

Summary
Sonic hedgehog (SHH) is a master developmental regulator. In 1995, the SHH crystal structure predicted that SHH-E176 (human)/E177 (mouse) regulates signaling through a Zn2+-dependent mechanism. While Zn2+ is known to be required for SHH protein stability, a regulatory role for SHH-E176 or Zn2+ has not been described. Here, we show that SHH-E176/177 modulates Zn2+-dependent cross-linking in vitro and is required for endogenous signaling, in vivo. While ectopically expressed SHH-E176A is highly active, mice expressing SHH-E177A at endogenous sites (ShhE177/−) are morphologically indistinguishable from mice lacking SHH (Shh−/−), with patterning defects in both embryonic spinal cord and forebrain. SHH-E177A distribution along the embryonic spinal cord ventricle is unaltered, suggesting that E177 does not control long-range transport. While SHH-E177A association with cilia basal bodies increases in embryonic ventral spinal cord, diffusely distributed SHH-E177A is not detected. Together, these results reveal a novel role for E177-Zn2+ in regulating SHH signaling that may involve critical, cilia basal-body localized changes in cross-linking and/or conformation.
Graphical abstract
Introduction
Sonic hedgehog (SHH) is a highly conserved signaling protein required for ventral fate specification throughout vertebrate embryonic development (Dessaud et al., 2008; Echelard et al., 1993; Ingham and Placzek, 2006). SHH exerts its cellular patterning effects through the combined action of short-and long-range signaling (Dessaud et al., 2008). Early in neural development, SHH secreted from the notochord induces differentiation of the overlying floor plate in a contact-dependent manner (Jessell, 2000; Roelink et al., 1995). Subsequently, SHH acts at a distance to specify more dorsally positioned cell types in the developing spinal cord (Briscoe and Ericson, 2001). In anterior regions, where the notochord is absent, SHH expression from the midline specifies developing forebrain neurons (Kohtz et al., 1998).
SHH undergoes a series of processing events before it is secreted from the cell (Bumcrot et al., 1995). The active SHH protein fragment is a 19kD signaling domain generated by signal sequence cleavage and subsequent autoproteolysis of the 45kD precursor (Lee et al., 1994). During autoproteolysis, the 19kD fragment can be modified at its C-terminus by cholesterol (Porter et al., 1995; Porter et al., 1996) and by a palmitate at the N-terminal amino acid (Pepinsky et al., 1998). The roles of N-terminal palmitate and C-terminal cholesterol (N-and C-lipid) post-translational modifications in SHH activity and transport are region-and species-specific (Ingham, 2001; Ingham and McMahon, 2001). A combination of different N-and C-lipid modified SHH protein forms can be isolated from mouse embryonic brain and limbs (Feng et al., 2004). In cell culture and explant assays, the N-lipid increases SHH-induced cellular differentiation compared to the unmodified form (Kohtz et al., 1998; Kohtz et al., 2001; Pepinsky et al., 1998; Williams et al., 1999). This is consistent with the finding that both mutant mice lacking palmitoylation and Shh−/− mice display similar ventral patterning defects (Chen et al., 2004; Chiang et al., 1996).
The role of the C-lipid in SHH activity is less well defined. Cell culture studies demonstrate that the SHH C-lipid is required for cell surface association (Bumcrot et al., 1995; Lee et al., 1994), while the C-lipid plays an important role in SHH long-range signaling in mice (Huang et al., 2007; Lewis et al., 2001). Interestingly, forebrain neural explant assays indicate that the C-lipid reduces SHH activity (Feng et al., 2004).
The role of N-and C-lipids in Shh multimerization in vitro has previously been reported (Chen et al., 2004; Feng et al., 2004; Zeng et al., 2001). N-and C-lipid content determine the extent of multimerization, with multimerization increasing SHH activity in forebrain neural explant assays (Feng et al., 2004) as well as cell lines (Chen et al., 2004; Zeng et al., 2001). Previous reports that the largest multimers, formed in the presence of both N-and C-lipids, have reduced signaling capacity (Feng et al., 2004), led to idea that soluble SHH multimers exhibit different activities based on size. Together, these data led to the proposal that SHH multimerization hides hydrophobic side chains enabling long-range transport (Zeng et al., 2001), while the C-lipid maintains the SHH protein in an inactive state during long-range transport (Feng et al., 2004), thereby requiring an activation mechanism. Conversion between active and inactive SHH states was proposed, but not shown.
In addition to lipid modifications, SHH binds heparin through an N-terminal Cardin-Weintraub sequence (Rubin et al., 2002), that also contains a conserved site required for SHH multimerization (Goetz et al., 2006). The Cardin-Weintraub motif was predicted to play a role in Shh multimerization through a heparin sulfate (HS) dependent mechanism (Dierker et al., 2009a). In these studies, specific HS sulfations are required for metalloprotease-mediated release of lipidated SHH from the cell membrane (Dierker et al., 2009b). In Drosophila, a critical Lys residue is essential for the formation of Hh multimers, which localize to HS-proteoglycans and participate in long-range transport (Vyas et al., 2008). These studies further support a role for SHH multimerization in signaling in vivo.
Despite extensive work on the relationship between SHH biochemistry and signaling activity in vivo, a major question still remains regarding the role of Zn2+ in SHH signaling, as predicted by crystal structure analysis of the SHH amino terminal fragment (Hall et al., 1995). Based on Zn2+ coordination that is structurally homologous to the active site in known Zn2+ hydrolases, SHH glutamate 177 (human, E176; mouse E177) was proposed to be the catalytic amino acid required for proteolytic activity (Hall et al., 1995). Site-specific mutations in human (Day et al., 1999) and mouse (Fuse et al., 1999) SHH proteins show that mutation of two (H140 and D147) of four Zn2+ coordinating residues results in significant loss of the bound Zn2+ and reduced protein stability (Day et al., 1999). However, SHH-E177A/E176A mutant proteins not only bind Zn2+, but are equivalent to wild-type SHH (wtSHH) in signaling assays, as well as patched 1 (PTC1) receptor binding (Day et al., 1999; Fuse et al., 1999). Additional experiments failed to identify SHH-mediated proteolytic activity (Day et al., 1999) or a requirement of E177/E176 in metalloprotease-dependent secretion (Dierker et al., 2009b).
To date, all published experiments testing SHH-E177A/E176A activity have been performed in cell lines, spinal cord explants, or Drosophila embryos (Day et al., 1999; Fuse et al., 1999). Because of the lack of differences in activity between SHH-E177A/E176A and wtSHH proteins, it is presently thought that E177 does not play a role in SHH signaling.
This report demonstrates for the first time that SHH-E177/E176 is required for SHH signaling at endogenous, but not ectopic sites in the mouse embryo. We show that E176/E177 modulates a Zn2+-mediated conformational change, detected by formation of cross-linked dimers in mutated SHH-E176A proteins. In ShhE177A/− mouse mutant spinal cord, SHH-E177A accumulates near cilia basal bodies (BBs), but fails to signal. Therefore, we propose a model whereby E176/E177-Zn2+ is critical for SHH activity near cilia BBs in the mouse embryo, affecting SHH conformation/cross-linking during pre-and/or post-signaling steps.
Results
A conformation-specific antibody recognizes SHH cross-linked dimers and basal body-associated SHH in the mouse embryonic spinal cord
Previous studies demonstrated the presence of cross-linked forms of SHH in embryonic protein extracts by Western analysis (Feng et al., 2004). It was not known whether cross-linking occurred during SHH protein preparation or whether this reflected a biologically significant event that plays a role in SHH activity. In order to detect cross-linked SHH in vivo, we used α-SHHCL/P, an antibody generated against cross-linked human SHH N-terminal 197 amino acids. Anti-SHHCL/P recognizes SDS-resistant, cross-linked wtSHH (wtSHHCL), but not soluble monomers of wtSHH (N-and C-lipid containing SHH purified from C17 cells) or uSHHNM (recombinant unmodified human SHH purified from E. coli, lacking both N-and C-lipids (Kohtz et al., 2001), and also lacking a 6-histidine tag, removed by proteolytic cleavage, (Kohtz et al., 2001; Williams et al., 1999)) (Figure 1A).
Figure 1. Conformation-specific antibody (α-SHHCl/P) recognizes cross-linked wtSHH in vitro and punctate, cilia BB-associated SHH in embryonic spinal cord ventricles.

A. uSHHN (lipid lacking N terminal fragment), cross-linked wtSHH (wtSHHCL, N-and C-Lipid containing), and soluble monomeric wtSHH (wtSHH monomer, N-and C-lipid containing) are separated by SDS-PAGE and probed by Western blot analysis using α-SHHCl/P and H160 (α-SHH, Santa Cruz). α-SHHCl/P recognizes wtSHHCL, but not soluble monomeric wtSHH or monomeric uSHHN. H160 recognizes monomeric uSHHN and monomeric wtSHH, but not cross-linked wtSHHCL. Schematics for generating wtSHHCL,mono (N-terminal palmitate (Palm) and C-terminal cholesterol (Chol) containing) and uSHHN (lipid lacking) are shown on the right. B. Immunocytochemistry using α-SHHCl/P (green) and DAPI stain for nuclei (blue) detects SHH in C17 cell lines stably transfected with wtSHH, but not C24S-SHHN (N-and C-lipid lacking), and non-transfected control cells (C17). α-SHHCl/P detects wtSHH, but does not detect C24S-SHHN, as it is not expected to remain associated with the membrane. No staining is detected in non-transfected control cells, C17. C. Schematic of coronal section through the E9.5 spinal cord and notochord, with dashed boxes highlighting the regions shown in each subsequent image. D–L. Confocal analysis of immunofluorescent staining in E9.5 spinal cord using α-SHHCl/P (green; D-G′, I, L) or H160 (green; H, J, K) with γ-tubulin (cilia BBs, red; D–F, F′, H–L) or α-acetylated α-tubulin (axonemes, α-tubulin, red; G, G′). Notochord staining using γ-tubulin and α-SHHCl/P (I) or H160 (J). Shh−/− spinal cord staining using H160 (K) and α–SHHCl/P (L). DAPI (nuclei, blue). (F′–G′) Threedimensional reconstructions of ten confocal images taken sequentially along the z-axis from images F and G, respectively (AMIRA software). M. Quantification of the numbers of SHHCL/P puncta (green bars) or SHHCL/P/γ-tubulin double labeled puncta (yellow bars) comparing 2000μm2 regions in INT or VENT regions. *p<0.05, Student’s t-test. N. Schematic of the ventral spinal cord depicting the location of SHHCL/P detected by α-SHHCl/P (cilia BB-associated, black puncta) and H160 (non-BB associated, soluble, diffuse SHH, gray area). Scale bars = 5 μm (D–G, H–L) and 0.5 μm (F′-G′).
