

Abstract
Background
Endoplasmic reticulum stress is associated with podocyte apoptosis in the pathogenesis of diabetic nephropathy (DN). A previous study has demonstrated that emodin has a protective effect in the kidney by suppressing proliferation of mesangial cells and inhibiting the renal tubular epithelial-to-mesenchymal transition. However, the effects of emodin on the podocyte apoptosis in DN and its mechanisms are unknown.
Aim
This study aimed to explore the effect of emodin on DN model KK-Ay mice and high glucose induced podocytes apoptosis via the PERK–eIF2α pathway.
Methods
KK-Ay mice model of DN were treated with emodin at dose of 40 and 80 mg/kg/day for 8 weeks. Urine albumin, serum creatinine, blood urea nitrogen levels and the renal histopathology in mice were performed. In vitro, conditionally immortalized mouse podocytes exposed to HG (30mM) were incubated with emodin. Cell viability was measured by CCK-8 assay. Additionally, we performed RNA interference and measured the apoptosis in cultured podocytes treated with emodin. Immunohistochemistry, immunofluorescence, western blot, and real-time PCR were used to detect gene and protein expression both in vivo and in vitro.
Results
The results showed that emodin treatment ameliorated urine albumin, serum creatinine, and blood urea nitrogen of DN mice. The pathological damage of kidney tissue was also improved after treatment with emodin. Moreover, emodin increased nephrin expression. Podocytes apoptosis and endoplasmic reticulum stress markers (GRP78) were significantly reduced upon emodin treatment. Furthermore, emodin treatment decreased the expression of phosphorylated protein kinase RNA-like endoplasmic reticulum kinase (P-PERK), phosphorylated P-eIF2α, ATF4, and CHOP. In vitro, emodin treatment was further found to decrease the GRP78 level induced by high glucose or tunicamycin (TM). Besides, emodin and PERK knockdown inhibited the apoptosis of podocytes cultured in high glucose by counteracting the upregulation of phosphorylated PERK, phosphorylated eIF2α, ATF4, and CHOP.
Conclusion
Overall, the findings indicate that emodin mitigates podocytes apoptosis by inhibiting the PERK-eIF2α signaling pathway in vivo and in vitro, and, therefore, exerts a protective action on podocytes in DN.
Keywords: emodin, diabetic nephropathy, endoplasmic reticulum stress, podocyte apoptosis, PERK-eIF2α
Introduction
Diabetic nephropathy (DN) is a progressive microvascular complication occurring among people with diabetes mellitus and has become a prime cause of end-stage renal disease (ESRD) in the USA.1 This disease is initially characterized by the appearance of moderately increased albuminuria, followed by overt proteinuria and a gradual decline in glomerular filtration rate.2,3 Accumulating evidence has demonstrated that podocytes apoptosis is an important factor in the pathogenesis of proteinuria in DN,4–6 and inhibition of podocytes apoptosis is a potential therapeutic target in DN.7 However, the mechanisms of podocytes apoptosis in DN have not been fully clarified yet.
The endoplasmic reticulum (ER) is a central organelle in which proteins are synthesized, folded, and modified, and where calcium, a crucial factor for cellular homeostasis, is stored.8–10 Within the ER, an imbalance between protein folding and transfer of properly folded proteins to other organelles can cause the accumulation of unfolded or misfolded proteins, triggering the activation of the unfolded protein response (UPR) and ER stress.11,12 Sustained or aberrant ER stress is cytotoxic, and might lead to cell injury or death.13–15 A growing body of evidence demonstrates that ER stress has a main function in the progression of diabetes complications, including DN.16–19 Some studies also indicate that the reduction of ER stress with 4-phenylbutyric acid (PBA) or tauroursodeoxycholic acid (TUDCA) can protect the kidneys from functional degradation by inhibiting the overexpression of lipocalin-2 (LCN2) in DN.10 In addition, metformin treatment can ameliorate ER stress and renal fibrosis induced by high glucose in the renal tubular epithelial cell line HK-2, through the activation of the adenosine monophosphate-activated protein kinase (AMPK) pathway.20
The anthraquinone emodin (1,3,8-trihydroxy-6-methylanthraquinone) is an orange crystalline phenolic compound extracted from the roots and rhizome of several plants, such as the traditional Chinese herbs Rheum palmatum, Polygonum cuspidatum, and Cascara buckthorn.21,22 Emodin has been widely used to reduce inflammation, to inhibit cell proliferation and ER stress, and to prevent obesity.23–26 This study investigates the therapeutic use of emodin in DN. Previous studies have indicated that emodin can protect the cells from oxidative stress in HK-2 cells by regulating the mitogen-activated protein kinases (MAPK) signaling pathway.27 Additionally, in podocytes, emodin ameliorates the tubular epithelial-to-mesenchymal transition (EMT) induced by high glucose, through the inhibition of integrin-linked kinase (ILK) and desmin in streptozotocin (STZ)-induced diabetic rats.21 However, the effect of emodin on ER stress-triggered podocytes apoptosis remains unclear. This study investigates whether emodin suppresses the ER stress-induced podocytes apoptosis in DN and highlights the possible therapeutic use of emodin in DN. Additionally, we explored the mechanism of the effect of emodin and tested if emodin can inhibit the protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2α (eIf2α) signaling pathway, which is activated during the UPR.28
Methods
Chemicals
Emodin (Figure 1, C15H10O5, molecular weight=270.23, purity by high-performance liquid chromatography [HPLC]≥98%) was purchased from Pufei De Biotech. Co. Ltd (Chengdu, China).
Figure 1.
Chemical structure of emodin.
