

Abstract
Cell-free protein synthesis (CFPS) can be used in many applications to produce polypeptides and to analyze mechanisms of mRNA translation. Here we describe how to make and use a CPFS system from the model filamentous fungus Neurospora crassa. The extensive genetic resources available in this system provide capacities to exploit robust CFPS for understanding translational control. Included are procedures for the growth and harvesting of cells, the preparation of cell-free extracts that serve as the source of the translational machinery in the CFPS and the preparation of synthetic mRNA to program the CFPS. Methods to accomplish cell-free translation and analyze protein synthesis, and to map positions of ribosomes on mRNAs by toeprinting, are described.
1. Introduction
Cell-free protein synthesis (CFPS) systems derived from prokaryotes and eukaryotes provide powerful methods for producing polypeptides [1] and for understanding mechanisms underlying protein synthesis. We have developed a CFPS system using whole-cell extracts prepared from the model filamentous fungus Neurospora crassa for determining mechanisms regulating protein synthesis at the levels of initiation, elongation and termination [2–12]. We have previously described in detail some of the methods to prepare and use N. crassa CFPSs [13,14]. Since then, we have developed additional assays for which this system is suited, including real-time analyses of the kinetics of the programmed synthesis of firefly luciferase [10,11], the incorporation of unnatural amino acids into nascent peptides [7], and analyses of the peptidyltransferase center reactivity to puromycin [6]. Furthermore, the availability of a full collection of targeted gene-deletion mutants of N. crassa enables the use of mutants to produce CFPSs to assess how changes in cellular factors affect translationally-related processes [11,15]. We have also observed that many who are interested in using this system are not familiar with the growth and maintenance of this filamentous fungus for production of the cell mass needed to produce CFPSs. Here we provide procedures to prepare and use the N. crassa CFPS system.
2. Background for Neurospora crassa
Neurospora is a haploid filamentous fungus that has well-defined asexual (vegetative) and sexual life-cycles that has an extensive and important history as a model organism starting from the foundational work establishing biochemical genetics through the Nobel Prize-winning ‘‘one-gene one-enzyme” studies of Beadle and Tatum in 1941 [16–19]. N. crassa wild-type and mutant strains can be obtained from the Fungal Genetics Stock Center (FGSC), Manhattan KS (http://www.fgsc.net/). In addition to mutant strains obtained through classical genetic approaches [20], a complete set of N. crassa gene-deletion strains [21] is available as individual strains, or a complete set, from the FGSC. Because N. crassa grows asexually as a multinuclear syncytium of filamentous hyphae, a lethal deletion-mutation in a nuclear genome can be sheltered by other nuclei in the syncytium.
Laboratory wild-type N. crassa strains are typically grown asexually (vegetatively) on Vogel’s minimal medium and mutants are grown on appropriately supplemented minimal medium (additional information concerning methods for growing N. crassa can be found at http://www.fgsc.net/Neurospora/neurospora.html) and in [22]. Biosafety level 1 is generally considered sufficient for laboratory handling of N. crassa strains containing recombinant DNA. N. crassa forms multinuclear asexual spores called macroconidia (typically referred to as conidia) profusely on aerial hyphae; typically, more than 1010 spores can be harvested from wild-type cultures grown on solid medium in a Fernbach flask [22] and 107 spores can be easily removed from a slant culture by dipping in a sterile moist stick. Conidia are hydrophobic and easily dispersed in air; it is important to harvest conidia in a hood, not on the benchtop, to minimize airborne dispersal of conidia in the lab. Properly handled, N. crassa does not pose a problem for airborne contamination.
Because N. crassa vegetative cells, including macroconidia, are generally multinuclear, to assure genetically pure cultures, it is important to bring N. crassa through a stage where there is a uninucleate phase. For vegetatively growing cells, the simplest way to accomplish this is to enhance the formation of uninucleate microconidia and to purify microconidia through filtration [23] as described below.
To obtain colonial growth of N. crassa on plates, spreading of the vegetative hyphae (the hyphal mass is called a mycelium) is inhibited by adding sorbose to the growth medium. While N. crassa is typically grown with sucrose as a carbon source, it cannot be grown on sucrose in the presence of sorbose because sorbose is a noncom-petitive inhibitor of invertase [24], which cleaves sucrose. Thus, for growing N. crassa as colonies on plates, fructose + glucose has historically been used as a combined carbon source when sorbose is added to the media [25]. Another crucial aspect of using FGS plates is that, for reasons that remain obscure, the plates must be prepared fresh (we typically prepare them within 24 h of use) or the efficiency of plating is drastically reduced.
3. Methods
All of the water used (we use Milli-Q purified water) in reactions, and reagent solutions used which do not already contain Tris, are treated with diethylpyrocarbonate (DEPC) to inactivate ribonucleases. DEPC is added to a concentration of 0.1% to water or reagent solutions, and after at least 12 h have elapsed, the DEPC is inactivated by autoclaving.
3.1. Reagents and stocks
Trace Element Solution
| Component | Final concentration | Stock | Amount stock/100 ml |
|---|---|---|---|
| Citric acid·H2O | 5 g | ||
| ZnSO4·7H2O | 5 g | ||
| Fe(NH4)2(SO4)2·6H2O | 1 g | ||
| CuSO4·5H2O | 0.25 g | ||
| MnSO4·H2O | 0.05 g | ||
| H3BO3 | 0.05 g | ||
| Na2MoO4·2H2O | 0.05 g |
Components are dissolved consecutively to water. Adjust to a final volume of 100 ml with water and store at 4 °C.