In Figure 1A, wtSHHCL migrates at ~52kD, presumably dimers, whereas wtSHH monomers migrate ~20kD (Feng et al., 2004). Since wtSHHCL and wtSHH are identical in primary sequence, α-SHHCL/P specifically recognizes SHH after cross-linking. Failure of α-SHHCL/P to recognize dual lipid-modified wtSHH monomers (wtSHHM), suggests that α-SHHCL/P is not a lipid-specific antibody. The commercially available α-SHH antibody H160 (generated against SHH N-terminal 160 amino acids, Santa Cruz) recognizes soluble monomeric uSHHNM and wtSHH monomers, but not wtSHHCL (Figure 1A). Therefore, α-SHHCL/P and H160 differentially recognize SHH based on dimer/monomer formation.
We next determined α-SHHCL/P specificity in cell lines that express different SHH proteins, wtSHH (N-and C-terminal lipid-containing SHH protein made) and C24S-SHHN (N-and C-terminal lipid-lacking SHH protein) Fig 1B. Mutation of C24S prevents N-terminal lipid modification in vivo, while expression of the N-terminal fragment of SHH prevents addition of the C-terminal lipid, generating C24S-SHHN that lacks both N and C-terminal lipids in C17 cells, as previously described (Feng et al., 2004). WtSHH was previously shown to form both soluble multimers and cross-linked aggregates, and remains predominately membrane bound (Feng et al., 2004). In contrast, C24S-SHHN fails to form soluble multimers, and is predominately secreted (Feng et al., 2004). The ability of α-SHHCL/P to recognize wtSHH expressed by C17 neural cells supports recognition of SHH in its native conformation in vivo.
It is now well established that essential SHH signaling components localize to primary cilia (Corbit et al., 2005; Haycraft et al., 2005; Rohatgi et al., 2007; Zimmerman and Yoder, 2015). In addition, GFP fusion of SHH has shown close association with γ-tubulin at cilia BBs (Chamberlain et al., 2008). Therefore, we next determined whether α-SHHCL/P identifies SHH associated with cilia BBs (γ-tubulin) and/or cilia axonemes (acetylated α-tubulin). In E9.5 mouse spinal cord (Fig 1C), •C), CL/Pidentifies SHH in puncta restricted to the apical domain of neuroepithelial cells lining the ventricle (Fig 1D–G). Therefore, α-SHHCL/P identifies an epitope shared between SHHCL proteins in vitro, and punctate, cilia BB-associated SHH proteins in embryonic spinal cord ventricles, in vivo. This protein is referred to as SHHCL/P. In E9.5 spinal cord, SHHCL/P ventricular accumulation is greater in ventral (Figures 1F–G) and intermediate regions (Fig 1E), compared to dorsal (Fig 1D). Cilia BBs (γ-tubulin) and cilia axonemes (acetylated α-tubulin) are also found in puncta along the apical spinal cord (Figures 1D–L). Three-dimensional surface rendering of sequential z-axis images (AMIRA software) magnifies SHHCL/P association near cilia BBs (Fig 1F′) and cilia axonemes (Fig 1G′). The H160 antibody detects non-BB associated SHH, (referred to as diffuse SHH) in the floor plate (Fig 1H) and notochord (Fig 1J). H160 staining of SHH is distinct in both appearance and localization when compared to α-SHHCL/P. Staining with α-SHHCL/P detects puncta in the notochord (Fig 1I), but not in the basal region of the floor plate.
Quantification of SHHCL/P puncta in E9.5 spinal cord shows that the number of puncta does not differ between ventral and intermediate regions (Figure 1M, green bars). However, the number of SHHCL/P puncta associated with cilia BBs increases in intermediate compared to ventral spinal cord (Fig 1M, yellow bars), in agreement with results reported for SHH-GFP (Chamberlain et al., 2008). Quantitative analysis at E9.5 supports that SHHCL/P association with cilia BBs is increased in the intermediate spinal cord compared to ventral regions. Figure 1N is a schematic of the Shh+/+ ventral spinal cord showing the relative location of puncta identified by α-SHHCL/P (dark circles) and diffuse SHH identified by H160 (gray region). The absence of staining in C24S-SHHN and C17 cells lacking SHH supports α-SHHCL/P specificity for SHH (Fig 1B). In addition, Shh−/− mice do not stain for H160 (Figure 1K) or α-SHHCL/P (Figure 1L), supporting antibody specificities. Together, these experiments support that α-SHHCL/P specifically recognizes a SHH conformation shared by cross-linked SHH in vitro and basal body co-localized SHH in the embryonic ventral spinal cord. This is referred to as cross-linked conformation.
Key regulators of SHH signaling control SHHCL/P distribution in the embryonic ventral spinal cord
In order to further characterize the relationship between SHHCL/P and cilia BB association in vivo, we determined the role of regulators of the SHH pathway (SHH receptor patched (PTC1), SHH cholesterol modification, and intraflagellate transport protein 172, IFT172) on SHHCL/P distribution in the embryonic ventral spinal cord. The SHH receptor PTC1 binds and sequesters SHH (Incardona et al., 2000; Marigo et al., 1996) and localizes to cilia axonemes (Rohatgi et al., 2007). Mice lacking Ptc1 exhibit neural tube defects and die beginning from E9.0 (Goodrich et al., 1997). Therefore, we next examined SHHCL/P localization in Ptc1− /− E8.75 spinal cord, comparing diffusely distributed SHH with SHHCL/P (Figure 2A–F). In Ptc+/+, SHHCL/P associates with γ-tubulin (Figure 2B, B′) and α-tubulin (Figure 2C, C′) at cilia BBs and axonemes, respectively, along the ventricle. Upon loss of Ptc1, both diffuse SHH and SHHCL/P are affected, with less restriction to the floorplate (Figure 2D–F, 2D′–F′). SHHCL/P forms large aggregates lining the ventricle and at a distance from the ventricle (Figure 2E, E′, F, F′), indicating greater diffusion and loss of discrete puncta in the absence of PTC1. Increased SHH protein expression is expected, as increased Shh RNA expression is observed in Ptc1−/− mice (Goodrich et al., 1997). However, increased SHH expression in Ptc1−/− ventral spinal cord cannot account for the extensive nature of SHHCL/P aggregation and diffusion, supporting that PTC1 sequesters both diffuse SHH and punctate SHHCL/P in vivo, with a significant role in localizing SHHCL/P to cilia BB’s.
Figure 2. SHHCL/P localization in the embryonic ventral spinal cord of Ptc1−/−, ShhN/, and Ift172/Wimple mice.

SHHCl/P co-localization with cilia BBs is determined using immunofluorescence microscopy in embryonic ventral spinal cord sections of mice lacking PTC1, Shh cholesterol modification, and IFT172 mutation (Wimple). E8.75 ventral spinal cord: Shh+/+ (A–C, A′–C′), Ptcl−/− (D–F, D′–F′). E9.5 ventral spinal cord, Shh+/+ (G, H, G′, H′), ShhN/−, lacking the C-terminal cholesterol modification, (I,J, I′,J′), Ift172/Wimple (K, L, K′, L′). White dotted lines outline ventral spinal cord ventricles and notocord. Anti-SHH antibodies (α-SHHCL;P and α-SHHCL;M/D) are green. Anti-γ-tubulin detects cilia BBs in red (A, B, D, E, A′, B′, D′, E′, G–L, G′–L′, M)). Anti-a-tubulin detects cilia axonemes in red (C, C′, F, F′). Loss of PTC1 causes increased diffusion and aggregation of SHHCL/P (H, H′, I, I′), and diffusion of SHHCL;M/D (G, G′). α-SHHCL;M/D recognizes floorplate and notochord localized SHH, but not cilia BB associated SHH puncta. M. Anti-SHHCL;M/D (green) does not recognize targets in Shh−/− E9.5 ventral embryonic spinal cord, γ-tubulin (red). N. Western analysis using α-SHHCL;M/D recognizing an epitope shared by both wtSHHCL and uSHHN proteins. Scale Bars: 10μm (A–M), 5μm (A′–L′).
Given the role of the SHH C-terminal cholesterol (C-lipid) in membrane retention (Porter et al., 1995; Porter et al., 1996), we next compared the effect of C-lipid loss on diffuse and SHHCL/P localization in ShhN/ E9.5 ventral spinal cord (Figure 2I, I′, J, J′). In ventricles and floorplate of ShhN/, SHHCL/P and diffuse SHH are either reduced or absent. The absence of diffuse SHH in floorplate, but not in the notochord agrees with previously reported effects on SHH localization in ShhN/ spinal cord (Huang et al., 2007). Loss of the SHH cholesterol does not completely eliminate SHHCL/P –cilia BB association (Figure 2J′), suggesting that the SHH C-lipid facilitates, but is not required for SHHCL/P –cilia BB association.
We next asked whether an intraflagellate transport (IFT) protein known to affect SHH signaling through ciliary defects, affects SHHCL/P localization in the embryonic spinal cord. Ift172/Wimple mice contain a null mutation in IFT172, lack nodal cilia, and show reduced SHH signaling (Huangfu and Anderson, 2005; Huangfu et al., 2003). In Ift172/Wimple spinal cord, diffuse SHH is detected in the notochord, while diffuse SHH and SHHCL/P are not detected in the floorplate or spinal cord ventricles (Figure 2K, K′, L, L′), consistent with reduced SHH signaling. Thus, defects in primary cilia resulting from mutating IFT172 prevent detection of both diffuse SHH and SHHCL/P in the floorplate and spinal cord ventricles. In order to detect diffuse SHH in these studies, we used α-SHHCL;M/D, which stains diffusely distributed SHH in E8.75 and E9.5 floorplate and notochord (Figure 2A, A′, G, G′), similar to that found for H160 at E9.5 (Figure 1H, J). α -SHHCL;M/D fails to detect SHH proteins in Shh−/− E9.5 spinal cord (Figure 2M), supporting anti-SHH specificity, expected for an affinity purified antibody. By Western analysis, α-SHHCL;M/D recognizes both wtSHHCL and uSHHN monomers, showing recognition of a shared epitope in vitro, that is likely masked in vivo.
