested that animals expressing reduced levels of N-cadherin might exhibit an intermediate phenotype. Indeed, even in the absence of structural heart disease, mice heterozygous for the N-cadherin-null allele exhibit a reduced number of large Cx43-containing plaques and increased arrhythmic susceptibility [39]. Gap junctions were further decreased in number and size in N-cad+/–;Cx43+/– compound heterozygous mice, which exhibit increased arrhythmogenicity compared to the single mutants. These double mutant mice also show an increase in dephosphorylated Cx43. The cytoskeletal scaffold protein zona occludens-1 (ZO-1) binds to the carboxy tail of Cx43 [40] and it was shown to regulate plaque size [41]. The smaller gap junctions in the N-cad+/– cardiomyocytes might be explained by the reduced ZO-1 at the ICD in these myocytes. In future studies it will be important to understand how myocardial stress leads to uncoupling of the N-cadherin/Cx43 macromolecular complex and subsequent gap junction remodeling in diseased myocardium. In this regard, a recent study suggests that oxidative stress disrupts the forward trafficking of Cx43 by interfering with N-cadherin–microtubule interactions at the ICD [42]. Further investigation of the cytoskeletal changes associated with heart disease may provide clues into the underlying mechanism(s) responsible for gap junction remodeling and arrhythmogenesis.
III. Cooperation between the mesenchymal cadherins, N-cadherin and cadherin-11
N-cadherin is first expressed in the nascent mesoderm migrating from the primitive streak in the gastrulating mouse embryo [14]. A day later (neurula stage), there is widespread expression of N-cadherin in the neural tube, notochord, somites, and precardiac mesoderm. Despite the widespread expression at this stage, embryos lacking N-cadherin undergo gastrulation, neurulation and somitogenesis, but the neural tube and somites often are malformed [14]. N-cadherin plays an important role in the coalescence of smaller presomitic clusters of cells into a well-organized epithelial somite structure [43, 44]. The expression of other cell adhesion molecules, including cadherin-11, might partially substitute for lack of N-cadherin in the paraxial mesoderm [45]. Cadherin-11 KO embryos have normal epithelial somite structure [43]. However, in embryos lacking both N-cadherin and cadherin-11, somites are further fragmented into smaller clusters than in the N-cad-null mice, suggesting that these two cadherins cooperate in the maintenance of epithelial somites [43].
Osteoblasts are the skeletal cells responsible for the synthesis, deposition, and mineralization of the bone matrix. Cells of the osteoblast lineage express both N-cadherin and cadherin-11. Germline deletion of the cadherin-11 gene (Cdh11) does not affect skeletal development, but there is a modest reduction in bone density and cranial sutures show minor calcification defects [46]. Interestingly, cadherin-11 is required for the maintenance of the synovial lining of joints and has been implicated in the destructive tissue response in inflammatory arthritis [47]. Expression of a dominant-negative N-cadherin protein in differentiated osteoblasts results in delayed peak bone mass acquisition, impaired osteogenic differentiation, and an osteogenic-to-adipogenic shift in bone marrow stromal cell precursors [48]. The more severe skeletal phenotype in mice expressing dominant-negative N-cadherin suggested that another cadherin might compensate for the lack of cadherin-11. N-cadherin haploinsufficiency by itself does not alter postnatal skeletal growth, but it accentuates ovariectomy-induced bone loss, the result of attenuated activation of bone formation following estrogen deprivation [49]. Conditional deletion of the N-cadherin gene in differentiated osteoblasts results in modest trabecular osteopenia with age [50]. However, double-mutant mice (N-cad+/–;Cad11–/–) exhibit a more severe skeletal phenotype of smaller bones, osteopenia and reduced bone strength relative to Cad11–/– mice, demonstrating that N-cadherin and cadherin-11 cooperate to maintain bone homeostasis (Fig. 5). Functional overlap between N-cadherin and cadherin-11 likely explains the ability of N-cadherin-null limb buds to undergo mesenchymal cell condensation and chondrogenesis, resulting in skeletal structures [51].
Figure 5.
