

Abstract
The synthesis of new families of stable or at least spectroscopically observable gold(III) hydride complexes is reported, including anionic cis-hydrido chloride, hydrido aryl, and cis-dihydride complexes. Reactions between (C^C)AuCl(PR3) and LiHBEt3 afford the first examples of gold(III) phosphino hydrides (C^C)AuH(PR3) (R = Me, Ph, p-tolyl; C^C = 4,4′-di-tert-butylbiphenyl-2,2′-diyl). The X-ray structure of (C^C)AuH(PMe3) was determined. LiHBEt3 reacts with (C^C)AuCl(py) to give [(C^C)Au(H)Cl]−, whereas (C^C)AuH(PR3) undergoes phosphine displacement, generating the dihydride [(C^C)AuH2]−. Monohydrido complexes hydroaurate dimethylacetylene dicarboxylate to give Z-vinyls. (C^N^C)Au pincer complexes give the first examples of gold(III) bridging hydrides. Stability, reactivity and bonding characteristics of Au(III)–H complexes crucially depend on the interplay between cis and trans-influence. Remarkably, these new gold(III) hydrides extend the range of observed NMR hydride shifts from δ −8.5 to +7 ppm. Relativistic DFT calculations show that the origin of this wide chemical shift variability as a function of the ligands depends on the different ordering and energy gap between “shielding” Au(dπ)-based orbitals and “deshielding” σ(Au–H)-type MOs, which are mixed to some extent upon inclusion of spin–orbit (SO) coupling. The resulting 1H hydride shifts correlate linearly with the DFT optimized Au–H distances and Au–H bond covalency. The effect of cis ligands follows a nearly inverse ordering to that of trans ligands. This study appears to be the first systematic delineation of cis ligand influence on M–H NMR shifts and provides the experimental evidence for the dramatic change of the 1H hydride shifts, including the sign change, upon mutual cis and trans ligand alternation.
Introduction
Transition metal hydrides are key components of many catalytic reactions and constitute one of the most important classes of coordination complexes.1 Hydrides of gold,2 by comparison, have been conspicuous by their absence and were long regarded as unstable or hypothetical. The first experimental evidence for their existence was reported by Andrews et al. in 2001, who generated binary hydrides such as AuH, (H2)AuH, (H2)AuH3, and [AuH4]− in frozen gas matrices below 5 K.3 The first example of an isolable gold(I) hydride was obtained in 2008, using an N-heterocyclic carbene (NHC) as stabilizing ligand.4 There are only two types of structurally characterized gold(III) hydrides known so far, both based on stabilization by C^N^C pincer ligands: (C^N^C)AuH (Chart 1, structure A) first described in 2012,5,6 and Bezuidenhout’s cation B, generated by protonation of the corresponding T-shaped Au(I) bis-carbene complex.7 In both cases the hydride ligands are trans to N-donors, which exert a weak trans-influence and thus increase the Au–H bond enthalpy while reducing its reactivity. The thermally stable hydride A was subsequently detected in a number of catalysis-relevant reactions, notably the water–gas shift reaction.8,9 Given that gold and hydrogen have very similar electronegativities, Au–H bonds are highly covalent. The reactivity of gold(III) hydrides reflects this; whereas B can be deprotonated only by very strong bases,7 complexes of type A undergo homolytic Au–H bond cleavage and insertion reactions with alkenes and alkynes via bimolecular pathways involving (C^N^C)AuII radical intermediates.6,10
Chart 1. Structures of Previously Reported Gold(III) Hydrides A and B and of the Starting Materials 1–3.

Gold hydrides have been postulated numerous times as part of catalytic cycles and a variety of coordination environments have been assumed,11−14 although experimental evidence for the structural diversity that is accessible to such species is as yet very limited. In square-planar Au(III) complexes the trans effect plays a fundamental role in determining chemical behavior, and the presence of strong electron-donating substituents trans to the hydride can dramatically increase the reactivity of the Au–H bond. Some examples in the recent literature showed that trans-carbon donors facilitate β-hydride elimination from gold(III) alkyls or formate complexes. Highly reactive gold hydride intermediates were postulated, which can undergo insertion or reductive elimination processes.11,14 However, until now these types of Au(III) hydrides have not been directly observed.
For this reason, we designed an experimental study aimed at trapping new families of Au(III) hydrides featuring a C-donor in the form of a cyclometalated aryl ligand in trans position, in order to investigate analogies and differences with the previously reported hydrides A and B. To achieve this goal, we explored complexes with different types of ligand environments (Chart 1, compounds 1–3). First, we investigated the reactivity of the C^C^N pincer complex 1(15) toward LiHBEt3 at low temperature, with the purpose of observing the corresponding hydride. Second, we extended this strategy to bidentate biphenyl-based C^C chelating ligands (2),16 where the fourth coordination position is occupied by different, nontethered Lewis bases. Finally, we investigated the C^N chelate complexes 3, which can be straightforwardly obtained by reacting (C^N^C)Au species with the strong Brønsted acid [H(OEt2)2][H2N{B(C6F5)3}2] (HAB2). In complexes of type 3,17 the dangling aromatic substituent acts as a steric protection for the site trans to the cyclometalated aryl group, and this was envisaged to offer the possibility of stabilizing the hydride against reductive elimination.
Using the starting materials 1–3, we report here several new classes of gold(III) hydrides, including (i) hydrido phosphine complexes, (ii) anionic hydrides, (iii) dihydrides, and (iv) bridging gold(III) hydrides. For the first time it has been possible to synthesize gold(III) complexes with the hydride ligand trans to a strong trans-effect C-donor ligand. We also show that the coordination geometry has an unexpectedly strong influence on the Au–H 1H NMR chemical shift, which can cover a range of ∼15 ppm. Computational studies show that the 1H NMR shifts of gold hydrides are strongly influenced by the spin–orbit coupling as a function of the ligand environment and depend on the nature of both cis and trans ligands. These ligand combinations also affect the thermal stability, leading, for example, to gold(III) phosphine hydrides suitable for crystallographic characterization. The investigation of the reactivity of these new hydrides reveals that they differ distinctly from compounds of type A.
Results and Discussion
(C^C^N) Complexes
C^C^N pincer complexes are coordination isomers of the much more widely studied C^N^C systems9,18 and show interesting reactivity and photophysical properties.15 In bonding terms C^C^N is complementary to C^N^C since strong and weak trans-effect ligands have swapped positions and dissociation of the pyridine moiety is less constrained, so that gold(III) hydrides with enhanced reactivity might be expected. With this aim, we explored the reactivity of (C^C^N) gold chloride 1 with 1.0 equiv of LiHBEt3 at 198 K. The reaction was monitored by 1H NMR spectroscopy in THF-d8 gradually raising the temperature from 203 to 243 K. The 1H NMR spectrum recorded at this temperature showed the clean formation of a single gold-containing species 4, which retained the typical pattern of a cyclometalated (C^C^N) system (Scheme 1). While no precipitate was seen in THF, when the reaction was performed in CD2Cl2, the formation of solid of LiCl was observed and free BEt3 was detected in the reaction mixture (δB = +85.1 ppm).
Scheme 1. Generation of Monohydride 4 and the Dihydride 5 (R = p-ButC6H4), Showing the Numbering Scheme Used for NMR Assignments.
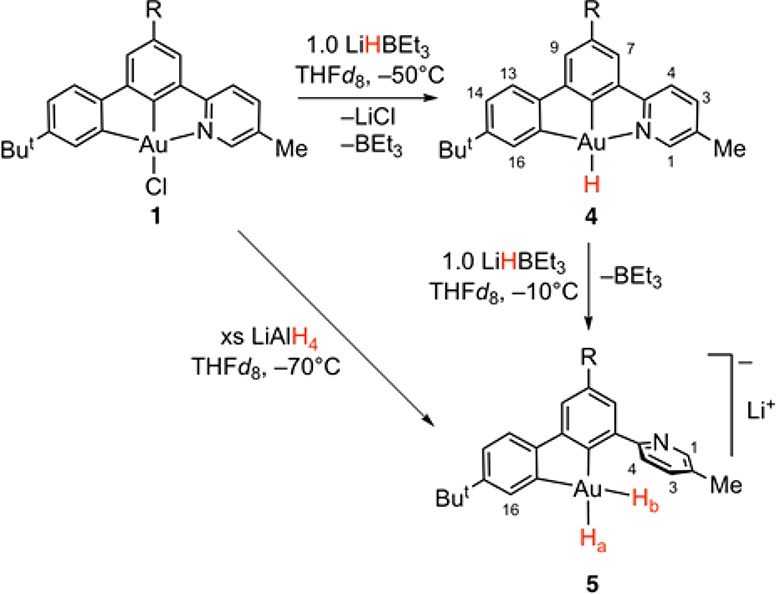
Multinuclear and multidimensional NMR experiments performed at 253 K in THF-d8 were consistent with the formulation of 4 as the gold hydride (C^C^N)AuH.19 The 1H NMR chemical shifts of metal hydrides are subject to relativistic spin–orbit (SO) effects (“heavy atom effect on the light-atom shielding”, HALA),20,21 which in the case of transition metal complexes with d8 electron configuration are typically shielding,20a as exemplified by shifts of δH = −6.58 (CD2Cl2) and −8.34 ppm (THF-d8) for A and B, respectively.5,7 In sharp contrast, the signal for the Au–H moiety in 4 is high-frequency shifted at δH = +6.33 ppm. An analysis of the dipolar contacts in the 1H NOESY NMR spectrum revealed selective interactions of the hydride signal with both ortho-protons of the coordinated pyridyl and of the cyclometalated aryl, indicating that the pyridine coordination remains intact upon exchange of chloride by hydride. No interactions with BEt3 groups were observed, excluding the formation of a gold-borohydride Au-HBEt3 complex. This was further confirmed by diffusion NMR experiments, which showed that 4 and BEt3 diffuse with different hydrodynamic dimensions. The carbon atom in trans position to the hydride is high-frequency shifted compared with the chloride complex and resonates at δC = 184.2 ppm; this shift can be attributed to the strong trans-influence of the H– ligand and concomitant SO-induced deshielding.21,22 On the other hand, the second metalated carbon atom is found at δC = 145.3 ppm, being trans to the weaker pyridine donor. The NMR peak assignments are also confirmed by relativistic quantum-chemical calculations (cf. Figure S49 in Supporting Information for 13C NMR data) and 1H hydride shifts are analyzed in detail (see Computational analysis below).
In THF solution at room temperature 4 is stable only for a few minutes, hampering its successful crystallization. When an NMR sample was warmed from 253 to 298 K, broadening of the Au–H and pyridine signals was observed, likely due to pyridine decoordination which may open decomposition pathways. Other solvents, such as benzene-d6 or CD2Cl2, can be used for generating 4, but no improvements in stability were observed. The 1H NMR signal of the Au–H moiety is marginally affected by the change of solvents and resonates at δH = 6.99 and 6.09 ppm in C6D6 and CD2Cl2, respectively.
Complex 4 reacts smoothly at room temperature with dimethyl acetylene dicarboxylate (DMAD) under trans-hydroauration to give a gold vinyl product (Z/E = 95:5). A similar vinyl product has previously been obtained by the thermal decomposition of the gold(III) formate complex (C^C^N)AuO2CH at 100 °C in the presence of di-tert-butyl acetylenedicarboxylate, presumably via 4 as the intermediate.14 On the other hand, 4 proved unreactive toward unactivated alkynes such as 2-butyne or 1-phenyl-1-propyne. This behavior contrasts with that of hydrides of type A, which insert a wide range of alkynes stereoselectively by a radical-mediated outer-sphere mechanism.6
When a solution of 4 in THF-d8 is treated with 1 or more equivalents of LiHBEt3 at 263 K, the monohydride is quantitatively converted into the unprecedented anionic dihydride complex 5. In striking contrast with what was observed in 4, both the hydride signals of 5 are shielded and located at δH = −0.59 (Ha) and −0.31 ppm (Hb). Ha appears as a doublet with a coupling constant 2JHH = 4.2 Hz, while Hb is a pseudotriplet due to the simultaneous coupling with Ha and the α proton of the cyclometalated aryl. The assignment of the hydride signals was confirmed by 1H NOESY spectroscopy, which shows selective dipolar interactions between Hb and the H3/H4 pair of the dangling pyridine, while Ha interacts specifically with H16 (Figure 1). Both carbon atoms attached to gold are high-frequency shifted due to the high trans-influence of the hydride ligands and resonate at δC = 172.3 (C trans to Hb) and 171.1 ppm (C trans to Ha). The dihydride 5 is stable in THF solution for days at room temperature but has so far escaped crystallization. Interestingly, 5 can also be prepared quantitatively by using an excess of LiAlH4 as hydride transfer agent, with no sign of reduction. The reaction of 5 with DMAD gives a complex mixture, likely arising from possible double insertions and/or vinyl elimination reactions; it did not prove possible to isolate a clean product.
Figure 1.

Left: overlay of two sections of the 1H NMR spectra of (a) 1 (203 K, THF-d8); (b) 4 (253 K, THF-d8); and (c) 5 (263 K, THF-d8). Right: a section of the 1H NOESY NMR spectrum of 4 (top, 253 K, THF-d8) and 5 (bottom, 263 K, THF-d8).
