

Abstract
Parkinson’s disease is the second most common neurodegenerative disorder without effective treatment. It is generally sporadic with unknown etiology. However, genetic studies of rare familial forms have led to the identification of mutations in several genes, which are linked to typical Parkinson’s disease or parkinsonian disorders. The pathogenesis of Parkinson’s disease remain largely elusive. Here, we report a novel genetic locus for an autosomal dominant, clinically typical and Lewy body confirmed Parkinson’s disease on the short arm of chromosome 20 (20pter-p12) and TMEM230 as the disease-causing gene. We show that TMEM230 encodes a transmembrane protein of secretory/recycling vesicles, including synaptic vesicles in neurons. The disease-linked TMEM230 mutants impair synaptic vesicle trafficking. Our data provide the first genetic evidence that a mutant transmembrane protein of synaptic vesicles in neurons is etiologically linked to Parkinson’s disease, with novel implications in understanding the pathogenic mechanism of Parkinson’s disease and for developing rational therapies.
Parkinson’s disease (PD) is a neurodegenerative disorder affecting approximately 1% of the population over age 60, with a lifetime incidence of over 2%. PD is characterized clinically by resting tremor, bradykinesia, rigidity, postural instability and a variety of other motor and non-motor symptoms, and pathologically by the loss of dopamine-producing neurons in the substantia nigra pars compacta in association with the presence of Lewy bodies in some surviving neurons1. Most of PD cases are sporadic with unknown etiology. Rare PD families have provided unique resources for identification of PD-linked genetic defects, and for the development of cellular and animal models for a better understanding of its pathogenesis2,3. Since the identification of α-synuclein gene (SNCA) as the first PD-linked gene4, several other causative or risk genes for PD or other parkinsonian disorders have been discovered5-22. Among the disease-causing genes, mutations in SNCA and LRRK2, or an increase in SNCA copy number are linked to Lewy body confirmed PD. A perspective on this topic was comprehensively reviewed by Langston et al1,23. However, despite extensive ongoing studies of the previously identified molecule targets, the pathogenic mechanisms of PD remain largely elusive1,24. Here, we show that mutations in TMEM230 lead to clinically typical and Lewy body confirmed PD. Our findings suggest synaptic vesicle trafficking defects underlie PD pathogenesis, with implications in therapeutic development.
RESULTS
A novel locus of dominant and Lewy body confirmed PD on 20p
We have followed and studied a large North American PD family (#9853) of Northern European ancestry for over two decades. This family included 81 members, among whom 15 were affected (Supplementary Fig. 1). Detailed clinical information was obtained from 14 affected individuals, who exhibited variable symptoms of typical PD, including bradykinesia, resting tremor, rigidity, and postural instability. The symptoms responded well to levodopa therapy in most cases. The mean age at the disease onset was 67.0 ± 9.5 (SD) years (range: 48-85). Death occurred after an average of 13.2 ± 4.1 (SD) years. Pathological analysis of autopsy samples from three affected individuals (II-4, II6 and II-14) showed PD pathology, including marked loss of dopaminergic neurons and the presence of typical Lewy bodies and Lewy neurites in the substantia nigra pars compacta.
We have collected DNA samples from 65 members, including 13 affected individuals from this family (Supplementary Fig. 1). We performed genome-wide linkage analysis using over 300 microsatellite polymorphic markers, and subsequently a dense panel of 21 microsatellite markers on the 20p. Two-point LOD score over 3.3 and multi-point score over 3.8 were obtained for several informative markers on the tip of chromosome 20 (Supplementary Table 1). We constructed a genetic haplotype that was shared by all the 13 affected members, but not by the two older unaffected individuals (II-9 and II-12) in the second generation (Supplementary Figs. 1 and 2). The individual II-9 died at the age of 87 years without any signs of PD. The individual II-12 died at age of 78 years without PD symptoms and there was no PD pathology at autopsy. Both unaffected members in the second generation did not carry the disease-linked haplotype. A crossover between D20S901 and D20S894 observed in affected individuals II-1 and III-1 defined the lower border of the shared haplotype (Supplementary Fig. 1), thus, establishing a novel locus for this autosomal dominant, clinically typical and Lewy body confirmed PD in a minimum candidate region (MCR) of 10.7Mb in 20pter-p12, above D20S894 on the short arm of chromosome 20.
Mutations in TMEM230 in PD
There are a total of 141 known genes in this MCR. We initially analyzed exons of 17 protein-coding genes in one affected individual (II-11) by Sanger sequencing, but we did not find any novel variants (Supplementary Table 2). We then employed a whole-exome sequencing approach. We selected one unaffected individual (II-9), who lived to 87 years old without PD, and four distantly related affected individuals (II-4, III-1, III-20 and III-26), so that the number of the co-segregating, but non-PD-linked variants could be reduced.