SHH-E176 regulates Zn2+ dependent cross-linking
In order to characterize SHH cross-linking that causes differential recognition by α-SHHCL/P, we developed an assay for detecting cross-linking, in vitro. While wtSHH requires extensive storage at −80°C (≥6 months) in order to form the cross-linked form detected in Fig 1A, uSHHN forms cross-linked dimers (uSHHN-CLdimers) within 30 minutes at 37°C, in a pH-dependent manner (Figure 3A, compare lane 1, pH 6.5, lane 3, pH 7.0, and lane 5, pH 7.5), with pH 7.5 providing optimal cross-linking conditions.
Figure 3. SHH-E176 controls Zn2+-dependent cross-linking and recognition by conformation-specific antibodies.

Factors affecting the formation of SHH non-reducible cross-linked dimers (CLdimer) A. uSHHN (lanes 1–9), lanes 1–2, pH 6.5, lanes 3–4, pH 7.0, lanes 5–9, pH 7.5, lanes 2, 4, 6, 9, 1 mM Zn2+, lane 7, 1 mM Mg2+, lanes 8 and 9, 1 mM EDTA. Western analysis was performed using anti-SHH antibody H160. B. uSHHN (lanes 1–5), lane 1, Zn2+ addition 30 minutes after cross-linking reaction starts, lane 2, minus Zn2+, lane 3, Zn2+ addition from the beginning of the cross-linking reaction, lanes 4–5, 100ng heparin, lane 5, 1 mM Zn2+. Western analysis was performed using anti-SHH antibody H160. Solid arrows indicate CLdimer and monomer, and dashed arrow indicates CLintra. C. SHHN proteins affinity purified from secreted cell culture supernatants, affinity purified on a 5E1 column (5E1, monoclonal anti-SHH, Developmental Hybridoma Studies Bank), and assayed for cross-linking. SHHN (lanes 1–2, 1′–2′), SHHN-E176A (lanes 3–4, 3′–4′), wtSHH (lanes 5–6, 5′–6′), and SHH-E176A (lanes 7–8, 7′–8′) were incubated in the presence (lanes 2, 4, 6, 8) or absence (lanes 1, 3, 5, 7) of 1mM Zn2+. Western analysis was performed using α-SHHCL/P (lanes 1′–8′), stripped and reprobed with H160 (lanes 1–8). Arrows indicate CLdimer and monomer. D. Description of recombinant proteins used in A–D: *Full-length construct, produces N-and C-lipid modified SHH proteins. SHH proteins are affinity purified from culture supernatants of transfected C17 neural cells, as previously described for wtSHH (Feng et al., 2004), **N-terminal construct produces N-lipid containing, C-lipid lacking SHH proteins. SHH proteins are affinity purified from supernatants of transfected C17 neural cells, previously described for (Feng et al., 2004), §N-terminal construct lacks both N-and C-lipid modifications. uSHHN is produced in E.coli, Ni-Agarose affinity purified, histidine tag cleaved, as previously described in (Williams et al., 1999). + (antibody recognizes), − (antibody does not recognize), weak (antibody weakly recognizes), ND (not determined), +ç (recognizes wtSHH crosslinked form after storage at −80°C for 6 months). Note: Recombinant proteins (1–5) do not contain tags at the N-or C-termini. Proteins 1–4 were purified by affinity chromatography on a 5E1 anti-SHH-antibody column, while the 6Xhis-tag on uSHHN (protein 5) was removed prior to crosslinking assays.
Crystal structure and mutational analysis previously showed that uSHHN stability requires binding to Zn2+ (Day et al., 1999; Hall et al., 1995). Therefore, we next tested the possibility that Zn2+ influences SHH cross-linking. Zn2+ inhibits uSHHN cross-linking at pH 7.0 (Figure 3A, compare lanes 3 and 4), and pH 7.5 (Figure 3A, compare lanes 5 and 6). Cross-linking reactions performed at pH 6.5 do not result in uSHHN-CLdimer formation (Figure 3A, lane 1), even in the absence of Zn2+, showing that lowering to pH 6.5 abrogates the need for Zn2+.
Zn2+, but not Mg2+ prevents formation of uSHHN-CLdimers (compare Figure 3A, lanes 6 and 7), supporting Zn2+ specificity. Addition of 10 mM EDTA, a Zn2+ chelator, allows uSHHN-CLdimers to form in the presence of Zn2+ (Figure 3, lane 9), indicating that Zn2+ chelation prevents cross-linking inhibition. Furthermore, Zn2+ added 30 minutes after uSHHN-CLdimers are formed, cannot disassemble CLdimers (Figure 3B, lane 1), showing that Zn2+ alone cannot reverse uSHHN cross-linking, in vitro. Other labs have shown that uSHHN becomes unstable in the absence of Zn2+, degrading when Zn2+ is removed. Therefore, in the cross-linking assays, it is expected that uSHHN is bound to Zn2+. Adding more Zn2+, rather than being required for stability, may then play a cross-linking role. This is supported by the finding that addition of the Zn2+ chelator EDTA, prevents Zn2+ inhibition of crosslinking, without degrading SHH. However, it should be noted that 1 mM Zn2+ used in these experiments is well above physiological concentrations in the picomolar range.
Given the significant role of the SHH heparin-binding domain in signaling (Rubin et al., 2002), we next tested whether heparin affects formation of uSHHN-CLdimers. Addition of heparin does not prevent formation of uSHHN-CLdimers (Figure 3B, lane 4), or cross-linking inhibition by Zn2+ (Figure 3B, lane 5). However, heparin increases the formation of a faster migrating uSHHN cross-linked intermediate that may represent intramolecular cross-linking, rather than dimerization (CLintra, as indicated by the dashed arrow in Figure 3B, lane 4). Therefore, Zn2+ can modulate the effect of the SHH-heparin binding on SHH cross-linking.
In order to further investigate SHH cross-linking control, we used results obtained from the crystal structure of uSHHN, predicting that there are three Zn2+ coordinating residues (Hall et al., 1995). While two of these bind Zn2+, and are necessary for protein stability (Day et al., 1999), one (human, SHH-E176, mouse, SHH-E177) was predicted to be the catalytic amino acid in metalloprotease activity. However, SHH metalloprotease activity was never reported. Given the ability of 1 mM Zn2+ to prevent uSHHN cross-linking (Figure 3A, B), we tested the possibility that SHH-E176-Zn2+ coordination regulates SHH cross-linking. Crosslinking experiments (Figure 3C) using four different SHH proteins that contain different combinations of N-and C-terminal lipids (see schematics in Figure 3D), and/or the E176A mutation, were performed to test whether E176 regulates SHH cross-linking. In these assays, preparation of SHHN-E176A and SHH-E176A from transfected C17 neural cell supernatants was similar to that previously described for SHHN and wtSHH (Feng et al., 2004). Unlike uSHHN, SHHN-CLdimers and wtSHH-CLdimers are not detected after 30-minute incubations at 37°C and pH 7.5 (Figure 3C, lanes 1, 5, 1′, 5′). However, mutation of E176 to A (SHHN-E176A, SHH-E176A) dramatically changes SHH cross-linking, producing both SHHN-E176A CLdimers and SHH-E176A CLdimers (Figure 3C, lanes 3′ and 7′). In contrast to uSHHN (Figure 3A, B), Zn2+ increases the formation of SHHN-E176A-CLdimers (Figure 3C, compare lanes 3′ and 4′), and SHH-E176A-CLdimers (Figure 3C, compare lanes 7′ and 8′). Detection with α-SHHCL/P reveals the following important differences between E176A mutated (Figure 3C, lanes 3′, 4′, 7′, 8′) and wildtype SHH proteins (Figure 3C, lanes 1′, 2′, 5′, 6′): 1. E176A mutation causes SHH to form CLdimers not detected in wildtype proteins, 2. Zn2+ increases cross-linking of E176A mutant proteins, without affecting wildtype proteins. H160 does not detect wtSHHCL (Fig 1A), but Figure 3C shows that H160 faintly recognizes SHHN-E176A-CLdimers (Figure 3C, lanes 3 and 4) and SHH-E176A-CLdimers (Figure 3C, lane 8). In addition, H160, but not α-SHHCL/P, recognizes monomers of SHHN, SHHN-E176A and wtSHH (Figure 3C, lanes 16), while monomers of SHH-E176A are not detected (Figure 3C, lanes 7 and 8). Therefore, cholesterol prevents E176A mutant proteins (SHH-E176A) from forming monomers that are recognized by H160 (compare Figure 3C lanes 3, 4 (cholesterol lacking) and lanes 7, 8 (cholesterol containing)). Together, these data support that SHH-E176 and the C-lipid modulates the effects of Zn2+ on SHH cross-linking, and recognition by conformation-specific antibodies, H160 and α-SHHCL/P.
The properties of different SHH proteins forms and recognition by H160 and α-SHHCL/P are summarized in Figure 3D. It is important to note that these are properties of secreted, soluble proteins. When cell lysates from C17 cells transfected with ShhE176A are probed with H160, monomeric forms are detected (Figure S1), distinguishing between the role of E176 in cross-linking secreted and membrane bound proteins.