50× Vogel’s minimal salt stock
| Component | Final concentration | Stock | Amount stock/L |
|---|---|---|---|
| Na3citrate·2H2O | 125 g | ||
| KH2PO4 | 250 g | ||
| NH4NO3 | 100 g | ||
| MgSO4·7H2O | 10 g | ||
| CaCl2·2H2O | 5 g | ||
| Trace element solution | 5 ml | ||
| Biotin solution | 0.1 mg/ml | 2.5 ml |
Components are dissolved consecutively and completely, in order, in 735 ml of water in a large Erlenmeyer flask, with stirring. Moderate heating can be applied if there is difficulty. CaCl2 is dissolved separately in 20 ml of water and added slowly to the stock. Biotin solution is prepared by dissolving 5 mg biotin in 50 ml of water and the solution is stored at −20 °C as 2.5 ml aliquots. Alternatively, 5 mg biotin is dissolved in 50 ml of 50% ethanol, and the solution can be stored at 5 °C. Add water to 1 L, mix, add 1 ml of chloroform as preservative, store at room temperature.
2× Synthetic Cross (SC) salt stock
| Component | Final concentration | Stock | Amount stock/3 L |
|---|---|---|---|
| KNO3 | 6 g | ||
| K2HPO | 4.2 g | ||
| KH2PO4 | 3 g | ||
| MgSO4·7H2O | 3 g | ||
| NaCl | 0.6 g | ||
| H3BO3 | 0.6 g | ||
| CaCl2·2H2O | 0.05 g | ||
| Trace element Solution | 0.6 ml | ||
| Biotin solution | 0.1 mg/ml | 0.3 ml |
Components are dissolved consecutively in a large Erlenmeyer flask, with stirring and moderate heating. CaCl2 is dissolved separately in 20 ml of water and added slowly to the stock. Add water to 3 L, mix, add 2 ml of chloroform as preservative, store at 4 °C.
10× FGS
| Component | Final concentration | Stock | Amount stock/100 ml |
|---|---|---|---|
| Fructose | 0.5% | 0.5 g | |
| Glucose | 0.5% | 0.5 g | |
| Sorbose | 10% | 10 g |
Add water to 100 ml, sterilize by autoclaving, store at room temperature.
Vogel’s sucrose medium
| Component | Final concentration | Stock | Amount stock/L |
|---|---|---|---|
| Vogel’s minimal salt | 1× | 50× | 20 ml |
| Sucrose | 2% | 20 g |
Add water to 1 L, sterilize by autoclaving. For solid medium, add 2% agar, and then autoclave.
Microconidiation medium
| Component | Final concentration | Stock | Amount stock/100 ml |
|---|---|---|---|
| SC salt | 0.1× | 2× | 5 ml |
| Sucrose | 0.5% | 0.5 g | |
| Agar | 2% | 2 g | |
| Sodium iodoacetate | 1 mM | 100 mM | 1 ml |
Add water to 99 ml, sterilize by autoclaving. Sodium iodoacetate is freshly prepared in water, sterilized by filtration, and added to the autoclaved medium cooled to 55 °C
FGS colonial growth medium
| Component | Final concentration | Stock | Amount Stock/250 ml |
|---|---|---|---|
| Vogel’s minimal salt | 1× | 50× | 5 ml |
| Sucrose | 2% | 5 g | |
| Agar | 2% | 5 g | |
| FGS | 1× | 10× | 2.5 ml |
Add water to 247.5 ml, sterilize by autoclaving. FGS is added to the autoclaved medium cooled to 55 °C.
Buffer A
| Component | Final concentration | Stock | Amount stock/L |
|---|---|---|---|
| HEPES-KOH, pH 7.6 | 30 mM | 1 M | 30 ml |
| KOAc, pH 7.0 | 100 mM | 4 M | 25 ml |
| Mg(OAc)2, pH 7.0 | 3 mM | 0.3 M | 10 ml |
| DTT | 2 mM | 2 M | 1 ml |
Add DEPC-treated water to 1 L, mix, store at 4 °C. DTT is added immediately prior to use.
50× Protease Inhibitor Cocktail (PIC)
| Component | Final concentration | Stock | Amount stock/ml |
|---|---|---|---|
| APMSF | 1.25 mg/ml | 25 mg/ml | |
| Pepstatin A | 0.25 mg/ml | 5 mg/ml | |
| Antipain | 0.25 mg/ml | 5 mg/ml | |
| Chymostatin | 0.25 mg/ml | 5 mg/ml | |
| Elastatinal | 0.05 mg/ml | 1 mg/ml | |
| Leupeptin | 0.25 mg/ml | 5 mg/ml |
Dissolve 25 mg APMSF, 5 mg Pepstatin A, 5 mg Antipain, 5 mg Chymostatin, and 1 mg Elastatinal in DMSO to make 1 ml 1000× stock. Dissolve 5 mg leupeptin in DEPC-treat water to make 1 ml 1000× stock. Store both stocks at −80 °C. To make 50 × stock before use, take 50 μl of each stock, add DEPC-treated water to 1 ml. The remaining 50× stock is stored at −80 °C.
5× Transcription Buffer
| Component | Final concentration | Stock | Amount stock/ml |
|---|---|---|---|
| HEPES-KOH, pH 7.6 | 400 mM | 1 M | 400 μl |
| MgCl2 | 80 mM | 1 M | 80 μl |
Add DEPC-treated water to 1 ml, mix, prepare 200 μl aliquots, freeze in liquid nitrogen, store at °80 °C.
Nucleoside triphosphates (NTPs) for capped synthetic RNA synthesis
| Component | Final concentration | Stock | Amount stock/ml |
|---|---|---|---|
| ATP | 30 mM | 100 mM | 30 μl |
| CTP | 30 mM | 100 mM | 30 μl |
| GTP | 6 mM | 100 mM | 4 μl |
| UTP | 30 mM | 100 mM | 30 μl |
Add DEPC-treated water to 0.1 ml, mix, freeze in liquid nitrogen, store at −80 °C.
2× RNA loading buffer
| Component | Final concentration | Stock | Amount stock/ml |
|---|---|---|---|
| NEB 6 × DNA loading buffer | 2× | 6× | 0.33 ml |
| Urea | 12% | 0.12 g |
Add DEPC-treated water to 1 ml, store at 4 °C.