Across the five individuals, we identified a total of 90,289 variants. We excluded the variants with an average heterozygosity of >0.01 in multiple databases, including the dbSNP (v130), HapMap and 1000 Genome databases. After filtering for functional significance (i.e., nonsense, missense, splice site) and removing the tolerated variants, a single missense variant (c.422G>T, and p.Arg141Leu encoded by TMEM230) was left. This variant was present in four PD patients, but not in the unaffected individual. We performed Sanger sequencing and co-segregation analysis using all additional samples available to us from this family. The Arg141Leu mutation fully co-segregated with the disease in this large kindred (Supplementary Figs. 1 and 3a). All 13 affected individuals whose DNA samples were available for sequencing analysis carried this mutation. This mutation was also presumably present in the other two affected deceased individuals (I-1 and II-8), due to transmission of this mutation to their next generations. This mutation was not present in any SNP databases with a total of 15,184 sequenced alleles (Supplementary Table 4). We further sequenced a control cohort of 1,238 samples from North America, and we did not find this mutation (Supplementary Table 4). Consistent with our linkage data, TMEM230 is located on 20p13, about 5.1Mb from 20pter, in the middle of our mapped 10.7Mb-MCR.
In addition to the TMEM230-Arg141Leu mutation, two other novel variants were identified on the affected chromosome in our unfiltered exome sequencing data. These two variants were also located in the MCR, but in the intronic regions without apparent functional significance (Supplementary Table 5). To make sure that all the exons in the MCR were fully and sufficiently covered, we further Sanger-sequenced all the 10 exons that had a relatively lower coverage of ≤ 20X (2-20X) in our exome sequencing data. We did not find any new variants in the MCR on the affected chromosome. Thus, the TMEM230-Arg141Leu mutation appeared to be the only missense mutation shared by all the patients in this large PD family in this MCR.
To examine whether TMEM230 mutations are present in other PD patients, we analyzed 832 PD DNA samples collected from North America, including 433 familial PD and 399 sporadic PD cases. We found two novel mutations in TMEM230. A c.551A>G was identified in a familial PD patient. This mutation changes the stop codon (X) to a Trp (X184Trp), adding six amino acids (WHPPHS) at the C-terminal (Supplementary Fig. 3b). This male patient had a disease onset at the age of 33 years. His maternal male cousin was described to have PD, with the disease onset at the age of 35 years. No additional information and DNA samples from this family were available for further analysis. The other mutation, c.275A>G (p.Tyr92Cys), was identified in a sporadic male PD patient with the disease onset at age 34 years (Supplementary Fig. 3c). His mother carries this mutation without PD signs when evaluated at 57 years old. Similarly, the X184Trp and Tyr92Cys mutations were not present in SNP databases with a total of 15,184 alleles sequenced and in over 1,000 in-house controls (Supplementary Table 4). The X184Trp and Tyr92Cys were identified in a single patient each, their pathogenicity to PD remains to be further examined. Through our screening, we also found three other missense variants, which are known SNPs, in both PD and control cohorts with similar frequencies (Supplementary Table 4), and obviously these known SNPs did not co-segregate with PD in respective families (Supplementary Fig. 4).
To test if mutations in TMEM230 cause PD in other ethnic PD patients, we extended our genetic study to a Chinese PD cohort of 574 cases, including 225 familial index cases and 349 sporadic cases. We identified a single mutation, c.550_552delTAGinsCCCGGG, in seven familial index cases from seven unrelated families, but not in the sporadic cases (Supplementary Fig. 3d and Supplementary Figs. 5-11). In this mutation, the stop codon TAG is replaced by six nucleotides CCCGGG, resulting in the replacement of the stop codon (X) by proline and glycine (p.X184ProGly) at the protein level, adding seven amino acids (PGHPPHS) at the C-terminal (Supplementary Fig. 3d). Notably, the last five amino acids (HPPHS) are identical in both X184Trp and X184ProGly mutants (Fig. 1a). In two families with additional DNA samples available from other PD patients, we could confirm the co-segregation of disease with this mutation (Supplementary Figs. 8 and 11). In these nine PD patients from seven families with the X184ProGly mutation, five patients from four families are homozygotes (Supplementary Fig. 1d and Supplementary Figs. 5, 6, 9 and 11). To test if mutations in other known PD-linked genes, such as SNCA, LRRK2 and VPS35, or other parkinsonism-linked genes, are present in these Chinese PD patients, we further performed whole-exome sequencing using the DNA samples from these nine Chinese PD patients with the X184ProGly mutation. We did not find new variants in these genes. We Sanger-sequenced DNA samples from our 528 in-house Chinese controls, and we did not find this mutations. Moreover, this mutation was not present in the Chinese exome-sequencing database, including 9,906 neurologically normal Chinese controls, from the BGI (Beijing Genomics Institute)-Shenzhen. Notably, neither of the four TMEM230 mutations identified in PD patients in this study, including Arg141Leu, Tyr92Cys, X184Trp and X184ProGly, was present in the Exome Aggregation Consortium database from a total of 60,706 unrelated individuals.
Figure 1.