SHH-E176/E177 is required for endogenous, but not ectopic SHH signaling
The differential cross-linking properties between E176A mutated proteins and wildtype proteins observed in Figure 3C raised the possibility that the E176 site plays a role in signaling activity. However, previous experiments failed to detect differences in activity between wtSHH and SHH-E176A mutant proteins (Day et al., 1999; Fuse et al., 1999). Despite extensive characterization of E176A mutant protein signaling, the effects of SHH-E176A on ectopic signaling in developing mouse brain have not been reported. Therefore, we used a gain-of-function approach to assay ectopic SHH activity by retroviral injection into the embryonic forebrain (Feng et al., 2006; Gaiano et al., 1999; Himmelstein et al., 2010; Kohtz et al., 2001; Olsson et al., 1997)
Retroviruses expressing human wtSHH and SHH-E176A N-and C-lipid modified proteins were made using the retroviral backbone shown in Figure 4A. Western analysis of C17 cells infected with retroviruses show that both wtSHH and SHH-E176 proteins are expressed (Figure 4B, lanes 2, 3). It is important to note that Figure 4B analyzes cell lysates, where H160 can detect monomers of membrane-associated SHH-E176A (Figure 4B, lane 3), differing from the secreted forms used in the cross-linking assays (Figure 3C, lanes 5, 7, 5′, 7′). WtShh and Shh-E176A retrovirus-injected brains have similar forebrain dorsal midline morphological defects, with cortical thinning, and enlarged ventricles, as previously described after wtShh injection (Gaiano et al., 1999)(Figure 4G, wtShh, and 4J, Shh-E176A). In addition, Dlx2, a gene normally restricted to ventral regions (Figure 4E), is ectopically expressed in the cortex, in both wtShh and Shh-E176A infected brains (arrows, Figure 4H and 4K). Furthermore, retroviral expression of SHHN-E176A also shows dorsal midline morphological and gene expression defects in 8/8 embryos analyzed (Figure S2). In line with previous reports that SHH-E176A is active in multiple assays, these results show that SHH-E176A is also active when ectopically expressed in the embryonic mouse forebrain.
Figure 4. SHH-E176A is highly active when ectopically expressed in the developing mouse forebrain.

A. Diagram of retroviral backbone (pCLE) used to express the human SHH protein. Full length human cDNA sequence was inserted into cloning site (CS), producing N-and C-lipid modified SHH proteins. Placental alkaline phosphatase (PLAP) is bicistronic with the Shh cDNA, allowing detection of virally infected cells (Gaiano et al., 1999; Kohtz et al., 2001). B. Whole cell extracts of C17 cells infected with wtSHH and SHH-E176A expressing viruses are Western-blotted and probed with H160 antibody, showing that both viruses express monomeric, cell associated SHH proteins. C–K. Analysis of embryos injected with retroviruses at E9.5 and analyzed at E12.5 (C–E) uninfected littermates, (F–H) wtSHH, (I–K) Shh-E176A. Whole embryos (C, F, I), and sections (D, E, G, H, J, K) of E12.5 mouse brains 3 days after infection with SHH expressing. Infected viral clusters are visualized by alkaline phosphatase staining in the dorsal midline of the telencephalon (G, J). Uninfected littermate control does not contain alkaline phosphatase-expressing clusters of cells (D). In situ hybridization of adjacent sections probed for ectopic expression of Dlx2 (E, H, K). wtSHH, n=8/8, SHH-E176A, n=4/4 display gross morphological defects. Scale bar in D, (same for E, G, H, J, K) is 300 μm. Arrows indicate ventralization of dorsal telencephalon by SHH overexpression and ectopic expression of Dlx2.
In order to determine whether SHH-E177 plays a role at endogenous sites of SHH signaling, we generated mice containing an E177A point mutation, as described in Fig 5A. ShhE177A/+ mice were crossed to Shh heterozygous mice (Shh+/−) to generate ShhE177A/− embryos for analysis (Figure 5A). A glutamate (E) to alanine (A) point mutation was made at amino acid 177 in exon 2, and verified by sequencing of cDNA from ShhE177A/− embryos (Figure 5B). At the same time, an Mlu1 restriction site was introduced (Figure 5B, TA*C) to distinguish between Shh+/+ and ShhE177A/−, by restriction analysis. Sequencing of the full-length ShhE177A cDNA verifies that no other mutations are present (not shown).
Figure 5. E177 is required for Shh signaling in vivo.

A. ShhE177A/− embryos display gross morphological defects compared to Shh+/+ at E12.5. Schematic showing homologous recombination in embryonic stem cells. The mouse bacterial artificial chromosome 429m20 contains a 16 kb fragment spanning the Shh genomic region. A glutamate to alanine point mutation at amino acid 177 was introduced in exon 2, and a conditional triple polyadenylation signal was inserted in the 5′ UTR. Schematic representation shows homologous recombination between the BAC targeting vector and the mouse Shh (mShh) genomic locus to generate the floxed transcription stop E177A targeted allele (ShhTS-E177A). ShhTS-E177A/+ mice were bred with EllaCre mice that express Cre recombinase under the control of the adenovirus Ella promoter to excise the floxed transcription stop. The resulting allele contains only one loxP site and the E177A mutation (ShhE177A). Black rectangles represent exons, lines represent introns. Correct targeting in ES cells is shown by Southern blotting. ApaLI and HindIII restriction sites are indicated. Stars represent the location of probes. B. RNA isolated from Shh+/−, ShhE177A/+ and ShhE177A/− embryos is reverse transcribed and cDNA is amplified by PCR. The E177A point mutation results in the generation of an MluI restriction enzyme site. MluI digest of an intron-spanning PCR on cDNA shows that the E177A mutation is expressed and the transcript is correctly spliced. Sequencing of cDNAs isolated from E9.5 Shh+/+ and ShhE177A/− embryos confirms introduction of E177A, and Mlu1 restriction site. C. X-galstaining for Ptc-lacZ expression in whole E9.5 embryos shows Ptc1 target gene expression is lost in ShhE177A/−PtclacZ/+ and Shh−/−PtclacZ/+ embryos. D–L. RNA in situ hybridization analysis using Ptc1 (D–F), Gli1 (G–I) and Shh (J–L) probes on E9.5 lumbar spinal cord sections in Shh+/+ (D,G,J), ShhE177A/− (E,H,K), and Shh−/− (F,I,L). M–a. Immunofluorescence staining of spinal cord progenitor markers HNF3β (M–O), Nkx2.2 (P–R), Nkx6.1 (S–U), Pax6 (V–X) and Pax7 (Y–a) on Shh+/−, ShhE177A/−, and Shh−/− E9.5 lumbar spinal cord sections, as indicated. (b–d) Nkx2.1 RNA situ hybridization of E12.5 forebrain sections from Shh+/+ (b), and anterior sections from ShhE177A/−(c) and Shh−/− (d). Scale bars: 500 μm (A,C) and 50 μm (D–a).
ShhE177A/− embryos have gross deformities in presumptive midbrain and forebrain structures; by E12.5, ShhE177A/− embryos have a nasal proboscis (Figure 5A), similar to Shh−/− embryos (Chiang et al., 1996). ShhE177A/− embryos have shortened limbs and lack digits, which are present in their Shh+/+ and Shh+/− littermates at this age (Figure 5A). Additionally, ShhE177A/− embryos die before birth.
In order to verify that Shh RNA is expressed and correctly spliced from the targeted allele containing E177A mutated sites, RT-PCR was performed on RNA isolated from E9.5 Shh+/−, ShhE177A7/+, and ShhE177A/− embryos (Figure 5B), and MluI digested (Figure 5B). Detection of SHH-E177A protein with α-SHHCL/P in E9.5 ShhE177A/− spinal cord sections provides additional support that targeting does not disrupt Shh mRNA or protein expression (see below).
In order to determine the extent of the severity of SHH signaling loss in ShhE177A/− embryos, expression of the SHH receptor Ptc1 was determined, providing a direct readout of SHH pathway activity (Goodrich et al., 1997; Marigo et al., 1996). We generated ShhE177A/−PtclacZ/+ embryos to examine Ptc1 expression (Goodrich et al., 1997). X-gal staining for β-galactosidase activity shows loss of Ptc1 expression in the ShhE177A/− spinal cord (Figure 5C). RNA in situ hybridization analysis confirms the absence of Ptc1 in ShhE177A/− spinal cord (Figure 5E). The expression of Gli1, a transcriptional target and mediator of SHH signaling (Lee et al., 1997; Matise and Joyner, 1999; Walterhouse et al., 1993) is also absent in ShhE177A/−spinal cord (Figure 5H). Despite the loss of SHH activity, Shh RNA is detected in the ShhE177A/− notochord (Figure 4K), but absent in Shh−/− embryos (Figure 5L). Shh RNA expression is not detected in the ShhE177A/− floor plate, as would be expected if E177A SHH proteins were inactive in vivo.
In order to further investigate the role of E177 in SHH signaling, we analyzed dorsal/ventral patterning in E9.5 ShhE177A/− mutant spinal cord. Consistent with the absence of a defined floor plate by Shh RNA (Figure 5 K), HNF3β+ floor plate cells are not observed (Figure5 N). Class II factors Nkx2.2 and Nkx6.1, induced by SHH and expressed in ventral progenitor domains (Ericson et al., 1997) are also not detected in the ShhE177A/− mutant (Nkx2.2, Figure 5Q, Nkx6.1, Figure 5T). Pax6 and Pax7 are class I factors that are repressed by SHH (Ericson et al., 1997). In the ShhE177A/− spinal cord Pax6 and Pax7 expression domains shift ventrally compared to Shh+/+ expression (Pax6, Figure 5W, Pax7, Figure 5Z). This is similar to the Shh−/− spinal cord (Figures 5X, a), although subtle differences in the expression pattern gradient may be present in the ShhE177A/− mutant.
We next asked whether E177A forebrain morphological defects are accompanied by ventral forebrain gene expression defects. Figure 5(b–d) shows that Nkx2.1, a ventral forebrain gene activated by SHH in vitro (Kohtz et al., 1998) and absent in Shh−/− (Chiang et al., 1996), is not detected in ShhE177A/− in anterior sections. Together, these data show that SHH-E177 is required for patterning of embryonic spinal cord and brain.