10× Energy Mix
| Component | Final concentration | Stock | Amount stock/ml |
|---|---|---|---|
| HEPES-KOH, pH 7.6 | 200 mM | 1 M | 200 μl |
| ATP | 10 mM | 100 mM | 100 μl |
| GTP | 1 mM | 100 mM | 10 μl |
| Phosphocreatine | 200 mM | 500 mM | 400 μl |
| DTT | 20 mM | 2 M | 10 μl |
Add DEPC-treated water to 1 ml, mix, prepare 200 μl aliquots, freeze in liquid nitrogen, store at −80 °C.
10 U/μl Creatine Phosphokinase (CPK) solution
| Component | Final concentration | Stock | Amount stock/1.75 ml |
|---|---|---|---|
| HEPES-KOH, pH 7.6 | 10 mM | 1 M | 17.5 μl |
| DTT | 1 mM | 2 M | 0.875 μl |
| Creatine phosphokinase | 10 U/μl | Sigma C3755 | 17,500 U |
| Glycerol | 50% | 875 μl |
Add DEPC-treated water to 1.75 ml, mix, prepare 25 μl aliquots, freeze in liquid nitrogen, store at −80 °C.
Luciferin/Coenzyme A solution
| Component | Final concentration | Stock | Amount stock/10 ml |
|---|---|---|---|
| D-Luciferin, potassium salt | 25 mM | Gold Biotechnology | 80 mg |
| Coenzyme A | 5 mM | Nanolight Technology | 38 mg |
Dissolve in 50% ethanol, prepare 1 ml aliquots, freeze in liquid nitrogen, store at −80 °C.
Gel fixing solution
| Component | Final concentration | Stock | Amount Stock/100 ml |
|---|---|---|---|
| Water | 45 ml | ||
| Acetic acid | 10% | 10 ml | |
| Methanol | 45% | 45 ml |
We prepare fresh before use.
5× Reverse Transcription Buffer (RT)
| Component | Final concentration | Stock | Amount stock/ml |
|---|---|---|---|
| Tris-HCl, pH 8.0 | 250 mM | 1 M | 250 μl |
| KCl | 375 mM | 1 M | 375 μl |
| MgCl2 | 50 mM | 1 M | 50 μl |
Add DEPC-treated water to 1 ml, mix, prepare 200 μl aliquots, freeze in liquid nitrogen, store at °80 °C.
2× sequencing gel loading buffer
| Component | Final concentration | Stock | Amount stock/40 ml |
|---|---|---|---|
| Bromophenol Blue | 0.05% | 2 mg | |
| Xylene Cyanol FF | 0.05% | 2 mg | |
| EDTA, pH 8.0 | 20 mM | 1 M | 0.8 ml |
| Formamide | 91% | 36.4 ml |
Add DEPC-treated water to 40 ml, store at 4 °C.
6% polyacrylamide gel
| Component | Final concentration | Stock | Amount stock/100 ml |
|---|---|---|---|
| Sterile water | 40 ml | ||
| Urea | 46.8% | 1 M | 46.8 g |
| 19:1 (mono:bis) Acrylamide Solution | 6% | 40% | 15 ml |
| TBE | 1× | 10× | 10 ml |
After urea completely dissolves in the acrylamide and TBE solution with the help of a heat block (be careful not to boil the solution), 100 μl of 50% APS solution and 20 μl of TEMED are added to the gel solution. Then the solution is poured between the wrapped glass plates using a 60-ml syringe.
3.2. Purification of N. crassa homokaryons
The multinucleate macroconidia used for vegetative propagation of conidiating N. crassa strains can be heterokaryotic (containing nuclei with different genotypes) if they form from heterokaryotic cultures [22]. In order to obtain genetically pure (homokaryotic) strains, uninucleate microconidia can be obtained, purified and used to establish vegetative cultures. To accomplish this, macroconidia from the parental strains are inoculated into slants prepared with microconidiation medium. Cultures are grown at room temperature for 10 days to obtain viable homokaryotic, uninucleate microconidia [23]. These culture conditions promote the formation of microconidia and suppress the formation of macroconidia. The microconidia are harvested and separated from the hyphae and macroconidia by making a suspension by adding 2 ml of sterile water to the slant tubes, vortexing, and passing the cell-suspension through 5 μm filters (Millipore SLSV025LS). The number of microconidia is counted using a hemocytometer slide under a compound microscope and the concentration is calculated. These microconidia are spread and germinated at 30 °C on freshly prepared FGS colonial growth medium. Alternatively, the microconidia can be germinated on Vogel’s sucrose plates supplemented with 0.005% Tergitol NP-10 (added to autoclaved medium when the medium has cooled to 55 °C) (http://www.fgsc.net/Neurospora/NeurosporaProtocolGuide.htm). Note that colony spreading can become rapid after several days on tergitol-containing medium. Colonies obtained after 2–3 days incubation are generally homokaryotic if the growth of homokaryons is possible and these colonies are transferred individually to Vogel’s sucrose slants for vegetative growth. Taking a plug of the colonies with a sterile Pasteur pipet is an ideal way to transfer colonies from plates to slants; the Pasteur pipet can be resterilized and reused for this purpose.
3.3. Growth of N. crassa for extract preparation
Conidia are removed from a slant using a sterile moist wood stick (touching the stick to the conidia is sufficient to obtain enough conidia) and are transferred to a tube containing 5 ml of sterile water to make a suspension. The entire suspension of conidia is then spread on the surface of 250 ml of solid Vogel’s sucrose medium in a 2.5 L Fernbach flask. Air promotes conidia formation and we find it easiest to cap the flask with sterile 541 filter paper rubber-banded around the neck of the flask. The culture is incubated at room temperature for 10 to 14 days. To harvest the conidia, 70 ml of Vogel’s sucrose medium is added to the flask to make a suspension by shaking vigorously. The suspension is passed through two layers of sterile cheesecloth held by rubber bands across the opening of the receiving sterile 250 ml beaker to remove mycelial fragments. The cheesecloth is fitted loosely into the opening to allow a pool of suspended conidia to drain slowly into the beaker. Rinse the flask with an additional of 70 ml of Vogel’s sucrose medium and combine. The number of conidia is counted using a hemacytometer slide under a compound microscope and the concentration is calculated. Typically we dilute a small aliquot of the conidia suspension 1:200 prior to counting.