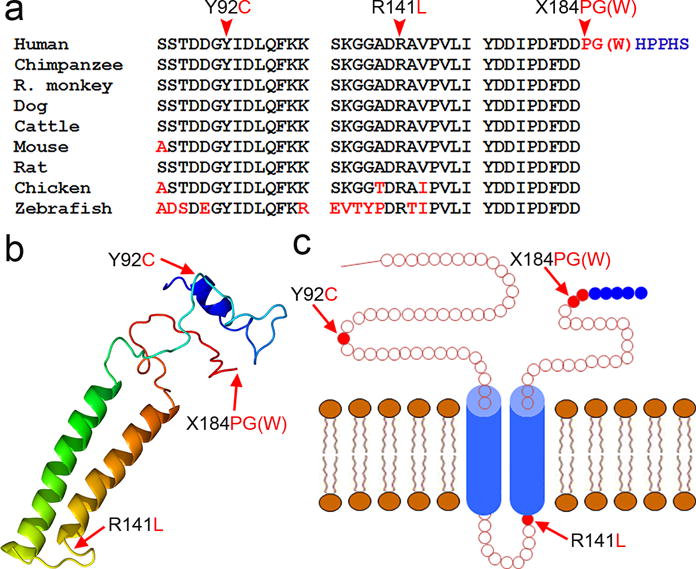
Mutations of TMEM230 in patients with PD. (a) Evolutionary conservation of mutated amino acids in the TMEM230 protein in different species. Amino acids identical to human TMEM230 are in black letters, and non-identical ones are denoted in red letters. The mutated amino acids are indicated by arrowheads on the top. (b) Predicted putative secondary and tertiary structures. The predominant isoform 2 of TMEM230 is shown. Image is colored by rainbow from N (blue) → C (red) terminus. Two major alpha helix regions are shown on the left. Mutation sites are indicated by arrows. (c) Schematic diagram showing the topology of the TMEM230 protein. Two transmembrane segments (aa 109-129, and aa 142-162) are shown in blue, with the N- and the C-terminal regions exposed to the cytosol. Mutations are indicated by arrows. The X184W and X184PG mutations lead to the C-terminal addition of six and seven amino acids, respectively, with the last five amino acids being identical (HPPHS, blue).
The apparent aggregation of the X184ProGly homozygotes in the Chinese familial PD cohort may suggest an increased penetrance of this homozygous mutation in the late-onset PD due to a dose effect. The obligate heterozygous carrier of a homozygous patient had not developed PD before she died at 96 years old (Supplementary Fig. 6), which may suggest an incomplete penetrance of this mutation in heterozygous condition in some mutation carriers.
To test whether the high prevalence of the X184ProGly mutation in the Chinese PD patients is due to a founder effect, we analyzed three TMEM230 intragenic SNPs closely flanking the mutation site in five homozygous patients (Supplementary Fig. 12). The presence of heterogeneity for these SNPs in four out of these five homozygous patients suggests that the X184ProGly arise from independent mutation events, or a very ancient mutation (Supplementary Fig. 12). The X184Trp and X184ProGly were identified in different ethnic populations, but both mutations occur at the stop codon, suggesting a hotspot for the PD-linked mutations. The X184ProGly mutation accounts for 7/225 (3.1%) of Chinese familial PD index cases in our cohort, appearing to be the most prevalent single genetic defect in the Chinese familial PD cases. Notably, the amino acids or codon at Arg141, Tyr92 and X184 are highly evolutionally conserved (Fig. 1a).
TMEM230 in synaptic vesicles in neurons
TMEM230, also known as C20orf30, is a gene yet to be experimentally characterized. There is no apparent sequence homology to any other known genes or proteins. There are four major alternatively spliced mRNA variants from five exons in humans (Supplementary Fig. 13a). The mRNA variant 1 encodes protein isoform 1, a polypeptide of 183 amino acids. The other three mRNA variants (2-4) encode protein isoform 2, a polypeptide of 120 amino acids (Supplementary Fig. 13a). Isoform 2 lacks the N-terminal 63 amino acids of the isoform 1 in humans. Across species spanning zebra fish to most mammals, there is only a single protein isoform of 120 amino acids that is equivalent to human isoform 2. The longer isoform 1 appears to be present only in primates. The N-terminal polypeptides of isoform 1 in the primates are divergent in size and homology. However, the C-terminal polypeptides of 120 amino acids, which are identical to isoform 2, are highly conserved, suggesting the functional importance of isoform 2.
We characterized the transcriptional and translational profiles of human TMEM230. Our data suggest that isoform 2 accounts for >95% of the total protein isoforms (Supplementary Fig. 13).
TMEM230 encodes a transmembrane protein (TMEM230) of unknown location and function. Computer programs for protein structure and function prediction suggested that this protein has two major α-helical domains (Fig. 1b) and two transmembrane segments, with the N- and the C-terminal regions exposed to the cytosol25-27 (Fig. 1c). Notably, all four PD-associated mutations are located in the loop regions (Fig. 1c). The Tyr92Cys, X184Trp and X184ProGly were not predicted to remarkably alter the structure. However, the positively charged amino acid residual Arg141 in TMEM230-Wt was predicted to be located on the luminal site, adjacent to the second transmembrane segment. When the Arg141 is mutated to Leu141, the Leu141 appeared to be a part of the second transmembrane segment and buried in the membrane.