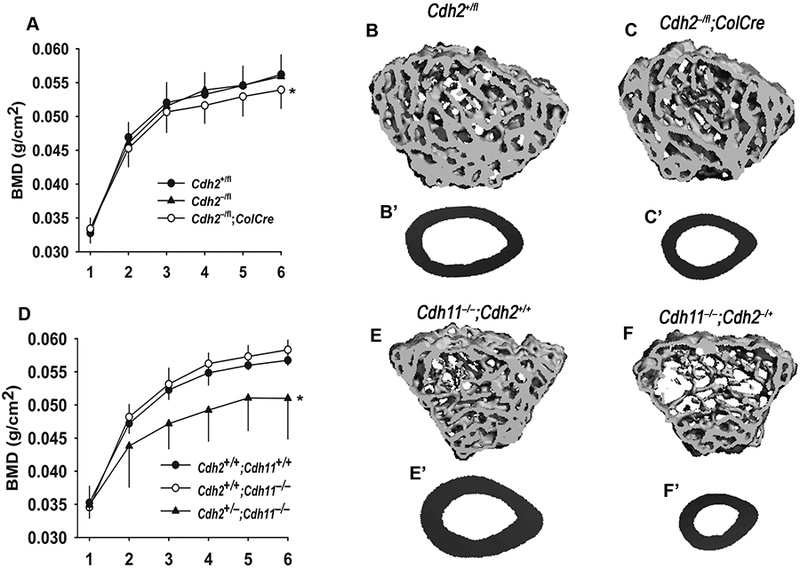
Osteopenia in cadherin-deficient mice. (A) Whole-bone and mineral density (BMD), monitored by DXA bone scans at monthly intervals, was lower in Cdh2–/fl;ColCre than in Cdh2+/fl and wild type littermates from the age of 4 months on (*p<0.05 for genotype, 2-way ANOVA; n = 4–5). (B) MicroCT reconstruction of the tibial epiphysis and (B’) cross-section of the mid-diaphysis of control Cdh2+/fl mice, contrasting with more rarefied trabecular pattern (C) and smaller cortical cross-sectional area (C’) of Cdh2–/fl;ColCre mice. (D) BMD was significantly lower in Cdh2+/–;Cdh11–/– mice than in the other two genotype groups from the age of 2 months on (*p<0.05 for genotypes, 2-way ANOVA; n = 4–7). Relative to Cdh11–/– mice (E, E’), Cdh2+/–;Cdh11–/– mice exhibit severely disrupted trabecular microarchitecture (F) and severely diminished cortical cross-section (F’). Reprinted with permission from Ref. [50]
IV. N-cadherin-mediated signaling in cancer
IV.1. Cadherin switching and EMT
Changes in the expression of cell adhesion receptors accompany the transition from benign tumors to invasive, malignant cancer and the subsequent metastatic dissemination of tumor cells. E-cadherin is expressed in epithelial tissues, where it maintains cell–cell adhesion and tissue architecture, and it is often downregulated during malignant progression. In contrast, de novo expression of N-cadherin is often observed in tumors of epithelial origin. This so-called ‘cadherin switch’ from E-cadherin to N-cadherin is associated with epithelial–mesenchymal transition (EMT) observed during cancer invasion [52, 53] (see also chapter by Gheldof and Berx). As a result of EMT, epithelial cells lose their normal cell polarity and become spindle-shaped and more motile. Cadherin switching is a common occurrence in most cancers, including breast, prostate, and pancreatic cancer, as well as melanoma. N-cadherin is not required for the morphological changes associated with TGF-β1-induced EMT in mammary epithelial cells, but rather it is important for the change in cell behavior (i.e. increased motility) [54]. However, recent genetic data (N-cad knock-in model discussed later in this chapter) indicate that ectopic expression of N-cadherin in mammary epithelial cells is sufficient to promote EMT in vivo [55].
The mechanisms by which N-cadherin promotes cell motility are not completely understood. However, cross-talk with growth factor receptors likely contributes to this cellular behavior. Fibroblast growth factor receptors (FGFR) comprise a subfamily of receptor tyrosine kinases (RTK) that are master regulators of a broad spectrum of cellular and developmental processes, including apoptosis, proliferation, migration and angiogenesis [56]. Upregulation of N-cadherin in breast cancer cells was shown to enhance cell motility, invasion, and metastasis [57–60]. Importantly, N-cadherin attenuates the MAPK-ERK pathway by stabilizing fibroblast growth factor receptor-1 (FGFR-1) on the cell surface, thus enhancing receptor signaling [60]. Interestingly, the matrix metalloprotease, MMP-9, was upregulated in N-cadherin-expressing mammary tumors cells in association with increased invasion [57, 60]. In a PyMT mouse model, N-cadherin was shown to mediate tumor cell migration by suppressing Akt3 function, whereas Akt1 and Akt2 were unaffected [61]. Hence, N-cadherin may influence multiple signaling pathways that cooperate to mediate invasion and metastasis.
Pancreatic ductal adenocarcinoma (PDA) is an aggressive and deadly disease that is characterized by invasive, metastatic progression and profound resistance to conventional therapeutics. The ability of the KPC (LSL-K-rasG12D;LSL-p53R17H;Pdx-1/Cre) murine model to recapitulate –histologically and genetically– the cognate features of human pancreatic cancer [62] has allowed investigators to directly test the requirement for N-cadherin in this highly lethal disease. Remarkably, KPC mice with reduced levels of N-cadherin (i.e. N-cad+/–) survive 25% longer than animals expressing two wild-type N-cadherin (Cdh2) alleles [63]. The survival benefit is likely due to a cumulative effect of N-cadherin’s role in different aspects of tumorigenesis, including tumor-cell survival, growth, migration and invasion. The matrix metalloproteinase MMP-7, the expression of which is associated with poor survival in PDA patients [64], is decreased in the KPC;N-cad+/– tumors. Aberrant FGF signaling is associated with poor prognosis of pancreatic cancer patients [65, 66], and activation of FGF signaling induces cell migration and invasion of pancreatic cancer cells in vitro [67]. KPC;N-cad+/– tumor cells exhibit a decrease in FGF-stimulated ERK1/2 activation and a decrease in migration and invasion, consistent with N-cadherin’s ability to promote FGFR signaling in PDA [63]. The survival benefit observed in the KPC model is consistent with previous results showing that knockdown of N-cadherin leads to smaller tumors in an orthotopic model of pancreatic cancer [68].
Paradoxically, KPC N-cad–/– tumors devoid of N-cadherin did not result in a survival advantage, whereas partly reducing N-cadherin levels prolonged survival of the animals [63]. As the N-cadherin gene is presumably deleted before tumor initiation, the KPC N-cad–/– tumors must rely on an alternative mechanism for the development and progression of PDA. The Ig superfamily member NCAM is a potential candidate to mediate FGFR signaling in the absence of N-cadherin. Indeed, NCAM was upregulated in KPC;N-cad–/– tumor cells and these cells had a similar response to FGF-stimulation as N-cad+/+ tumor cells. Additional studies will be necessary to determine how KPC Ncad–/– tumor cells overcome the requirement for N-cadherin in tumorigenesis.