The C^C Ligand System
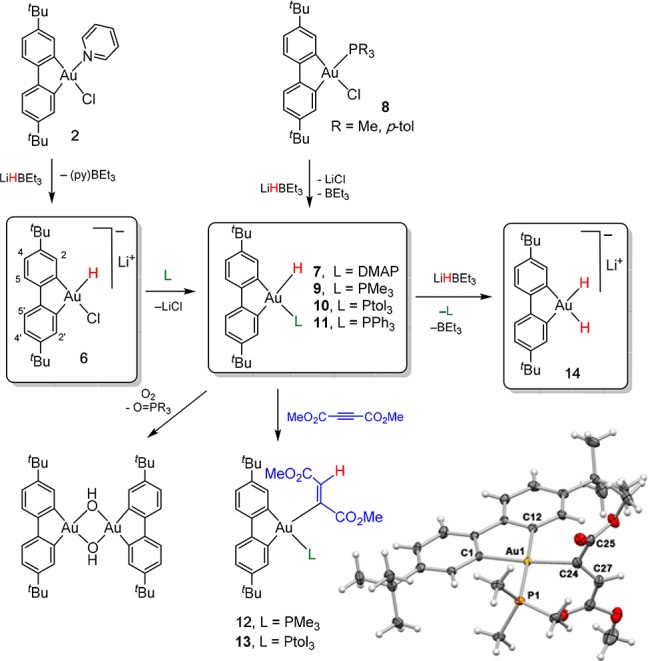
The formation of 5 shows that even anionic gold(III) dihydrides can be made supported by a dianionic C^C chelate backbone, rather than by tridentate pincer ligands. We therefore explored similar ligands which lack the tethered pyridine donor. The complex (C^C)AuCl(py) 2 (C^C = 4,4′-di-tert-butylbiphenyl-2,2′-diyl) was combined with 2.0 equiv of LiHBEt3 in THF-d8 at 198 K and the reaction was monitored by 1H NMR spectroscopy at 223 K. Soon after mixing the reagents, the solution turned bright yellow, and the first NMR spectrum showed the complete consumption of the starting material to give a single clean species showing two different sets of signals for the C^C ligand, suggesting the formation of an asymmetric gold complex. 1H NOE and diffusion NMR spectroscopy showed that the py → BEt3 Lewis adduct had formed. These data are consistent with pyridine displacement by the hydroborate and elimination of the borane-pyridine adduct, leading to the formation of the chloro hydride anion, [(C^C)Au(H)Cl]− (6). The reaction of 2 is therefore quite different from that of the (C^C^N) ligand system 1, where the pyridine was not initially displaced. The Au–H moiety in 6 was identified as a broad singlet at δH = +2.43 ppm, which showed selective dipolar interactions with only one α proton of the ligand at δH = 7.72 ppm. As observed for 4, the two metalated carbon atoms resonate at quite different frequencies, due to the different trans-influences of the chloride and hydride ligands, such that C trans to H resonates at δC = 174.8 ppm, while C trans to Cl is found at 148.4 ppm. Product 6 is stable in solution for hours at 213 K, but upon raising the temperature to 253 K it is converted to an undefined secondary species showing a broad hydride signal at δH = +0.9 ppm. Diffusion NMR experiments in CD2Cl2 at 228 K suggested the formation of aggregates containing at least four gold centers. This species undergoes reductive decomposition above 253 K, and its identity could not be ascertained. Since this reactivity was likely due to the facile chloride elimination from 6, we explored more strongly coordinating bases to stabilize the hydride products.
In order to intercept a (C^C)Au hydride of a structure comparable to 4, the chloro hydride 6 was reacted with 1 mol equiv of p-dimethylaminopyridine (DMAP) at 195 K and the resulting mixture was monitored by 1H NMR spectroscopy at 253 K. Although at a 1:1 stoichiometry DMAP does not bind to gold but displaces pyridine from the py → BEt3 adduct, with 4.0 equiv of DMAP 6 is converted quantitatively to (C^C)AuH(DMAP) (7) (Scheme 2). This complex shows a hydride signal at δH = +3.25 ppm, which exhibits NOE interactions with one α proton of the C^C ligand and with the ortho protons of the coordinated DMAP (δH = 8.10). 1H NOESY NMR reveals that at 253 K free and coordinated DMAP are in chemical exchange. Likely for this reason, 7 decomposes at room temperature within about 30 min.
Scheme 2. Synthesis and Reactions of (C^C)Gold(III) Hydrides, Showing the Atom Numbering Scheme Used for NMR Assignments, and the Molecular Structure of the Insertion Product 12.
Selected bond distances [Å] and angles [°]: Au1–C1 2.079(7), Au1–C12 2.070(6), Au1–P1 2.353(2), Au1–C24 2.078(7), C1–Au1–C12 81.1(3), C1–Au1–P1 96.8(2), P1–Au1–C24 89.7(2), C24–Au1–C12 92.3(3), C1–Au1–C24 173.4(3), C12–Au1–P1 177.3(2), torsion C1–Au1–C24–C25 91(2), torsion C1–Au1–C24–C27–88(3).
By contrast, thermally stable hydrides are straightforwardly obtained by reacting the phosphine complexes (C^C)Au(Cl)PR38 (R = Me, p-tolyl) with LiHBEt3 in toluene at room temperature to give spectroscopically clean samples of 9 and 10, respectively, which were isolated by filtration and vacuum drying. The same complexes, as well as the PPh3 derivative 11, are also accessible from the chloro hydride 6 on reaction with phosphines. However, in that case purification and crystallization of the products was hampered by the presence of the py → BEt3 byproduct, which displays similar solubility characteristics to 9–11 in organic solvents.
The complexes 9–11 are stable in THF solution at room temperature for several days. The 31P NMR spectra showed high-frequency shifted signals at δP = −8.2, δP = 32.6, and δP = 34.9 ppm for 9, 10, and 11, respectively, confirming phosphine coordination. The 1H NMR hydride signals at 1.53 (9), 2.33 (10), and 2.40 ppm (11) appear as doublets due to their coupling with the phosphorus atoms. The 2JPH values (32.6 Hz for 9, 33.0 for 10 and 32.3 Hz for 11) are compatible with H and P in cis-positions. 2JPC values are in agreement with the proposed structures: trans couplings fall between 130 and 140 Hz, while cis2JPC constants are about 4–5 Hz.
Single crystals of 9 were obtained by the slow evaporation of a dry diethyl ether solution under an N2 atmosphere. The structure contains four independent molecules in the asymmetric unit, with very similar structural parameters. Figure 2 gives the geometric parameters of just one of these molecules. A complete description of the structure is given in the Supporting Information. Locating the position of the hydride next to a heavy atom correctly by crystallography is intrinsically difficult; however, the Fourier map for the Au coordination plane unequivocally reveals the presence of electron density at a distance of about 1.4 Å (see Supporting Information, Figure S45). The other structural parameters are as expected for a square planar Au(III) complex. From the differences between the two Au–C bonds of the C^C ligand, the hydride exerts a larger trans influence than the phosphine (Au1–C1 2.047(4) Å vs Au1–C12 2.115(4) Å).
Figure 2.
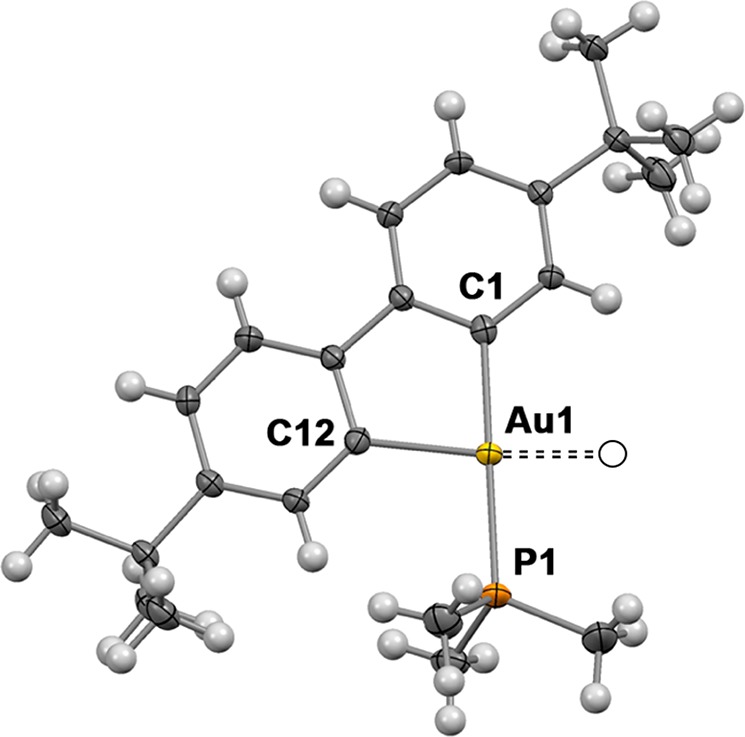
Molecular structure of (C^C)AuH(PMe3) 9; the approximate hydride position is indicated by an open circle. Selected bond distances [Å] and angles [°]: Au1–C1 2.047(4), Au1–C12 2.115(4), Au1–P1 2.325(1), C1–Au1–C12 81.5(2), C12–Au1–P1 98.7(1), C1–Au1–P1 179.6(1).
Although solid samples of 9–11 are thermally stable under ambient conditions, crystallization attempts of 9 in toluene also revealed that over time reductive C–H elimination and ligand rearrangements are possible, as indicated by the isolation of the gold(I) complex [(tBuC6H3)2(AuPMe3)2]x as a byproduct (see Supporting Information, Figure S46). Furthermore, unlike the (C^N^C)AuH hydrides of type A which are stable to air, moisture and weak acids, solutions of 9–11 are sensitive to exposure to air. For example, a sample of 10 left to crystallize without a protective atmosphere afforded the crystallographically identified hydroxide [(C^C)Au(μ–OH)]2, together with (p-tol)3P=O (see SI, Figure S47). The formation of these products suggests O2 insertion into the Au–H bond23 to give an Au(OOH)(PR3) intermediate, which decomposes to the gold hydroxide with oxidation of the phosphine. This reactivity was also observed upon reacting 10 with dry O2.
As in the case of 4, the hydrides 9 and 10 insert DMAD to give the corresponding Z-vinyl complexes 12 and 13. However, whereas 4 reacts instantaneously with DMAD at room temperature, the same reaction with 9 and 10 proceeds more slowly. When 1–2 mol equiv of DMAD are used, 9 is consumed within 2 h, while the reaction of 10 takes about 12 h, likely due to the increased steric repulsion of the phosphine ligands. The reaction affording 12 is quantitative, whereas 13 is formed in 80% yield together with unidentified side products.
The stereochemistry of the DMAD hydroauration products was proved by 1H NOE NMR spectroscopy, which showed the presence of dipolar contacts between the vinylic CH and both the methoxy groups. No signs of Z/E isomerization were observed after 2 days in solution. Single crystals of 12 suitable for X-ray diffraction were obtained by slow evaporation of a toluene/dichloromethane solution. The crystal structure confirmed the trans-orientation of the two methylcarboxylate moieties. The vinyl group adopts a perpendicular orientation with respect to the coordination plane of Au (torsion angle C1–Au1–C24-C27 88(3)°). The planar disposition of the C28(O)OMe carboxylate group and the vinyl moiety is indicative of π-delocalization; the other carboxylate residue adopts a twisted geometry. The orientation of the carboxylate groups is stabilized in the solid state by a network of intermolecular O···H interactions.
The addition of a second molar equivalent of LiHBEt3 to 9 and 10 leads to the substitution of the phosphine ligand by H–, to give the anionic cis-dihydride 14 (Scheme 2). For example, upon reacting 10 with 1.0 equiv LiHBEt3 at room temperature in THF-d8, 31P NMR spectroscopy revealed the disappearance of the signal at δP = 32.6 and the concomitant formation of free para-tolyl phosphine (δP = −8.0). The 1H NMR spectrum indicated a C2-symmetric C^C ligand, together with a new signal at δH = 0.09 which accounts for two protons and is assigned to Au–H (Supporting Information, Figures S23–S24). The hydride signal appears as a pseudotriplet, due to coupling with the two C^C ring protons in 2 and 2′ positions; these protons also show selective NOE interactions with the hydride signal. In agreement with these observations, the Au–C carbon atoms resonate at δC = 170.7 in the 13C NMR spectrum. Complex 14 can be also generated in toluene-d8, for which the hydride resonance of the AuH2 moiety was found at δH = 1.00 ppm. Given that for cis-(Ph3P)AuClH(Ph) and cis-(Ph3P)AuClH2 the calculated barriers for C–H reductive elimination are only 7.5 and 6.5 kcal mol–1, respectively,24 and considering the lack of orbital directionality of H, the observed thermal stability of gold(III) hydrido aryls 9–11 and of the dihydride 14 is remarkable and unexpected. As was seen for 10, eventually reductive decomposition does take place, and although 14 appears stable at room temperature for hours in THF and toluene solutions, any isolation attempts led to reductive decomposition, affording free biphenyl and metallic gold.
Other gold cis-hydrido aryl complexes resistant to reductive elimination are similarly accessible, by treatment of the pentafluorophenyl complex [NBu4][(C^C)Au(Cl)C6F5] (15) at room temperature with 1.2 equiv LiHBEt3. The anionic hydride 16 is formed in quantitative yield, according to 1H NMR spectroscopy (Scheme 3). The chloride-to-hydride exchange in THF-d8 is comparatively slow and complete within 24 h, whereas in toluene the formation of 16 is instantaneous. The Au–H signal resonates at δH = 1.15 ppm in THF-d8, partially overlapping with a tert-butyl signal, and was identified by means of its selective dipolar interaction with the H2 proton of the cyclometalated aryl ring at δH = 8.06 ppm. As was seen previously for neutral C^C hydrides, the cyclometalated carbon atom trans to the hydride is high-frequency shifted to δC = 170.3, while the one trans to C6F5 resonates at δC = 160.1 ppm. Complex 16 reacts readily with DMAD to give the corresponding trans-vinyl complex 17. In contrast with the phosphine hydrides 9 and 10, the reaction with 16 is instantaneous at room temperature. The stereochemistry was confirmed by 1H NOESY NMR spectroscopy and by single crystal X-ray diffraction of 17 (Scheme 3). The structural parameters of the anionic vinyl 17 are very similar to those found for 12.
Scheme 3. Synthesis of NBu4[(C^C)AuH(C6F5)] (16) and the Formation and Molecular Structure of the Alkyne Insertion Product 17 (NBu4+ Omitted for Clarity).
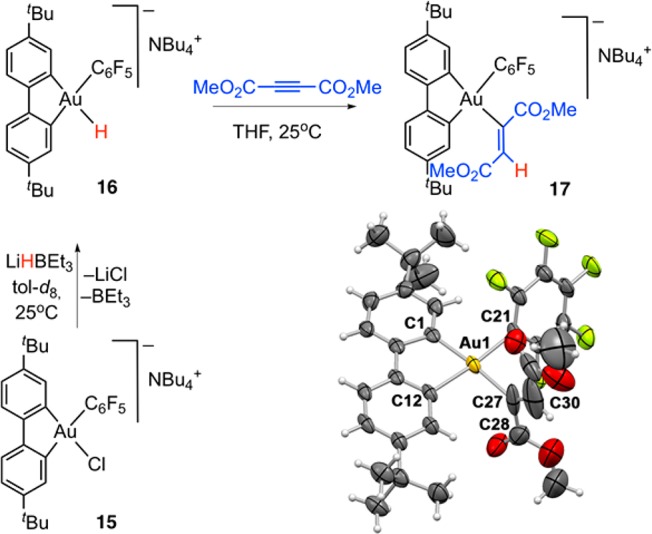
Selected bond distances [Å] and angles [°]: Au1–C1 2.046(8), Au1–C12 2.063(6), Au1–C27 2.093(8), Au1–C21 2.088(6), C27–C28 1.57(2), C27–C30 1.22(2), C1–Au1–C12 80.8(3), C1–Au1–C21 93.7(3), C21–Au1–C27 89.9(3), C27–Au1–C12 95.7(3), C1–Au1–C27 174.4(3), C12–Au1–C21 174.4(3).