Animals and treatment
Male C57BL/6J and KK-Ay mice (8 weeks old) were purchased from Beijing Huafukang Bioscience Co. Inc (Beijing, China). All experiments were performed in accordance with the National Institutes of Health guideline (NIH Pub. No. 85-23, revised 1996). The study protocol was approved by the Institutional Animal Care and Use Committee of the Capital Medical University (Permission number: AEEI-2016-124). All animals were housed at a constant temperature (24°C±1°C) and humidity (50%–60%), with a regular 12-h light and 12-h dark cycle in a specific-pathogen-free (SPF) room. C57BL/6J mice were fed with common forage, and KK-Ay mice were fed with research forage for 4 weeks, as previously described.29 Random blood glucose (RBG) levels were measured in blood obtained from the caudal vein of each animal with a portable glucometer (One Touch® Ultra, LifeScan, Johnson & Johnson, CA, USA). KK-Ay mice with RBG higher than 16.7 mM and a urine albumin creatinine ratio (ACR) over 300 μg/mg showed kidney damage closely resembling human DN30 and represented a DN model, according to a previous study.29
C57BL/6J mice were considered as the normal (control) group (n=8). KK-Ay mice were randomly divided into three groups: DN mice (model group, n=8), DN mice treated with low-dose emodin (n=8, 40 mg/kg/day), and DN mice treated with high-dose emodin (n=8, 80 mg/kg/day). This dose of emodin has been proved to be effective and safe.25,31 Emodin was dissolved in 0.5% carboxymethyl cellulose-Na (CMC-Na, Solarbio, Beijing, China) and administered by stomach perfusion. Normal and DN mice were orally administrated with the same volume of 0.5% CMC-Na. During the trial period, mice were fed with the diet mentioned above and had free access to water.
Cell culture and transfection
A conditionally immortalized mouse podocyte cell line (BNCC337685) was purchased from BeNa Culture Collection (Beijing, China). The podocytes were cultured in RPMI 1640 medium (Genview, League City, TX, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA) and recombinant interferon gamma (IFN-γ, PeproTech, London, UK) at 33°C under an atmosphere of 5% CO2. Podocytes were cultured without IFN-γ for more than 7 days to induce their differentiation. The differentiated podocytes at 80% confluency were cultured for 24 h in RPMI 1640 medium without FBS and divided into the following groups: 1) the normal group, in which podocytes were treated with RPMI 1640 containing 5.5 mM glucose; 2) the mannitol group, in which podocytes were treated with RPMI 1640 containing 5.5 mM glucose and 24.5 mM D-mannitol; 3) the model group, in which podocytes were treated with RPMI 1640 containing 30 mM glucose; 4) the low-dose emodin group, in which podocytes were treated with RPMI 1640 containing 30 mM glucose and 20 μM emodin; 5) the high-dose emodin group, in which podocytes were treated with RPMI 1640 containing 30 mM glucose and 40 μM emodin; 6) the TUDCA group, in which podocytes were treated with RPMI 1640 containing 30 mM glucose and 5 mM TUDCA (Selleck, Houston, TX, USA);32 7) the tunicamycin (TM) group, in which podocytes were treated with RPMI 1640 containing 5 μg/mL TM (Aladdin, Shanghai, China);33 and 8) the PERK silencing group, in which podocytes were treated with RPMI 1640 containing 30 mM glucose and transfected with a plasmid expressing PERK short hairpin RNA (shRNA). Cells were treated as described for 24 h. The emodin concentrations and incubation time were chosen based on the results of the proliferation assays described below.
Three PERK (Gene Bank ID: NM_010121) shRNA were introduced in the GV102 expression plasmid (GeneChem, Shanghai, China). The shRNA target sequences were as follows: PERK-shRNA1:TCGGAGACAGTGTTTGGCTTA, PERK-shRNA2:GATGCCGAGAATGATGGGAAA, PERK-shRNA3:CGTGGAGGACGCCGAGGCATT. Podocytes were transiently transfected with the PERK shRNA expression vectors or a control vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Non-infected podocytes (parental) was also used. The highest efficiency of PERK knockdown by shRNA vectors in podocytes was selected by quantitative PCR (qPCR). The primer sequences for PERK were (forward) 5′-CAAGGTCGGGCAGGAAGAG-3′ and (reverse) 5′-TAATACGACTCACTATAGGG-3′.
Proliferation assays
Podocytes were seeded in 96-well plates with RPMI 1640 plus 10% FBS. Before experiments, the medium was then replaced with serum-free RPMI 1640 for 24 h. The podocytes were then induced with 30 mM glucose in the absence or presence of 0, 10, 20, 40, 80, 160 μM emodin for 24 h or 48 h. Then 10 μL of the cell counting kit-8 (CCK-8, Beyotime, Shanghai, China) solutions were added to each well and incubated for 1 h at 37°C and 5% CO2. The absorption values (5 wells/group) were then measured at 450 nm with a microplate reader (Thermo Multiskan MK3, Thermo Fisher Scientific, Atlanta, GA, USA). The results are shown as relative cell viability compared with normal group podocytes, considering that the cell viability of normal group podocytes was 100%.
Biochemical assays
After being treated for 8 weeks, all mice were sacrificed, and blood samples were collected to measure blood urea nitrogen (BUN) and serum creatinine (SCr) with an automatic chemistry analyzer (AU2700+ISE, Olympus, Tokyo, Japan). Timed (24 h) urine was collected from mice at 8, 12, 16, and 20 weeks of age in individual metabolic cages. Urine albumin was measured using the ALB Elisa Kit (Meichenlianchuang, Beijing, China), and the value obtained represented the urine albumin excretion (UAE).
Morphological studies
Fresh kidneys from each mouse were fixed in 4% paraformaldehyde, and embedded in paraffin. Then 4–6 μm-thick sections were transferred to glass slides, and processed for periodic acid Schiff (PAS) and Masson’s staining. Morphologic alterations were examined by light microscopy (Leica DM60008, Kyoto, Japan). Sections of the renal cortex were harvested and fixed in 2% glutaraldehyde, and ultrathin sections were then collected for staining. The samples were observed with a transmission electron microscope (JEM-1400 plus, JEOL, Tokyo, Japan). The images were analyzed quantitatively by NIS-Elements BR 3.2 software (Nikon Instruments, Tokyo, Japan).
Immunohistochemical analysis
The paraffin sections were processed via routine dewaxing and hydration. Antigen retrieval was performed with a microwave, and non-specific antibody binding sites were blocked with 3% H2O2 (ZSGB-BIO, Beijing, China) for 10 min. The samples were then incubated with an anti-nephrin antibody (1:500, Santa Cruz Biotech, Santa Cruz, CA, USA) at 4°C overnight, and with polyperoxidase-anti-mouse IgG (ZSGB-BIO, Beijng, China). Immunoreactive signals were developed upon incubation with 3,3-diaminobenzidine (DAB, ZSGB-BIO). The sections were observed with the Leica DM60008 microscope at 400× magnifications, and the quantitative analysis of the immunohistochemical images was done with the Image-Pro plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Flow cytometry analysis
Podocytes were briefly digested with 0.25% trypsin without ethylenediaminetetraacetic acid (EDTA, Solarbio, Beijing, China), and then washed with ice-cold phosphate-buffered saline (PBS). The percentage of apoptotic podocytes was determined using flow cytometry, using the Annexin V-FITC/PI Apoptosis Kit (Keygen Biotech, Nanjing, China), following the manufacturer’s recommendations. Data collection and analysis were performed using a flow cytometer (LSRFortessa™ SORP, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and FACS scan software (Becton, Dickinson and Company).