N-cadherin has also been implicated in tumor cell–endothelial cell interactions critical for the transit of tumor cells into and out of blood vessels. Transendothelial migration is a dynamic process that involves the constant breaking and remaking of intercellular contacts and is accompanied by drastic changes in cell shape and cytoskeletal reorganization in both the tumor cell and its neighboring endothelial cells. Two groups demonstrated that N-cadherin function is important for transendothelial migration of melanoma cells [69] and prostate cancer cells [70]. In melanoma cells, tyrosine phosphorylation of the N-cadherin cytoplasmic domain by Src family kinases resulted in β-catenin dissociation leading to translocation of β-catenin to the nucleus and activation of gene transcription [71]. Collectively, these in vitro and in vivo studies provide compelling evidence that N-cadherin plays a critical role at one or more steps in cancer progression.
IV.2. N-cadherin as a therapeutic target for cancer
The ability of N-cadherin antagonists to act as anti-cancer drugs has gained considerable attention in recent years. Synthetic peptides containing the sequence His-Ala-Val (HAV), which is found in the first extracellular (EC1) domains of classical cadherins, were initially shown to inhibit compaction of mouse embryos at the eight-cell stage [72]. Cadherin antagonists have since been refined, including a cyclic pentapeptide containing HAV (known as ADH-1) that inhibits N-cadherin but not E-cadherin function [73]. ADH-1 recently received orphan drug designation from the United States Food and Drug Administration (FDA) for its use in conjunction with melphalan for the treatment of Stage IIB/C, III, and IV malignant melanoma. ADH-1 has shown promise in controlling metastatic disease in pre-clinical and initial clinical trials. In animal studies, ADH-1 reduced tumor growth in an orthotopic mouse model of pancreatic cancer using N-cadherin-overexpressing BxPC-3 cells [74]. In a melanoma xenograft model, the combination therapy of ADH-1 and melphalan significantly slowed growth of N-cadherin-positive tumor cells [75]. Additionally, monoclonal antibodies against the ectodomain of N-cadherin slowed growth, invasion, and metastasis of prostate cancer cells in a xenograft model [76]. Whether an N-cadherin antagonist can block metastatic dissemination of tumor cells in the aggressive pancreatic cancer model described above (i.e. KPC mice) remains to be determined.
V. Functional overlap and distinct functions for N- and E-cadherin
V.1. Are N- and E-cadherin functionally interchangeable?
Changes in cadherin expression are often associated with changes in cellular morphology and tissue architecture. E- and N-cadherin exhibit unique and mostly mutually exclusive spatio-temporal expression patterns during embryonic morphogenesis [77]. E-cadherin is present in the unfertilized egg as maternal mRNA and protein and is essential for preimplantation development [78]. Specifically, E-cadherin is required for the compaction of the blastomeres at the morula stage and the formation of the trophectoderm, the first polarized epithelial cell layer in the mouse embryo. N-cadherin is initially expressed at the gastrulation stage when epiblast cells downregulate E-cadherin and undergo an epithelial–mesenchymal transition (EMT). De novo expression of N-cadherin occurs at this time in the nascent mesoderm. N- and E-cadherin share several structural and functional features related to their adhesive activity. The extracellular domains of N- and E-cadherin both contain five cadherin domains, but these are less conserved (46%) than their respective cytoplasmic domains (62%), which interact with the catenins [79]. The differences in the extracellular domains of the cadherins may provide cell-context-dependent functions via specific interactions with distinct growth factor receptors besides homophilic interactions with like cadherins.
One of the central questions in mammalian developmental biology is why the organism uses N- and E-cadherin differentially during specific developmental processes. Studies manipulating cadherin expression in vivo are beginning to provide insight into the functional similarities and differences between these two classical cadherins. The ability of a cardiac-specifically expressed E-cadherin transgene to rescue early cardiac morphogenesis (i.e. looping stage) in the N-cadherin-null embryo was the first example of functional replacement of a cadherin subtype during organogenesis [15]. A limitation of this study was the restricted period of expression of the transgene, so it was not possible to assess E-cadherin function later in cardiac development. In contrast to the above rescue experiment, it was discovered that misexpression of E-cadherin together with N-cadherin in the adult heart is not compatible with normal cardiac function, as such animals develop dilated cardiomyopathy and succumb to sudden cardiac death [80]. Although the cellular mechanism responsible for this phenotype is unknown, it was proposed that the E-cadherin/catenin complex at the intercalated disc might interfere with the normal transmission of the contractile force across the sarcolemma, leading to cardiomyopathy. Interestingly, cyclin D1 expression and DNA synthesis were increased in E-cadherin transgenic hearts compared to N-cadherin-overexpressing animals, suggesting a specific proliferative signal from the epithelial cadherin. Of potential relevance to these transgenic studies, E-cadherin was reported to be upregulated in the heart of hypertensive rats, suggesting that inappropriate E-cadherin expression might indeed play a role in natural heart disease [81].