While phosphine dissociation and creation of a coordination site cis to the hydride could in principle be considered a possible pathway in the reactions of phosphine hydrides 9 and 10 with DMAD, the facile alkyne insertion into the Au–H bond of 16, where no such ligand dissociation is possible, shows that alkyne π-coordination to Au(III)–H is not part of the insertion process. We also established that, unlike the insertion reactions of (C^N^C)AuH (A) with alkyl and aryl acetylenes,6 a radical chain reaction via Au(II) intermediates is not involved.
While a detailed study of this insertion mechanism is beyond the scope of this work, it is apparent that only alkynes with a low reduction potential insert into the gold hydrides described here. A similar observation was made with the gold(I) hydride (IPr)AuH (IPr = 1,3-bis(diisopropylphenyl)imidazol-2-ylidene).4 DMAD has a reduction potential of only −0.8 V (compared to a value of −2.1 V for diphenylacetylene).25 It seems likely therefore that the insertion of DMAD follows an electron transfer mechanism, which would generate an alkyne radical anion within the solvent cage. Alkyne radical anions are known to exhibit trans geometry,26 and such an intermediate, followed by H abstraction and Au–C bond formation, would explain the observed Z-vinyl stereochemistry of DMAD hydroauration (Scheme 4). The reactivity of our gold(III) hydrides seems to resemble therefore the DMAD insertion process observed for platinum dihydrides trans-H2Pt(PR3)2 (but not for trans-PtH(Cl)(PEt3)2, which follows a coordination–cis insertion pathway).27
Scheme 4. Proposed Mechanism of DMAD Hydroauration with (C^C)Au Hydrides.

Reactivity of (C^C)Gold Vinyl Complexes
As we observed previously, (C^N^C)Au(III) vinyl complexes obtained by alkyne trans-hydroauration are thermally stable and are not protodeaurated by strong acids.6 This might be ascribed to the effect of the weak pyridine donor trans to vinyl, which strengthens the Au–C bond. In 12, 13, and 17, the vinyl ligands are trans to anionic carbon, which exerts a much stronger trans effect and thus labilize the Au-vinyl bond. In order to investigate whether this structural modification has an impact on the reactivity of these complexes, we explored photoisomerization, protodeauration, and reduction with LiHBEt3 of these gold vinyls.
Photoisomerization of 12 and 17 was performed by irradiating samples in THF-d8 at 365 nm for 2 h at room temperature. 12 does not photoisomerize easily under these conditions and gives a thermodynamic E/Z ratio of 20:80, whereas the anionic vinyl complex 17 leads to an equilibrium E/Z ratio of 50:50. This supports the hypothesis that the Z isomer obtained upon hydroauration of DMAD is the kinetic product and that the structure of the ancillary ligand has an impact on the isomer distribution in the thermodynamic mixture.
The reactivity of (C^C)Au vinyl complexes toward acids and hydrides was exemplified for 12. On treatment with 2.5 equiv of LiHBEt3, 12 reacts quantitatively under selective reduction of one of the two ester functions to give the alkoxyvinyl product 18 (Scheme 5). The identity of 18 was confirmed by multinuclear and multidimensional NMR spectroscopy (Supporting Information). This reactivity demonstrates the facile postsynthesis derivatization of alkyne hydroauration products and their remarkable resistance to reducing conditions.
Scheme 5. Photoisomerization, Ester Reduction, and Reductive C–C Bond Formation of Gold Vinyl Complexes.
Reductive elimination of a C–C cross-coupling product can be induced by acids. When a solution of 12 in THF-d8 was treated with the strong Brønsted acid [H(OEt2)][H2N{B(C6F5)3}2]28 (1 equiv) at room temperature, the orange color of the solution faded immediately and a dark precipitate of metallic gold was observed. The 1H NMR spectrum of the supernatant revealed the formation of the 2-vinylbiphenyl 19 in about 65% yield. Protodeauration of the Au-vinyl bond to give MeO2CH=CHCO2Me was not detected. The appearance of an AX system at δH = 7.20 and 7.34 ppm suggests that the acid protodeaurates selectively one Au–C bond of the cyclometalated C^C ligand, likely trans to vinyl, to give a cationic aryl-vinyl intermediate capable of reductive C–C coupling. This Au-aryl bond cleavage kinetically outperforms Au-vinyl protonation. The stereochemistry of the vinyl group is retained in 19.
The C^N–CH Ligand System
Protodeauration of (C^N^C) Au(III) pincer complexes with HAB2 affords cleaved (C^N–CH)Au(III) cations 3 (Scheme 6), where a Et2O molecule is weakly coordinated to gold in trans position to the remaining cyclometalated aryl.17 We therefore envisioned that this particular ligand environment could offer the possibility to intercept yet another family of Au(III) hydrides which could also have the hydride ligand trans to a carbon atom but, being supported by a C^N rather than a C^C chelate, would constitute the cationic analogues of the (C^C)AuH(L) species described above. Moreover, the presence of the dangling aryl group might offer extra steric protection to the hydride moiety.
Scheme 6. Formation of Binuclear C^N Bonded Gold(III) Hydrides.
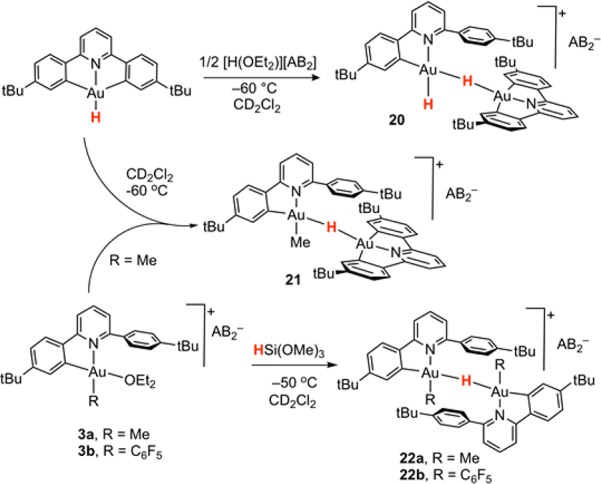
As a first control experiment, we reinvestigated the reaction of (C^N^C)AuH with HAB2 at a 1:1 molar ratio. As previously reported, the Au–H bonds in pincer gold(III) hydrides are covalent and do not liberate H2 upon reaction with acids. Instead, one Au–C bond is protolytically cleaved, a process that can be expected to enable fast reductive C–H elimination.29 To our surprise, when the protodeauration reaction was performed at 213 K in CD2Cl2, the starting material was quantitatively converted to the new species 20. This complex shows two scalarly coupled hydride signals, both at much lower frequency than the ones seen for C^C complexes, δH = −4.88 and δH = −8.39 ppm. In the aromatic region of the 1H NMR spectrum there are two sets of signals in a 1:1 ratio, with one set showing the typical fingerprint of a cleaved (C^N–CH) ligand, while the second set was that of an intact (C^N^C)Au fragment. In agreement with this, only 50% of the added acid had in fact reacted, with the other 50% remaining unchanged (δH = 16.7 ppm). These findings were consistent with protodeauration of one pincer Au–C bond to generate a cationic gold hydride, followed by coordination of a molecule of neutral (C^N^C)AuH to the vacant coordination site on the [(C^N–CH)AuH]+ cation to give the H-bridged binuclear dihydride 20 (Scheme 6). Whereas in gold(I) chemistry H-bridged complexes are not uncommon,4 this is the first example of a bridging hydride in gold(III) chemistry. In agreement with the reaction sequence outlined above, the same product can be generated using only 0.5 equiv of acid. The geminal coupling constants for the dihydride was found to be 8.2 Hz, much larger than the geminal coupling in the anionic mononuclear dihydride 6 (2JHH = 4.2 Hz). 1H NOESY methods allowed us to assign the signals at δH = −4.88 and δH = −8.39 ppm to the bridging and the terminal hydride, respectively. Unfortunately, the dihydride 20 is thermally unstable and at 213 K decomposes over a period of 1 h, hampering full 13C NMR characterization.
In analogy to complex 20, the cationic gold(III) methyl complex 3a reacts with 1 equiv of (C^N^C)AuH at 213 K to give the bridging hydride 21 (δH = −5.14 ppm). Product 21 proved to be rather unstable and decomposed at low temperature within hours, in line with the low barrier of reductive methane elimination calculated for (Ph3P)AuH(CH3)Cl (11.1 kcal mol–1).24 Nevertheless, it seems remarkable that gold hydrido methyl complexes are so readily accessible.
In the search for milder hydride donors, the reactions of the cleaved pincer complexes 3a and 3b with HSi(OMe)3 were tested. Complexes 22a (R = Me) and 22b (R = C6F5) proved to be more stable than 20 and 21 and could be fully characterized by multinuclear NMR at low temperature. The hydride signal appeared as singlets at δH = −2.53 and δH = −2.21 ppm for 22a and 22b, respectively (Supporting Information, Figure S41). The 1H NMR spectra of complexes 22a,b show the presence of four different signals for the dangling aryl substituents, suggesting that dimerization of two gold units blocks the free rotation of the aromatic ring about the C–C bond. 1H NOESY NMR spectroscopy revealed the presence of selective dipolar interactions between the bridging hydride and only one proton of the protodeaurated ring, confirming that the Au–H–Au moiety is located in the pocket formed by the two paired cleaved pincer complexes. In the case of complex 22a, the same interaction was observed for the Au–Me moiety.
Although complexes 22a,b are stable at 253 K, they decompose at room temperature, following reaction pathways which depend on the substituents on gold. Warming a sample of 22b to 297 K generates C6F5H quantitatively within minutes; in this case the reductive coupling of H with C6F5 is preferred. The methyl complex 22a is slightly more thermally stable and survives 297 K for about 30 min before decomposing. However, there is no elimination of CH4; instead, the methyl migrates to the C^N^C ligand, while the hydride is likely eliminated through reductive deprotonation to give protonated pyridinium salts.30
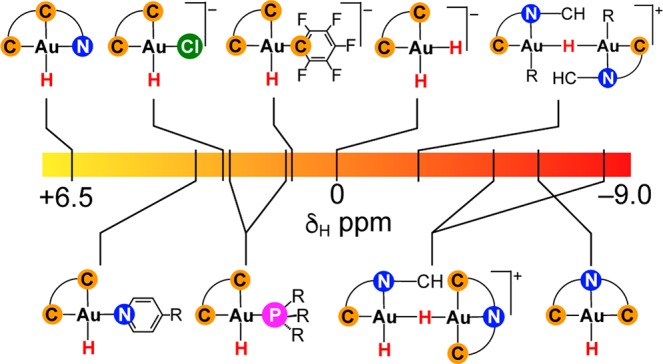
Connection between Gold Hydride 1H NMR Chemical Shifts and Electronic Structure
The gold(III) hydride complexes described above are characterized by a spread of 1H NMR chemical shifts covering an unexpectedly wide range of about 15 ppm. Table 1 summarizes the relevant structural, calculated and experimental data. Particularly striking is the difference between the regioisomeric pincer complexes (C^N^C)AuH and (C^C^N)AuH, which show resonances of δ −6.51 and +6.09 ppm, respectively (both in CD2Cl2). While the former features an upfield (low-frequency) shift often seen for diamagnetic hydride complexes with a partially filled d-shell,19 the latter displays a notable downfield 1H shift, resembling that in a gold(I) hydride, (IPr)AuH,4 although both (C^N^C)AuH and (C^C^N)AuH contain the metal center in the same +III oxidation state. To rationalize the observed trends and to gain insight into the electronic structures and the factors that result in these spectroscopic differences, we turned to relativistic quantum-chemical calculations of structures and 1H NMR hydride shifts at the two-component ZORA-SO level, including spin–orbit coupling (see Supporting Information for computational details).31,32
Table 1. DFT Optimized Au–H Bond Lengths (in Å) and Computed and Experimental 1H NMR Hydride Chemical Shifts of Gold Hydridesa.

In ppm vs TMS. All calculations done for PBE0-D3(BJ)/ECP/def2-TZVP optimized structures (see SI).
Chemical shifts computed at the 2c-ZORA(SO)/PBE0-XC/TZ2P level; SO-induced contributions to hydride shifts, δSO, are given in parentheses.
For comparative purposes, the hydride shifts were also computed at the four-component, fully relativistic 4c-mDKS/PBE0/Dyall-VDZ/IGLO-II level.
Solvents used in NMR measurements are given in parentheses.
This work.
All investigated structures were optimized at the PBE0-D3(BJ)/ECP/def2-TZVP level using a quasi-relativistic small-core pseudopotential for gold, along with atom-pairwise corrections for dispersion forces. First, we note an excellent agreement between X-ray and DFT optimized structures, with differences in Au–L bond-lengths of less than 0.02 Å, except for the Au–H bonds, where values determined by X-ray diffraction are often much larger (by up to 0.15 Å) than found computationally (cf. Figure S48 in Supporting Information). The precise detection of the hydride position by X-ray crystallography, however, suffers from weak scattering of X-rays by light H atoms and these data should be taken with caution. The reliability of DFT-determined Au–H bond-lengths is demonstrated by the excellent agreement between computed and experimental 1H hydride shifts for the optimized structures, with a standard deviation of 0.4 ppm (see Figure S50 in Supporting Information). We also note a very good match between experimental and calculated 2JHP coupling constants in the hydrido phosphine complexes 9 (2Jexpt = 32.6 Hz; 2Jcalcd = 31.4 Hz) and 10 (2Jexpt = 32.3 Hz; 2Jcalcd = 30.9 Hz).