Western blot
Mice renal cortexes and cultured podocytes were homogenized with radio-immunoprecipitation assay buffer (RIPA, Solarbio, Beijing, China) on ice, and total proteins were extracted. Equal amounts of proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes (0.45 μm, Millipore, Burlington, MA, USA). The membranes were then blocked with 5% non-fat dry milk in Tris-buffered saline with Tween 20 (TBST 10×, Solarbio) at room temperature for 1 h, washed with TBST 3 times and incubated with the primary antibodies at 4°C overnight, respectively. After three washes of 10 min with TBST, the membranes were incubated with an anti-mouse/rabbit IgG horseradish peroxidase HRP-conjugated antibody (1:2,000, Cell Signaling Technology, Danvers, MA, USA) at room temperature for 1 h and washed with TBST. Immunoreactive signals were developed with the enhanced chemiluminescent (ECL) method (Millipore), using the gel imaging system FUSION FX6 XT (Vilber Lourmat, Marne-la-Vallée, France). The primary antibodies used were: 78 kDa glucose-regulated protein (GRP78), phosphorylated (P)-eIf2α, Bcl-2-associated X (Bax), activating transcription factor (ATF6), and phosphorylated(P)-inositol-requiring enzyme 1 (IRE1) (1:1,000, Abcam, Cambridge, UK), B-cell lymphoma 2 (Bcl-2, 1:500, Abcam), Nephrin, P-PERK (1:500, Santa Cruz Biotech), PERK, CCAAT-enhancer-binding protein homologous protein (CHOP), and activating transcription factor 4 (ATF4) (1:1,000, Cell Signaling Technology). β-actin (1:500, Dingguo, Beijing, China) was used as the internal control.
qRT-PCR analysis
Total RNA was isolated from the renal cortex and cultured podocytes using TRIzol Reagent (Keygen Biotech) and retrotranscribed using the PrimeScript™ RT reagent Kit with a gDNA Eraser (Takara, Tokyo, Japan), following the manufacturers’ recommendations. qPCR was performed in triplicate using the SYBR® Premix Ex Taq™ II (Takara) and the ABI PRISM 7500 FAST Real-TIME PCR System (ABI, Vernon, CA, USA). The relative expression of mRNA was calculated using the 2−ΔΔCt method. The primers were designed and synthesized as follows: GRP78: forward 5′-GAAGGAGGATGTGGGCACG-3′ and reverse 5′-CGCATCGCCAATCAG-ACG-3′, ATF4: forward 5′-CCT-GAACAGCGAAGTGTTGG-3′ and reverse 5′-TGGAGAACCCATGAGGTTTCAA-3′, CHOP: forward 5′-CAGCGACAGAGCCAGAAT-3′ and reverse 5′-CAAGGTGAAAG-GCAGGGA-3′.
Immunofluorescence and phalloidin staining
Podocytes were cultured on coverslips in 24-well plates for 24 h. Cells were washed 3-times with PBS, fixed with 4% paraformaldehyde on ice for 10 min, permeabilized with 0.1% Triton X-100 (Dingguo) for 15 min, and blocked with 10% normal goat serum for 1 h at room temperature. Podocytes were then incubated with the primary antibodies at 4°C overnight, washed with PBS, and incubated with Cy3- or fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG antibodies (Beyotime, Shanghai, China) in the dark for 1 h at 37°C. The nuclei were then stained with 4′,6-diamidino-2-phenylindole (DAPI, Genview) for 5 min in the dark. Finally, the coverslips were mounted on slides with an anti-fluorescence quencher, and the stained podocytes were observed with a Leica DM60008 microscope. The following primary antibodies were used: GRP78 antibody (1:500, Abcam) and phosphorylated eukaryotic initiation factor 2α (P-eIF2α) (1:500, Abcam).
Alternatively, podocytes were permeabilized with Triton X-100 and washed 3-times with PBS. Subsequently, podocytes were stained with 100 μg/mL phalloidin-conjugate working solution (AAT Bioquest, Sunnyvale, CA, USA) for 60 min at room temperature. Cells were washed 3-times with PBS and observed with a laser scanning confocal microscope (TCS SP8 STED, Leica, Wetzlar, Germany).
Statistical analysis
The SPSS 19.0 software (IBM, Armonk, New York, USA) was used to perform statistical analyses. All values are presented as the mean±SD. The t-test was used to assess the differences between two groups, and one-way analysis of variance (ANOVA) was used to assess differences among more than two groups. P<0.05 was considered as statistically significant.
Results
Effect of emodin on the levels of urinary albumin, blood urea, and creatinine
Severe albuminuria was detected in the model group compared with the normal group. However, treatment with emodin significantly decreased the UAE levels of DN mice in a dose-dependent and time-dependent way (Figure 2A, P<0.05). The levels of BUN and SCr in the model mice were obviously higher than those in normal mice. Both low-dose (40 mg/kg/d) and high-dose (80 mg/kg/d) emodin caused a significant decrease in the levels of BUN and SCr compared with the model group (Figure 2B and C, P<0.05). Together, these results suggest that emodin improves the renal function and ameliorates albuminuria in KK-Ay mice.
Figure 2.

Effect of emodin on UAE (A), BUN (B), and SCr (C) of KK-Ay mice. Values are the mean±SD, n=8, ##P<0.001 versus the normal group; *P<0.05, **P<0.001 versus the model group.
Abbreviations: UAE, urine albumin excretion; BUN, blood urea nitrogen; SCr, serum creatinine.
Effect of emodin on the renal histopathology of DN mice
Kidney tissues were collected for PAS and Masson staining at the end of the experiment. Both PAS and Masson staining demonstrated that model mice presented obvious pathological changes compared to the normal group, including mesangial matrix expansion and marked glycogen storage. However, treatment with emodin dose-dependently ameliorated these changes (Figure 3A and B). The relative pathological integral in PAS staining was higher in model mice than normal mice, whereas emodin treatment significantly decreased the integral (Figure 3D, P<0.05). Masson stained sections of kidney tissues revealed more collagen fibers in model mice than normal mice. After the 8-week treatment, collagen fibers in KK-Ay mice were obviously improved (Figure 3E, P<0.05). Additionally, electron microscopy showed a thicker basement membrane and focal fusion foot processes in model mice, while emodin treatment improved the ultrastructure of the renal tissue in a dose-dependent way (Figure 3C and F, P<0.05). Overall, these data indicate that emodin alleviates the pathological changes of the kidneys in DN mice.