Conidia are inoculated to Vogel’s sucrose medium at a final concentration of 1 × 107 conidia/ml. The volume change from adding the conidia suspension is not considered with calculation. Specifically, a 1-liter culture in a 2-liter Erlenmeyer flask is incubated at 32 °C with orbital shaking (180–200 rpm) until 90% of the conidia show germ tubes (typically 5–6 h). The germlings are harvested by vacuum filtration onto 9 cm Whatman 541 filter paper using a Büchner funnel. The filter paper with the germlings is placed in a sterile 50 ml conical screwcap tube with 40 ml of fresh growth medium, the germlings are resuspended by vigorous handshaking, and transferred to 1-liter of fresh pre-warmed growth medium in a 2-liter flask, incubated an additional 1 h at 32 °C with orbital shaking, and harvested again. For reasons that remain to be determined, this ‘‘medium-switch” results in CFPS systems being more dependent on the addition of exogenous amino acids [2]. Following harvesting, germling pads are rinsed on the filter with ice-cold buffer A, then peeled from the filter and weighed; a typical yield from the wild-type strain is 10g per flask. The pads are cut into small pieces, placed in a sterile 50 ml conical screwcap tube, and frozen in liquid nitrogen. The frozen germling pads can be used immediately or stored at −80 °C until ready to use.
3.4. Preparation of N. crassa cell-free extract
Freshly prepared Buffer A (1:1 vol Buffer A (ml):cell weight (g)) is dripped directly into liquid nitrogen by using a sterile Pasteur pipette to form small frozen beads. Frozen buffer beads and germling pads (10–20 g) from the previous step are transferred into a pre-chilled 6801 grinding vial of a SPEX SamplePrep 6850 Freezer/Mill along with the Spex-supplied iron rod. The vial is then sealed and placed in the liquid nitrogen filled tank of the Freezer/Mill. The sample is cooled for 10 min and then powdered by three 2-min grind cycles with a speed setting at 10 with 1-min rest between each cycle. The powdered cells are transferred to pre-chilled 50 ml polycarbonate centrifuge tubes, allowed to thaw in an ice water bath for about an hour, and then centrifuged at 4 °C for 15 min at 16,000 rpm (30,600 μg) in a Sorvall SS34 rotor. The supernatant is carefully transferred to a second pre-chilled poly-carbonate centrifuge tube, using a sterile Pasteur pipette, avoiding both the pellet and the fatty upper layer. The cell-free extract is centrifuged again with the same settings. The supernatant is carefully collected and placed in a fresh 50 ml conical tube that is maintained on ice. Small molecules are removed from the extract by chromatography at 4 °C through 10 ml Zeba spin desalting columns (Pierce) as instructed by the manufacturer (4 ml extract per column). Buffer A is used to preequilibrate the columns. Protease inhibitor cocktail is then added to 1× final concentration to the purified extracts. For storage, aliquots of extract (100 μl) are put into 0.6 ml Eppendorf tubes, frozen with liquid nitrogen, and stored at −80 °C.
3.5. Preparation of synthetic mRNAs to program the CFPS
The mRNA templates used to program cell-free translation reactions are synthesized from linear DNA templates using T7 RNA polymerase (New England Biolabs) in vitro. The N. crassa CFPS system is highly cap- and poly(A)-dependent [2].The DNA templates typically contain a stretch of 30 A residues after the mRNA 3′UTR at the mRNA 3′ end to function as a poly(A) tail following transcription. Alternatively, the poly(A) tail can be added after transcription is completed using E.coli poly(A) polymerase (New England Biolabs) as instructed by the manufacturer. Each 100 μl in vitro transcription reaction to synthesize capped mRNA contains:
| 0.5 μg/μl linear DNA template | 12 μl |
| 5 × transcription buffer | 20 μl |
| 2NTPs | 10 μl |
| 1 M DTT | 2 μl |
| 0.1 M spermidine | 2 μl |
| 10 mM cap analog | 28 μl |
| RNasin plus (Promega) | 2 μl (80 U) |
| T7 RNA polymerase | 2.4 μl (120 U) |
| Total | 100 μl |
Reaction mixtures are incubated for 2 h at 37 °C. The DNA template is then removed by adding 1 unit of RQ1 DNase I (Promega) and incubation at 37 °C for an additional 15 min. DEPC-treated water (35 μl) and 5 M NH4OAc (15 μl) are added, and each reaction mixture is extracted with 150 μl of phenol:chloroform (1:1), and then with 150 μl of chloroform. The aqueous phase is transferred to a fresh 1.6 ml Eppendorf tube and the mRNA is precipitated by adding 1 vol of isopropanol, chilling for at least 20 min at –20 °C, and centrifugation at 4 °C for 20 min in an Eppendorf 5415R minicentrifuge at 13,200 rpm (16,000g). The supernatant is carefully removed and the pellet is washed once with 70% ethanol at room temperature, briefly vacuum dried, and then resuspended in 50 μl of DEPC-treated water. To check the quality and quantity of the synthesized mRNA, 1 μl of mRNA solution is diluted with 3 μl of DEPC-treated water and mixed with an equal volume of 2× RNA loading buffer. The samples are then heated in a 65 °C water bath for 3 min, chilled on ice, and then loaded on a nondenaturing 1% TAE agarose gel containing 0.5 μg/ml ethidium bromide alongside marker lane containing 0.25 μg of 1 kb DNA ladder (New England Biolabs) markers. After electrophoresis, the gel is wrapped with foodservice film and scanned with a GE Typhoon Trio + imager. The image is analyzed with ImageQuantTL software and the RNA amount calculated relative to the DNA markers. This method is not perfectly accurate because RNA and DNA stain differently with ethidium bromide but it is precise and reproducible. The final concentration of mRNA is adjusted to 60 ng/μl and stored in aliquots at −80 °C. In our experience, multiple freeze–thaw cycles adversely affect the translatability of the mRNA.