The experimental 1H NMR shifts of terminal hydride atoms are found to correlate well with DFT optimized Au–H distances (Figure 3a), which suggests the potential use of 1H NMR spectroscopy in refining precise hydride positions and explains, inter alia, the striking downfield 1H shift in (C^C^N)AuH (4). Interestingly, complexes in Table 1 with longer and more ionic Au–H bonds feature high-frequency (downfield) hydride shifts, which contrasts with organic C–H compounds.33 To explore the generality of the linear δ(1H) vs d(Au–H) relationship (Figure 3) and to understand the trends in light of electronic structure, a more thorough analysis of 1H hydride shifts for a larger set of Au(III) hydrides was performed (see Table 1 and Tables S1–S4 in the SI for numeric data).
Figure 3.
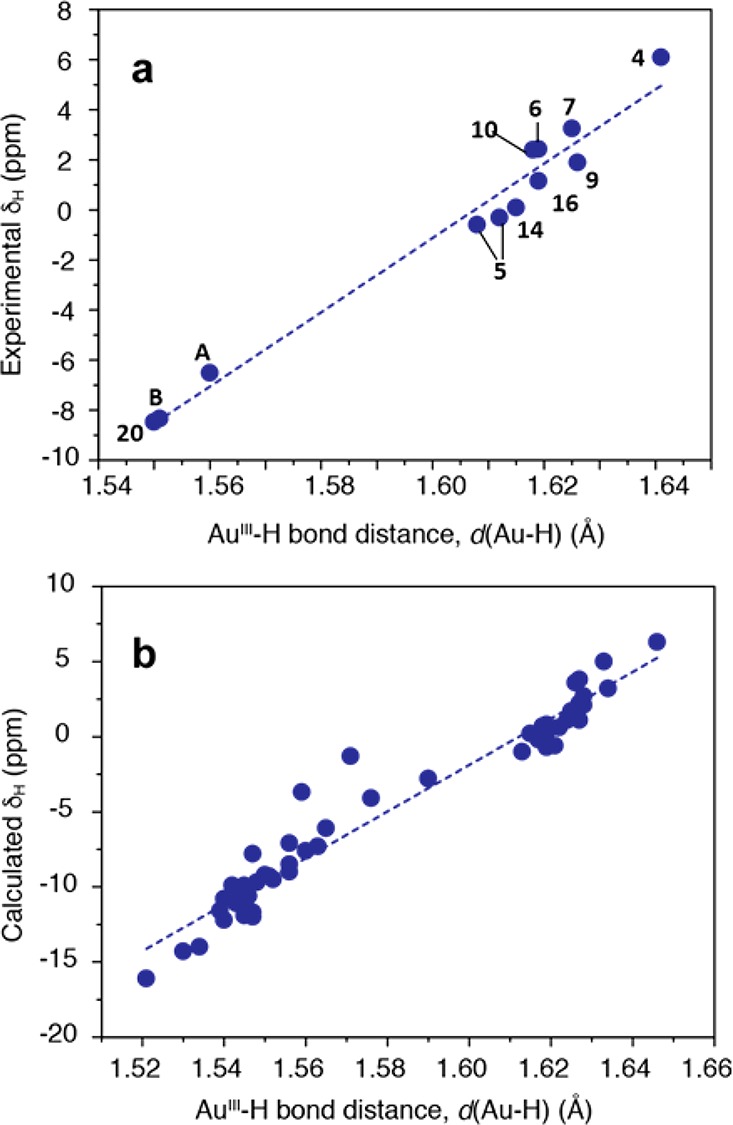
Dependence of experimental (a) and computed (b) 1H NMR chemical shifts of terminal Au–H hydride atoms (in ppm vs TMS) on the Au–H bond-length (Å) for gold(III) complexes with diverse ligand environments. The Au–H bond distances were obtained computationally at the DFT (PBE0-D3(BJ)/def2-TZVP) level. (b) contains both experimentally characterized and hypothetical Au(III) hydride complexes (cf. Table 1 and Tables S2–S4 in SI for numerical data). Linear regressions: (a) δH = 149.67 d(Au–H) −240.57, R2 = 0.970; (b) δH = 155.1 d(Au–H) −249.97, R2 = 0.950.
Decomposition of the computed 1H shieldings into diamagnetic, paramagnetic and spin–orbit contributions shows that the trends in isotropic hydride shifts are dominated by the δSO term (Table 1 and Tables S1–S4 in the SI). Artificial elongation/shortening of Au–H bonds by a value of 0.05 Å from their optimized values in selected gold(III) hydrides causes only a small change in 1H hydride shifts (<1 ppm, data not shown), and thus the surprisingly good correlation between δ(1H) and d(Au–H) has an indirect rather than direct origin.20c The opposite sign of the 1H hydride shifts of the coordination isomers (C^C^N)AuH and (C^N^C)AuH should thus be attributed to different electronic structures (cf. Figure 4) and is rationalized below in terms of molecular orbitals contributing dominantly to δSO.
Figure 4.

Relevant frontier MOs at scalar-relativistic (SR) level and their mixing to two-component spinors responsible for the overall differences in the 1H hydride shifts of (C^N^C)AuH (left) and (C^C^N)AuH (right). Only one of two degenerate spinors is shown. The corresponding MOs are shown as isosurface plots (0.03 au), with the hydride ligand generally being placed at the bottom. The mixing percentage of SR MOs in spinors is indicated above the corresponding SR MOs.
Analysis of the δSO contributions at the 2c-ZORA level show that the hydride deshielding in (C^C^N)AuH, with the strong C-anionic trans ligand, is dominated by two spinors, HOMO–1 and HOMO–3, both composed from the σ(Au–H)-type SR HOMO–1 (with an appreciable gold 6p character) and a primarily ligand-centered π-type orbital with little metal character, SR HOMO–3 (note that both the occupied MOs are magnetically coupled mainly with an antibonding σ*(Au–H)-type orbital, LUMO+3). Since “shielding” Au(dπ)-type MOs (SR HOMO–8, SR HOMO–10) lie energetically far below the σ(Au–H) MO (by more than 2 eV), these two types of scalar relativistic MOs do not mix with each other upon inclusion of SO coupling, that hinders hydride shielding and results in the high-frequency hydride shift. In contrast, the 1H shielding for the (C^N^C)AuH complex, bearing a weak trans N-donor ligand, is dominated by magnetic couplings between the Au(dπ)-type occupied and σ*(Au–H) virtual MOs. The highest σ(Au–H)-type MO is energetically close to the Au(dπ) orbitals, with ΔE ∼ 0.5 eV, and the deshielding effect of the former diminishes upon SO mixing with Au(dπ)-based MOs.34
As the comparison between the tethered (C^C^N) and nontethered (C^C)(N) ligand systems in complexes 4 and 7 shows, the steric constraint imposed by the C^C^N pincer ligand also has an important effect. In C^C^N the trans-influence of C is increased, leading to a significant lengthening of the Au–H bond (1.641 Å in 4 vs 1.625 Å in 7) and a further deshielding of the hydride NMR resonance by about 3 ppm.
Theoretical papers relating 1H NMR hydride shifts as a function of the trans ligand strength have appeared very recently.20c,21,22b However, the influence of ligands in cis-position has so far not been explored. Since in square-planar complexes both cis and trans effects are operative, we performed calculations for three series: trans-[HAu(C6H5)2L]q, cis-[HAu(bph)L]q and cis-[HAu(ppy)L]q+1 (bph = 2,2′-biphenyl dianion, ppy = 2-phenylpyridine anion; q = 0 or −1) in order to study the influences of cis and trans ligands separately for a wide range of common σ-donor and π-acceptor ligands (see SI, Tables S2–S4). The ligand effects on the 1H hydride shifts are depicted in Figure 5.
Figure 5.
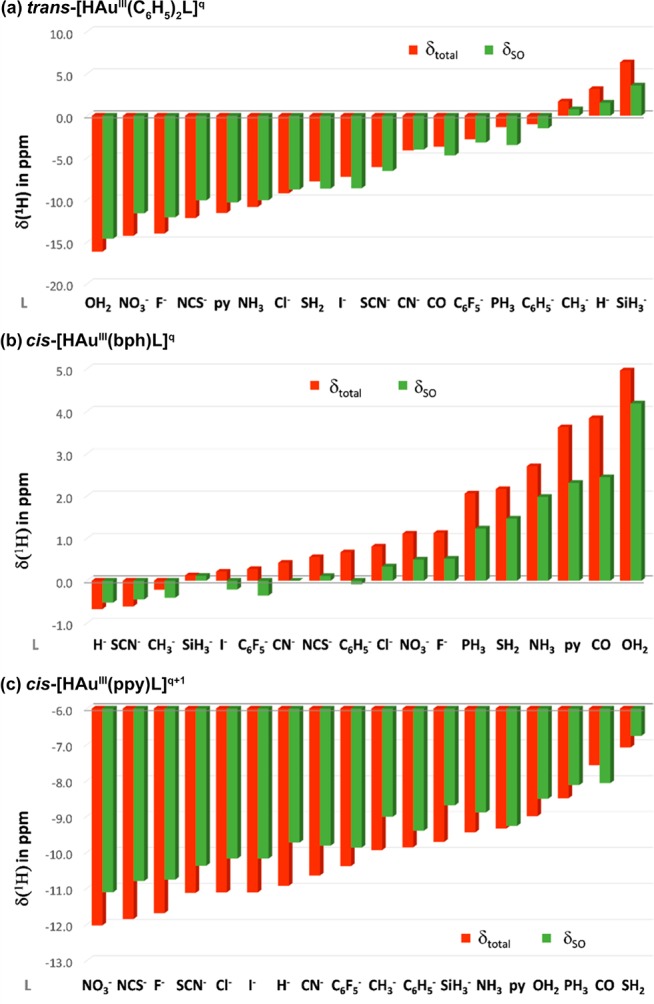
Dependence of the computed 1H hydride shifts (δtotal) and spin–orbit-induced shift contributions (δSO) on (a) the trans ligand L in the trans-[HAuIII(C6H5)2L]q; (b) the cis-ligand in the cis-[HAuIII(bph)L]q; and (c) the cis-ligand in the cis-[HAuIII(ppy)L]q+1 series (2c-ZORA(SO)/PBE0-XC/TZ2P results; see Tables S2–S4 in SI for numerical data).
Trans-ligands produce chemical shift changes that are roughly four times larger than cis-ligand effects, consistent with the much larger variation in Au–H distances in the trans case and the strikingly different ordering and relative energies of σ(Au–H)-type and Au(dπ) orbitals. The influence of trans ligands on the 1H shift spans about 22.5 ppm, with H2O as the weakest donor investigated here showing the highest SO shielding contribution, and SiH3– the highest SO deshielding. Hydride shifts and Au–H bond-lengths in gold(III) complexes follow a trend similar to the one observed for linear LAuIH hydrides (see SI, Figure S51 and Tables S1 and S2), with the 1H shifts in the Au(III) series being systematically shifted upfield.35
Ligands in cis-positions have a weaker influence on hydride shifts (spanning about 6 ppm) but show the opposite trend: the weakest σ-donors (and strongest π-acceptors) exhibit the highest deshielding (the most positive 1H hydride shifts). As demonstrated for the cis-[HAu(bph)L]q series with L = NH3, PH3, CH3 and SiH3– (cf. Figure 5), the cis ligand influence has the same qualitative explanation as postulated for the trans-influence above, but ligands with stronger trans-influence located in the cis-position decrease the energy gap between the highest-lying σ(Au–H) bonding MO and Au(dπ)-based MO, just the opposite of what is seen for the trans-influence (see SI, Figures S52–S53).
According to the data for the cis-[HAu(bph)L]q and cis-[HAu(ppy)L]q+1 series, the order of increasing cis ligand influence depends also on the trans ligand. However, in general the strongest cis-influence (reflected in the longest Au–H bonds and the most downfield 1H shifts) is observed for neutral weak σ-donors (such as S, N and O-based ligands) and CO, while anionic ligands exert the weakest cis-influence, consistent with our experimental NMR data collected in Table 1.
To conclude, the observed correlation between measured 1H hydrides shifts and Au–H bond-lengths has an indirect origin, but works surprisingly well for a wider set of studied mononuclear Au(III) hydrides (Figure 3). This can be attributed to decisive SO-shift contribution and only marginal changes in paramagnetic shielding. Since Au–H distances correlate roughly with QTAIM delocalization indices of the Au–H bond, a reasonable relationship is also established between 1H NMR hydrides shifts and Au–H bond covalency (see Figure 6): the more ionic the Au–H bond, the more positive is the hydride shift.36,37 Note that the differences in Au–H bond covalency could also explain the similar reactivities of gold(III) hydrides with C-anionic trans ligands (with δH ranging from −0.6 to +7.0 ppm) and of gold(I) hydride (IPr)AuH (δH = 3.38 ppm/CD2Cl2) toward alkynes: both these compound types insert DMAD but not less activated alkynes, whereas the gold(III) pincer hydride (C^N^C)AuH (δH = −6.51 ppm/CD2Cl2) with a much shorter and more covalent Au–H bond behaves differently and adopts a different mechanism.
Figure 6.
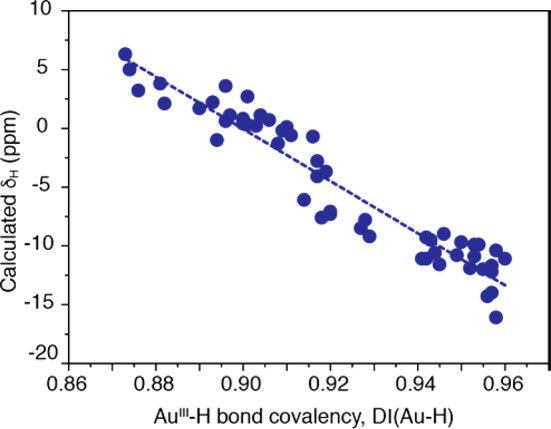
Correlation of computed 1H NMR hydride shifts (in ppm vs TMS) with QTAIM delocalization indices, DI(Au–H), as a measure of the bond covalency, for a series of mononuclear Au(III) hydride complexes (cf. SI, Tables S2–S4 for numerical data).