Only cilia BB-associated SHH-E177A is detected, in vivo
ShhE177A/− mutants phenocopy Shh−/− embryos, and indicate that E177 is critical for SHH activity at endogenous signaling sites. This is unexpected given that SHH-E176A/E177A mutations do not eliminate activity in a variety of in vitro and in vivo assays (Figure 4), (Day et al., 1999; Fuse et al., 1999). Given that ShhE177A/− embryos express correctly spliced Shh mRNAs, we hypothesized that altered SHH-E177A protein localization may be responsible for loss of signaling. Given differential recognition of SHH-E176A secreted proteins by α-SHHCL/P and H160 in vitro (Figure 3C, D), and differential detection of SHH punctate and diffuse forms in vivo, we next used immunohistochemistry to determine whether E177A mutation affects SHH localization in ShhE177A/− ventral spinal cord. Consistent with SHH protein localization shown in Figure 1H, H160 detects wtSHH in a diffuse pattern in the floor plate of Shh+/+ spinal cord (Figure 6A, A′). However, in ShhE177/− spinal cord, H160 does not detect SHH-E177A proteins (Figure 6B, B′). This is consistent with the results shown by Western analysis, where H160 fails to recognize secreted SHH-E176A proteins (Figure 3C, lane 7). α-SHHCL/P detects cilia BB-associated wtSHH (Figure 6D, D′) and SHH-E177A (Figure 5E, E′), with increased accumulation of cilia BB-associated SHH-E177A proteins compared to wtSHH (compare Figure 6D′ and E′). Detection of SHH-E177A in cilia BBs is also consistent with Western analysis showing that α-SHHCL/P recognizes wtSHHCL (Fig 1A) and SHH-E176ACL (Figure 3C). H160, and a-SHHCL/P do not detect proteins in Shh−/− spinal cord, supporting SHH antibody specificities (Figure 6C, C′, F, F′). Therefore, SHH-E177A proteins co-localize with cilia BBs along spinal cord ventricles, but are not detected in floorplate region, outside of cilia BBs.
Figure 6. Increased SHHCl/P cilia BB association and SHH cross-linking in ShhE177A/− embryonic spinal cord.

- E176A/E177A modulates Zn2+-mediated conformational change, detected by increased formation of cross-linked dimers.
- E176A/E177A is active at ectopic, but not endogenous sites in vivo.
- In ShhE177A/− mutant spinal cord, increased accumulation of SHH-E177A occurs near cilia BBs.
- SHHCL/P is still present in the ventral spinal cord even in the absence of the SHH receptor PTC1, supporting a pre-signaling role of SHHCL/P.
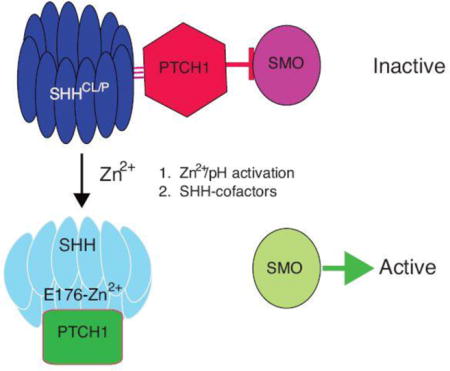
We propose that SHH signaling is regulated by a Zn2+-induced cross-linked/conformational change that occurs at cilia BBs. The E176/E177 site is required for this cross-linked/conformational change at cilia BBs, explaining why E177A mutation eliminates SHH signaling at endogenous sites. Co-factors that regulate the availability/concentration of Zn2+, pH, the C-terminal cholesterol modification, and PTC1 are likely to contribute to SHH signaling at endogenous sites, an environment that is not recapitulated in ectopic signaling events. The dotted arrow indicates the possibility of a dynamic state of conversion between inactive and active states. Proof of such conversions will rely on the development of tools to study these events in vivo.
We next compared the numbers of SHH puncta along the spinal cord ventricles of Shh+/+ and ShhE177A/− (Figure 6G). The schematic in Figure 6G shows an embryonic spinal cord section, with dashed boxes highlighting the intermediate (Int) and ventral regions (Vent). Puncta containing SHH were counted from 2000μm2 areas in the Int and Vent regions of Shh+/+ and ShhE177A/− lumbar spinal cord sections. The number of SHH puncta are not statistically different between the Int and Vent regions of Shh+/+ spinal cord (black bars, Figure 6G), or the Int and Vent regions of the ShhE177A/− spinal cord (gray bars, Figure 6G). However, there are significantly greater numbers of SHH-E177A puncta in both Int and Vent ShhE177A/− regions than in the corresponding Shh+/+ spinal cord regions. Analysis of SHH-cilia BB co-localization indicates that SHH-E177A increases association with cilia BBs in ventral, but not intermediate regions, where SHH-cilia BB already occurs in the majority regions (Figure 6G, graph on the right). Increased numbers of SHH-E177A puncta compared to wtSHH puncta may result from the inability of SHH to signal, and therefore increased accumulation in proteasomes located near cilia BBs. Alternatively, E177A mutation may allow better visualization of SHH-cilia BB puncta.
In order to resolve issues of detection, we next used Western analysis of E10.5 membrane extracts, and α-SHHCL/P and H160 to compare the profile of SHH proteins in Shh+/+ and ShhE177A/−. Figure 6H shows that E177A mutation in vivo shifts monomeric SHH to form dimers, but that monomers are still detected. Detection of both SHH-E177A dimers and monomers is consistent with results found in transfected cells where H160 to detects cell-associated SHH-E176A monomers (Figure S1) and α-SHHCL/P detects secreted SHH-E176A dimers (Figure 3C).
Figure 6I contains schematics of Shh+/+ and ShhE177A/− spinal cords comparing wtSHH and SHH-E177A protein distribution. Diffusible wtSHH is present in the floor plate and notochord, whereas cilia BB-associated wtSHHCL/P concentrates in puncta along the ventricle. SHH-E177ACL/P is only detected associated with cilia BBs along ventricles, in higher concentrations than wtSHHCL/P.
Discussion
In this paper, we show that mice containing a glutamate (E) to alanine (A) point mutation in E177 (ShhE177A/−) phenocopy Shh−/− mice with respect to embryonic morphological and gene expression defects in spinal cord and forebrain. In contrast to ectopic assays, where SHH-E176A is highly active, SHH-E177A is inactive at endogenous sites in vivo. In addition, SHH-E177A proteins are only detected near cilia BBs, with greater accumulation at cilia BBs compared to wtSHH. Several lines of evidence in this report support that zinc-dependent SHH cross-linking and/or conformational changes are responsible for the discrepancy between SHH-E176A ectopic and endogenous activities. While alternative mechanisms are also possible, based on the cumulative in vivo and in vitro data, we propose a model in which SHH-E176/E177 modulates the effects of Zn2+ on SHH protein cross-linking and/or conformation, an event that is required for signaling at cilia BBs (Figure 6J).
SHH cross-linking indicates conformational change
Factors controlling SHH multimerization have been studied in detail, specifically the role of N-and C-lipids (Chen et al., 2004; Feng et al., 2004; Ingham, 2001; Zeng et al., 2001). However, direct studies of multimerization in vivo have not been possible, mainly due to the lack of reagents to distinguish between multimers and monomers. Therefore, the biological significance of SHH multimerization has only been inferred from properties of recombinant SHH proteins, in vitro, and corresponding activities of these proteins in ectopic assays and mouse models. In order to directly visualize SHH multimers in vivo, we made several efforts to generate an N-lipid modified specific SHH antibody (using palmitoylated SHH peptides in vitro). These antibodies were unsuccessful in recognizing SHH proteins in embryonic spinal cord sections or denatured SHH proteins by Western analysis.
In contrast, we were able to generate a conformation-specific antibody (α-SHHCL/P) that recognizes cross-linked wtSHHCL in Westerns (Fig 1A), and cilia BB-associated wtSHHP in embryonic spinal cord sections (Fig 1E, F, F′, I). It is important to note that the ability of α-SHHCL/P to detect a SHH conformation that is shared between SHHCL proteins in vitro and cilia BB-associated SHHP in vivo, supports the biological significance of the SHHCL conformation formed by crosslinking. Despite being able to detect several cross-linked forms of SHH in embryonic tissues by western analysis (Feng et al., 2004), without shared detection of the cross-linked conformation in vivo, the SHH cross-linked conformation could be the result of SHH misfolding during in vitro manipulations, with little or no biological significance. Thus, shared recognition of SHHCL and SHHP reinforces the validity of using cross-linking and recognition by conformation-specific antibodies to study SHH conformational changes, with the advantage that factors that control cross-linking in vitro can shed insight into the regulation of SHH conformation in vivo.
The ability of conformation-specific antibodies to distinguish between cilia BB-associated SHHP and non-BB associated SHHD (diffusible SHHD) supports the hypothesis that changes in SHH conformation occur at cilia BBs either during pre-signaling or post-signaling events (Figure 6J). Given that cilia BBs are the site of active SHH signaling (Zimmerman and Yoder, 2015), it is not surprising that SHH conformational change critical for signaling would occur at cilia BBs. The finding that SHH-E177/E176 controls SHH cross-linking in vitro, as detected by α-SHHCL/P (Figure 3C), and is also critical for signaling in vivo (Figure 4) further supports a critical role of SHH cross-linking and/or conformational change during signaling. Thus, the use of conformation-specific antibodies (α-SHHCL/P, H160) and in vitro cross-linking assays to detect conformational changes, combined with characterization of SHH-E176A protein localization in spinal cord cilia BBs of ShhE177A/− mice reveals a previously unknown role for cross-linking/conformational control of SHH during signaling.
Zinc activation mechanism and concentration gradient
SHH is secreted by the notochord and the floor plate and is thought to act in a graded manner to induce expression of a unique combination of homeodomain transcription factors in the developing spinal cord (Jessell, 2000). However, the graded distribution of the SHH protein along the dorsal/ventral spinal cord has never been convincingly shown. While the numbers of SHHCL/P containing puncta do not change between ventral and intermediate regions (Fig 1M), cilia BB-association increases in intermediate compared to ventral spinal cord. In addition, analysis of ShhE177A/− mutant spinal cord indicates increased accumulation of SHH-E177ACL/P puncta in both intermediate and ventral regions (Figure 6G), suggesting that E177 is required for SHH activation, but not necessarily gradient formation.