To examine the subcellular localization of TMEM230 protein, we performed immunohistochemical staining of mouse brain sections. TMEM230 appeared to localize to vesicle structures in neurons, including dopaminergic neurons in the substantia nigra (Supplementary Fig. 14). To characterize these structures, we engineered 25 TMEM230 constructs in three expression vectors and performed confocal microscopy using transiently transfected SH-SY5Y and Neuro-2a cells. TMEM230 showed a similar pattern of membrane-bound distribution in both cell types, but Neuro-2a cells revealed better morphological details due to their relatively larger size (Supplementary Fig. 15). Therefore, we used Neuro-2a cells for further characterization. We observed that the vesicle structures labeled by TMEM230 were predominantly located in the perinuclear region as clusters and in other cytosolic areas as isolated vesicles or vesicle clusters (Supplementary Fig. 15a-o). Both isoforms 1 and 2 showed a similar pattern of distribution (Supplementary Fig. 15a-o). An eGFP or DsRed tag to the C-terminus of this protein did not affect its subcellular distribution (Supplementary Fig. 15a-o).
To characterize the vesicle structures labeled by TMEM230, we tested several subcellular organelle markers. TMEM230 did not show apparent co-localization with markers for mitochondria, lysosomes or endoplasmic reticulum (Supplementary Fig. 16a-l). The perinuclear clusters labeled by TMEM230 were in close proximity to the Golgi apparatus, but were distinct from GOLGA2, a marker for the cis-Golgi matrix protein (Supplementary Fig. 16m-p’). We then tested syntaxin 6 (STX6), a transmembrane protein predominantly enriched in the trans-Golgi network (TGN). We found that STX6 co-localized with TMEM230 in these perinuclear clusters and some vesicles (Supplementary Fig. 16q-t), suggesting that the perinuclear structures labeled by TMEM230 represent TGN, and possibly secretory budding and recycling vesicles.
To test whether these secretory vesicles evolve into synaptic vesicles that are the most abundant secretory/recycling vesicles in neurons, we examined synaptophysin (SYP), vesicle-associated membrane protein 2 (VAMP2) and vesicular monoamine transporter 2 (VMAT2), three known transmembrane proteins of synaptic vesicles. We observed co-localization of TMEM230 and these synaptic vesicle markers (Supplementary Fig. 17a-l, and data not shown). The PD-linked mutations did not alter the localization pattern of the protein (Supplementary Fig. 17i-l, and data not shown). We confirmed the co-localization of TMEM230 with mouse endogenous Syp and Vmat2 in mouse embryonic primary neurons, including dopaminergic neurons (Fig. 2a-h). The mouse endogenous Tmem230 was also shown to co-localize to the vesicles with endogenous Syp and Vmat2 in the non-transfected mouse primary neurons (Fig. 2i-p). Immunoblot data from synaptosome fractionation revealed that Tmem230 had an enrichment pattern similar to Syp (Fig. 2q), and immunogold electron microscopy indicated that this protein is located in the synaptic vesicle pool region in the presynapse (Fig. 2r). These data, therefore, indicate that the TMEM230 is primarily a transmembrane protein of synaptic vesicles in neurons.
Figure 2.
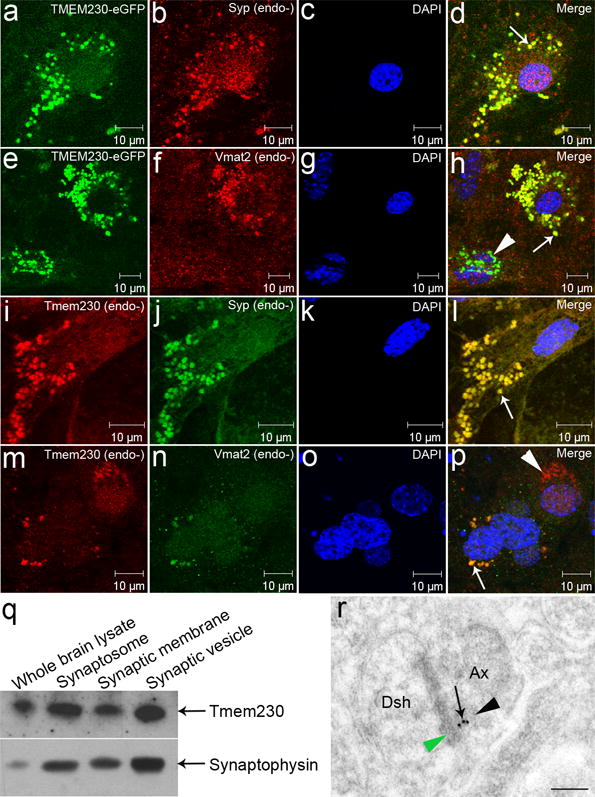
Localization of TMEM230 to synaptic vesicles in neurons. (a-p) Confocal microscopy was performed using transfected (a-h) and non-transfected (i-p) primary neurons from the mouse midbrain. Mouse endogenous (endo-) Tmem230, Syn and Vmat2 were detected with respective antibodies. Representative co-localization signals are indicated by arrows. (e-h) A TMEM230 transfected primary neuron (arrow) shows TMEM230 co-localization with endogenous Vmat2 (dopaminergic neuron); the other transfected neuron (arrowhead) that is TMEM230-positive, but vmat2-negative, suggesting a non-dopaminergic neuron. (m-p) A mouse endogenous Tmem230-positive, but Vmat2-negative (non-dopaminergic neuron) is indicated by an arrowhead. (q) Enrichment of Tmem230 in synaptosomes and synaptic vesicles. Western blot was performed using four fractions isolated from rat brains as indicated. Tmem230 shows a similar enrichment pattern to synaptophysin. (r) Localization of Tmem230 in the presynaptic vesicle pool region. Immunogold electron microscopy was performed using rat brain sections. A representative perforated synapse in rat CA1 region is shown in the center of the image. Presynaptic axonal terminal (Ax) and dendritic spine head (Dsh) are shown. Tmem230-positive gold particles are indicated by an arrow. The presynaptic vesicle pool region and postsynaptic density are indicated by black and green arrowheads, respectively.