V.2. Knock-in of N-cadherin into the E-cadherin locus
Differences in the ectodomain structure of the different cadherins might affect their strength of adhesion. Using a quantitative dual pipette assay, the adhesive strength of E-cadherin was determined to be 3–4 × higher than that of N-cadherin [82, 83]. In the above-mentioned transgenic experiments, E-cadherin was expressed in heart muscle, which normally expresses only N-cadherin [80]. In a reverse experiment, Kemler and colleagues generated a gene replacement of N-cadherin into the endogenous E-cadherin locus in mice, referred to as N-cad knock-in (KI) [84]. In this approach, N-cadherin is subjected to the same transcriptional regulation as E-cadherin. Somewhat surprisingly, heterozygous mice co-expressing E- and N-cadherin in all epithelial tissues of the body are viable with no gross phenotypic abnormality. The lack of an obvious aberration in these mice is in contrast to the transgenic model discussed above, where co-expression of E- and N-cadherin in the working myocardium leads to cardiomyopathy [80]. This suggests that heart muscle in particular is sensitive to changes in the cadherin expression profile. However, homozygous N-cad KI mice have a similar lethal phenotype as the E-cad null embryos, demonstrating that E-cadherin provides a specific function(s) that cannot be replaced by N-cadherin in the preimplantation embryo [84]. N-cad KI embryos genetically depleted of maternal E-cadherin undergo compaction at the morula stage, but they are unable to maintain a polarized trophectoderm layer and die at the blastocyst stage. The adhesive force generated by N-cadherin might not be sufficient to maintain the trophectoderm, which must withstand the pressure generated by the blastocoel fluid. In contrast, when embryonic stem cells derived from the N-cad KI blastocysts are injected subcutaneously, they can form teratomas containing various epithelial cell lineages, demonstrating that N-cadherin can support epithelial cell differentiation. In regard to cell-context-dependent adhesion, the mechanical stress on the tumor epithelium is likely much less than on the highly specialized trophectoderm, consistent with the idea that the adhesive force generated by the N-cadherin/catenin complex is less than in the case of E-cadherin.
V.3. N-cadherin function in intestinal epithelium
The consequences of replacing E-cadherin with N-cadherin in specific epithelial lineages later in development and in the adult have provided some unexpected phenotypes. This experiment can be performed by combining the N-cad KI allele, a floxed E-cad allele and a tissue-specific Cre transgene to delete the wild-type E-cadherin allele, leaving only N-cadherin expressed from the E-cadherin locus. In one study, it was shown that N-cadherin structurally substitutes for E-cadherin in the intestinal epithelial cell layer, but mutant pups are malnourished and die around weaning age [85]. The presence of N-cadherin replacing E-cadherin was sufficient to induce hyperplasia of the intestinal epithelium, leading to polyps that presumably interfered with the animals’ ability to feed normally. The self-renewing intestinal epithelium is divided into two functional compartments: the crypts, and the villi that protrude into the lumen and absorb nutrients. Previous studies demonstrated the requirement for the canonical Wnt signaling pathway for homeostasis of the intestinal epithelium [86]. For reasons that are not clear, N-cad KI mice exhibit increased nuclear localization of β-catenin in the crypt cells, providing a possible explanation for the hyperplastic phenotype of the small intestine. Additionally, BMP signaling might play a role in the pathophysiology of the mutant intestine. Although the molecular explanation for the mutant phenotype is unknown, this experiment clearly demonstrates that N-cadherin elicits signals in the stem cell compartment of the intestine that are distinct from those elicited by E-cadherin.
V.4. N-cadherin function in the mammary epithelium
In another intriguing study using the N-cad KI allele, expression of N-cadherin in the mammary epithelium of pregnant dams interfered with lactation due to precocious involution [55]. As in the gut, this phenotype appears to be due to aberrant signaling and not to a structural defect. However, in the mammary gland N-cadherin enhances FGF signaling, not Wnt/β-catenin signaling. Of interest, phospho-Stat3, a key player in the involution process, is strongly activated in the N-cad KI alveolar epithelial cells. Furthermore, a functional relationship between N-cadherin and p53 may exist, as heterozygous deletion of the p53 gene rescues the lactation defect in N-cad KI mice. Multiple rounds of lactation led to accumulation of fibrotic tissue and cysts in N-cad KI mice and to invasive tumors when in addition one p53 allele was deleted in the mammary gland. Most surprisingly, using the Rosa26 reporter allele, N-cad KI cells were visualized as they disseminated from the epithelial duct and invaded the surrounding stroma, a process likely driven by activation of FGF signaling. Collectively, these genetic studies show that N-cadherin is functionally interchangeable with E-cadherin as a cell adhesion molecule in epithelial cells but that it stimulates different signaling pathways, which can lead to hyperplasia (intestine) or invasive growth (mammary gland), depending on the cellular context.
VI. Cooperation between N-cadherin and integrins
Cell-cell and cell-extracellular matrix (ECM) adhesions represent highly integrated networks of protein interactions that are crucial for tissue homeostasis and the responses of individual cells to their microenvironments [87]. Through the actin cytoskeleton cadherins and integrins cooperate to form an interdependent functional network that translates mechanical inputs into intracellular signals. It is generally accepted that N-cadherin promotes cell migration, however it has also been reported that interfering with N-cadherin expression can increase cell motility in neural crest cells [88], endothelial cells [23], and tumor cells [89]. It is possible that increased β1-integrin function may positively influence cell motility in N-cadherin-deficient endothelial cells [23]. Depending on the type of migration assay used, subtle differences in cell behavior may not be appreciated. For example, although N-cadherin-null neural crest cells were shown to migrate faster in culture, their directionality was abnormal so that the total distance they traveled was less than in case of wild-type cells [88]. Moreover, using time-lapse imaging of ex vivo whole guts, directionality of migration was perturbed in N-cadherin-deficient enteric neural crest cells, although the speed of locomotion of the mutant cells was not affected [90]. In the same study, it was shown that depletion of both N-cadherin and β1-integrin in enteric neural crest cells led to a failure to colonize the distal part of the gut and there was more severe aganglionosis compared to the single mutants thus demonstrating that these cell-cell and cell-ECM adhesion molecules cooperate to mediate cell migration.