A conformational change that is required for signaling raises the possibility that SHHCL/P is inactive in vivo, and requires conversion to become active (see model in Figure 6J–I). A conversion model was also proposed based on the finding that the C-lipid required for long range signaling, reduces SHH activity in forebrain explant assays (Feng et al., 2004). If conversion is involved, then the gradient of SHH activity would depend on factors contributing to SHH activation, rather than the concentration of SHH ligand. Analysis of ShhE177A/− mice is consistent with in vitro properties of E177A/E176A, and support a Zn2+ -dependent model of SHH activation (Fig 6J–I). The simplest Zn2+ activation model is shown in Fig 6J–I, where cilia BB-localized SHH-E176-Zn2+ binding changes SHH conformation, enabling proper binding to PTC1, release of Smoothened (SMO) and downstream signaling. The uniform distribution of both wtSHHCl/P and SHH-E177ACl/P from ventral to intermediate spinal cord argues against a gradient distribution of cilia BB-associated SHH; instead, the ability of E176/E177 to bind Zn2+, as determined by Zn2+ availability, or co-factors of SHH signaling, may regulate activity at a specific location.
The proposed Zn2+ activation model not only predicts that wtSHHCl/P conformation does not prevent association with PTC1, but also that PTC1 sequesters wtSHHCL/P. Both are supported by the following experimental evidence: (1) wtSHHCl/P is detected in the absence of PTC1, but requires PTC1 for proper localization, (2) SHHN and SHHN-E176A directly bind PTC1 with similar binding affinities (Day et al., 1999), (3) E176A/E177A mutant proteins exhibit increased cross-linking (Figure 3C), while retaining potency in ectopic assays (Figure 4, Fig S2). Taken together, the activation model predicts that SHH-E176/E177 −Zn2+ conformational change is required during PTC1 association, for subsequent PTC1 release of SMO (Figure 6J-I).
An alternative model is that E176 is required for signaling, but SHHCL/P forms during a post-signaling event (on the right in Figure 6J-II). While the ability to detect SHHCL/P in the absence of the SHH receptor PTC1 argues against post-signaling, the inability to reversibly cross-link SHH in vitro argues against a conversion model. Additional evidence against wtSHHCl/P or SHH-E177ACl/P as part of a post-signaling complex is the presence of SHH-E177ACl/P along spinal cord ventricles even in the absence of signaling. Given that E176 modulates pH -dependent SHH crosslinking in vitro, and that endosomes allow pH-specific compartmentalization, it is possible that the enodocytic pathway may be involved, unifying both pre-and post signaling mechanisms.
It is important to note that H160 fails to detect SHH-E177A in ShhE177A/− mutant spinal cord sections, but detects cell-associated SHH-E176A monomers by Western analysis. Thus, it is possible that loss of the bioactive form (as detected by H160) of SHH is responsible for loss of SHH-E177A activity. Therefore, alternative mechanisms may be involved in generating the bioactive form of SHH. Clearly, future experiments that require developing novel tools will be necessary to distinguish between these possibilities.
Differential requirement of E176/E177 in endogenous vs. ectopic SHH signaling
Previous studies comparing SHH signaling activity of soluble multimers in forebrain neural explants assays, showed that medium sized, but not large multimers (>669kD) were active. This led us to propose that inactive SHH is transported, requiring an activation step after long-range transport (Feng et al., 2004). More recently, Zn2+ mediated metalloprotease cleavage by ADAM (Ohlig et al., 2012), resulting in N-terminal lipid removal, was proposed to be required for SHH activation. However, neither report was able to show that SHH activation occurs through multimer disassembly or cleavage events, in vivo, or to determine in vivo sites where such activation occurs.
If SHHCl/P is inactive, its presence in Shh+/+ spinal cord supports the likelihood that inactive SHH is a normal part of SHH signaling at cilia BBs, and does not only appear as a result of E177A mutation, or in vitro manipulation of SHH proteins.
Although our data shows that E177 is required for SHH activity at endogenous sites in mice, previous and present studies (mouse embryonic forebrain, Figure 4C) show that E176/E177 is not required in culture or at ectopic sites (C3H10T1/2 cells, chick neural plate explants, Drosophila embryonic cuticle). In addition, previous experiments demonstrated that E176A/E177A mutation does not affect protein stability (Day et al., 1999).
One of the major questions raised by our results is: why is E176/E177 required for endogenous, but not ectopic assays? One possibility is that context-dependent regulation by Zn2+ occurs at endogenous sites, but not at ectopic sites; for instance, Zn2+ activation may be regulated by changes in pH, and co-factors, as proposed in the models shown in Figure 6J.
In vitro experiments support the involvement of pH in regulating E176/E177-Zn2+ cross-linking, as Zn2+ is required to inhibit cross-linking at pH 7.0 and 7.5, but not pH 6.5. One possibility is that accessibility of E176/E177 to Zn2+ is regulated by the arrangement or size of multimers. Additionally, co-factors of SHH signaling, specifically components of the extracellular matrix (ECM), may distinguish endogenous and ectopic roles of E176/E177 and the requirement for conformational regulation. While early studies showed that SHH interacts with heparin (Bumcrot et al., 1995; Lee et al., 1994), later work showed that ECM heparin sulfate proteoglycans and SHH interactions with proteoglycans influence the formation of the Hh morphogen gradient in Drosophila and SHH signaling activity in mice (Yan and Lin, 2009). Site-specific SHH mutations in the Cardin-Weintraub interaction domain (R34A/K38A) disrupt interactions with proteoglycans, specifically interfering with proliferative but not patterning activities in mice (Chan et al., 2009). In vitro uSHHN cross-linking assays suggest that heparin, in the absence, but not presence of Zn2+, increases formation of a faster migrating cross-linked intermediate uSHHN-CLintra (Figure 3B). Therefore, it will be important to determine how the SHH-heparin binding domain or SHH-proteoglycan interactions contribute to Zn2+-dependent cross-linking/conformational control.
The identification of transmembrane proteins HIP1 (Chuang and McMahon, 1999) and fibronectin III domain-containing proteins IHOG/CDO/BOC indicate both negative and positive regulation of Hh/SHH activity (Beachy et al., 2010; Tenzen et al., 2006; Yao et al., 2006). Together, these data raise the possibility that differential expression of SHH binding proteins, factors that regulate pH, and Zn2+ availability (referred to as co-factors, Figure 6J models) at endogenous sites compared to ectopic sites, regulates SHH signaling through cross-linking/conformational control.
Materials and Methods
Antibody Generation
The SHH N-terminal 197 amino acids fused to GST, was cross-linked to KLH (keyhole limpet hemocyanin, Pierce) and injected into rabbits for polyclonal antibody production (Pocono Farms). Several different bleeds were affinity purified on a column containing SHH N-terminal 197aa protein fragment. Conformation-specific, affinity-purified antibodies were tested for recognizing cross-linked wtSHH (N-and C-lipid containing SHH), but not soluble monomers, by Western analysis, generating α-SHHCL/P. WtSHH is affinity purified on a 5E1 antibody column from stably transfected C17 cell culture supernatants, as previously described (Feng et al., 2004). 5E1 purified antibodies were obtained from Developmental Studies Hybridoma Bank (DSHB, University of Iowa); 5E1 is a well-characterized mouse monoclonal antibody that recognizes SHH (Ericson et al., 1996). α-SHHCL;M/P (rabbit sera raised against SHH aa24–197, gift from Jessell lab) was made by affinity purification on uSHHN column (aa24–197), and specificity verified by western and immunohistochemistry on Shh−/− spinal cord sections. Cross-linked wtSHH (wtSHHCL) is spontaneously generated by extended storage in DMEM containing 20% fetal bovine serum (~6 months, − 80°C). uSHHN is recombinant human SHH purified from E. coli and lacks both N-and C-lipids (Kohtz et al., 2001).
Cross-linking Assays
50 ng of uSHHN (Figure 3A) or 3 ng of SHHN, SHHN-E176A, wtSHH, SHH-E176A (Figure 3C) were incubated with 1 mM ZnCl2, 1 mM MgCl2, or alone in crosslinking buffer (20 mM Hepes-KOH pH 7.5, 15 mM NaCl, 0.9 mM KCl, 0.5 mM MgCl2, 0.02 mM EDTA, 0.03% Triton X-100, 0.15% Tween 80, Aprotinin 0.625 μg/ml, 3 mM PMSF and 1 mM DTT) for 30 min at 37°C. For incubations at pH 6.5 and pH 7.0, 20 mM Hepes-KOH was replaced with 20 mM Pipes pH 6.5 and 20 mM Tris-Cl pH7.0, respectively. For samples containing EDTA, 10 mM EDTA was added prior to incubation. Westerns were probed with H160 or α-SHHCL/P. WtSHHCL (Fig 1A) was generated by storage of wtSHH containing supernatants from stably transfected C17 cell line at −80 for ≥ 6 months, followed by 5E1 monoclonal anti-SHH antibody affinity chromatography.
Immunofluorescent Staining and Confocal Microscopy
The HNF3β (clone 4C7, dilution 1:8), Nkx2.2 (clone 74.5A5, dilution 1:8), Nkx6.2 (clone F64A6B4, dilution 1:6), Pax6 (1:11), Pax7 (1:8) monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa), and mouse anti-acetylated α-tubulin (Zymed, 1:500) and anti γ-tubulin (Sigma, 1:700) were detected by anti-mouse Cy3 (Jackson, 1:250) and DAPI 1:1000 to label nuclei. H160 (Santa Cruz, dilution 1:400) and anti-SHHCL (1:1400), were detected using tyramide amplification, according to manufacturer’s instructions (TSA, AlexaFluor 488, Molecular Probes).
Zeiss confocal 510 Meta microscope with Plan-Neofluar 10×/0.3, Plan-Apochromat 63×/1.4 Oil DIC, and Alpha-Plan-Apochromat 100×/1.46 Oil DIC objectives and AIM software was used; Z-stacks were taken at 100× objective by merging between 8 and 11 images taken at the optimal interval. Each image was 8 μm thick, and was overlaid 50% with the preceding and following image using AIM software.