TMEM230 in recycling endosomes
Synaptic vesicles are continuously recycled through endocytic mechanism following exocytosis in order to replenish the reserve vesicle pool28. RAB GTPases and other signal proteins coordinate the sorting, trafficking and recycling of these vesicles. Most early endocytic steps rely on RAB5A, which mediates fusion of endocytic vesicles to form the early endosomes29. To test if TMEM230 is present in early endosomes, we co-transfected cells with TMEM230 and RAB5A constructs, and found that TMEM230 and RAB5A co-localized together in early endosomes (Supplementary Fig. 18a-d).
Early endosomes have three distinct destinations, depending on the sorting signals on the early endosomes: degradation in the lysosomes, recycling to the plasma membrane or retrieval to the Golgi apparatus. We tested several representative RAB and other proteins. We found that the TMEM230 signals partially and weakly co-localized with RAB7 (Supplementary Fig. 18e-h), a protein that contributes to the biogenesis of late endosomes and autophagosomes, and subsequent trafficking to lysosomes. Similarly, the TMEM230 signals also partially and weakly co-localized with MAP1LC3A (Supplementary Fig. 18i-l), an autophagosome marker. We observed that majority of the RAB11A-labeled vesicles were consistently TMEM230-positive (Supplementary Fig. 18m-p), suggesting that a major fraction of the TMEM230-labeled synaptic vesicles evolves into recycling endosomes, leading to the direct reuse of the synaptic vesicles as expected.
Endosome retrieval to the TGN is mediated by retromer complex. VPS35 is a component of the retromer. A D620N mutation in VPS35 has been previously reported in several autosomal dominant PD families10,11. To test whether TMEM230 is involved in the vesicles retrieval to TGN by retromers, we transfected Neuro-2a cells with both TMEM230 and VPS35 constructs, and we observed a remarkable fraction of the TMEM230 signals overlapped with VPS35 signals (Fig. 3a-d). We further confirmed this finding using primary neurons (Fig. 3e-p). TMEM230 and VPS35 have distinct physiological localizations, with TMEM230 to synaptic vesicles, and VPS35 to retromers. The convergence of these two PD-linked proteins in the endosomal trafficking to TGN pathway suggests that the PD caused by mutant VPS35 and TMEM230 may share a common pathogenic pathway that is related to defects in synaptic vesicle trafficking and recycling.
Figure 3.
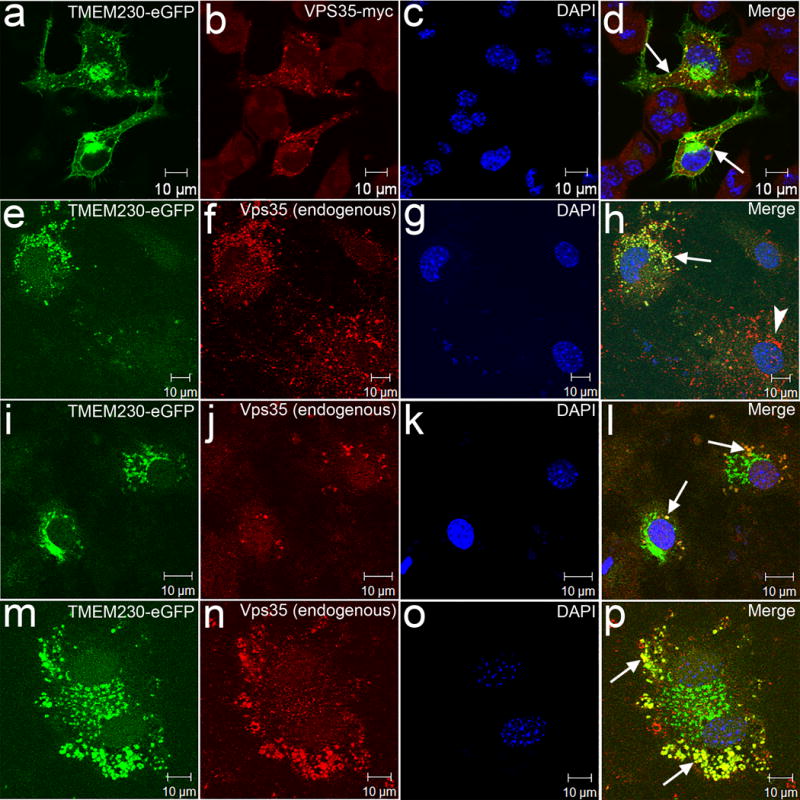
Convergence of TMEM230 and VPS35 in vesicle trafficking/recycling to TGN. (a-d) Confocal microscopy was performed using Neuro-2a cells transfected with TMEM230-eGFP construct together with VPS35-myc as indicated. (e-p) Confocal microscopy was performed using mouse primary neurons transfected with TMEM230-eGFP. The mouse endogenous Vps35 was detected with an antibody. Representative images showing the overlap of the signals (arrows) of TMEM230 and mouse endogenous Vps35. (e-h) A less TMEM230 transfected neuron showing Vps35, but only marginal TMEM230-eGFP is indicated by an arrowhead.