Conclusions
In summary, this work shows that a rather diverse range of gold(III) hydride complex types is accessible using C^C chelate ligands, and that stabilization by tridentate pincer ligands is neither a requirement nor a guarantee for thermal stability. Suitable combinations of cis and trans donors provide access to the first examples of stable Au(III) hydrido phosphine complexes, as well as to anionic hydrido halide, hydrido aryl, and even dihydrido complexes. The biphenyl-based C^C chelate imparts a remarkable stability toward reductive elimination and successfully suppresses H–C and even H–H coupling. C^N chelate complexes derived from protodeauration of (C^N^C) pincer precursors proved to be a convenient platform for intercepting the first examples of bridging gold(III) hydrides. The chemistry of the new C^C- and C^C^N-based gold hydrides, with strong C-donors trans to H, was found to differ significantly from that of the C^N^C (weak trans donor) coordination isomers; e.g., the former react only with DMAD but not less activated alkyl or aryl acetylenes, while for the latter the opposite is found. Chemical reactivity correlates with the hydride 1H NMR chemical shift, since both are subject to trans and cis ligand effects. On the basis of detailed quantum-chemical calculations and analysis including relativistic spin–orbit coupling effects, a linear relationship could be established between the computed Au–H bond distances, the hydridic character of the Au–H bond, and the hydride NMR chemical shifts. Strong trans effect ligands, such as C– in (C^C^N)AuH, raise the deshielding σ(Au–H) orbital and increase the energetic separation from orbitals with shielding Au(dπ)-type MO, resulting in deshielding (positive chemical shift), whereas in (C^N^C)AuH σ(Au–H) and Au(dπ) are close in energy, so that the hydride shift is dominated by the shielding SO contribution. Whereas trans-ligand influence calculated for a sequence of hypothetical complexes cover a range of over 22 ppm, cis-ligand influence is more limited, about 6 ppm, although both these effects are mutually interdependent. The effect of cis-ligands follows an approximate inverse order to trans-ligands, with the strongest cis-influence (and the longest Au–H bonds) being observed for the weakest neutral σ-donors. This appears to be the first systematic evaluation of cis-ligand effects on 1H chemical shifts in square-planar d8 systems.
Experimental Section
General Considerations
When required, manipulations were performed using standard Schlenk techniques under dry argon or using a nitrogen-filled MBraun Unilab glovebox equipped with a high capacity recirculator (<1.0 ppm of O2 and H2O). Argon was purified by passing through columns of supported P2O5 with moisture indicator and of activated 4 Å molecular sieves. Anhydrous solvents were freshly distilled from the appropriate drying agents and degassed. Triphenylphosphine (99%), tris(p-tolyl)phosphine (98%), trimethylphosphine (1.0 M in THF), dimethylaminopyridine (DMAP, >99%), lithium triethylborohydride (1.0 M in THF), dimethyl acetylenedicarboxylate (DMAD, 99%) trimethoxysilane (95%), were obtained by Sigma-Aldrich and dried, when necessary. Solid LiAlH4 was obtained by vacuum-drying a commercial solution in THF (Sigma-Aldrich). CD2Cl2 (Apollo Scientific), THF-d8 and toluene-d8 (Fluorochem Ltd.) were freeze–pump–thaw degassed over CaH2, distilled and stored over activated 4 Å molecular sieves. C6F5Ag(CH3CN),38 [(C^C)AuCl]2,16 (C^C)AuCl(py) (2),16 (C^N^C)AuH,5 (C^N^C)AuMe,39 (C^N^C)AuC6F5,40 [H(OEt2)2][H2N{B(C6F5)3}2]28 (HAB2) were synthesized according to literature procedures. Protodeaurated species [(C^N–CH)AuMe][AB2] (3a) and [(C^N–CH)AuC6F5][AB2] (3b) were generated within the glovebox, as reported previously.17
Low-temperature in situ NMR experiments were performed under anhydrous and anaerobic conditions by using screw cap NMR tubes equipped with a PTFE septum. In a typical experiment, the gold precursor was loaded into the NMR tube inside a glovebox and dissolved in the appropriate solvent. Successively, the sample was inserted into a cold bath at 195 K and the desired reagents were injected through the septum by using a micrometric gastight syringe. The cold sample was then inserted into the precooled NMR probe and characterized. 1H, 1H PGSE, 1H inversion recovery, 19F, 13C{1H}, 1H COSY, 1H NOESY, 1H,13C HMQC, and 1H,13C HMBC NMR experiments were recorded on a Bruker DPX–300 spectrometer equipped with a 1H,BB smartprobe and Z-gradients. 1H NMR spectra were referenced to the residual protons of the deuterated solvent. 13C NMR spectra were referenced to the D-coupled 13C signals of the solvent. 19F NMR spectra were referenced to an external standard of CFCl3. 31P NMR spectra were referenced to an external standard of H3PO4 85%. Photoisomerization experiments were performed by using a Blak-Ray B-100 Series high-powered UV lamp and by irradiating solutions contained in J-Young NMR tubes.
Synthesis and Characterization of New Gold Precursors
(C^C^N)AuCl (1)
This complex was synthesized in 89% yield by following literature procedures,15 using 2-bromo-5-methylpyridine as ligand precursor. 1H NMR (300.13 MHz, CD2Cl2, 297 K, J values in Hz) δ 8.78 (brs, 1H, H1), 7.80 (d, 4JHH = 1.6, H16), 7.72 (AB system, 2H, H3+H4), 7.61 (d, 3JHH = 8.4, 2H, H19), 7.52 (d, 3JHH = 8.4, 2H, H20), 7.39 (d, 4JHH = 1.5, H7), 7.34 (d, 4JHH = 1.5, H9), 7.23 (AB system, 2H, H13+H14), 2.33 (s, 3H, Me), 1.38 (s, 9H, C(21)CMe3), 1.32 ppm (s, 9H, C(15)CMe3). 13C{1H} NMR (75.47 MHz, CD2Cl2, 297 K) δ 164.7 (s, C11), 160.1 (s, C5), 151.0 (s, C15), 150.8 (s, C21+C12), 150.4 (s, C17), 149.4 (s, C10), 148.7 (s, C1), 141.2 (s, C3+C6), 140.0 (s, C8), 137.9 (s, C18), 135.2 (s, C2), 131.4 (s, C16), 126.9 (s, C19), 125.8 (s, C20), 124.4 (s, C14), 121.5 (s+s, C13+C9), 120.2 (s, C7), 119.8 (s, C4), 35.0 (s, C(15)CMe3), 34.5 (s, C(21)CMe3), 31.1 (s, C(15)CMe3 or C(21)CMe3) 31.0 (s, C(21)CMe3 or C(15)CMe3), 18.2 ppm (s, Me). Calcd for C32H33AuNCl (found) C, 57.86 (57.69); H, 5.01 (4.94); N, 2.11 (2.18) %.
(C^C)AuCl(Ptol3)
[(C^C)AuCl]2 (200 mg, 0.2 mmol) was stirred in dichloromethane (30 mL) with excess tris-para-tolylphosphine (120 mg, 0.4 mmol) until a clear solution was obtained. The reaction mixture was filtered through Celite and the solvent removed under reduced pressure. Hexane was added, the mixture sonicated briefly and the white solid powder filtered and washed with hexane. It was then recrystallized from Et2O/hexane to afford (C^C)AuCl(Ptol3) (290 mg, 90%). 1H NMR (300.13 MHz, CD2Cl2, 297 K, J values in Hz) δ 8.38 (dd, 4JHP = 9.5, 4JHH=1.8, 1H, H2), 7.53–7.59 (m, 6H, o-tol3), 7.24–7.32 (m, 8H, m-tol3+H5+H5′), 7.19 (dd, 3JHH = 8.0 4JHH = 1.6, 1H, H4), 7.03 (dd, 3JHH = 8.0 4JHH = 1.8, 1H, H4′), 6.92 (dd, 4JHP = 3.4, 4JHH = 1.8, 1H, H2′), 2.39 (s, 9H, Me), 1.31 (s, 9H, CMe3), 0.67 ppm (s, 9H, CMe3′). 31P{1H} NMR (121.49 MHz, CD2Cl2, 297 K) 40.2 ppm. 13C{1H} NMR (75.47 MHz, CD2Cl2, 297 K, J values in Hz) δ 166.1 (d, 2JCP = 129.6, C1), 154.0 (d, 2JCP = 7.3, C1′), 151.3 (d, 3JCP = 4.5, C6′), 149.7–149.6 (m, C6+C3), 149.2 (d, 4JCP = 2.7, C3), 142.3 (d, 4JCP = 2.4, CMe tol3), 135.1 (d, 2JCP = 11.5, o-tol3), 134.2 (d, 3JCP = 10.2, C2′), 130.5 (d, 3JCP = 1.8, C2), 129.4 (d, 4JCP = 11.2, m-tol3), 125.1 (d, 1JCP = 51.0, ipso–C tol3), 124.5 (br s, C4), 123.8 (s, C4′), 120.7 (s, C5′), 120.4 (d, 4JCP = 7.6, C5), 35.3 (s, CMe3), 35.4 (s, CMe3′), 31.2 (s, CMe3), 30.4 (s, CMe3′), 21.2 ppm (s, Me tol3). Calcd for C41H45AuClP (found) C, 61.44 (61.36); H, 5.66 (5.67) %.
(C^C)AuCl(PMe3)
[(C^C)AuCl]2 (270 mg, 0.27 mmol) was stirred in THF (10 mL) under Ar. Excess trimethylphosphine (0.5 mL of 1 M solution in THF, 0.5 mmol) was added at 25 °C and the mixture was stirred until a clear solution was obtained. The reaction mixture was filtered through Celite and the solvent removed under reduced pressure. Hexane was added, the mixture sonicated briefly and the white solid powder filtered and washed with hexane to afford (C^C)AuCl(PMe3) (250 mg, 81%). 1H NMR (300.13 MHz, CD2Cl2, 297 K, J values in Hz) δ 8.29 (br d, 1H, H2), 7.43–7.13 (br m, 5H, H4+H4′+H5+H5′+H2′), 1.85 (d, 2JHP = 10.8, PMe3), 1.34 ppm (s, 18H, CMe3+CMe3′). 31P{1H} NMR (121.49 MHz, CD2Cl2, 297 K) 3.4 ppm. 13C{1H} NMR (75.47 MHz, CD2Cl2, 297 K, J values in Hz) δ 165.2 (d, 2JCP = 138.4, C1), 153.2 (br s, C1′), 151.6 (s, C6′), 149.6 (m, C3+C3′), 149.2 (s, C6), 130.0 (s, C2+C2′), 124.6 (s, C4 or C4′), 124.4 (s, C4′ or C4), 121.5 (s, C5′), 120.4 (brd, 5JCP = 6.6, C5), 35.2 (s, CMe3 or CMe3′), 31.2 (s+s, CMe3+CMe3′), 14.2 ppm (d, 1JCP = 30.4, PMe3). Calcd for C23H33AuClP (found) C, 48.22 (48.43); H, 5.81 (5.89) %.
(C^C)Au(C6F5)(py)
C6F5Ag(CH3CN) (93 mg, 0.3 mmol) in Et2O (5 mL) was added to a solution of 2 (170 mg, 0.3 mmol) in dichloromethane (5 mL) in a centrifuge tube, with immediate formation of AgCl. After 5 min, the tube was centrifuged. The clear solution was decanted and the solvent removed under reduced pressure to afford the product as a white solid (144 mg, 60%). 1H NMR (300.13 MHz, CD2Cl2, 297 K, J values in Hz) δ 8.78 (d, 3JHH = 5.2, 2H, o-py), 8.04 (t, 3JHH = 7.8, 1H, p-py), 7.68 (t, 3JHH = 6.7, 2H, m-py), 7.37 (d, 3JHH = 8.0, 1H, H5), 7.32 (d, 3JHH = 8.0, 1H, H5′), 7.18–7.22 (m, 2H, H4+H4′), 6.84 (d, 4JHH = 1.2, 1H, H2), 6.34 (d, 4JHH = 1.2, 1H, H2′), 1.50 (s, 9H, CMe3), 1.08 ppm (s, 9H, CMe3′). 19F NMR (282 MHz, CD2Cl2, 297 K) δ −120.8 (m, o-F), −159.6 (t, p-F), −161.9 (m, m-F) ppm. 13C{1H} NMR (75.47 MHz, CD2Cl2, 297 K) δ 151.5 (s, C1′), 150.1 (s, o-py), 149.9 (s, C1), 149.5 (s, C6+C6′), 144.5 (s, C3+C3′), 140.1 (s, p-py), 132.4 (s, C2), 127.7 (s, C2′), 127.1 (s, m-py), 124.5 (s, C5), 124.1 (s, C5′), 121.1 (s, C4′), 120.3 (s, C4), 34.6 (s, CMe3), 34.4 (s, CMe3′), 30.92 (s, CMe3), 30.89 ppm (s, CMe3′). Calcd for C31H29Au F5N (found) C, 52.6 (51.96); H, 4.13 (4.55); N, 1.98 (2.01) %.
[nBu4N][(C^C)AuCl(C6F5)] (15)
(C^C)Au(C6F5)(py) (50 mg, 71 μmol) was stirred in dichloromethane (5 mL) in the presence of excess tetrabutylammonium chloride (30 mg, 0.1 mmol) for 1 h. The solvent was removed under reduced pressure. Dichloromethane (5 mL) was added and the mix stirred again for 1 h. The solvent was removed under reduced pressure. The residue was sonicated in Et2O and the white powder filtered to yield 15 (45 mg, 98%). 1H NMR (300.13 MHz, CD2Cl2, 297 K, J values in Hz) δ 8.24 (d, 4JHH = 2.0, 1H, H2), 7.25 (d, 3JHH = 8.0, 1H, H5), 7.24 (d, 3JHH = 8.0, 1H, H5′), 7.12 (dd, 3JHH = 8.0, 4JHH = 1.9, 1H, H4), 7.08 (dd, 3JHH = 8.0, 4JHH = 1.9, 1H, H4′), 6.81 (d, 4JHH = 1.6, 1H, H2′), 2.99–3.06 (m, 8H, CH3(CH2)2CH2N), 1.41–1.52 (m, 8H, CH3CH2CH2CH2N), 1.33 (s, 9H, CMe3), 1.26–1.32 (m, 8H, CH3CH2(CH2)2N), 1.10 (s, 9H, CMe3′), 0.92 ppm (t, 3JHH = 7.6, 12H, CH3(CH2)3N). 19F NMR (282 MHz, CD2Cl2, 297 K) −119.6 (m, o-F), −162.3 (t, p-F), −163.2 (m, m-F) ppm. 13C{1H} NMR (75.47 MHz, CD2Cl2, 297 K) δ 151.1 (s, C2′), 150.2 (s, C2), 149.2 (s, C5′), 148.7 (s, C5), 131.3 (s, C4′), 130.0 (s, C4), 129.9 (s, C1′), 123.4 (s, C3′), 123.1 (s, C3), 120.3 (s, C6′), 120.1 (s, C1), 119.5 (s, C6), 58.6 (s, CH3(CH2)2CH2N), 35.0 (s, CMe3′), 34.3 (s, CMe3), 31.4 (s, C8′), 30.9 (s, C8), 23.8 (s, CH3CH2CH2CH2N), 19.6 (s, CH3CH2(CH2)2N), 13.4 ppm (s, CH3(CH2)3N). Calcd for C42H60AuClF5N (found) C, 55.64 (55.64); H, 6.68 (6.58); N, 1.55 (1.68) %.