Figure 3.

Real histopathology after emodin treatment. (A) PAS staining. (B) Masson staining (original magnification ×400, n=4). (C) Electron microscopy (original magnification ×10,000, n=4). (D) Relative pathological integral in PAS staining. (E) Relative collagen fibers in Masson staining. (F) Relative basement membrane thickness in electron microscopy. In (A) and (B), the normal group presented normal glomeruli. Compared with the model group, emodin mitigated mesangial matrix expression and glycogen storage (black arrows, A). In (C), the GBM is shown in the yellow box. The GBM was thicker and the effacement of podocyte foot processes was evident in the model group. Emodin significantly improved these morphological changes. Values are the mean±SD; n=5, ##P<0.001 versus the normal group; *P<0.05, **P<0.001 versus the model group.
Abbreviations: PAS, Periodic Acid-Schiff; GBM, glomerular basement membrane.
Emodin downregulated the expression of GRP78 associated with ER stress and protected podocytes in DN mice
The upregulated expression of GRP78 is considered a major hallmark of ER stress. The ER stress is a process in which podocytes reduced their marker protein nephrin expression. Insufficient expression of nephrin negatively affects the integrity of the glomerular filtration membranes and causes urinary albumin effusion.34,35 To investigate the effects of emodin on ER stress, GRP78 expression was detected by Western blot and qPCR. Relative to the normal group, GRP78 protein and mRNA were more expressed in the kidney cortex of the model group (P<0.001). A decline in GRP78 expression was associated with emodin treatment, and the highest dosage (80 mg/kg/d) had the biggest effect (P<0.05, Figure 4B, D, and G). The nephrin protein level was also significantly decreased in the model mice (P<0.001), and emodin upregulated nephrin protein in a dose-dependent manner by Western blot (P<0.05, Figure 4A and C). Similar results were obtained when we analyzed the expression of nephrin in the renal cortex by immunohistochemistry (Figure 4E and F). These findings indicated that emodin protects podocytes from injury induced by ER stress in DN mice.
Figure 4.

Effect of emodin on nephrin and GRP78 expression of kidney in KK-Ay mice. (A–D) The protein levels and quantification of nephrin and GRP78 in the mice renal cortexes were detected by Western blot, nephrin, or GRP78 expression was normalized to β-actin expression. (E, F) Representative immunohistochemical staining and quantification of nephrin expression in KK-Ay mice. Brownish or yellowish deposits were regarded as positive immunostaining (original magnification ×400, n=4). (G) The mRNA levels of GRP78 in the mice renal cortexes were detected by RT-PCR. The results were normalized to the normal group, and data are shown as the mean±SD; n=3. #P<0.001 versus the normal group; *P<0.05 and **P<0.001 versus the model group.
Abbreviation: GRP78, 78 kDa glucose-regulated protein.
Emodin alleviated the cell apoptosis by suppressing the activation of the PERK signaling pathway in DN mice
The activation of PERK is in the center position in ER stress. In our study, we investigated the effects of emodin on the expression of PERK and downstream components of the ER stress response. The levels of phosphorylated PERK, phosphorylated eIF2α, ATF4, and CHOP were dramatically inhibited by high-dose (80 mg/kg/d) emodin administration compared with the model mice (P<0.05, P<0.001, Figure 5A–C, E, and F). Similar results were obtained when we analyzed the expression of ATF4 and CHOP mRNA in the renal cortex of mice (P<0.05, P<0.001, Figure 5D). In addition, we detected the effect of emodin on ATF6 and P-IRE expression in KK-Ay mice. Western blot showed that emodin had no overt effect on the level of ATF6 or P-IRE (Figure 5G–I). Because ER stress was associated to increased apoptosis, we investigated the effect of emodin on the cell apoptosis of kidney. Western blot assays indicated that the expression of antiapoptotic protein Bcl-2 decreased and that of proapoptotic protein Bax increased in model mice compared with normal mice. Low-dose (40 mg/kg/d) emodin had no significant effects on the levels of these proteins. However, high dose (80 mg/kg/d) emodin significantly increased the expression of Bcl-2 expression and reduced that of Bax (P<0.05, Figure 5J–L). These data implied that emodin alleviated the cell apoptosis by suppressing the activation of the PERK signaling pathway in DN mice.
Figure 5.

Effect of emodin on P-PERK, P-eIF2α, ATF4, ATF6, P-IRE1, CHOP, Bax, and Bcl-2 in KK-Ay mice kidneys. (A–C, E, F) Representative Western blot and quantification of P-PERK, P-eIF2α, ATF4, and CHOP in KK-Ay mice kidneys. (D) The mRNA level of ATF4, CHOP treated with emodin in KK-Ay mice kidneys. (G–I) The protein level of ATF6, P-IRE1 detected by Western blot in KK-Ay mice kidneys. (J–L) Western blot and quantification for Bax and Bcl-2 in KK-Ay mice kidneys. The results were normalized to the normal group, and data are shown as the mean±SD; n=3. #P<0.001 versus the normal group; *P<0.05 and **P<0.001 versus the model group.
Abbreviations: P-PERK, phosphorylated protein kinase RNA-like endoplasmic reticulum kinase; P-eIF2α, phosphorylated eukaryotic initiation factor 2α; ATF4, activating transcription factor 4; P-IRE1, phosphorylated(P)-inositol-requiring enzyme 1; CHOP, CCAAT-enhancer-binding protein homologous protein; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2.
Effect of emodin on the viability of podocytes cultured in the presence of high glucose
To determine the optimal emodin concentration and incubation time on the podocyte cell line, the cells were exposed to high glucose (30 mM),35,36 and treated with different concentrations of emodin for 24 or 48 h; the viability was measured with the CCK-8 assay. As shown in Figure 6, treatment with emodin for 48 h was more cytotoxic than the treatment for 24 h. Additionally, emodin enhanced podocyte viability in a dose-dependent manner up to 40 μM (P<0.05). At higher emodin concentrations, the viability of the cells began to decrease, and the dose of 160 μM significantly inhibited cell viability (P<0.001). Thus, to avoid cytotoxicity, we chose to perform our experiments on podocytes with 20 or 40 μM emodin for 24 h.