TMEM230 in Lewy bodies
Lewy bodies and Lewy neurites in surviving neurons of the affected brain regions are pathological hallmarks of idiopathic PD, autosomal dominant PD and dementia with Lewy body (DLB), suggesting a shared pathogenic pathway30. SNCA, a dominant PD-linked protein, is a major component of the Lewy bodies and Lewy neurites31. We tested whether TMEM230 is also involved in the formation of the Lewy bodies and Lewy neurites. We analyzed midbrain and neocortex sections from 10 sporadic PD cases and 7 DLB cases, respectively. We found that SNCA-positive Lewy bodies and Lewy neurites in these 17 cases were also consistently TMEM230-positive, suggesting that TMEM230 is a constitutive component of Lewy bodies and Lewy neurites (Supplementary Fig. 19).
Impairment of synaptic vesicle trafficking by mutations
Synaptic vesicle transmembrane proteins are of two obligatory classes: (I) transport proteins that are involved in the uptake of neurotransmitters and other components into synaptic vesicles; and (II) trafficking proteins that mediate the intracellular traffic of synaptic vesicles. The transport proteins of synaptic vesicles are generally large transmembrane proteins with 8-12 transmembrane segments. The much smaller molecular size and fewer transmembrane segments of TMEM230 compared to the known neurotransmitter transport proteins indicate that TMEM230 is likely to be a trafficking protein, rather than a neurotransmitter-transport protein of the synaptic vesicles. In the transfected primary neurons, the movement of the synaptic vesicles could be readily observed with live imaging microscopy. The dispersive pattern of this movement in the soma may be related to vesicle trafficking to their functional destinations32,33. We analyzed the synaptic vesicle movement by live imaging approach and found that all four PD-linked mutants led to significantly slower movement of the synaptic vesicles, as shown by reduced transport speed, track length and displacement length when compared to wild-type (Fig. 4).
Figure 4.
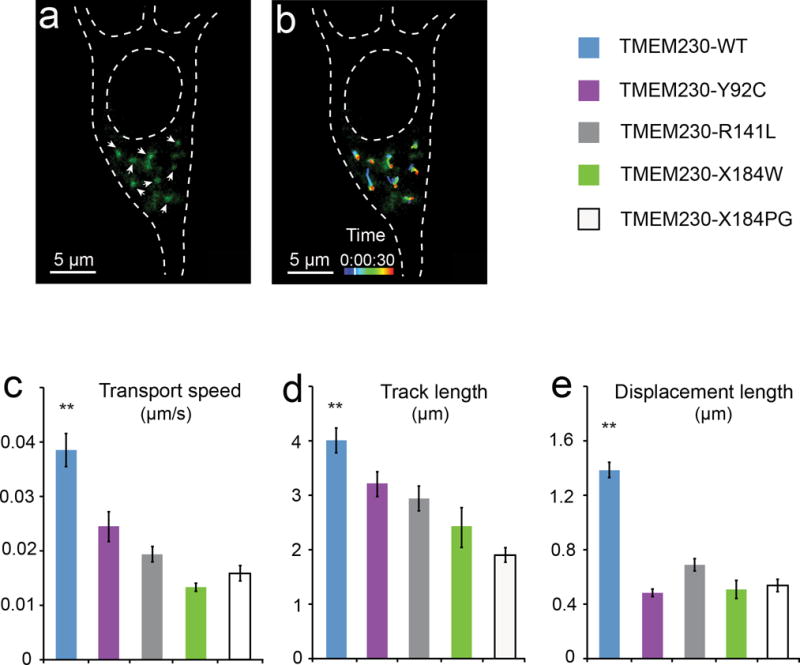
Impairment of synaptic vesicle trafficking by PD-linked mutant TMEM230. (a) Fluorescent image of synaptic vesicles in transfected primary neurons. Cultured mouse midbrain primary neurons were co-transfected with the GFP-tagged VAMP2 expressing vector (pEGFP-VAMP2) and each of the TMEM230 tag-free constructs in the pIRES2-ZsGreen1 vector. The pEGFP-VAMP2 was used to label the synaptic vesicles (Supplementary information). White arrowheads mark representative GFP-VAMP2-labeled vesicles quantified in panels b-e. (b) Movement track of the GFP-VAMP2-labeled vesicles. The trafficking of the vesicles in live primary neurons is monitored by confocal microscope and quantified using the Imaris software package. The movement tracks of eight representative vesicles from time-lapse live confocal images (30 frames with 5-second intervals) are shown, with pseudocolored movement tracks marking the position of vesicles at different time points during the 150 seconds. Images shown in panels (a) and (b) are taken at the time point marked on the pseudocolored time scale. (c-e) Quantification of the mean transport speed, track length and displacement length of GFP-VAMP2-labeled vesicles. Vesicles in neurons expressing PD-linked mutants (TMEM230-Y92C, TMEM230-R141L, TMEM230-X184W and TMEM230-X184PG) show impaired movement, as measured by transport speed (c), track length (d) and displacement length (e), when compared to the wild-type (TMEM230-WT). Data are from four independent experiments (1055 moving vesicles in 27 neurons). ** p < 0.01 (Oneway ANOVA test). Error bars, mean ± s.e.m.