In Situ NMR Experiments and Data
(C^C^N)AuH (4)
(C^C^N)AuCl (1) (15 mg, 0.0226 mmol) was loaded into a screw cap NMR within the glovebox and approximately 0.7 mL of dry THF-d8 were added under Ar. The resultant suspension was sonicated at room temperature for 10 s to obtain a pale-yellow solution. The NMR tube was then inserted in a cold bath at 195 K and, using a microsyringe, LiHBEt3 (1 equiv, 22.6 μL of a 1.0 M solution in THF) was injected through the septum. The sample was quickly shaken to obtain a bright yellow solution and inserted into the NMR probe and analyzed at 253 K. 1H NMR (300.13 MHz, THF-d8, 253 K, J values in Hz) δ 8.86 (s, 1H, H1), 8.00 (d, 1H, 3JHH = 8.3, H4), 7.89 (d, 1H, 3JHH = 8.3, H3), 7.70 (s, 2H, H7+H16), 7.65 (d, 2H, 3JHH = 8.3, H19), 7.58 (s, 1H, H9), 7.50 (d, 2H, 3JHH = 8.3, H20), 7.37 (d, 1H, 3JHH = 8.1, H13), 7.17 (dd, 1H, 3JHH = 8.1 4JHH = 1.8, H14), 6.33 (s, 1H, Au–H), 2.36 (s, 3H, Me), 1.38 (s, 9H, C(21)CMe3), 1.34 ppm (s, 9H, C(15)CMe3). 13C{1H} NMR (75.47 MHz, THF-d8, 253 K) δ 184.2 (s, C11), 165.9 (s, C5), 156.0 (s, C1), 154.9 (s, C12), 151.8 (s, C10), 150.8 (s, C15), 150.2 (s, C21), 145.3 (s, C17), 144.3 (s, C6), 141.5 (s, C3), 140.8 (s, C8), 139.9 (s, C18), 138.4 (s, C16), 136.3 (s, C2), 127.4 (s, C19), 126.1 (s, C20), 123.8 (s, C14), 121.9 (s, C13), 120.9 (s, C7+C9), 120.8 (s, C4), 35.1 (s, C(15)CMe3), 35.0 (s, C(21)CMe3), 31.5 (s+s, C(15)CMe3+C(21)CMe3), 17.9 ppm (s, Me). T1 (253 K, THF-d8) Au–H 1.30 s.
Li[(C^C^N)AuH2] (5)
Procedure (a): In the glovebox under N2, 1 (10 mg, 0.015 mmol) was loaded into a screw-cap NMR tube, and approximately 0.7 mL of dry THF-d8 were added. The resultant suspension was sonicated at room temperature for 10 s to obtain a pale-yellow solution. The NMR tube was then inserted in a cold bath at 195 K and 2.0 equiv LiHBEt3 (30.2 μL of a 1.0 M solution in THF) were injected through the septum by using a microsyringe. The sample was quickly shaken to give a colorless solution and analyzed at 263 K. Procedure (b): In the glovebox under N2, 15 mg of 1 and 3.0 equiv of solid LiAlH4 (2.5 mg) were loaded into a screw cap NMR tube. The NMR tube was inserted in a cold bath at 195 K and approximately 0.7 mL of dry THF-d8 were added. The resultant dark suspension was shaken and the NMR tube was inserted into the precooled NMR probe and analyzed at 203 K. 1H NMR (300.13 MHz, THF-d8, 263 K, J values in Hz) δ 8.26 (s, 1H, H1), 8.08 (m, 1H, H16), 7.72 (s, 1H, H9), 7.58 (m, 4H, H3+H4+H19), 7.43 (m, 4H, H7+H13+H20), 6.98 (dd, 3JHH = 8.1 4JHH = 2.1, 1H, H14), 2.37 (s, 3H, Me), 1.35 (s, 9H, (C21) CMe3), 1.31 (s, 9H, (C15) CMe3), −0.31 (ps t, 1H, 2JHH = 4.3, Au–Hb), −0.59 ppm (d, 1H, 2JHH = 4.3, Au–Ha). 13C{1H} NMR (75.47 MHz, THF-d8, 263 K) δ 172.3 (s, C17), 171.1 (s, C11), 167.2 (s, C5), 161.1 (s, C10), 155.4 (s, C12), 149.6 (s, C21), 149.2 (s, C6), 148.8 (s, C15), 147.5 (s, C1), 140.7 (s, C18), 140.4 (s, C16), 138.1 (s, C8), 137.0 (s, C3), 130.7 (s, C2), 126.9 (s, C19), 126.8 (s, C4), 125.9 (s, C20), 124.3 (s, C7), 120.7 (s, C14), 120.4 (s, C13), 118.6 (s, C9), 34.8 (s, C(15)CMe3), 34.7 (s, C(21)CMe3), 32.0 (s, C(15)CMe3), 31.6 (s, C(21)CMe3), 18.4 ppm (s, Me). T1 (−20 °C, THF-d8) Au–Ha 0.80 s, Au–Hb 0.82 s.
Li[(C^C)Au(H)Cl] (6)
In a glovebox under N2, 2 (15 mg, 0.026 mmol) was loaded into a screw-cap NMR tube and dissolved in approximately 0.7 mL of dry THF-d8. The NMR tube was then inserted in a cold bath at 195 K and 2.0 equiv LiHBEt3 (52.0 μL of a 1.0 M solution in THF) were injected through the septum, using a microsyringe. The sample was quickly shaken to obtain a bright yellow solution and inserted into the NMR probe and analyzed at 213 K. 1H NMR (300.13 MHz, THF-d8, 213 K, J values in Hz) δ 8.01 (m, 1H, H2′), 7.72 (overlapped with py signals, 1H, H2), 7.20 (d, 1H, 3JHH = 8.2, H5), 7.17 (d, 1H, 3JHH = 8.2, H5′). 6.96 (br d, 1H, 3JHH = 8.2, H4), 6.90 (dd, 1H, 3JHH = 8.2 4JHH = 1.8, H4′), 2.43 (br s, 1H, Au–H), 1.27 (s, 9H, CMe3), 1.25 ppm (s, 9H, CMe3′). 13C{1H} NMR (75.47 MHz, THF-d8, 213 K) δ 174.8 (s, C1′), 153.0 (s, C6), 151.4 (s, C6′), 149.3 (s, C3), 148.4 (s, C1), 147.1 (s, C3′), 139.1 (s, C2), 129.9 (s, C2′), 121.9 (s, C4), 121.8 (s, C4′), 120.1 (s, C5), 118.9 (s, C5′), 34.7 (s, CMe3), 34.1 ppm (s, CMe3′), 31.2 + 31.1 (s+s, CMe3+CMe3′).
(C^C)AuH(DMAP) (7)
A sample of 6 was generated as described above from 15 mg of 2 and kept at 195 K. Successively, 4.0 equiv of DMAP (12.7 mg) were dissolved in 0.3 mL of dry THF-d8 and injected through the septum by using a gastight syringe to obtain a yellow solution. The sample was then transferred into the precooled NMR probe and analyzed at 253 K. 1H NMR (300.13 MHz, THF-d8, 253 K, J values in Hz) δ 8.32 (d, 3JHH = 7.1, 2H, H7), 7.74 (ps t, 4JHH = 2.0, 1H, H2), 7.27 (m, overlapped with py signals, H5+H5′), 7.03 (dd, 3JHH = 8.1 4JHH = 2.0, 1H, H4), 6.99 (dd, 3JHH = 8.1 4JHH = 2.0, 1H, H4′), 6.92 (dd, 4JHH = 4.5 4JHH = 2.0, 1H, H2′), 6.89 (d, 3JHH = 7.1, 2H, H8), 3.25 (brs, 1H, Au–H), 3.13 (s, 6H, NMe2), 1.29 (s, 9H, CMe3), 1.17 ppm (s, 9H, CMe3′). 13C{1H} NMR (75.47 MHz, THF-d8, 253 K) δ 174.6 (s, C1′), 155.3 (s, C9), 153.3 (s, C6), 151.8 (s, C6′), 151.1 (s, C7), 148.8 (s, C3), 148.1 (s, C3′), 144.1 (s, C1), 139.9 (s, C2), 129.0 (s, C2′), 122.8 (s+s, C4+C4′), 119.9 (s, C5 or C5′), 119.2 (s, C5′ or C5), 108.5 (s, C8), 39.0 (s, NMe2), 35.1 (s, CMe3′), 34.7 (s, CMe3), 31.8 ppm (s, CMe3+CMe3′).
(C^C)AuH(PMe3) (9)
Procedure (a): A sample of 6 was generated as described above from 15 mg of 2 and kept at 195 K. Successively, 1.0 equiv of PMe3 (26 μL of a 1.0 M solution in THF) was injected through the septum by using a gastight syringe to obtain a pale-yellow solution. The sample was then transferred into the precooled NMR probe and analyzed at 253 K. Procedure (b): In the glovebox, 20 mg of (C^C)AuCl(PMe3) were loaded into a screw-cap NMR tube and dissolved in approximately 0.7 mL of toluene-d8. Outside the glovebox, 1.0 equiv LiHBEt3 (35 μL of a 1.0 M solution in THF) were injected through the septum by a microsyringe at room temperature to give a pale-yellow solution and a white precipitate. The solution was filtered over Celite into a J Young NMR tube and dried under vacuum to afford a pale-yellow powder, which was redissolved in dry toluene-d8 and analyzed at room temperature. 1H NMR (300.13 MHz, THF-d8, 253 K, J values in Hz) δ 7.94 (dt, 4JHP = 10.0 4JHH = 1.7, 1H, H2), 7.58 (m, 1H, H2′), 7.32 (d, 3JHH = 8.2, 1H, H5′), 7.28 (dd, 3JHH = 8.2 5JHP = 4.1, 1H, H5), 7.06 (dd, partially overlapped with H4, 1H, H4′), 7.04 (dd, partially overlapped with H4′, 1H, H4), 1.89 (d, 2JHP = 10.8, 9H, PMe3), 1.53 (brd, 2JHP = 32.6, 1H, Au–H), 1.31 (s, 9H, CMe3′), 1.28 ppm (s, 9H, CMe3). 13C{1H} NMR (75.47 MHz, THF-d8, 253 K, J values in Hz) δ 168.4 (d, 2JPC = 4.6, C1′), 160.6 (d, 2JPC = 136.2, C1), 154.8 (d, 3JPC = 5.5, C6), 154.1 (s, C6′), 149.3 (d, 4JPC = 10.0, C3), 148.4 (s, C3′), 140.8 (d, 3JPC = 5.4, C2), 133.0 (d, 3JPC = 6.6, C2′), 123.0 (s+s, C4+C4′), 120.5 (m, H5+H5′), 35.0 (s, CMe3′), 34.8 (s, CMe3), 31.7 (s+s, CMe3+CMe3′), 15.9 ppm (d, 1JCP = 32.5, PMe3). 31P{1H} NMR (121.49 MHz, THF-d8, 253 K) −8.2 ppm (s, PMe3). 1H NMR (300.13 MHz, Toluene-d8, 298 K, J values in Hz) δ 8.49 (dt, 4JHP = 10.6 4JHH = 2.0, 1H, H2), 7.58 (d, 3JHH = 8.2, 1H, H5′), 7.54 (m, 2H, H2′+H5), 7.22 (m, 2H, H4+H4′), 1.86 (dm, 2JHP = 34.7, 1H, Au–H), 1.39 (s+s, 18H, CMe3+CMe3′), 1.03 ppm (d, 2JHP = 10.2, 9H, PMe3).
(C^C)AuH(Ptol3) (10)
This complex was prepared by following the procedures described for 9. Procedure (a) was followed by using 15 mg of 2 and 7.9 mg of P(p-tol)3 (dissolved in 0.2 mL of THF-d8). Procedure (b) was followed by using 15 mg of (C^C)AuCl(Ptol3) and 19 μL of a 1 M LiHBEt3 solution in THF. 1H NMR (300.13 MHz, THF-d8, 213 K, J values in Hz) δ 8.00 (br d, 4JHP = 9.3, H2), 7.49 (m, 6H, H8), 7.34 (m, 8H, H5+H5′+H9), 7.08 (dd, 3JHH = 8.1 4JHH = 8.1, 1H, H4), 6.97 (m, 1H, H2′), 6.90 (dd, 3JHH = 8.1 4JHH = 8.1, 1H, H4′), 2.38 (s, 9H, Me), 2.33 (brd, 2JPH = 33.0, Au–H), 1.28 (s, 9H, CMe3), 0.77 (s, 9H, CMe3′). 13C{1H} NMR (75.47 MHz, THF-d8, 213 K, J values in Hz) δ 168.4 (d, 2JPC = 4.7, C1′), 159.8 (d, 2JPC = 129.0, C1), 153.7 (d, 3JPC = 5.0, C6), 153.3 (s, C6′), 148.8 (d, 4JPC = 9.0, C3), 147.7 (s, C3′), 142.1 (s, C10). 140.3 (d, 3JPC = 4.0, C2), 136.2 (br d, 3JPC = 4.3, C2′), 134.6 (d, 3JPC = 12.5, C8), 129.8 (d, 4JPC = 11.0, C9), 126.8 (d, 1JPC = 56.0, C7), 122.5 (s, C4), 121.5 (s, C4′), 120.0 (d, 4JPC = 6.5, C5), 119.4 (s, C5′), 34.3 (s, CMe3), 34.1 (s, CMe3′), 31.0 (s, CMe3), 30.4 (s, CMe3′), 20.6 ppm (s, Me). 31P{1H} NMR (121.49 MHz, THF-d8, 213 K) 32.6 ppm (s, Ptol3). 1H NMR (300.13 MHz, Toluene-d8, 298 K, J values in Hz) δ 8.60 (br d, 4JHP = 9.7, 1H, H2), 7.58 (m, 2H, H5+H5′), 7.51 (dd, 3JHP = 12.3 3JHH = 8.2, 6H, H8), 7.25 (m, 2H, H2′+H4), 7.15 (dd, 3JHH = 8.2 4JHH = 1.5, 1H, H4′), 6.79 (d, 3JHH = 8.2, 6H, H9), 2.94 (br d, 2JHP = 32.4, 1H, Au–H), 1.98 (s, 9H, Me), 1.38 (s, 9H, CMe3), 1.02 ppm (s, 9H, CMe3′).