3.6. CFPS reactions
By measuring the activity of luciferase synthesized in Neurospora cell-free extracts, the effects of cis- and trans-acting elements in translation can be analyzed. The following example shows how arginine (Arg) negatively regulates the synthesis of luciferase reporter through the arginine attenuator peptide (AAP) encoded by the N. crassa arg-2 uORF (upstream open reading frame). The wild-type AAP, but not a mutated AAP in which Asp-12 is substituted by Asn (D12N), stalls ribosomes in response to Arg. For every twenty 10-μl translation reactions, prepare the following mixture on ice:
| DEPC-treated water | 35.4 μl |
| 10× Energy mix | 20 μl |
| 10 U/μl CPK solution | 1.2 μl |
| 2 M KOAc | 7 μl |
| 0.1 M Mg(OAc)2 | 2.4 μl |
| 1 mM amino acids mix | 2 μl |
| RNasin Plus (Promega) | 2 μl (80 U) |
| Total | 70 μl |
The mixture is divided into two halves. To one-half, 5 μl of DEPC-treated water is added. To the other half, 5 μl of 40 mM Arg is added. 4 μl of each mixture is then aliquoted to individual 0.6 ml Eppendorf tubes held on ice and 1 μl of 6 ng/μl mRNA or 1 μl of water (no RNA control) is added to each tube. 5 μl of cell-free extract is added to each tube and the contents are gently mixed to start the translation reactions. Translation reactions are incubated at 26 °C for 30 min. To stop translation, 20 μl of passive lysis buffer (prepared from Promega’s 5× stock) is added to each reaction to the final concentration of 1× and mixed well. 15 μl from each reaction is then transferred to a nontransparent 96-well OptiPlate (Perkin Elmer), and luciferase activity is measured using lab-made luciferase assay reagent [26] or a Promega commercial luciferase assay kit with a Victor3-V multilabel counter (Perkin-Elmer). The results of such an analysis, in which the wild-type AAP shows its capacity to confer Arg-specific regulation on luciferase reporter synthesis when present as an uORF or when fused directly to the reporter, but a mutant AAP that does not stall ribosomes does not regulate reporter synthesis in either configuration, is shown in Fig. 1.
Fig. 1.
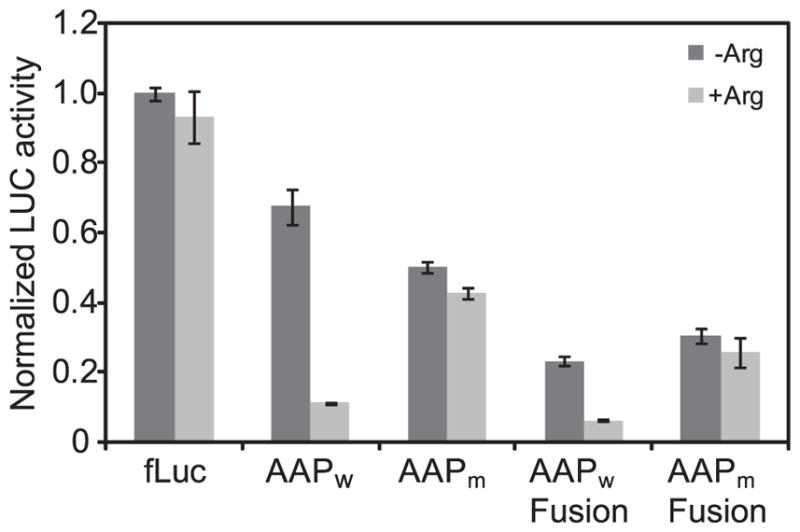
AAP-mediated regulation of mRNA translation in response to Arg in the N. crassa CFPS examined by measurement of luciferase reporter activity. Triplicate reactions were programmed with the indicated mRNAs and luciferase activity levels measured after 30 min of translation as described in the text. Reactions contained low Arg (black) or high Arg (gray). Amounts of luciferase produced, normalized to the amount produced from the control fLuc mRNA (lacking any known 5′-regulatory sequences) in low Arg, are shown. Plasmids used [27–29] to generate the mRNAs were: fLuc, pPQ101 (originally called pHLucNFS); AAPw, pPR301 (wild- type AAP as uORF); AAPm, pPS301 (non-regulatory D12N mutant AAP as uORF); AAPw Fusion, pKL105 (wild-type AAP fused in frame with luciferase); AAPm Fusion, pKLS105 (D12N mutant AAP fused in frame with luciferase). Error bars represent SD values.
Modifications can be made to the above protocol to monitor in real-time the first appearance of luciferase activity, which is indicative of the rate of translation elongation through the luciferase coding region [10,11]. The setup of translation reactions is similar to the one described above. However, the total volume of each reaction is increased to 50 μl, and includes 1 μl of 25 mM luciferin/5 mM coenzyme A. After all components are added, the translation reaction mixture is aliquoted in triplicates of 50 μl and transferred to a nontransparent 96-well OptiPlate. The translation reactions are incubated inside the Victor3-V multilabel counter. The counter is programmed to measure luminescence from each well every 10 s for 30 min. The relative luminescence units obtained are then plotted against time of reaction.