Figure 6.
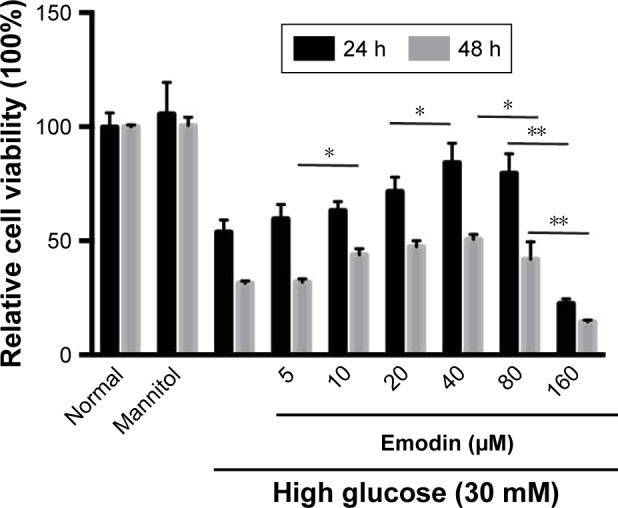
Effect of emodin on podocyte viability. Podocytes were cultured with high glucose (30 mM) and emodin at different concentrations (5, 10, 20, 40, 80, 160 μM) for 24 or 48 h. Relative cell viability was assessed with CCK-8 and expressed as percent podocytes survival relative to that of the normal cells. Values are the mean±SD, n=3, *P<0.05, **P<0.001.
Abbreviation: CCK-8, cell-counting kit-8.
Emodin increased nephrin expression and protected podocytes from injury exposed to high glucose
In the podocyte cell line, high glucose inhibited the expression of nephrin compared with the normal group. Treatment with mannitol was used as a control, to confirm that the effect of the glucose was not due to the high osmolarity of the treatment solution. Notably, treatment with emodin significantly increased nephrin levels in a dose-dependent way compared with the treatment with high glucose (P<0.05, Figure 7A and B). In normal podocytes, the cytoskeleton, as observed upon phalloidin staining, appears as a robust network of actin stress fibers. In the model group, intracellular actin stress fibers were collapsed or absent, leading to a polygonal cellular shape. Contrarily, emodin treatment was associated with remodeling of the intracellular actin stress fibers and rescue of the podocytes shape (Figure 7C). These findings indicated that emodin significantly protects podocytes from injury, and up-regulates the expression of nephrin.
Figure 7.

Effect of emodin on nephrin expression and podocytes cytoskeleton cultured with high glucose. (A, B) The protein levels and quantification of nephrin in podocytes cultured with high glucose were detected by Western blot. (C) Staining of podocytes cytoskeleton with phalloidin (original magnification ×630, scale bars: 25 μm, n=4). Actin stress fibers were arranged regularly or parallel to each other in the normal and mannitol-treated group. However, the actin stress fibers collapsed and appeared as cortical actin web, leading to a polygonal cellular shape in cells treated with high glucose. Emodin reversed the effects of high glucose. In the histograms, the results were normalized to the normal group, and data are shown as the mean±SD; n=3. #P<0.001 versus the normal group; *P<0.05 versus the model group.
Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole.
Emodin blocked podocytes ER stress induced by high glucose
In cultured podocytes, the levels of GRP78 were low in normal and mannitol-treated cells, and increased upon exposure with glucose. TUDCA, a well-known ER stress inhibitor, can attenuate podocyte injury and diabetic kidney disease.32,37 We found that emodin and TUDCA remarkably reduced GRP78 levels triggered by high-glucose (P<0.05, P<0.001). Once more, the higher dose (40 μM) of emodin had a bigger effect than the lower dose (20 μM). Also, there was no significant difference between emodin and TUDCA. In addition, we also found that emodin reversed the increased GRP78 induced by TM treatment (P<0.05, Figure 8A and B), TM has been widely used to trigger ER stress. These results were confirmed by immunofluorescence (Figure 8C and D). These data indicate that emodin significantly inhibits the activation of ER stress in podocytes cultured with high glucose.
Figure 8.

The induction of ER stress marker GRP78 was attenuated by emodin in podocytes. (A, B) The protein levels and quantification of GRP78 in podocytes were detected by Western blot. (C, D) Representative immunofluorescent staining and quantification of the fluorescence intensity of GRP78. In all histograms, the results were normalized to the normal group, and the data are shown as the mean±SD; n=3. #P<0.001 versus the normal group; *P<0.05 and **P<0.001 versus the model group. ΔP<0.05 versus TM group.
Abbreviations: ER, endoplasmic reticulum; GRP78, 78 kDa glucose-regulated protein; TM, tunicamycin; HG, high glucose; TUDCA, tauroursodeoxycholic acid; DAPI, 4′,6-diamidino-2-phenylindole.
Selection of the most effective PERK-shRNA expression vector targeting PERK
To estimate the comparative silencing effects of the PERK-shRNA expression vectors on the level of PERK, podocytes cultured with high glucose were transfected transiently with the recombinant plasmids. The PERK protein and mRNA levels were measured by Western blot and RT-PCR after PERK-shRNA expression vector transfection for 24 h. The results presented that transfection of PERK-shRNA1, PERK-shRNA2, and PERK-shRNA3 significantly decreased PERK expression in the podocytes cultured with high glucose (P<0.05). Yet, control transfection did not downregulate PERK expression. The gene silencing effect was nearly 75.0%, 62.9%, and 69.1%, respectively, in the PERK-shRNA1, PERK-shRNA2, and PERK-shRNA3 groups, when compared to the high glucose. Noticeably, the most obvious gene silencing effect was observed in the PERK-shRNA1 group (Figure 9A–C). Thus, we selected PERK-shRNA1 (hereafter referred to as PERK-RNAi) for subsequent stable transfection in the study.
Figure 9.

Effect of PERK-shRNA expression vectors on PERK expression in podocytes exposed to high glucose. (A, B) Protein level and quantification of PERK in podocytes after transient transfection of different PERK-shRNA expression vectors (PERK-shRNA1, PERK-shRNA2, and PERK-shRNA3). (C) Relative mRNA of PERK in podocytes after transient transfection of different PERK-shRNA expression vectors. The results were normalized to the high glucose group, and data are shown as the mean±SD; n=3. #P<0.05 versus the high glucose.