(C^C)AuH(PPh3) (11)
This complex was synthesized by following the procedure reported for (C^C)AuH(PMe3), by using 15 mg of 2, 26 μL of LiHBEt3 and 1.0 equiv of PPh3 (6.8 mg) in dry THF-d8. NMR characterization was carried out at 298 K. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 8.02 (brd, 4JHP = 9.7, 1H, H2), 7.66 (m, overlapped with py signals, H8), 7.50 (m, 9H, H9+H10), 7.30 (m, 2H, H5+H5′), 7.09 (brd, 3JHH = 8.3, 1H, H4), 7.05 (brs, 1H, H2′), 6.92 (brd, 3JHH = 8.3, 1H, H4′), 2.40 (brd, 2JHP = 32.3, 1H, Au–H), 1.29 (s, 9H, CMe3), 0.81 ppm (s, 9H, CMe3′). 13C{1H} NMR (75.47 MHz, THF-d8, 298 K, J values in Hz) δ 169.3 (d, 2JPC = 5.1, C1′), 160.5 (d, 2JPC = 129.5, C1), 154.5 (d, 3JPC = 5.2, C6), 154.1 (s, C6′), 149.8 (d, 4JPC = 9.4, C3), 148.8 (s, C3′), 140.9 (d, 3JPC = 4.7, C2), 136.5 (d, 3JPC = 5.5, C2′), 135.4 (d, 3JPC = 12.6, C8), 132.2 (br s, C10), 131.0 (d, 1JCP = 49.9, C7), 129.7 (d, 4JPC = 10.9. C9), 123.3 (s, C4), 122.5 (s, C4′), 120.8 (d, 4JPC = 7.2, C5), 120.2 (s, C5′), 34.9 (s, CMe3), 34.6 (s, CMe3′), 31.7 (s, CMe3), 31.4 ppm (s, CMe3′). 31P{1H} NMR (121.49 MHz, THF-d8, 298 K) 34.9 ppm (s, PPh3).
(C^C)Au(Z-C2H(CO2Me)2)(PMe3) (12)
A sample of 9 was generated from 15 mg of 2 in THF-d8 and treated with 2 equiv DMAD at room temperature. The conversion to the vinyl complex was complete in 2h. NMR characterization was performed in mixture with py-BEt3, at 298 K. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 7.54 (m, 3H, H2+H2′+H8), 7.32 (d, 3JHH = 8.2, 1H, H5′), 7.23 (dd, 3JHH = 8.2 5JHP = 3.8, 1H, H5), 7.11 (dd, 3JHH = 8.2 4JHH = 1.9, 1H, H4′), 7.01 (dd, 3JHH = 8.2 4JHH = 1.7, 1H, H4), 3.68 (s, 3H, H10), 3.59 (buried under THF, H12), 1.78 (buried under THF, PMe3), 1.34 (s, 9H, CMe3′), 1.24 ppm (s, 9H, CMe3). 13C{1H} NMR (75.47 MHz, THF-d8, 298 K, J values in Hz) δ 184.1 (d, 2JCP = 14.8, C7), 173.4 (s, C9), 170.0 (s, C11), 159.5 (d, 2JCP = 138.0, C1), 158.7 (s, C1′), 153.3 (d, 3JCP = 5.1, C6), 152.2 (s, C6′), 148.1 (d, 4JCP = 9.4, C3), 147.8 (s, C3′), 131.7 (d, 3JCP = 3.5, C2′), 131.6 (d, 3JCP = 9.0, C2), 127.1 (d, 3JCP = 3.0, C8), 122.6 (s, C4+C4′), 120.2 (s, C5′), 119.9 (d, 4JCP = 7.4, C5), 51.0 (s, C10), 50.6 (s, C12), 34.5 (s, CMe3), 34.3 (s, CMe3′), 31.1 (s, CMe3′), 30.8 (s, CMe3), 13.1 ppm (d, 1JCP = 31.7, PMe3). 31P{1H} NMR (121.49 MHz, THF-d8, 298 K) −5.6 ppm (s, PMe3).
(C^C)Au(Z-C2H(CO2Me)2)(Ptol3) (13)
A sample of 10 was generated from 15 mg of 2 in THF-d8 and treated with 2 equiv DMAD at room temperature. The conversion into the vinyl complex was complete in 12h, affording 13 in 80% yield. NMR characterization was performed in mixture with py-BEt3, at 298 K. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 7.65 (br, overlapped with pyridine signals, H14), 7.42 (dd, 1H, 4JHP = 9.5, 4JHH = 1.6, H2′), 7.2 (m, 6H, H15), 7.06 (m, 2H, H4′+H8), 6.95 (br t, 1H, H2), 6.88 (dd, 3JHH = 8.0, 4JHH = 1.7, H2′), 3.46 (s, 3H, H10), 3.43 (s, 3H, H12), 2.35 (s, 9H, PhMe), 1.20 (s, 9H, CMe3), 0.65 ppm (s, 9H, CMe3′). Partial 13C{1H} NMR (75.47 MHz, THF-d8, 298 K, J values in Hz) δ 186.0 (d, 2JCP = 13.3, C7), 172.1 (s, C9), 169.2 (s, C11), 159.6 (d, 2JCP = 126.3, C1), 159.3 (d, 2JCP = 6.3, C1′), 152.8 (d, 4JCP = 4.0, C6′), 152.6 (s, C6), 148.2 (d, 4JCP = 8.7, C3), 147.2 (s, C3′), 141.5 (s, C16), 137.0 (d, 3JCP = 8.8, C2′), 135.3 (br, C14), 131.7 (d, 3JCP = 2.4, C2), 129.1 (brs, C8), 128.7 (d, 3JCP = 12.4, C15), 126.8 (d, 1JPC = 49.8, C7), 122.7 (s, C4), 121.5 (s, C4′), 119.9 (d, 4JCP = 6.9, C5), 119.2 (s, C5′), 50.5 (s, C10), 50.2 (s, C12), 34.5 (s, CMe3), 33.8 (s, CMe3′), 30.8 (s, CMe3), 30.3 (s, CMe3′), 20.4 ppm (s, (C16)Me). 31P{1H} NMR (121.49 MHz, THF-d8, 298 K) 33.2 ppm (s, Ptol3).
Li[(C^C)AuH2] (14)
A solution of 10 in THF-d8 was generated from 15 mg of 2 as described above. Successively, a second equivalent of LiHBEt3 was added to the solution at room temperature and the slow conversion of the monohydride to the dihydride anion was observed, with liberation of free Ptol3. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 8.00 (m, 2H, H2), 7.17 (d, 3JHH = 8.1, 2H, H5), 6.86 (dd, 3JHH = 8.1 4JHH = 2.2, 2H, H4), 1.27 (s, 18H, CMe3), 0.09 ppm (ps t, 4JHH = 2.3, 2H, Au–H). 13C{1H} NMR (75.47 MHz, THF-d8, 298 K) δ 170.7 (s, C1), 156.2 (s, C6), 147.7 (s, C3), 140.2 (s, C2), 120.3 (s, C4), 119.2 (s, C5), 34.6 (s, CMe3), 32.0 ppm (s, CMe3). 1H NMR (300.13 MHz, Toluene-d8, 298 K, J values in Hz) δ 8.36 (m, 2H, H2), 7.55 (d, 3JHH = 8.1, 2H, H5), 7.20 (dd, 3JHH = 8.1 4JHH = 2.0, 2H, H4), 1.41 (s, 18H, CMe3), 1.00 ppm (ps t, 4JHH = 2.3, 2H, Au–H).
[NBu4][(C^C)Au(H)C6F5] (16)
Procedure (a): In the glovebox, 15 mg of [NBu4][(C^C)Au(Cl)C6F5] (15) were loaded into a screw cap NMR tube and dissolved in approximately 0.7 mL of THF-d8. Successively, 1.2 equiv of LiHBEt3 were added at 195 K and the sample was then allowed to warm up at room temperature. The reaction went to completion in 24h at room temperature. Procedure (b): In the glovebox, 20 mg of 15 were loaded into a screw-cap NMR tube and suspended in approximately 0.7 mL of toluene-d8. Successively, 1.2 equiv LiHBEt3 were added at room temperature to afford a pale-yellow solution with an off-white precipitate. The solution was filtered into a J-young NMR tube, dried under vacuum and redissolved in THF-d8. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 8.07 (m, 1H, H2), 7.26 (d, 3JHH = 8.1, 1H, H5), 7.23 (d, 3JHH = 8.1, 1H, H5′), 7.09 (m, 1H, H2′), 7.00 (dd, 3JHH = 8.1 4JHH = 2.2, 1H, H4), 6.94 (dd, 3JHH = 8.1 4JHH = 2.2, 1H, H4′), 2.91 (m, 8H, Hα), 1.35 (br m, 8H, Hβ), 1.32 (s, 9H, CMe3), 1.23 (q, 3JHH = 7.8, Hγ), 1.15 (overlapped with CMe3′, Au–H), 1.14 (s, 9H, CMe3′), 0.90 ppm (t, 12H, 3JHH = 7.8, Hδ). 13C{1H} NMR (75.47 MHz, THF-d8, 298 K) δ 170.3 (s, C1′), 160.1 (brs, C1), 155.3 (s, C6′), 154.3 (s, C6), 148.8 (s, C3′), 148.0 (s, C3), 141.1 (s, C2′), 133.1 (s, C2), 121.8 (s+s, C4+C4′), 119.9 (s, C5′), 119.6 (s, C5), 58.8 (s, Cα), 34.8 (s, CMe3+CMe3′), 31.8 (s, CMe3), 31.7 (s, CMe3′), 24.2 (s, Cβ), 20.2 (s, Cγ), 13.8 ppm (s, Cδ).19F NMR (282.4 MHz, THF-d8, 298 K, J values in Hz) −116.6 (m, 3JFF = 23.0, 2F, o–F), −165.6 (m, 3F, p–F+m–F). 1H NMR (300.13 MHz, Toluene-d8, 298 K, J values in Hz) δ 8.31 (s, 1H, H2), 7.41 (br s, 1H, H2′), 7.34 (m, 2H, H5+H5′), 7.04 (m, partially overlapped with toluene, H4+H4′), 2.47 (br m, 8H, Hα), 1.67 (brs, 1H, Au–H), 1.39 (s, 9H, CMe3), 1.32 (brm, partially overlapped with THF and BEt3, Hβ), 1.23 (s, 9H, CMe3′), 0.99 (brm, partially overlapped with BEt3, Hγ), 0.83 ppm (brt, 3JHH = 7.3, 9H, Hδ).
[NBu4][(C^C)Au(Z-C2H(CO2Me)2)C6F5] (17)
A sample of [NBu4][(C^C)Au(H)C6F5] was generated from 10 mg of (C^C)AuCl(py) in THF-d8 and treated with 2 equiv of DMAD at room temperature to give a bright orange solution. Conversion into the vinyl product was instantaneous, as indicated by 1H NMR. The THF solution was then passed through silica, dried and washed with light petroleum ether to remove most of borane and DMAD impurities. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 7.59 (d, 4JHH = 2.1, 1H, H2), 7.26 (s, 1H, H8), 7.21 (m, 2H, H5+H5′), 6.97 (m, 3H, H2′+H4+H4′), 3.52 (s, 3H, H10), 3.48 (s, 3H, H12), 3.02 (m, 8H, Hα), 1.43 (m, 8H, Hβ), 1.23 (s, 9H, CMe3), 1.19 (q, 8H, 3JHH = 7.4, Hγ), 1.13 (s, 9H, CMe3′), 0.9 ppm (t, 9H, 3JHH = 7.4, Hδ). 13C{1H} NMR (75.47 MHz, THF-d8, 298 K) δ 184.7 (s, C7), 175.0 (s, C9), 169.2 (s, C11), 159.8 (s, C1′), 159.2 (brs, C1), 153.3 (s, C6′), 152.7 (s, C6), 147.6 (s, C3), 147.2 (s, C3′), 132.8 (s, C2′), 132.4 (s, C2), 126.4 (s, C8), 121.6 (s, C4), 121.1 (s, C4′), 119.2 (s, C5′), 118.8 (s, C5), 57.7 (s, Cα), 50.1 (s, C10), 49.8 (s, C12), 34.4 (s, CMe3), 34.0 (s, CMe3′), 31.0 (s, CMe3), 30.9 (s, CMe3′), 23.6 (s, Cβ), 19.4 (s, Cγ), 13.1 ppm (s, Cδ). 19F NMR (282.4 MHz, THF-d8, 298 K, J values in Hz) −116.4 (dm, 3JFF = 34.1, 1F, o–F), −117.2 (dm, 3JFF = 34.1, 1F, o–F), −165.2 (t, 3JFF = 19.8, 1F, p–F), −166–0 (m, 2H, o–F). 1H NMR (300.13 MHz, CD2Cl2, 298 K, J values in Hz) δ 7.58 (d, 4JHH = 1.6, 1H, H2), 7.28 (m, 3H, H5+H5′+H8), 7.04 (dd, 3JHH = 8.1 4JHH = 1.8, 2H, H4+H4′), 3.6 (s, 3H, H12), 3.54 (s, 3H, H10), 2.85 (m, Hα), 1.33 (m, Hβ), 1.24 (s, 9H, CMe3), 1.18 (m, Hγ), 1.13 (s, 9H, CMe3′), 0.91 ppm (t, 3JHH = 7.3, Hδ).