To visualize the radiolabeled luciferase or other polypeptides synthesized from added mRNA in CFPS, the cell-free extract needs be treated with nuclease to remove endogenous mRNAs before being added to the translation reaction mixtures. 100 μl of extract is incubated with 1 μl of 100 mM CaCl2 and 5 U of micrococcal nuclease (Sigma) at 26 °C for 10 min. Then, 1.5 μl of 170 mM EGTA (pH 7.0) is added to chelate the calcium and inactivate the Ca2+-dependent nuclease. The extract is placed back on ice and is ready to use. [35S]Met (>1000 Ci/mmol, 10 mCi/ml; MP Biomedicals) is added to translation reaction mixtures at a final concentration of 0.5 μCi/μl. The 1 mM stock solution of 20 amino acids is replaced by one that contains no methionine.
To visualize peptidyl-tRNA by [35S]Met labeling, for example, to assess the effects of arginine and AAP on the function of ribosome peptidyl transferase center (PTC), in vitro translation reactions (10 μl) are programmed with 60 ng of RNA [6]. The AAP encoding mRNAs are truncated within the coding region. When this is done, the ribosomes translating the mRNA are expected to remain at the 3′ end of the mRNA with the last codon of the truncated mRNA in the ribosome P site, and with the nascent peptide attached to the P site tRNA. Capped truncated transcripts lack a poly(A) tail and thus do not translate as efficiently as capped and polyadenylated transcripts; this deficit can be partially overcome by putting an A30-stretch in the capped mRNA’s 5′-leader region [7]. After the translation reactions are incubated for 5 min at 26 °C, puromycin is added at a final concentration of 1 mM to release nascent peptide from the peptidyl-tRNA. 5 μl are taken from each reaction immediately before the addition of puromycin and at different time points following puromycin addition and mixed with an equal volume of 2× NuPAGE LDS sample buffer (Invitrogen). Samples are then loaded onto 12% NuPAGE gels (Invitrogen), electrophoresed with morpholineethanesulfonic acid (MES) running buffer. After being fixed in 100 ml of gel fixing solution for 30 min, the gels are placed on 2 layers of filter paper, dried with a vacuum gel dryer, and exposed to a phosphor screen overnight. The screens are scanned with a GE Typhoon Trio + imager and the obtained images are analyzed with ImageQuantTL software. Furthermore, tRNA molecules can be identified by northern blot with appropriately designed probes.
3.7. Toeprinting (primer extension inhibition) assay
To understand translational mechanisms, the positions of ribosomes and associated factors on mRNAs can be precisely mapped using a technique called primer extension inhibition or ‘‘toeprinting”. Radiolabeled oligonucleotide primers used in toeprinting assay are prepared as follows.
| DEPC-treated water | 19 μl |
| 1 M Tris·HCl, pH 8.0 | 5 μl |
| 2.5 μM oligo primer | 20 μl |
| 0.1 M MgCl2 | 10 μl |
| 40 mM spermidine | 5 μl |
| 0.1 M DTT | 10 μl |
| [γ-32P]ATP | 30 μl |
| T4 polynucleotide kinase | 1 μl (10 U) |
| Total | 100 μl |
First, the mixture containing water, Tris, and oligo primer is heated at 96 °C for 3 min and then chilled on ice for another 3 min. MgCl2, spermidine, DTT, [γ-32P]ATP (3000 Ci/mmol 10 mCi/ml; Perkin Elmer), and T4 PNK (New England Biolabs) are then added to the mixture and the reaction is incubated at 37 °C for 45 min. To inactivate the kinase, EDTA is added to a final concentration of 50 mM. Radiolabeled primers are purified with mini Quick Spin Oligo Columns (Roche), stored at −80 °C and are good to use for up to two weeks after labeling.
Reference lanes for mapping the toeprint signals are obtained by carrying out sequencing reactions using a Thermo Sequenase Cycle Sequencing kit (Fisher) with 1 μl of the same radiolabeled primer used for toeprinting and 0.5 μg of the same DNA templates (in either linearized or circularized form) used to synthesize mRNAs. After sequencing reactions are completed, each sample is mixed with an equal volume of 2× sequencing gel loading buffer and stored at −20 °C until ready to use. 1 μl of each sequencing sample is then diluted 10-fold with gel loading buffer and 3 μl will be loaded onto the gel.
Before setting up the translation reactions, toeprinting reaction mixture for every 10 reactions is prepared as follows.
| DEPC-treated water | 12.5 μl |
| 5× Reverse transcription buffer | 20 μl |
| 0.1 M DTT | 10 μl |
| 2.5 mM dNTPs | 10 μl |
| RNasin Plus (Promega) | 2.5 μl (100 U) |
| Total | 55 μl |
5.5 μl of the toeprinting reaction mixture is aliquoted into individual 0.6 ml Eppendorf tubes and placed on ice. Translation reactions (10 μl) are performed as described above with 60 ng of mRNA in each. After different reaction times (e.g., 0, 5, 10, or 15 min), 3 μl is taken from the each translation reaction and transferred to the toeprinting reaction mixtures. The toeprinting mixtures are incubated at 55 °C for 3 min. This step is critical for the visualization of ribosomes by toeprinting, though the mechanism is unknown. 1 μl of radiolabeled primer is then added to each mixture and incubated at 37 °C for 5 min. Then, 0.5 μl Superscript III reverse transcriptase (100 U; Invitrogen) is added and the reactions are incubated at 37 °C for 30 min. The reactions are stopped by extraction with an equal volume of phenol:chloroform (1:1). Following extraction, the aqueous phase is mixed with an equal volume of 2× sequencing gel loading buffer. Toeprinting samples along with sequencing samples are heated at 80–85 °C for 5 min and then loaded on a 6% urea polyacrylamide sequencing gel (pre-run at 55 W for 45 min) and electrophoresed at 65 W until the bromophenol blue dye runs off the gel. The running time may be adjusted to optimize the resolution of products in different size ranges. Typically, the primers we use are 80–200 nucleotides downstream from the toeprint sites of interest. The gel is transferred to 2 layers of filter paper, dried with a vacuum gel dryer, and exposed to a phosphor screen overnight. The screen is scanned with a GE Typhoon Trio + imager and the image is analyzed with ImageQuantTL software.