Abbreviations: PERK, protein kinase RNA-like endoplasmic reticulum kinase; shRNA, short hairpin RNA.
Emodin treatment inhibited high glucose-induced activation of PERK signaling pathway
Next, we investigated the effects of emodin on the PERK pathway in podocytes cultured with high glucose. We knocked down PERK expression to investigate whether emodin alleviated the ER stress in DN through the PERK pathway. Western blot showed that the levels of phosphorylated PERK, phosphorylated eIF2α, ATF4, and CHOP were dramatically inhibited by emodin administration in a dose-dependent manner compared with the model group. Notably, PERK knockdown was associated with similar (but stronger) effects (P<0.05, P<0.001: Figure 10A–E). These results were further confirmed upon immunofluorescent staining of P-eIF2α (P<0.001: Figure 10F and G). These data demonstrated that emodin suppresses the activation of the PERK pathway triggered by high glucose.
Figure 10.


Effects of emodin on P-PERK, P-eIF2α, ATF4, and CHOP in podocytes. (A–E) Representative Western blot and quantification of P-PERK, P-eIF2α, ATF4, and CHOP. (F) Immunofluorescent for P-eIF2α in podocytes treated as indicated. (G) Quantification of the fluorescence intensity of P-eIF2α in each group. In all histograms, the results were normalized to the normal group, and data are shown as the mean±SD; n=3. #P<0.001 versus the normal group; *P<0.05 and **P<0.001 versus the model group.
Abbreviations: P-PERK, phosphorylated protein kinase RNA-like endoplasmic reticulum kinase; P-eIF2α, phosphorylated eukaryotic initiation factor 2α; ATF4, activating transcription factor 4; CHOP, CCAAT-enhancer-binding protein homologous protein; DAPI, 4′,6-diamidino-2-phenylindole.
Emodin protected podocytes from apoptosis induced by high glucose via the PERK pathway
Western blot assays indicated that the expression of antiapoptotic protein Bcl-2 decreased, and that of proapoptotic protein Bax increased in the model group compared with the normal group (P<0.05). Emodin significantly upregulated the expression of Bcl-2 and downregulated the level of Bax protein in a dose-dependent manner. Similarly, treatment with PERK-RNAi was associated with a decrease in the expression of Bax and an increase in the expression of Bcl-2 (P<0.05, P<0.001, Figure 11A–D). Annexin V-FITC/propidium iodide (PI) staining was performed to quantify the podocytes apoptosis via flow cytometry. The apoptotic rate of cells treated with high glucose was significantly higher than that of untreated podocytes, and the effects of the high-glucose treatment were suppressed by emodin or PERK-RNAi (P<0.05, Figure 11E and F). Therefore, emodin inhibits podocytes apoptosis induced by high glucose through PERK pathway.
Figure 11.


Effects of emodin on Bax, Bcl-2 expression, and apoptosis induced by high glucose. (A–D) Western blot and quantification for Bax and Bcl-2 in podocytes. (E) AV-FITC/PI staining was performed to quantify the apoptosis rates via flow cytometry. (F) Quantification of the results by AV-FITC/PI staining. In all histograms, the results were normalized to the normal group, and data are shown as the mean±SD; n=3. #P<0.001 versus the normal group; *P<0.05 and **P<0.001 versus the model group.
Abbreviations: Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; AV-FITC/PI, Annexin V-fluorescein isothiocyanate/propidium iodide; PERK, protein kinase RNA-like endoplasmic reticulum kinase.
Discussion
Emodin induces cell apoptosis or decreases cell proliferation in several tumors, including leukemia, breast and colon cancer, and lung carcinoma cells.38 Additionally, some studies indicate that emodin exerts an antiapoptotic effect to prevent myocardial cell injury in acute myocardial infarction39 and protects HK-2 human renal tubular cells from ischemia/reperfusion-induced apoptosis.27 In our study, we found that emodin protects podocytes from apoptosis via the suppression of ER stress in DN. Podocyte apoptosis is an important pathogenic alteration that leads to podocytes loss in DN. Emodin has a protective effect on renal dysfunction: it inhibits cell proliferation and fibronectin (FN) expression in mesangial cells exposed to high glucose by suppressing the p38 mitogen activated protein kinase (MAPK) pathway.40,41 Emodin also protects rat renal tubular cells against apoptosis by activating autophagy through the modulation of the AMPK/mammalian target of rapamycin (mTOR) signaling.42 In the present study, we have shown, for the first time, that podocyte apoptosis triggered by ER stress can be ameliorated by emodin treatment through the inhibition of the PERK/eIF2α axis in DN.
Clinical research has shown that elevated proteinuria is an important manifestation of glomerular injury and a hallmark of progressive kidney damage in patients with DN.3,5 The present study shows that, 12 weeks after induction of DN in a mouse model, marked urinary albumin, elevated SCr, and BUN were associated with a decrease in the podocyte-specific protein nephrin. Emodin treatment significantly and dose-dependently reduced urinary albumin and improved the renal function and the histopathology in DN mice. Importantly, we have found that emodin had no obvious toxicity on podocytes under the conditions used in our experiments. At the same time, emodin ameliorated foot process effacement and glomerular basement membrane (GBM) thickening, as well as the podocytes cytoskeleton. These results indicate that emodin might be an effective drug for treating DN-induced podocyte injury.
It has been shown that ER dysfunction is a key factor in the development of DN.7,17,20 In the present work, we proved that ER stress was induced in KK-Ay mice with DN. Our results also show that treatment with high glucose or TM induced the expression of the endoplasmic chaperone protein GRP78. GRP78 is a member of the heat shock protein-70 (HSP70) family, which dissociates from its three transducers (IRE1, PERK, and ATF6) when cells are under stress.43 Increased GRP78 protects misfolded proteins from aggregation, helps their correct translocation, and initiates the UPR pathway.44 In the present work, emodin treatment significantly attenuated GRP78 levels in DN KK-Ay mice, and similar effects were observed in podocytes treated with high glucose. To verify this hypothesis, we used the ER stress inhibitor, TUDCA; our results showed that emodin and TUDCA both downregulated the level of GRP78 and there was no difference between them. What’s more, emodin reversed the increased GRP78 induced by TM, the well-known ER stress inducer. These data suggest that emodin can be used in the treatment of DN because it suppresses ER stress.