Increased expression of SNCA by duplication and triplication has been shown to cause PD34-36. Our observations of the co-localization of SNCA and TMEM230 in some vesicles and vesicle clusters in the PD autopsy samples, together with the vesicle trafficking impairment by mutant TMEM230 raise the possibility that mutant TMEM230 may lead to impaired SNCA degradation and therefore, increased SNCA levels in cells. We explored this possibility in cellular models. Indeed, mild to moderate, but consistent increases in the SNCA level were observed in cells expressing the mutants when compared to those expressing the wild-type (Supplementary Fig. 20).
DISCUSSION
We report here a fully co-segregating mutation in TMEM230 in one of the largest families with autosomal-dominant, clinically typical and Lewy body confirmed PD. A variant Asn855Ser in DNAJC13 was recently reported in this family37. But this variant did not fully co-segregate with the disease, as shown by the absence of this variant in three affected individuals (II-1, III-1 and III-23)37. Moreover, this variant was present in the unaffected individual (II-9), who had not developed any signs of PD when she died at the age of 87 years. The inconsistency of the phenotype-genotype correlation in multiple individuals raised the concern that the variant Asn855Ser in DNAJC13 might not be the disease-causing mutation in this family, as the authors also noted37. We performed linkage analysis and established a new PD locus to 20pter-p12 using this family. Our whole-exome sequencing complemented with Sanger sequencing covered all the known transcripts in the MCR, and revealed the c.422G>T (p.Arg141Leu) mutation in TMEM230 to be the only missense mutation that fully co-segregates with the disease in the entire family. Moreover, we also found a highly prevalent TMEM230 mutation, X184ProGly, in nine PD patients from seven Chinese families. These data provide compelling genetic evidence that TMEM230 is the new PD-causing gene on the chromosome 20p locus. These data also indicate that although the disease modifying effect by the DNAJC13-Asn855Ser could not be excluded37, it is not the PD-causing mutation in this large PD family.
Among autopsy samples from four deceased patients (II-1, II-4, II-6 and II-14) in second generation, three had confirmed Lewy body pathology. The patient II-1 had all cardinal symptoms of PD, with a moderate response to levodopa. In addition, this patient had other clinical signs, including pseudo bulbar affect, dysphagia and dementia. Pathological analysis of autopsy sample from this patient revealed progressive supranuclear palsy (PSP) pathology. But his son (III-1) developed typical PD symptoms at the age of 49 years, with good response to levodopa. It remains to be further investigated why the same TMEM230-Arg141Leu mutation leads to PSP in one case, but PD in the others in this family. However, this phenotypic heterogeneity may not be an exception, because some patients with the Arg1441Cys or Arg1441His mutation in LRRK2, two well-established PD-causing mutations, have also been shown to present with clinical and pathological features of PSP7,38,39. These data may suggest an etiological link between PD and PSP.
PD and other parkinsonian disorders are typically sporadic, but disease-causing mutations in several genes have been identified in monogenically inherited forms. Among these genes, TMEM230 appears to be the only gene that encodes a transmembrane protein of the synaptic vesicles. Synaptic vesicles are the most abundant secretory/recycling vesicles in neurons. The precise functions of TMEM230, and pathogenic mechanism of the TMEM230-mediated PD remain to be further elucidated. Based on its molecular features, and its relationship with synaptic vesicle and endosomal markers tested in this study, we propose that TMEM230 is a trafficking protein of secretory/recycling vesicles, primarily involved in exocytosis, endocytosis and recycling of the synaptic vesicles in neurons; and dysfunction of the synaptic vesicles, such as impaired trafficking and recycling, underlies the pathogenesis of the TMEM230-linked PD.