In Situ Hybridization
In situ hybridization was performed by modification of Schaeren-Wiemers and Gerfin-Moser (Schaeren-Wiemers and Gerfin-Moser, 1993) as previously described (Kohtz and Fishell, 2004), with the exception of the rTTR probe, which was visualized using the method of Tekki-Kessaris et al. (Tekki-Kessaris et al., 2001). The Lhx5 in situ probe was made from pGEM-Teasy (Promega) after subcloning an RT-PCR fragment made from E12.5 mouse choroid plexus RNA using the following primers:
5′ Primer – 5′ACA TGA GGG TCA TTC AGG TGT GGT 3′
3′ Primer – 5′ TGT GCT TGG AAT CTC GAC CCT TCA 3′
RNA Isolation and Reverse Transcription
Total RNA was extracted from E9.5 brain tissue using the EZNA MicroElute Total RNA Kit (Omega). cDNA was synthesized using the qScript cDNA Synthesis Kit (Quanta Biosystems).
Membrane Extraction and Western Analysis
Wild-type (6), ShhE177A/−, (7) Shh−/− (7) anterior regions (E10.5) were dissected from the embryos, and tissue was frozen at −80°C for one week before use. Tissue was homogenized in buffer HE (5mM Hepes containing 1mM EDTA, 0.5mM DTT+protease inhibitor cocktail (Sigma)) using a micro-pestle in a microfuge tube prior to dispersing through a 25-gauge needle. Tissue was spun at 1000 rpm for 15 minutes. The cytoplasmic extract was removed from the nuclear pellet, and re-spun at 38,000 rpm for 30 minutes. The pellet was resuspended in buffer HE plus 0.2% Triton. This was spun at 38,000 rpm for 30 minutes. The membrane pellet was resuspended in SDS-PAGE sample buffer, denatured by boiling, separated by SDS-PAGE, and transferred to nitrocellulose. Western blots were cut (top, middle, bottom regions) and probed with 1μg/ml of anti-SHHCL/P, anti-β-actin antibody (1:20,000, SIGMA) or H160 (1:4000, Santa Cruz), followed by incubation with secondary antibody anti-rabbit peroxidase (1:5000 Sigma) and chemiluminescence kit detection (PerkinElmer).
Cell Line Generation and Protein Preparation
For recombinant protein production and viral constructs, a point mutation changing human SHH glutamate 176 to an alanine (GAG[E] changed to GCG[A] using the Stratagene QuikChange II Site-Directed Mutagenesis Kit), was obtained from Dr. E. Garber (Biogen), and verified by sequencing. Plasmids containing Shh cDNAs (wtSHH, SHH-E176A, SHHN, SHHN-E176A, and C24S-SHHN) were obtained from Dr. E. Garber (Biogen), and subcloned into the mammalian expression vector pZeo (Invitrogen), and transfected into neural stem cell line C17.2 (Snyder et al., 1992). Stably transfected cell lines were generated and preparation of proteins (wtSHH, SHH-E176A, SHHN, SHHN-E176A) was performed as previously described for wtSHH, and SHHN (Feng et al., 2004).
Generation of ShhE177A mouse mutants
The ShhE177A targeting construct was generated using lambda phage-based recombineering in E. coli as described previously (Liu et al., 2003). Using high-fidelity Taq (Roche), homology arms of approximately 500 bp were PCR amplified (with restriction sites added) from BAC DNA. Using a three-fragment ligation, homology arms were cloned into Nhe I and Bam HI sites of pBR322, with a KpnI site in between them. A 16 kb region was retrieved from Bac RPCI-23 429M20 (CHORI) into the retrieval plasmid using recombination-induced EL250 cells (Liu et al., 2003). Further targeting was performed on the retrieved plasmid. The polyadenylation targeting vector was constructed in PL452, a floxed-Neo-containing plasmid. The triple polyadenylation signal was cloned into EcoRI and SalI sites of PL452. Approximately 500 bp of targeting homology arms were cloned sequentially on either side of the polyA–floxed-Neo insert. Fragments were PCR amplified as described above and cloned into either Not I and BamH I or EcoR I and Kpn I sites. The triple polyA–floxed-Neo cassette was targeted into the retrieved 16 kb region using recombination-induced EL250 cells. The vector containing exon 2 was constructed in pKS, which also contained a Neo flanked by Frt sites. Targeting fragments were PCR amplified and cloned into either Hind III and Spe I, or Sfi I and Not I sites. The E177A point mutation was introduced using the Stratagene QuikChange II Site-Directed Mutagenesis Kit. Following mutagenesis, flpe recombination was induced and the cassette was targeted into the retrieval plasmid using recombination-induced EL250 cells. EL250 cells and recombineering plasmids PL253 and PL452 were provided by N. Copeland (National Cancer Institute). Successful targeting was confirmed by Southern blot analysis of the completed construct using internal probes (NEBlot kit, NEB).
Mouse ES cells were targeted by homologous recombination using standard procedures. Successful targeting in ES cells was confirmed by Southern blot, verifying proper recombination at both the 5′ and 3′ ends. Probes were generated both internal (5′) and external (3′) to the 16 kb homologous region. ApaLI sites yield a 10.9 kb (Shh+/+) and 13.5 kb (targeted) fragment that hybridizes with the 5′ probe. Hind III sites yield a 6.9 kb fragment that hybridizes with the 3′ probe. ShhTS-E177A/+ (floxed-Neo) mice were verified by Southern blot and crossed to EIIAcre mice (Jackson Labs) for two generations. Neo removal was verified by PCR (data not shown). Mice are kept on a 129 background.
Animals used in all studies were maintained according to protocols approved by Institutional Animal Care and Use Committee at Children’s Memorial Hospital Research Center.
Detection of β-galactosidase activity
Prior to staining, embryos were fixed with 4% paraformaldehyde for 5 min at room temperature, washed with 2mM MgCl2, 1% Igepal Ca-30, and .05% deoxycholate in PBS, and incubated overnight at 37°C in 5mM potassium ferrocyanide, 5mM potassium ferricyanide, and 1mg/ml X-gal.
Viral Expression and Embryonic Injections
WtSHH full-length cDNA and SHH-E176A point mutation (SHH-E176A) were inserted into the pCLE viral vector backbone, and pseudo-typed retrovirus was produced as previously described (Gaiano et al., 1999; Kohtz et al., 2001). The E176A point mutation was introduced using the Stratagene QuikChange II Site-Directed Mutagenesis Kit, and verified by sequencing.
Viruses were titered on the C17 neural cell line (Snyder et al., 1992). Viruses were diluted to 5×107 cfu/ml in PBS containing 80μg/ml polybrene (Sigma). 1 μl of virus was injected into the E9.5 mouse telencephalon using ultrasound-guided in utero injection, as previously described (Gaiano et al., 1999; Liu et al., 1998; Olsson et al., 1997). Embryos were harvested 3 days after injection. Sample preparation and analysis of viral infections was performed as described (Gaiano et al., 1999). Detailed animal care, preparation for surgery and the use of the ultrasound scanner (UBM scanner) have been previously described (Liu et al., 1998). Timed-pregnant Swiss Webster mice used for injections were obtained from Taconic breeding laboratories. Embryonic day 0.5 is defined as noon of the day a vaginal plug was found after overnight mating. Only virally infected brains, as detected by alkaline phosphatase staining, were analyzed. Virally-induced morphological changes are the same over a wide range of wtSHH viral titers, ranging from 5×107 cfu/ml to 1×109 cfu/ml (Kohtz et al., 2001). Dlx2 gene expression and morphological dorsal midline defects resulting from wtSHH overexpression in E9.5 embryos were previously reported (Himmelstein et. al. 2010, Kohtz et. al. 2001).
Primer Sequences
For gene targeting, we used the following primers for retrieval homology arms:
Nhel (5′-CTCTTTTCAGCCACAAATGTAGCTGGTG-3′)
Kpnl (5′-GGCTCTTTGGCTCCTCCTTGC-3′)
Kpnl (5′-GGGTTGTTTTTCTCCTCTCCTGGCT-3′)
BamHI (5′-ATGTGTGCGTTAACAGTCTTGGTTTCC -3′).
For targeting arms, we used the following primers:
Not I (5′-CCTCCCTCCCACCCACC-3′)
BamH I (5′-GCTCGTTCGTTCCCTCTGGC-3′)
EcoR I (5′-CGAGCGAGGAAGGGAGAGC-3′)
Kpn I (5′-TGACTGCTTGGTCACACGCA-3′)
Hind III (5′-CAATCAGGCCCCAGACAAGCC-3′)
Spe I (5′ -CCCCACCACC GAGATCTACTTATTTT CAAG-3′)
Sfi I (5′-ACATAGCTCATGACCCCTCCCCCAACC-3′)
Not I (5′-CGCCTTCAGCAATCCTGCCAGGGAC-3′)
For ES Southern probes we used the following primers:
5′ probes (5′-AACACAGAAGCAGCTTTCCCA-3′) and (5′-CAGGTGTGCGGGCTTTAGTC-3′)
3′ probes (5′-CCACGAGGATGGAGCCTGTA -3′) and (5′-GGCAGAGTCCTCTTGAACACAC-3′)
Supplementary Material
Highlights.
Conformation-specific anti-SHH antibodies distinguish between diffuse and punctate
SHH in the developing spinal cord.
SHH-E177/176, Zn2+, pH and heparin regulate SHH cross-linking.
SHH-E177 is required for endogenous but not ectopic signaling sites in developing
mice.
SHH basal body-association remains, while diffuse SHH is absent in ShhE177A/− spinal
cord.
SHH-E177-Zn2+ maintains SHH in an activation competent conformation.
Acknowledgments
We thank E. A. Garber (Biogen) for the human Shh-E176A and ShhN-E176A cDNA constructs, D. Baker (Biogen) for uSHHN protein, A.McMahon (Harvard) for ShhN/+ mice, T. Caspary (Emory) for Wim mice, D.S.H.’s thesis committee (J. Kessler, A. Chenn, and R. Miller, Northwestern University) for helpful comments throughout the course of this work, N. Copeland (National Cancer Institute) for EL250 cells and recombineering plasmids PL253 and PL452, D. Epstein (U Penn) for Shh in situ probe, J. Rubenstein (UCSF) for Dlx2 in situ probe, and the following lab members for technical expertise and advice: M. VanGompel (recombineering and plasmid design), J.C. Savage (initial characterization of α-SHHCL/P), and J. Feng (early charactrerization of SHH-E176A and SHHN-E176A proteins, including the Western in Figure S1). Antibodies shown in Figure 5M-a and 5E1 anti-SHH were obtained from the Developmental Studies Hybridoma Bank. This work is funded by NIMH R01MH094653 to J.D.K. The manuscript has been approved by all authors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
D.S.H. generated and characterized ShhE177A/− mice, performed experiments in Figs 1, 2, 5, 6, Figure S2 and contributed to manuscript preparation. I.C. and G.V.V. performed experiments in Figure 3. C. Bi and B.S.C. performed experiments in Figure 4. I.C. contributed to Figure 5. J.D.K. conceived and designed experiments, and wrote the manuscript.