Addition of cycloheximide (Cyh) to CFPS systems either before translation can be initiated (T0) or during steady-state translation can be used in conjunction with toeprinting to determine whether ribosomes reach an initiation codon by leaky scanning or by reinitiation [4]. More generally, addition of Cyh can increase toeprint signals to aid identification of initiation codons. The use of Cyh to examine initiation in N. crassa CFPS programmed with an mRNA containing the wild-type AAP uORF upstream of luciferase is shown in Fig. 2. When no Cyh is added and reactions are incubated for 15 min prior to toeprinting, a strong signal corresponding to ribosomes stalled at the AAP termination codon (the signal is ≈13 nt downstream from the termination codon in the ribosome A site) observed in response to Arg, but signals corresponding to AAP and luciferase initiation codons are weak or undetectable. When Cyh is added after 15 min of incubation and reactions immediately processed for toeprinting, the termination stall remains but in this example, there is only a slightly discernible effect of Cyh on signals corresponding to initiation codons. However, when Cyh is added at T0 and reactions incubated for 15 min prior to toeprinting, strong signals corresponding to the AAP and luciferase initiation codons are observed (the signals are ≈16 nt downstream from the initiation codons in the ribosome P site).
Fig. 2.
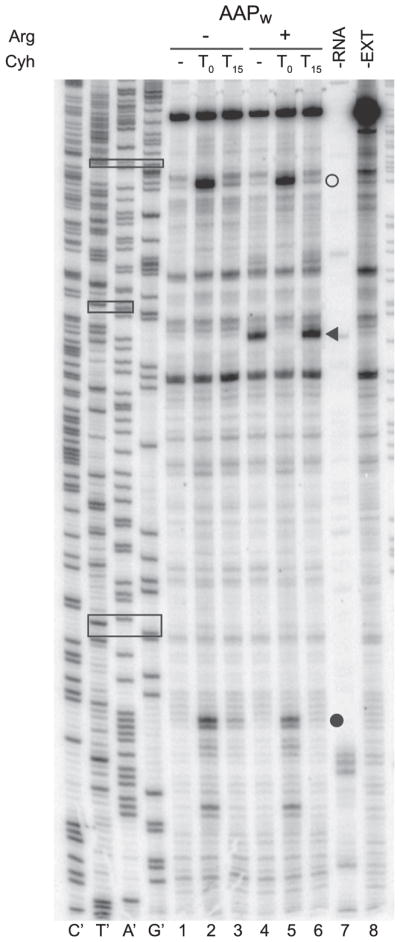
AAP-mediated regulation of mRNA translation in response to Arg in the N. crassa CFPS examined by toeprinting analysis of ribosome positions on mRNA. N.crassa CFPS was programmed with equal amounts (60 ng) of AAPw mRNA that contains the wild type AAP as a uORF upstream of luciferase. Cycloheximide (Cyh) was either not added to translation reactions (−), or was added either prior to starting the translation reactions (T0) or after 15 min of incubation (T15). Radiola-beled primer ZW4 [27] was used for primer extension analysis and for sequencing the pPR301 DNA template. The nucleotide complementary to the dideoxynu-cleotide added to each sequencing reaction is indicated below the corresponding lane to enable the sequence of the template to be directly deduced. Arrowhead, toeprints corresponding to ribosomes stalled at the termination codon of AAP; open circle, toeprints corresponding to ribosomes at AAP initiation codon; closed circle, toeprints corresponding to ribosomes stalled at the luciferase initiation codon. Boxes in the sequencing marker lanes (top to bottom) indicate AAP initiation, AAP termination, and luciferase initiation codons, respectively. –RNA, extract without RNA;-EXT, RNA without extract.
4. Perspective and conclusions
The N. crassa CFPS provides a robust platform for analyses of translation. It can be inexpensively and rapidly prepared and its activity is comparable to that of commercial systems available at much higher cost. By comparing luciferase activity produced by N. crassa CFPS in parallel with activity obtained from known quantities of purified recombinant luciferase, N. crassa CFPS programmed with near-saturating levels of capped and polyadenylated luciferase mRNA (120 ng RNA/10 μl CFPS) produces 2.5 μg luciferase/ml CFPS in 30 min. The availability of mutants that can be used to prepare CFPSs extends the utility of the system for analyzing critical translational processes including mechanisms of quality control [15].
Acknowledgments
National Institute of Health R01 GM058529.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.ymeth.2017.12.003.