Based on the nomenclature proposed by Langston et al1, TMEM230 appears to be the third disease-causing gene linked to clinically typical and Lewy body confirmed PD after SNCA and LRRK21. VPS35 is linked to autosomal dominant dopa-responsive parkinsonism. However, limited brain tissue analysis failed to reveal Lewy body pathology, thus did not allow definitive conclusions for Lewy body disease10,40. Our data suggest that TMEM230 and VSP35 may share a functional pathway in synaptic vesicle/endosome retrograde trafficking. Interestingly, LRRK2 may also be involved in vesicle trafficking processes. A recent study has shown that LRRK2 phosphorylates a subset of Rabs, which play important roles in intracellular vesicle trafficking; and PD-linked LRRK2 mutations increase the phosphorylation of these Rabs and inhibit their activity, which may disturb intracellular vesicle trafficking41. Thus, the defects of synaptic vesicle/endosome trafficking may be a convergent pathway in the pathogenesis of PD. Our findings also imply that improvement of the synaptic vesicle/endosome trafficking may have therapeutic effect. Indeed, NAB2, a small molecule that promotes endosomal trafficking, has recently been shown to be able to reverse PD-related pathological phenotypes in PD patient-derived neurons with a SNCA-A53T mutation42,43.
Supplementary Material
Acknowledgments
This study was supported by the American Parkinson’s Disease Association, the US National Institutes of Health (NS074366, NS37167, NS078287, NS094564, AG043970 and NS095972), National Natural Science Foundation of China (81271921, 81430023 and 81471300), the Les Turner ALS Foundation/Herbert and Florence C. Wenske Foundation Professorship, George Link, Jr. Foundation, the Les Turner ALS Foundation, and The Foglia Family Foundation. Whole exome sequencing was performed at the John P Hussman Institute for Human Genomics, University of Miami, Miller School of Medicine. Imaging work was performed at the Northwestern University Cell Image Facility supported by NIH (CA060553).
Footnotes
Author contribution
H.-X.D., A.H.R. and T.S. designed this study. H.-X.D., Y.S., Y.Y., K.B.A., M.A.P-V and T.S. performed linkage analysis. H.-X.D., Y.S., Y.Y., K.B.A., C.H., H.Z., S.D., K.N., H.D., B.T., Z. Y., Y.X., P.C., B.H., Z.S., Z.L., J.M.V and T.S. performed sequencing analysis. H.-X.D., Y.S., N.M., L.C., H.Z., F.F. and Y.-C.M. performed cell culture, immunoblot, immunohistochemistry and confocal microscopy. N.M. and Y.-C.M. performed primary neuron culture and trafficking assay. M.J.K. and D.K. performed the SNCA degradation assay. N.J.C. and D.A.N. performed immunogold electron microscopy. N.S. and A.H.R. collected family information and coordinated this study. B.T., Y.X., P.C., X.-P.G., Z.S., Z.L., J.J., J.M.V., O.M., A.H.R. and T.S. performed clinical studies. T.F., B.G., and A.H.R. performed pathological study and provided pathological samples. H.-X.D., A.H.R. and T.S. analyzed the data and wrote the paper.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Langston JW, Schule B, Rees L, Nichols RJ, Barlow C. Multisystem Lewy body disease and the other parkinsonian disorders. Nat Genet. 2015;47:1378–84. doi: 10.1038/ng.3454. [DOI] [PubMed] [Google Scholar]
- 2.Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–61. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Leroy E, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–2. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 6.Paisan-Ruiz C, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 7.Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Lautier C, et al. Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet. 2008;82:822–33. doi: 10.1016/j.ajhg.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strauss KM, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14:2099–111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 10.Vilarino-Guell C, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–7. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimprich A, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chartier-Harlin MC, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89:398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 14.Valente EM, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 15.Bonifati V, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 16.Paisan-Ruiz C, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shojaee S, et al. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet. 2008;82:1375–84. doi: 10.1016/j.ajhg.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krebs CE, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–7. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quadri M, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 2013;34:1208–15. doi: 10.1002/humu.22373. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 21.Edvardson S, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7:e36458. doi: 10.1371/journal.pone.0036458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mata IF, et al. The RAB39B p.G192R mutation causes X-linked dominant Parkinson’s disease. Mol Neurodegener. 2015;10:50. doi: 10.1186/s13024-015-0045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Key mendelian variants. Nat Genet. 2015;47:1371. doi: 10.1038/ng.3463. [DOI] [PubMed] [Google Scholar]
- 24.Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet. 2011;12:301–25. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 26.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kallberg M, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–22. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 29.Bucci C, et al. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–28. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- 30.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 31.Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 32.Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–55. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song AH, et al. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–60. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Fuchs J, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–22. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- 35.Ross OA, et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63:743–50. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 37.Vilarino-Guell C, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23:1794–801. doi: 10.1093/hmg/ddt570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wszolek ZK, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62:1619–22. doi: 10.1212/01.wnl.0000125015.06989.db. [DOI] [PubMed] [Google Scholar]
- 39.Spanaki C, Latsoudis H, Plaitakis A. LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology. 2006;67:1518–9. doi: 10.1212/01.wnl.0000239829.33936.73. [DOI] [PubMed] [Google Scholar]
- 40.Wider C, et al. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord. 2008;14:465–70. doi: 10.1016/j.parkreldis.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 41.Steger M, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5 doi: 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tardiff DF, et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science. 2013;342:979–83. doi: 10.1126/science.1245321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung CY, et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342:983–7. doi: 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.