References
- Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes & development. 2010;24:2001–2012. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Ericson J. Specification of neuronal fates in the ventral neural tube. Curr Opin Neurobiol. 2001;11:43–49. doi: 10.1016/s0959-4388(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Bumcrot DA, Takada R, McMahon AP. Proteolytic processing yields two secreted forms of sonic hedgehog. Molecular and cellular biology. 1995;15:2294–2303. doi: 10.1128/mcb.15.4.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain CE, Jeong J, Guo C, Allen BL, McMahon AP. Notochord-derived Shh concentrates in close association with the apically positioned basal body in neural target cells and forms a dynamic gradient during neural patterning. Development. 2008;135:1097–1106. doi: 10.1242/dev.013086. [DOI] [PubMed] [Google Scholar]
- Chan JA, Balasubramanian S, Witt RM, Nazemi KJ, Choi Y, Pazyra-Murphy MF, Walsh CO, Thompson M, Segal RA. Proteoglycan interactions with Sonic Hedgehog specify mitogenic responses. Nature neuroscience. 2009;12:409–417. doi: 10.1038/nn.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004;18:641–659. doi: 10.1101/gad.1185804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–13. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–621. doi: 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Day ES, Wen D, Garber EA, Hong J, Avedissian LS, Rayhorn P, Shen W, Zeng C, Bailey VR, Reilly JO, et al. Zinc-dependent structural stability of human Sonic hedgehog. Biochemistry. 1999;38:14868–14880. doi: 10.1021/bi9910068. [DOI] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–2503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- Dierker T, Dreier R, Migone M, Hamer S, Grobe K. Heparan sulfate and transglutaminase activity are required for the formation of covalently cross-linked hedgehog oligomers. J Biol Chem. 2009a;284:32562–32571. doi: 10.1074/jbc.M109.044867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierker T, Dreier R, Petersen A, Bordych C, Grobe K. Heparan sulfate-modulated, metalloprotease-mediated sonic hedgehog release from producing cells. The Journal of biological chemistry. 2009b;284:8013–8022. doi: 10.1074/jbc.M806838200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87:661–673. doi: 10.1016/s0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006;20:1470–1484. doi: 10.1101/gad.1416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, White B, Tyurina OV, Guner B, Larson T, Lee HY, Karlstrom RO, Kohtz JD. Synergistic and antagonistic roles of the Sonic hedgehog N-and C-terminal lipids. Development. 2004;131:4357–4370. doi: 10.1242/dev.01301. [DOI] [PubMed] [Google Scholar]
- Fuse N, Maiti T, Wang B, Porter JA, Hall TM, Leahy DJ, Beachy PA. Sonic hedgehog protein signals not as a hydrolytic enzyme but as an apparent ligand for patched. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:10992–10999. doi: 10.1073/pnas.96.20.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Kohtz JD, Turnbull DH, Fishell G. A method for rapid gain-of-function studies in the mouse embryonic nervous system. Nat Neurosci. 1999;2:812–819. doi: 10.1038/12186. [DOI] [PubMed] [Google Scholar]
- Goetz JA, Singh S, Suber LM, Kull FJ, Robbins DJ. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. The Journal of biological chemistry. 2006;281:4087–4093. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Hall TM, Porter JA, Beachy PA, Leahy DJ. A potential catalytic site revealed by the 1.7-A crystal structure of the amino-terminal signalling domain of Sonic hedgehog. Nature. 1995;378:212–216. doi: 10.1038/378212a0. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelstein DS, Bi C, Clark BS, Bai B, Kohtz JD. Balanced Shh signaling is required for proper formation and maintenance of dorsal telencephalic midline structures. BMC Dev Biol. 2010;10:118. doi: 10.1186/1471-213X-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Litingtung Y, Chiang C. Region-specific requirement for cholesterol modification of sonic hedgehog in patterning the telencephalon and spinal cord. Development. 2007;134:2095–2105. doi: 10.1242/dev.000729. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Incardona JP, Lee JH, Robertson CP, Enga K, Kapur RP, Roelink H. Receptor-mediated endocytosis of soluble and membrane-tethered Sonic hedgehog by Patched-1. Proc Natl Acad Sci U S A. 2000;97:12044–12049. doi: 10.1073/pnas.220251997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW. Hedgehog signaling: a tale of two lipids. Science. 2001;294:1879–1881. doi: 10.1126/science.1064115. [DOI] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes & development. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nature reviews Genetics. 2006;7:841–850. doi: 10.1038/nrg1969. [DOI] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Kohtz JD, Baker DP, Corte G, Fishell G. Regionalization within the mammalian telencephalon is mediated by changes in responsiveness to Sonic Hedgehog. Development. 1998;125:5079–5089. doi: 10.1242/dev.125.24.5079. [DOI] [PubMed] [Google Scholar]
- Kohtz JD, Lee HY, Gaiano N, Segal J, Ng E, Larson T, Baker DP, Garber EA, Williams KP, Fishell G. N-terminal fatty-acylation of sonic hedgehog enhances the induction of rodent ventral forebrain neurons. Development. 2001;128:2351–2363. doi: 10.1242/dev.128.12.2351. [DOI] [PubMed] [Google Scholar]
- Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development. 1997;124:2537–2552. doi: 10.1242/dev.124.13.2537. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Ekker SC, von Kessler DP, Porter JA, Sun BI, Beachy PA. Autoproteolysis in hedgehog protein biogenesis. Science. 1994;266:1528–1537. doi: 10.1126/science.7985023. [DOI] [PubMed] [Google Scholar]
- Lewis PM, Dunn MP, McMahon JA, Logan M, Martin JF, St-Jacques B, McMahon AP. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell. 2001;105:599–612. doi: 10.1016/s0092-8674(01)00369-5. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome research. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- Matise MP, Joyner AL. Gli genes in development and cancer. Oncogene. 1999;18:7852–7859. doi: 10.1038/sj.onc.1203243. [DOI] [PubMed] [Google Scholar]
- Ohlig S, Pickhinke U, Sirko S, Bandari S, Hoffmann D, Dreier R, Farshi P, Gotz M, Grobe K. An emerging role of Sonic hedgehog shedding as a modulator of heparan sulfate interactions. The Journal of biological chemistry. 2012;287:43708–43719. doi: 10.1074/jbc.M112.356667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Campbell K, Turnbull DH. Specification of mouse telencephalic and mid-hindbrain progenitors following heterotopic ultrasound-guided embryonic transplantation. Neuron. 1997;19:761–772. doi: 10.1016/s0896-6273(00)80959-9. [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- Porter JA, von Kessler DP, Ekker SC, Young KE, Lee JJ, Moses K, Beachy PA. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature. 1995;374:363–366. doi: 10.1038/374363a0. [DOI] [PubMed] [Google Scholar]
- Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, Jessell TM. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell. 1995;81:445–455. doi: 10.1016/0092-8674(95)90397-6. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Rubin JB, Choi Y, Segal RA. Cerebellar proteoglycans regulate sonic hedgehog responses during development. Development. 2002;129:2223–2232. doi: 10.1242/dev.129.9.2223. [DOI] [PubMed] [Google Scholar]
- Snyder EY, Deitcher DL, Walsh C, Arnold-Aldea S, Hartwieg EA, Cepko CL. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell. 1992;68:33–51. doi: 10.1016/0092-8674(92)90204-p. [DOI] [PubMed] [Google Scholar]
- Tekki-Kessaris N, Woodruff R, Hall AC, Gaffield W, Kimura S, Stiles CD, Rowitch DH, Richardson WD. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development. 2001;128:2545–2554. doi: 10.1242/dev.128.13.2545. [DOI] [PubMed] [Google Scholar]
- Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Developmental cell. 2006;10:647–656. doi: 10.1016/j.devcel.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Vyas N, Goswami D, Manonmani A, Sharma P, Ranganath HA, VijayRaghavan K, Shashidhara LS, Sowdhamini R, Mayor S. Nanoscale organization of hedgehog is essential for long-range signaling. Cell. 2008;133:1214–1227. doi: 10.1016/j.cell.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Walterhouse D, Ahmed M, Slusarski D, Kalamaras J, Boucher D, Holmgren R, Iannaccone P. gli, a zinc finger transcription factor and oncogene, is expressed during normal mouse development. Developmental dynamics : an official publication of the American Association of Anatomists. 1993;196:91–102. doi: 10.1002/aja.1001960203. [DOI] [PubMed] [Google Scholar]
- Williams KP, Rayhorn P, Chi-Rosso G, Garber EA, Strauch KL, Horan GS, Reilly JO, Baker DP, Taylor FR, Koteliansky V, et al. Functional antagonists of sonic hedgehog reveal the importance of the N terminus for activity. J Cell Sci. 1999;112(Pt 23):4405–4414. doi: 10.1242/jcs.112.23.4405. [DOI] [PubMed] [Google Scholar]
- Yan D, Lin X. Shaping morphogen gradients by proteoglycans. Cold Spring Harbor perspectives in biology. 2009;1:a002493. doi: 10.1101/cshperspect.a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S, Lum L, Beachy P. The ihog cell-surface proteins bind Hedgehog and mediate pathway activation. Cell. 2006;125:343–357. doi: 10.1016/j.cell.2006.02.040. [DOI] [PubMed] [Google Scholar]
- Zeng X, Goetz JA, Suber LM, Scott WJ, Jr, Schreiner CM, Robbins DJ. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature. 2001;411:716–720. doi: 10.1038/35079648. [DOI] [PubMed] [Google Scholar]
- Zimmerman K, Yoder BK. SnapShot: Sensing and Signaling by Cilia. Cell. 2015;161:692–692 e691. doi: 10.1016/j.cell.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.