References
- 1.Chong S. Overview of cell-free protein synthesis: historic landmarks, commercial systems, and expanding applications. Curr Protoc Mol Biol. 2014;108:1–11. doi: 10.1002/0471142727.mb1630s108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, Sachs MS. Arginine-specific regulation mediated by the Neurospora crassa arg-2 upstream open reading frame in a homologous, cell-free in vitro translation system. J Biol Chem. 1997;272:255–261. doi: 10.1074/jbc.272.1.255. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Sachs MS. Ribosome stalling is responsible for arginine-specific translational attenuation in Neurospora crassa. Mol Cell Biol. 1997;17(9):4904–4913. doi: 10.1128/mcb.17.9.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaba A, Wang Z, Krishnamoorthy T, Hinnebusch AG, Sachs MS. Physical evidence for distinct mechanisms of translational control by upstream open reading frames. EMBO J. 2001;20:6453–6463. doi: 10.1093/emboj/20.22.6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doronina VA, Wu C, De Felipe P, Sachs MS, Ryan MD, Brown JD. Site-specific release of nascent chains from ribosomes at a sense codon. Mol Cell Biol. 2008;28(13):4227–4239. doi: 10.1128/MCB.00421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei J, Wu C, Sachs MS. The arginine attenuator peptide interferes with the ribosome peptidyl transferase center. Mol Cell Biol. 2012;32:2396–2406. doi: 10.1128/MCB.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C, Wei J, Lin PJ, Tu L, Deutsch C, Johnson AE, Sachs MS. Arginine changes the conformation of the arginine attenuator peptide relative to the ribosome tunnel. J Mol Biol. 2012;416(4):518–533. doi: 10.1016/j.jmb.2011.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei J, Zhang Y, Ivanov IP, Sachs MS. The stringency of start codon selection in the filamentous fungus Neurospora crassa. J Biol Chem. 2013;288:9549–9562. doi: 10.1074/jbc.M112.447177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou M, Guo J, Cha J, Chae M, Chen S, Barral JM, Sachs MS, Liu Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013;495(7439):111–115. doi: 10.1038/nature11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell. 2015;59(5):744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caster SZ, Castillo K, Sachs MS, Bell-Pedersen D. Circadian clock regulation of mRNA translation through eukaryotic elongation factor eEF-2. Proc Natl Acad Sci USA. 2016;113(34):9605–9610. doi: 10.1073/pnas.1525268113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanov IP, Wei J, Caster SZ, Smith KM, Michel AM, Zhang Y, Firth AE, Freitag M, Dunlap JC, Bell-Pedersen D, Atkins JF, Sachs MS. Translation initiation from conserved non-AUG codons provides additional layers of regulation and coding capacity. mBio. 2017;8(3) doi: 10.1128/mBio.00844-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sachs MS, Wang Z, Gaba A, Fang P, Belk J, Ganesan R, Amrani N, Jacobson A. Toeprint analysis of the positioning of translational apparatus components at initiation and termination codons of fungal mRNAs. Methods. 2002;26(2):105–114. doi: 10.1016/S1046-2023(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 14.Wu C, Amrani N, Jacobson A, Sachs MS. The use of fungal in vitro systems for studying translational regulation. In: Lorsch J, editor. Translation Initiation: Extract Systems and Molecular Genetics. Elsevier; San Diego: 2007. pp. 203–225. [DOI] [PubMed] [Google Scholar]
- 15.Doamekpor SK, Lee JW, Hepowit NL, Wu C, Charenton C, Leonard M, Bengtson MH, Rajashankar KR, Sachs MS, Lima CD, Joazeiro CA. Structure and function of the yeast listerin (Ltn1) conserved N-terminal domain in binding to stalled 60S ribosomal subunits. Proc Natl Acad Sci USA. 2016;113(29):E4151–E4160. doi: 10.1073/pnas.1605951113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beadle GW, Tatum EL. Genetic control of biochemical reactions in Neurospora. Proc Natl Acad Sci USA. 1941;27:499–506. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horowitz NH. Fifty years ago: the Neurospora revolution. Genetics. 1991;127:631–636. doi: 10.1093/genetics/127.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis RH. Neurospora: contributions of a model organism. Oxford University Press; Oxford, Great Britain: 2000. [Google Scholar]
- 19.Davis RH, Perkins DD. Timeline: Neurospora: a model of model microbes. Nat Rev Genet. 2002;3(5):397–403. doi: 10.1038/nrg797. [DOI] [PubMed] [Google Scholar]
- 20.Perkins DD, Radford A, Sachs MS. The Neurospora Compendium: Chromosomal Loci. Academic Press; San Diego, CA: 2001. [Google Scholar]
- 21.Dunlap JC, Borkovich KA, Henn MR, Turner GE, Sachs MS, Glass NL, McCluskey K, Plamann M, Galagan JE, Birren BW, Weiss RL, Townsend JP, Loros JJ, Nelson MA, Lambreghts R, Colot HV, Park G, Collopy P, Ringelberg C, Crew C, Litvinkova L, DeCaprio D, Hood HM, Curilla S, Shi M, Crawford M, Koerhsen M, Montgomery P, Larson L, Pearson M, Kasuga T, Tian C, Basturkmen M, Altamirano L, Xu J. Enabling a community to dissect an organism: overview of the Neurospora functional genomics project. Adv Genet. 2007;57:49–96. doi: 10.1016/S0065-2660(06)57002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis RH, de Serres FJ. Genetic and microbiological research techniques for Neurospora crassa. Methods Enzymol. 1970;27A:79–143. [Google Scholar]
- 23.Ebbole D, Sachs MS. A rapid and simple method for isolation of Neurospora crassa homokaryons using microconidia. Fungal Genet Newsl. 1990;37:17–18. [Google Scholar]
- 24.Trevithick JR, Metzenberg RL. Kinetics of the inhibition of Neurospora invertase by products and aniline. Arch Biochem Biophys. 1964;107:260–270. doi: 10.1016/0003-9861(64)90328-5. [DOI] [PubMed] [Google Scholar]
- 25.Brockman HE, de Serres FJ. “Sorbose toxicity” in Neurospora. Am J Bot. 1963;50:709–714. [Google Scholar]
- 26.Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal Biochem. 2000;282(1):158–161. doi: 10.1006/abio.2000.4605. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Fang P, Sachs MS. The evolutionarily conserved eukaryotic arginine attenuator peptide regulates the movement of ribosomes that have translated it. Mol Cell Biol. 1998;18(12):7528–7536. doi: 10.1128/mcb.18.12.7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang P, Wang Z, Sachs MS. Evolutionarily conserved features of the arginine attenuator peptide provide the necessary requirements for its function in translational regulation. J Biol Chem. 2000;275:26710–26719. doi: 10.1074/jbc.M003175200. [DOI] [PubMed] [Google Scholar]
- 29.Fang P, Wu C, Sachs MS. Neurospora crassa supersuppressor mutants are amber codon-specific. Fungal Genet Biol. 2002;36(3):167–175. doi: 10.1016/s1087-1845(02)00014-2. [DOI] [PubMed] [Google Scholar]