An important component of the UPR is PERK, a transmembrane protein containing a stress-sensing domain that faces the ER lumen. The overload of misfolded proteins in the ER triggers the dissociation of PERK from GRP78, and the consequent oligomerization and autophosphorylation of PERK,45 resulting in the phosphorylation of eIF2α.46 Phosphorylated eIF2α selectively stimulates the translation of the ATF4 mRNAs and, therefore, increases the levels of ATF4, a critical transcription factor mediating the expression of many genes correlated with cell recovery and stress.45 Under prolonged ER stress conditions, ATF4 stimulates the expression of several transcription factors, including CHOP.46 PERK/ATF4 activation was detected in the renal tissue of DN, indicating that excessive ER stress was a manifest factor in modifying slit diaphragm protein synthesis.47 In the present study, in the DN model mice and in podocytes stimulated with high-glucose, we found elevated levels of phosphorylated PERK, phosphorylated eIF2α, and ATF4. Treatment with emodin significantly decreased the levels of these proteins compared with the model group. Meanwhile, treatment with emodin had no effect on the level of ATF6 and P-IRE1. These data clearly indicate that emodin suppresses the ER stress by inhibiting the PERK pathway. To verify this hypothesis, we knocked down PERK expression by RNAi in podocytes cultured with high glucose and found that the effects of PERK silencing and emodin treatment were comparable. Similarly, chrysin, a natural flavonoid, attenuates podocytes apoptosis in diabetic mice via the inhibition of the ER stress-mediated PERK-eIF2α pathway.48
CHOP is a major mediator of the ATF4 signaling. It has been shown that CHOP gene expression is involved in cell cycle arrest and/or apoptosis.49,50 Previous studies show that increased expression of CHOP decreases the expression of the antiapoptotic members of the Bcl-2 family.51 Conversely, in CHOP−/− mice the cells are protected from lipopolysaccharide (LPS)-induced apoptosis, associated with increased Bax expression and decreased Bcl-2 expression.50 Another study demonstrated that increasing the expression of CHOP could downregulate Bcl-2 and induce apoptosis in HK-2 cells.52 This study has demonstrated similar changes in the levels of these proteins in the renal glomeruli of DN mice, while emodin treatment significantly reversed these alterations. Similar results were obtained in vitro. In addition, treatment with emodin and PERK knockdown mitigated the increased apoptosis of podocytes exposed to high glucose. These observations strongly suggested that emodin protects podocytes from apoptosis by inhibiting the PERK pathway.
Conclusion
The data obtained in the present study indicate that emodin reduces the ER stress in renal tissues and podocytes exposed to high glucose via the inhibition of the PERK-elF2α pathway, thereby downregulating CHOP expression, and ultimately inhibiting podocytes apoptosis. Meanwhile, oral administration of emodin improves albuminuria and the renal function in KK-Ay mice. These findings point to the possible therapeutic use of emodin in DN. However, additional studies are needed to comprehensively understand the role of emodin in ameliorating DN.
Acknowledgments
This work was supported by grants from the Scientific Research Project of Beijing Educational Committee (No KZ201610025024) and the Major National Basic Research Program of China (973 Program, No 2012CB518602).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Fried LF, Emanuele N, Zhang JH, et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369(20):1892–1903. doi: 10.1056/NEJMoa1303154. [DOI] [PubMed] [Google Scholar]
- 2.Yasuda-Yamahara M, Kume S, Tagawa A, Maegawa H, Uzu T. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy. 2015;11(12):2385–2386. doi: 10.1080/15548627.2015.1115173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravindran S, Kuruvilla V, Wilbur K, Munusamy S. Nephroprotective effects of metformin in diabetic nephropathy. J Cell Physiol. 2017;232(4):731–742. doi: 10.1002/jcp.25598. [DOI] [PubMed] [Google Scholar]
- 4.Hakroush S, Cebulla A, Schaldecker T, Behr D, Mundel P, Weins A. Extensive podocyte loss triggers a rapid parietal epithelial cell response. J Am Soc Nephrol. 2014;25(5):927–938. doi: 10.1681/ASN.2013070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ZS, Xiong F, Xie XH, Chen D, Pan JH, Cheng L. Astragaloside IV attenuates proteinuria in streptozotocin-induced diabetic nephropathy via the inhibition of endoplasmic reticulum stress. BMC Nephrol. 2015;16:44. doi: 10.1186/s12882-015-0031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallipattu SK, He JC. The podocyte as a direct target for treatment of glomerular disease? Am J Physiol Renal Physiol. 2016;311(1):F46–F51. doi: 10.1152/ajprenal.00184.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yasuda M, Tanaka Y, Kume S, et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim Biophys Acta. 2014;1842(7):1097–1108. doi: 10.1016/j.bbadis.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Inagi R. RAGE and glyoxalase in kidney disease. Glycoconj J. 2016;33(4):619–626. doi: 10.1007/s10719-016-9689-8. [DOI] [PubMed] [Google Scholar]
- 9.Kawakami T, Gomez IG, Ren S, et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol. 2015;26(5):1040–1052. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Karoui K, Viau A, Dellis O, et al. Endoplasmic reticulum stress drives proteinuria-induced kidney lesions via lipocalin 2. Nat Commun. 2016;7:10330. doi: 10.1038/ncomms10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cybulsky AV. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 2010;77(3):187–193. doi: 10.1038/ki.2009.389. [DOI] [PubMed] [Google Scholar]
- 12.Madhusudhan T, Wang H, Dong W, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun. 2015;6:6496. doi: 10.1038/ncomms7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 14.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13(11):681–696. doi: 10.1038/nrneph.2017.129. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Dong H, Jia C, et al. Activation of mTORC1 in collecting ducts causes hyperkalemia. J Am Soc Nephrol. 2014;25(3):534–545. doi: 10.1681/ASN.2013030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu WK, Liu R, Pei H, Li B. Endoplasmic reticulum stress-related factors protect against diabetic retinopathy. Exp Diabetes Res. 2012;2012:507986. doi: 10.1155/2012/507986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu GC, Fang F, Zhou J, et al. Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia. 2012;55(9):2522–2532. doi: 10.1007/s00125-012-2586-1. [DOI] [PubMed] [Google Scholar]