VII. Conclusions/Future directions
The goal of this chapter was to highlight recent advances in our understanding of N-cadherin biology, particularly in relation to human disease. To overcome the requirement for N-cadherin in the early mouse embryo, it is necessary to utilize a conditional mutagenesis approach (i.e. Cre/loxP technology) to investigate N-cadherin function in specific cell lineages later in development as well as the adult organism. The ability to ablate N-cadherin in a tissue-specific and inducible manner is beginning to shed new light on the complex role of this cell adhesion receptor in both normal physiological and pathological conditions. N-cadherin function in particular appears to be cell-context-dependent, as it can mediate strong cell–cell adhesion in the heart, but induce changes in cell behavior toward a more migratory phenotype in other contexts. The importance of N-cadherin for maintaining mechano-electrical coupling in the heart is evident. However, how the N-cadherin/catenin complex and other ICD proteins are affected by heart disease is poorly understood. Recent studies suggest that oxidative stress can lead to uncoupling of N-cadherin from Cx43 though there is no obvious change in N-cadherin expression at the ICD [42]. The underlying cytoskeleton may be affected in cardiomyocytes experiencing stress [91], so that the normal protein interactions mediated by the N-cadherin/catenin complex are altered causing impaired gap junction trafficking to the ICD. The challenge is to understand the molecular changes that occur in the failing heart that might modify protein interactions with the cytoplasmic domain of N-cadherin, thus affecting linkage with the actin cytoskeleton and overall adhesion strength of the N-cadherin/catenin complex.
The KPC;N-cad+/– mice are an ideal model system to further investigate the role of N-cadherin in tumor development and progression. In contrast to its structural role in the heart, N-cadherin haploinsufficiency affects tumor-cell survival, growth, migration and invasion in the KPC model. It will be important to determine which signaling pathways are required for the survival benefit in the KPC;N-cad+/– animals. This could lead to a new combination therapy targeting different points in the same pathway or two different pathways (e.g. N-cadherin and FGF signaling pathways) to achieve maximum benefit. According to typical Boyden chamber assays, KPC;Ncad+/– tumor cells exhibit decreased migration and invasion [63]. Whether this in vitro result translates to a less invasive phenotype in vivo awaits further investigation using methods of cell lineage tracing in the KPC mouse model. Considering the wide distribution of N-cadherin expression, it is somewhat surprising that the N-cadherin antagonist ADH-1 is well tolerated by patients. This lack of toxicity might mean that the N-cadherin levels and/or molecular accessibility to the drug may be different on the surface of tumor cells in comparison to normal cells.
In conclusion, loss- and gain-of-function genetic analyses in the mouse have provided new insight into how N-cadherin can influence cell adhesion, cellular communication, and signaling in different cellular contexts. The identification of N-cadherin haploinsufficiency phenotypes further substantiates the importance of this cell adhesion receptor in mammalian physiology. The mouse models discussed in this chapter represent only the beginning as N-cadherin is implicated in various biological processes and in many organ systems. As most studies to date have examined N-cadherin function under normal physiological conditions, it will be particularly informative to manipulate N-cadherin levels in mouse models of human disease.
Acknowledgments
The research in the author’s laboratory is supported by National Institutes of Health grant (HL111788) and American Heart Association grant (GRNT12030343).
LITERATURE CITED
- 1.Grunwald GB, Pratt RS and Lilien J (1982) Enzymic dissection of embryonic cell adhesive mechanisms. III. Immunological identification of a component of the calcium-dependent adhesive system of embryonic chick neural retina cells. J Cell Sci. 55, 69–83 [DOI] [PubMed] [Google Scholar]
- 2.Volk T and Geiger B (1984) A 135-kd membrane protein of intercellular adherens junctions. Embo J. 3, 2249–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatta K, Okada TS and Takeichi M (1985) A monoclonal antibody disrupting calcium-dependent cell-cell adhesion of brain tissues: possible role of its target antigen in animal pattern formation. Proc Natl Acad Sci U S A. 82, 2789–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hatta K, Nose A, Nagafuchi A and Takeichi M (1988) Cloning and expression of cDNA encoding a neural calcium-dependent cell adhesion molecule: its identity in the cadherin gene family. J Cell Biol. 106, 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR and Hendrickson WA (1995) Structural basis of cell-cell adhesion by cadherins. Nature. 374, 327–337 [DOI] [PubMed] [Google Scholar]
- 6.Dahl U, Sjodin A, Larue L, Radice GL, Cajander S, Takeichi M, Kemler R and Semb H (2002) Genetic dissection of cadherin function during nephrogenesis. Mol Cell Biol. 22, 1474–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollnagel A, Grund C, Franke WW and Arnold HH (2002) The cell adhesion molecule M-cadherin is not essential for muscle development and regeneration. Mol Cell Biol. 22, 4760–4770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radice GL, Ferreira-Cornwell MC, Robinson SD, Rayburn H, Chodosh LA, Takeichi M and Hynes RO (1997) Precocious mammary gland development in P-cadherin-deficient mice. J Cell Biol. 139, 1025–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hynes RO (1996) Targeted mutations in cell adhesion genes: what have we learned from them? Dev Biol. 180, 402–412 [DOI] [PubMed] [Google Scholar]
- 10.Linask KK, Knudsen KA and Gui YH (1997) N-cadherin-catenin interaction: necessary component of cardiac cell compartmentalization during early vertebrate heart development. Dev Biol. 185, 148–164 [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Castro MI, Vielmetter E and Bronner-Fraser M (2000) N-Cadherin, a cell adhesion molecule involved in establishment of embryonic left-right asymmetry. Science. 288, 1047–1051 [DOI] [PubMed] [Google Scholar]
- 12.Shiraishi I, Takamatsu T and Fujita S (1993) 3-D observation of N-cadherin expression during cardiac myofibrillogenesis of the chick embryo using a confocal laser scanning microscope. Anat Embryol (Berl). 187, 115–120 [DOI] [PubMed] [Google Scholar]
- 13.Ong LL, Kim N, Mima T, Cohen-Gould L and Mikawa T (1998) Trabecular myocytes of the embryonic heart require N-cadherin for migratory unit identity. Dev Biol. 193, 1–9 [DOI] [PubMed] [Google Scholar]
- 14.Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M and Hynes RO (1997) Developmental defects in mouse embryos lacking N-cadherin. Dev. Biol. 181, 64–78 [DOI] [PubMed] [Google Scholar]
- 15.Luo Y, Ferreira-Cornwell M, Baldwin H, Kostetskii I, Lenox J, Lieberman M and Radice G (2001) Rescuing the N-cadherin knockout by cardiac-specific expression of N- or E-cadherin. Development. 128, 459–469 [DOI] [PubMed] [Google Scholar]
- 16.Kostetskii I, Moore R, Kemler R and Radice GL (2001) Differential adhesion leads to segregation and exclusion of N-cadherin-deficient cells in chimeric embryos. Dev Biol. 234, 72–79 [DOI] [PubMed] [Google Scholar]
- 17.Luo Y and Radice GL (2003) Cadherin-mediated adhesion is essential for myofibril continuity across the plasma membrane but not for assembly of the contractile apparatus. J Cell Sci. 116, 1471–1479 [DOI] [PubMed] [Google Scholar]
- 18.Goncharova EJ, Kam Z and Geiger B (1992) The involvement of adherens junction components in myofibrillogenesis in cultured cardiac myocytes. Development. 114, 173–183 [DOI] [PubMed] [Google Scholar]
- 19.Soler AP and Knudsen KA (1994) N-cadherin involvement in cardiac myocyte interaction and myofibrillogenesis. Dev Biol. 162, 9–17 [DOI] [PubMed] [Google Scholar]
- 20.Hertig CM, Eppenberger-Eberhardt M, Koch S and Eppenberger HM (1996) N-cadherin in adult rat cardiomyocytes in culture. I. Functional role of N-cadherin and impairment of cell-cell contact by a truncated N-cadherin mutant. J Cell Sci. 109 (Pt 1), 1–10 [DOI] [PubMed] [Google Scholar]
- 21.Luo Y, High FA, Epstein JA and Radice GL (2006) N-cadherin is required for neural crest remodeling of the cardiac outflow tract. Dev Biol. 299, 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarro P, Ruco L and Dejana E (1998) Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J Cell Biol. 140, 1475–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo Y and Radice GL (2005) N-cadherin acts upstream of VE-cadherin in controlling vascular morphogenesis. J Cell Biol. 169, 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D and Dejana E (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98, 147–157 [DOI] [PubMed] [Google Scholar]
- 25.Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M and Huber P (1999) Role of vascular endothelial-cadherin in vascular morphogenesis. Development. 126, 2093–2102 [DOI] [PubMed] [Google Scholar]
- 26.Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S and Franke WW (2006) The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol. 85, 469–485 [DOI] [PubMed] [Google Scholar]
- 27.Franke WW, Borrmann CM, Grund C and Pieperhoff S (2006) The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 85, 69–82 [DOI] [PubMed] [Google Scholar]
- 28.Pieperhoff S and Franke WW (2008) The area composita of adhering junctions connecting heart muscle cells of vertebrates. VI. Different precursor structures in non-mammalian species. Eur J Cell Biol. 87, 413–430 [DOI] [PubMed] [Google Scholar]
- 29.Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD and Radice GL (2005) Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 96, 346–354 [DOI] [PubMed] [Google Scholar]
- 30.Saffitz JE, Hames KY and Kanno S (2007) Remodeling of gap junctions in ischemic and nonischemic forms of heart disease. J Membr Biol. 218, 65–71 [DOI] [PubMed] [Google Scholar]
- 31.Saffitz JE (2009) Arrhythmogenic cardiomyopathy and abnormalities of cell-to-cell coupling. Heart Rhythm. 6, S62–65 [DOI] [PubMed] [Google Scholar]
- 32.Severs NJ, Bruce AF, Dupont E and Rothery S (2008) Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 80, 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beardslee MA, Laing JG, Beyer EC and Saffitz JE (1998) Rapid turnover of connexin43 in the adult rat heart. Circ Res. 83, 629–635 [DOI] [PubMed] [Google Scholar]
- 34.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN and Jan LY (2007) Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 128, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solan JL and Lampe PD (2009) Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 419, 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD and Radice GL (2005) Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 97, 474–481 [DOI] [PubMed] [Google Scholar]
- 37.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H and Fishman GI (2001) Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 88, 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng L, Yung A, Covarrubias M and Radice GL (2011) Cortactin is required for N-cadherin regulation of Kv1.5 channel function. J Biol Chem. 286, 20478–20489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Levin MD, Xiong Y, Petrenko N, Patel VV and Radice GL (2008) N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol. 44, 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M and Tada M (1998) Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 273, 12725–12731 [DOI] [PubMed] [Google Scholar]