- 18.Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. 2016;118(11):1808–1829. doi: 10.1161/CIRCRESAHA.116.306923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You YH, Quach T, Saito R, Pham J, Sharma K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol. 2016;27(2):466–481. doi: 10.1681/ASN.2015030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Moon SY, Kim JS, et al. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am J Physiol Renal Physiol. 2015;308(3):F226–F236. doi: 10.1152/ajprenal.00495.2014. [DOI] [PubMed] [Google Scholar]
- 21.Chen T, Zheng LY, Xiao W, Gui D, Wang X, Wang N. Emodin ameliorates high glucose induced-podocyte epithelial-mesenchymal transition in-vitro and in-vivo. Cell Physiol Biochem. 2015;35(4):1425–1436. doi: 10.1159/000373963. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Tian Y, Yu J, et al. iTRAQ-based quantitative proteomic analysis reveals multiple effects of Emodin to Haemophilus parasuis. J Proteomics. 2017;166:39–47. doi: 10.1016/j.jprot.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 23.Gao J, Wang F, Wang W, Su Z, Guo C, Cao S. Emodin suppresses hyperglycemia-induced proliferation and fibronectin expression in mesangial cells via inhibiting cFLIP. PLoS One. 2014;9(4):e93588. doi: 10.1371/journal.pone.0093588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Xu Z, Wang S, et al. Emodin ameliorates hepatic steatosis through endoplasmic reticulum-stress sterol regulatory element-binding protein 1c pathway in liquid fructose-feeding rats. Hepatol Res. 2016;46(3):E105–E117. doi: 10.1111/hepr.12538. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Ding L, Song B, et al. Emodin improves lipid and glucose metabolism in high fat diet-induced obese mice through regulating SREBP pathway. Eur J Pharmacol. 2016;770:99–109. doi: 10.1016/j.ejphar.2015.11.045. [DOI] [PubMed] [Google Scholar]
- 26.Wei G, Wu Y, Gao Q, et al. Effect of emodin on preventing postoperative intra-abdominal adhesion formation. Oxid Med Cell Longev. 2017;2017:1740317. doi: 10.1155/2017/1740317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H, Huang RS, Yu XX, et al. Emodin protects against oxidative stress and apoptosis in HK-2 renal tubular epithelial cells after hypoxia/reoxygenation. Exp Ther Med. 2017;14(1):447–452. doi: 10.3892/etm.2017.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 29.Wang JY, Gao YB, Zhang N, et al. miR-21 overexpression enhances TGF-β1-induced epithelial-to-mesenchymal transition by target smad7 and aggravates renal damage in diabetic nephropathy. Mol Cell Endocrinol. 2014;392(1–2):163–172. doi: 10.1016/j.mce.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Tomino Y. Lessons from the KK-Ay mouse, a spontaneous animal model for the treatment of human type 2 diabetic nephropathy. Nephrourol Mon. 2012;4(3):524–529. doi: 10.5812/numonthly.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song YP, Fan XF, Guo ZY, Fan YJ, Yang JL. Therapeutic effects of emodin in type 2 diabetes mellitus in KKAy mouse model. Int J Clin Exp Med. 2017;10(10):14408–14413. [Google Scholar]
- 32.Fan Y, Zhang J, Xiao W, et al. Rtn1a-mediated endoplasmic reticulum stress in podocyte injury and diabetic nephropathy. Sci Rep. 2017;7(1):323. doi: 10.1038/s41598-017-00305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng YC, Chang JM, Chen CA, Chen HC. Autophagy modulates endoplasmic reticulum stress-induced cell death in podocytes: a protective role. Exp Biol Med. 2015;240(4):467–476. doi: 10.1177/1535370214553772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21(2):212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui F, Zou D, Gao Y, et al. Effect of tongxinluo on nephrin expression via inhibition of notch1/snail pathway in diabetic rats. Evid Based Complement Alternat Med. 2015;2015:424193. doi: 10.1155/2015/424193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X, Gao Y, Xu L, et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci Rep. 2017;7(1):9371. doi: 10.1038/s41598-017-09907-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Liu CP, Xu KF, et al. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am J Nephrol. 2008;28(6):1014–1022. doi: 10.1159/000148209. [DOI] [PubMed] [Google Scholar]
- 38.Dumit VI, Zerbes RM, Kaeser-Pebernard S, et al. Respiratory status determines the effect of emodin on cell viability. Oncotarget. 2017;8(23):37478–37490. doi: 10.18632/oncotarget.16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Tu X, Lin G, et al. Emodin-mediated protection from acute myocardial infarction via inhibition of inflammation and apoptosis in local ischemic myocardium. Life Sci. 2007;81(17–18):1332–1338. doi: 10.1016/j.lfs.2007.08.040. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Huang H, Liu P, et al. Inhibition of phosphorylation of p38 MAPK involved in the protection of nephropathy by emodin in diabetic rats. Eur J Pharmacol. 2006;553(1–3):297–303. doi: 10.1016/j.ejphar.2006.08.087. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Liu W, Wang Q, et al. Emodin suppresses cell proliferation and fibronectin expression via p38MAPK pathway in rat mesangial cells cultured under high glucose. Mol Cell Endocrinol. 2009;307(1–2):157–162. doi: 10.1016/j.mce.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Gu LB, Tu Y, Hu H, Huang YR, Sun W. Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol Sin. 2016;37(2):235–245. doi: 10.1038/aps.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–277. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 44.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62(6):670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkins C, Liu Q, Minthorn E, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73(6):1993–2002. doi: 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- 46.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Sun Y, Cao Y, et al. HRD1 prevents apoptosis in renal tubular epithelial cells by mediating eIF2α ubiquitylation and degradation. Cell Death Dis. 2017;8(12):3202. doi: 10.1038/s41419-017-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang MK, Park SH, Kim YH, et al. Chrysin ameliorates podocyte injury and slit diaphragm protein loss via inhibition of the PERK-eIF2α-ATF-CHOP pathway in diabetic mice. Acta Pharmacol Sin. 2017;38(8):1129–1140. doi: 10.1038/aps.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 51.Guo J, Zhu J, Ma L, et al. Chronic kidney disease exacerbates myocardial ischemia reperfusion injury: role of endoplasmic reticulum stress-mediated apoptosis. Shock. 2018;49(6):712–720. doi: 10.1097/SHK.0000000000000970. [DOI] [PubMed] [Google Scholar]
- 52.Zhu S, Jin J, Wang Y, et al. The endoplasmic reticulum stress response is involved in apoptosis induced by aloe-emodin in HK-2 cells. Food Chem Toxicol. 2012;50(3–4):1149–1158. doi: 10.1016/j.fct.2011.12.018. [DOI] [PubMed] [Google Scholar]