- 41.Hunter AW, Barker RJ, Zhu C and Gourdie RG (2005) Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 16, 5686–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC and Shaw RM (2010) Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 120, 266–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horikawa K, Radice G, Takeichi M and Chisaka O (1999) Adhesive subdivisions intrinsic to the epithelial somites. Dev Biol. 215, 182–189 [DOI] [PubMed] [Google Scholar]
- 44.Linask KK, Ludwig C, Han MD, Liu X, Radice GL and Knudsen KA (1998) N-cadherin/catenin-mediated morphoregulation of somite formation. Dev Biol. 202, 85–102 [DOI] [PubMed] [Google Scholar]
- 45.Kimura Y, Matsunami H, Inoue T, Shimamura K, Uchida N, Ueno T, Miyazaki T and Takeichi M (1995) Cadherin-11 expressed in association with mesenchymal morphogenesis in the head, somite, and limb bud of early mouse embryos. Dev Biol. 169, 347–358 [DOI] [PubMed] [Google Scholar]
- 46.Kawaguchi J, Azuma Y, Hoshi K, Kii I, Takeshita S, Ohta T, Ozawa H, Takeichi M, Chisaka O and Kudo A (2001) Targeted disruption of cadherin-11 leads to a reduction in bone density in calvaria and long bone metaphyses. J Bone Miner Res. 16, 1265–1271 [DOI] [PubMed] [Google Scholar]
- 47.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M and Brenner MB (2007) Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 315, 1006–1010 [DOI] [PubMed] [Google Scholar]
- 48.Castro CH, Shin CS, Stains JP, Cheng SL, Sheikh S, Mbalaviele G, Szejnfeld VL and Civitelli R (2004) Targeted expression of a dominant-negative N-cadherin in vivo delays peak bone mass and increases adipogenesis. J Cell Sci. 117, 2853–2864 [DOI] [PubMed] [Google Scholar]
- 49.Lai CF, Cheng SL, Mbalaviele G, Donsante C, Watkins M, Radice GL and Civitelli R (2006) Accentuated ovariectomy-induced bone loss and altered osteogenesis in heterozygous N-cadherin null mice. J Bone Miner Res. 21, 1897–1906 [DOI] [PubMed] [Google Scholar]
- 50.Di Benedetto A, Watkins M, Grimston S, Salazar V, Donsante C, Mbalaviele G, Radice GL and Civitelli R (2010) N-cadherin and cadherin 11 modulate postnatal bone growth and osteoblast differentiation by distinct mechanisms. J Cell Sci. 123, 2640–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo Y, Kostetskii I and Radice GL (2005) N-cadherin is not essential for limb mesenchymal chondrogenesis. Dev Dyn. 232, 336–344 [DOI] [PubMed] [Google Scholar]
- 52.Huber MA, Kraut N and Beug H (2005) Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 17, 548–558 [DOI] [PubMed] [Google Scholar]
- 53.Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y and Johnson KR (2008) Cadherin switching. J Cell Sci. 121, 727–735 [DOI] [PubMed] [Google Scholar]
- 54.Maeda M, Johnson KR and Wheelock MJ (2005) Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 118, 873–887 [DOI] [PubMed] [Google Scholar]
- 55.Kotb AM, Hierholzer A and Kemler R (2011) Replacement of E-cadherin by N-cadherin in the mammary gland leads to fibrocystic changes and tumor formation. Breast Cancer Res. 13, R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eswarakumar VP, Lax I and Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149 [DOI] [PubMed] [Google Scholar]
- 57.Hazan RB, Phillips GR, Qiao RF, Norton L and Aaronson SA (2000) Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 148, 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen K and Hazan RB (2007) N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 67, 3106–3116 [DOI] [PubMed] [Google Scholar]
- 59.Nieman MT, Prudoff RS, Johnson KR and Wheelock MJ (1999) N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 147, 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suyama K, Shapiro I, Guttman M and Hazan RB (2002) A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2, 301–314 [DOI] [PubMed] [Google Scholar]
- 61.Chung S, Yao J, Suyama K, Bajaj S, Qian X, Loudig OD, Eugenin EA, Phillips GR and Hazan RB (2012) N-cadherin regulates mammary tumor cell migration through Akt3 suppression. Oncogene [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S and Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 7, 469–483 [DOI] [PubMed] [Google Scholar]
- 63.Su Y, Li J, Witkiewicz AK, Brennan D, Neill T, Talarico J and Radice GL (2011) N-cadherin haploinsufficiency increases survival in a mouse model of pancreatic cancer. Oncogene [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukuda A, Wang SC, Morris J. P. t., Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ and Hebrok M (2011) Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 19, 441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishiwata T, Friess H, Buchler MW, Lopez ME and Korc M (1998) Characterization of keratinocyte growth factor and receptor expression in human pancreatic cancer. Am J Pathol. 153, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wagner M, Lopez ME, Cahn M and Korc M (1998) Suppression of fibroblast growth factor receptor signaling inhibits pancreatic cancer growth in vitro and in vivo. Gastroenterology. 114, 798–807 [DOI] [PubMed] [Google Scholar]
- 67.Nomura S, Yoshitomi H, Takano S, Shida T, Kobayashi S, Ohtsuka M, Kimura F, Shimizu H, Yoshidome H, Kato A and Miyazaki M (2008) FGF10/FGFR2 signal induces cell migration and invasion in pancreatic cancer. Br J Cancer. 99, 305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shintani Y, Hollingsworth MA, Wheelock MJ and Johnson KR (2006) Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 66, 11745–11753 [DOI] [PubMed] [Google Scholar]
- 69.Qi J, Chen N, Wang J and Siu CH (2005) Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol Biol Cell. 16, 4386–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drake JM, Strohbehn G, Bair TB, Moreland JG and Henry MD (2009) ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell. 20, 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qi J, Wang J, Romanyuk O and Siu CH (2006) Involvement of Src family kinases in N-cadherin phosphorylation and beta-catenin dissociation during transendothelial migration of melanoma cells. Mol Biol Cell. 17, 1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blaschuk OW, Sullivan R, David S and Pouliot Y (1990) Identification of a cadherin cell adhesion recognition sequence. Dev Biol. 139, 227–229 [DOI] [PubMed] [Google Scholar]
- 73.Williams E, Williams G, Gour BJ, Blaschuk OW and Doherty P (2000) A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. 275, 4007–4012 [DOI] [PubMed] [Google Scholar]
- 74.Shintani Y, Fukumoto Y, Chaika N, Grandgenett PM, Hollingsworth MA, Wheelock MJ and Johnson KR (2008) ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int J Cancer. 122, 71–77 [DOI] [PubMed] [Google Scholar]
- 75.Augustine CK, Yoshimoto Y, Gupta M, Zipfel PA, Selim MA, Febbo P, Pendergast AM, Peters WP and Tyler DS (2008) Targeting N-cadherin enhances antitumor activity of cytotoxic therapies in melanoma treatment. Cancer Res. 68, 3777–3784 [DOI] [PubMed] [Google Scholar]
- 76.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, An J, Horvath S, Gleave M, Rettig MB, Wainberg ZA and Reiter RE (2010) Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 16, 1414–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takeichi M (1988) The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development. 102, 639–655 [DOI] [PubMed] [Google Scholar]
- 78.Larue L, Ohsugi M, Hirchenhain J and Kemler R (1994) E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A. 91, 8263–8267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miyatani S, Shimamura K, Hatta M, Nagafuchi A, Nose A, Matsunaga M, Hatta K and Takeichi M (1989) Neural cadherin: role in selective cell-cell adhesion. Science. 245, 631–635 [DOI] [PubMed] [Google Scholar]
- 80.Ferreira-Cornwell MC, Luo Y, Narula N, Lenox JM, Lieberman M and Radice GL (2002) Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J Cell Sci. 115, 1623–1634 [DOI] [PubMed] [Google Scholar]
- 81.Craig MA, McBride MW, Smith G, George SJ and Baker A (2010) Dysregulation of cadherins in the intercalated disc of the spontaneously hypertensive stroke-prone rat. J Mol Cell Cardiol. 48, 1121–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chu YS, Thomas WA, Eder O, Pincet F, Perez E, Thiery JP and Dufour S (2004) Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J Cell Biol. 167, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chu YS, Eder O, Thomas WA, Simcha I, Pincet F, Ben-Ze’ev A, Perez E, Thiery JP and Dufour S (2006) Prototypical type I E-cadherin and type II cadherin-7 mediate very distinct adhesiveness through their extracellular domains. J Biol Chem. 281, 2901–2910 [DOI] [PubMed] [Google Scholar]
- 84.Kan NG, Stemmler MP, Junghans D, Kanzler B, de Vries WN, Dominis M and Kemler R (2007) Gene replacement reveals a specific role for E-cadherin in the formation of a functional trophectoderm. Development. 134, 31–41 [DOI] [PubMed] [Google Scholar]
- 85.Libusova L, Stemmler MP, Hierholzer A, Schwarz H and Kemler R (2010) N-cadherin can structurally substitute for E-cadherin during intestinal development but leads to polyp formation. Development. 137, 2297–2305 [DOI] [PubMed] [Google Scholar]
- 86.Pinto D, Gregorieff A, Begthel H and Clevers H (2003) Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 17, 1709–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber GF, Bjerke MA and DeSimone DW (2011) Integrins and cadherins join forces to form adhesive networks. J Cell Sci. 124, 1183–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu X, Li WE, Huang GY, Meyer R, Chen T, Luo Y, Thomas MP, Radice GL and Lo CW (2001) Modulation of mouse neural crest cell motility by N-cadherin and connexin 43 gap junctions. J Cell Biol. 154, 217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deramaudt TB, Takaoka M, Upadhyay R, Bowser MJ, Porter J, Lee A, Rhoades B, Johnstone CN, Weissleder R, Hingorani SR, Mahmood U and Rustgi AK (2006) N-cadherin and keratinocyte growth factor receptor mediate the functional interplay between Ki-RASG12V and p53V143A in promoting pancreatic cell migration, invasion, and tissue architecture disruption. Mol Cell Biol. 26, 4185–4200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Broders-Bondon F, Paul-Gilloteaux P, Carlier C, Radice GL and Dufour S (2012) N-cadherin and beta1-integrins cooperate during the development of the enteric nervous system. Dev Biol. 364, 178–191 [DOI] [PubMed] [Google Scholar]
- 91.Smyth JW, Vogan JM, Buch PJ, Zhang SS, Fong TS, Hong TT and Shaw RM (2012) Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circ Res. 110, 978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piven OO, Kostetskii IE, Macewicz LL, Kolomiets YM, Radice GL and Lukash LL (2011) Requirement for N-cadherin-catenin complex in heart development. Exp Biol Med (Maywood). 236, 816–822 [DOI] [PubMed] [Google Scholar]
- 93.Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL and Takeichi M (2007) N-cadherin mediates cortical organization in the mouse brain. Dev Biol. 304, 22–33 [DOI] [PubMed] [Google Scholar]
- 94.Johansson JK, Voss U, Kesavan G, Kostetskii I, Wierup N, Radice GL and Semb H (2010) N-cadherin is dispensable for pancreas development but required for beta-cell granule turnover. Genesis. 48, 374–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pontoriero GF, Smith AN, Miller LA, Radice GL, West-Mays JA and Lang RA (2009) Co-operative roles for E-cadherin and N-cadherin during lens vesicle separation and lens epithelial cell survival. Dev Biol. 326, 403–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Himes AD, Fiddler RM and Raetzman LT (2011) N-cadherin loss in POMC-expressing cells leads to pituitary disorganization. Mol Endocrinol. 25, 482–491 [DOI] [PMC free article] [PubMed] [Google Scholar]