Li[(C^C)Au(Z-C2H(CO2Me)CH2OBEt3(PMe3)] (18)
A sample of 12 was generated in situ in THF-d8 and treated at room temperature with aliquots of LiHBEt3 until reaching a Au/hydride stoichiometric ratio of 2.5. Full conversion of 12 into 18 was observed immediately. 1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 7.93 (m, 1H, H8), 7.62 (dd, 4JPH = 9.7, 4JHH = 1.6, 1H, H2), 7.55 (brs, 1H, H2′), 7.32 (d, 3JHH = 8.1, 1H, H5′), 7.25 (dd, 3JHH = 8.0, 5JPH = 4.1, 1H, H5), 7.09 (dd, 3JHH = 8.1, 4JHH = 1.6, 1H, H4′), 7.03 (br d, 3JHH = 8.0, 1H, H4), 3.01 (br d, 3JHH = 4.3, 2H, H9), 3.59 (buried under THF, H11), 1.81 (partially overlapped with THF, PMe3), 1.33 (s, 9H, CMe3′), 1.24 (s, 9H, CMe3), 0.66 (br t, overlapped with BEt3, O–BCH2CH3), 0.00 ppm (br t, overlapped with BEt3, O–BCH2CH3). 13C{1H} NMR (75.47 MHz, THF-d8, 298 K, J values in Hz) δ 174.0 (s, C7), 163.2 (d, 2JPC = 6.2, C1′), 159.8 (d, 2JPC = 138.7, C1), 159.7 (d, 3JPC = 14.6, C10), 154.2 (d, 3JPC = 5.4, C6′), 153.6 (s, C6), 149.9 (s, C8), 148.6 (s, C3′), 148.5 (s, 4JPC = 9.9, C3), 132.5 (d, 3JPC = 7.1, C2′), 132.4 (s, C2), 123.4 (s, C4), 123.2 (s, C4′), 120.6 (m, C5+C5′), 64.7 (s, C9), 51.1 (s, C11), 35.2 (s, CMe3), 35.1 (s, CMe3′), 31.8 (s, CMe3′), 31.7 (s, CMe3), 14.0 (d, 1JPC = 31.6, PMe3), 13.9 (brs, overlapped with PMe3, O–BCH2CH3), 10.9 ppm (overlapped with BEt3, O–BCH2CH3).
Z-Dimethyl(3-tbutyl(6-(4-tbutylphenyl))phenyl)fumarate (19)
A sample of 12 was generated in THF-d8 and treated at room temperature with substoichiometric portions of [H(OEt2)2][H2N{B(C6F5)3}2] until reacting a stoichiometric ratio of acid/Au of 1.2. Upon addition of the acid, the deep orange color of 12 faded to give a pale-yellow solution and the formation of a dark precipitate. 1H NMR spectroscopy revealed the complete disappearance of 12 to give 19 in 65% yield. NMR characterization is given in mixture.
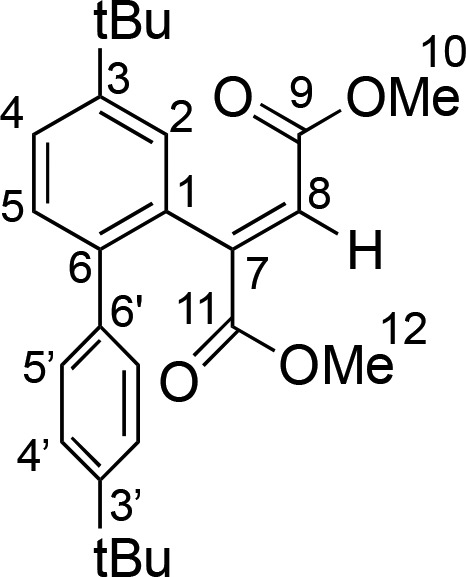
1H NMR (300.13 MHz, THF-d8, 298 K, J values in Hz) δ 7.43 (dd, 3JHH = 8.1, 4JHH = 2.0, 1H, H4), 7.34 (d, 3JHH = 8.5, 2H, H4′), 7.24 (m, 2H, H5+H8), 7.20 (d, 3JHH = 8.5, 2H, H5′), 6.83 (s, H8), 3.54 (s, 3H, H12), 3.3 (s, 3H, H10), 1.34 (s, 9H, CMe3), 1.32 ppm (s, 9H, CMe3′). Partial 13C{1H} NMR (75.47 MHz, THF-d8, 298 K) δ 165.8 (s, C9), 165.4 (s, C11), 149.7 (overlapped with AB2, C3′), 149.4 (s, C3), 138.8 (overlapped with AB2, C6′+C6), 133.0 (s, C1), 128.9 (s, C8), 128.8 (s, C5), 128.7 (s, C5′), 127.3 (s, C8), 125.4 (s, C4), 124.5 (s, C4′), 51.2 (s, C10), 50.8 (s, C12), 34.7 (s+s, CMe3+CMe3′), 30.7 ppm (s, CMe3+CMe3′).
[(C^N–CH)AuH(μ-H)Au(C^N^C)][AB2] (20)
Within the glovebox, 15 mg of (C^N^C)AuH were loaded into a screw-cap NMR tube and dissolved in approximately 0.3 mL of dry CD2Cl2. One equiv [H(OEt2)2][H2N{B(C6F5)3}2] (33.1 mg, 0.028 mmol) was dissolved in 0.4 mL of dry CD2Cl2 and loaded into a gastight syringe equipped with a rubber stopper. Outside the glovebox, the NMR tube was inserted into a cold bath at 198 K and the acid solution was slowly injected through the septum at low temperature. The sample was then shaken quickly, inserted in the precooled NMR probe and studied at 213 K. Extensive decomposition was observed after 1 h at 213 K. 1H NMR (300.13 MHz, CD2Cl2, 213 K, J values in Hz) δ 8.23 (t, 3JHH = 8.0, 1H, H1), 8.08 (d, 3JHH = 8.0, 1H, H2), 7.79 (m, 3H, H5+H2′+H1a), 7.60 (m, 2H, H6+H8), 7.38 (m, 8H, H5′+H2a+H5a+H6a), 7.17 (m, 4H, H6′+H8a), 5.56 (br s, NH2), 1.13 (s, 9H, CMe3), 1.03 (s, 18H, CMe3a), 0.81 (s, 9H,CMe3′), −4.93 (d, 2JHH = 8.6, 1H, Hb), −8.47 ppm (br d, 2JHH = 8.6, 1H, Ha).
[(C^N–CH)AuMe(μ-H)Au(C^N^C)][AB2] (21)
Ten mg of (C^N^C)AuMe and 1.0 equiv of [H(OEt2)2][H2N{B(C6F5)3}2] were loaded within a screw cap NMR and dissolved in approximately 0.5 mL of CD2Cl2. Outside the glovebox, the NMR sample was inserted in a cold bath at 195 K and 1.0 equiv (C^N^C)AuH (9.7 mg, 0.018 mmol, dissolved in 0.3 mL of CD2Cl2) were injected through the septum to give a bright yellow solution, which was quickly shaken and inserted into the precooled NMR probe at 213 K. 21 was observed in 65% yield, along with unreacted 3a. Partial 1H NMR (300.13 MHz, CD2Cl2, 243 K, J values in Hz) 8.21 (m, 2H, H1+H2), 7.89 (m, 2H, H1a+H2′), 7.72 (br d, overlapped with 3a, H5), 7.50 (br d, 3JHH = 8.1, 1H, H6), 7.43 (m, 6H, H2a+H5a+H5′), 7.34 (br s, 4H, H8a+H6′), 7.27 (s, overlapped with 3a, H8), 7.20 (d, 3JHH = 8.1, H6a), 5.62 (br s, NH2), 1.71 (s, 3H, Au–Me), 1.26 (s, 9H, CMe3), 1.08 (s, overlapped with Et2O, CMe3a), 0.76 (s, 9H, CMe3′), −5.14 ppm (s, 1H, Au–H–Au).
[(C^N–CH)AuMe(μ-H)MeAu(C^N–CH)][AB2] (22a)
Fifteen mg of (C^N^C)AuMe and 1.0 equiv of [H(OEt2)2][H2N{B(C6F5)3}2] were loaded within a screw cap NMR and dissolved in approximately 0.6 mL of CD2Cl2. Outside the glovebox, the NMR sample was inserted in a cold bath at 195 K and 5.0 equiv of HSi(OMe)3 were injected through the septum. The sample was quickly shaken and inserted in the precooled NMR probe at 243 K. 1H NMR (300.13 MHz, CD2Cl2, 243 K, J values in Hz) δ 8.18 (t, 3JHH = 7.8, 2H, H1), 8.06 (d, 3JHH = 7.8, 2H, H2), 7.76 (d, 3JHH = 8.2, 2H, H5), 7.46 (d, 3JHH = 7.8, 2H, H2′), 7.42 (d, 3JHH = 8.2, 2H, H6), 7.33 (br m, 4H, H8+H6′a), 7.21 (d, 3JHH = 8.4, 2H, H5′a), 6.77 (d, 3JHH = 8.2, 2H, H6′b), 6.55 (d, 3JHH = 8.2, 2H, H5′b), 5.66 (br s, 2H, NH2), 1.35 (s, 3H, Au–Me), 1.31 (s, 18H, CMe3), 0.75 (s, 18H, CMe3′), −2.53 ppm (s, 1H, Au–H–Au). 13C{1H} NMR (75.47 MHz, CD2Cl2, 223 K, J values in Hz) δ 161.2 (s, C3), 160.3 (s, C3′), 154.9 (s, C7), 154.1 (s, C9), 153.5 (s, C7′), 147.5 (br d, 1JCF = 238.5, o–F H2N[B(C6F5)3]2–), 141.8 (s, C1), 141.2 (s, C4), 138.9 (br d, 1JCF = 250.0, p–F H2N[B(C6F5)3]2–), 136.5 (br d, 1JCF = 250.0, m–F H2N[B(C6F5)3]2–), 137.2 (s, C4′), 130.7 (s, C5′a), 127.2 (s, C8+C5), 126.2 (s, C6), 125.7 (s+s, C5′b+C6′a), 125.2 (s, C6′b), 124.4 (s, C2′), 118.8 (s, C2), 35.7 (s, CMe3), 34.3 (s, CMe3′), 30.9 (s, CMe3), 30.2 (s, CMe3′), 10.0 ppm (s, Au–Me).
[(C^N–CH)AuC6F5(μ-H)Au(F5C6)(C^N–CH)][AB2] (22b)
Fifteen mg of (C^N^C)AuC6F5 and 1.0 equiv [H(OEt2)2][H2N{B(C6F5)3}2] were loaded within a screw-cap NMR and dissolved in approximately 0.6 mL of CD2Cl2. Outside the glovebox, the NMR sample was inserted in a cold bath at 195 K and 5.0 equiv HSi(OMe)3 were injected through the septum. The sample was quickly shaken and inserted in the precooled NMR probe at 223 K. 1H NMR (300.13 MHz, CD2Cl2, 223 K, J values in Hz) δ 8.34 (t, 3JHH = 7.8, 2H, H1), 8.13 (d, 3JHH = 7.8, 2H, H2), 7.78 (d, 3JHH = 8.2, 2H, H5), 7.56 (d, 3JHH = 7.8, 2H, H2′), 7.50 (m, 4H, H5′a+H6′a), 7.38 (d, 3JHH = 8.2, 2H, H6), 7.07 (d, 3JHH = 8.3, 2H, H6′b), 6.33 (d, 3JHH = 8.3, 2H, H5′b), 6.07 (s, 2H, H8), 5.64 (br s, 4H, NH2), 1.00 (s, 18H, CMe3), 0.82 (s, 18H, CMe3′), −2.21 ppm (s, 1H, Au–H–Au). 13C{1H} NMR (75.47 MHz, CD2Cl2, 223 K, J values in Hz) δ 162.2 (s, C3), 160.3 (s, C3′), 159.5 (s, C9), 156.5 (s, C7), 154.8 (s, C7′), 147.5 (br d, 1JCF = 239.8, o–F H2N[B(C6F5)3]2–), 143.3 (s, C1), 138.9 (s, C4), 138.8 (br d, 1JCF = 250.0, p–F H2N[B(C6F5)3]2–), 136.4 (br d, 1JCF = 250.0, m–F H2N[B(C6F5)3]2–), 134.9 (s, C4′), 130.0 (s, C5′a), 129.4 (s, C8), 128.2 (s, C5), 127.9 (s, C6′a), 127.5 (s, C6′b), 127.2 (s, C6), 126.2 (s, C2′), 125.7 (s, C5′b), 119.5 (s, C2), 35.5 (s, CMe3), 34.7 (s, CMe3′), 30.5 (s, CMe3), 29.9 ppm (s, CMe3′). 19F NMR (282.4 MHz, CD2Cl2, 223 K, J values in Hz) −118.2 (dq, 3JFF = 25.7 4JFF = 6.0, 1F, o–F Au–C6F5), −119.7 (d ps t, 3JFF = 27.7 4JFF = 6.4, 1F, o–F Au–C6F5), −133.2 (br s, 12F, o–F H2N[B(C6F5)3]2–), −154.8 (t, 3JFF = 21.0, 1F, p–F Au–C6F5), −159.4 (t, 3JFF = 21.1, 1F, p–F H2N[B(C6F5)3]2–), −160.1 (m, 1F, m–F Au–C6F5), −164.9 ppm (brm, 13F, m–F Au–C6F5 + m–F H2N[B(C6F5)3]2–).
Acknowledgments
This work was supported by the European Research Council. M.B. is an ERC Advanced Investigator Award holder (Grant No. 338944-GOCAT). P.H. gratefully acknowledges support from the Berlin DFG excellence cluster on Unifying Concepts in Catalysis (UniCat) as well as funding from the Slovak VEGA grant agency (Grants No. 1/0507/17 and 1/0712/18) and from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 752285. We thank Prof. Cristina Nevado for helpful discussions. We are grateful to the EPSRC National Crystallographic Service, Southampton, UK, for collection of crystallographic data sets.41
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b04478.
The authors declare no competing financial interest.
Supplementary Material
References
- a Norton J. R.; Sowa J. R. Chem. Rev. 2016, 116, 8315–8317. 10.1021/acs.chemrev.6b00441. [DOI] [PubMed] [Google Scholar]; b Chem. Rev. 2016, 116, 8315–9000 (themed issue).27506870 [Google Scholar]
- Schmidbaur H.; Raubenheimer H. G.; Dobrzańska L. Chem. Soc. Rev. 2014, 43, 345–380. 10.1039/C3CS60251F. [DOI] [PubMed] [Google Scholar]
- a Wang X.; Andrews L. J. Am. Chem. Soc. 2001, 123, 12899–12900. 10.1021/ja012261u. [DOI] [PubMed] [Google Scholar]; b Wang X.; Andrews L. Angew. Chem., Int. Ed. 2003, 42, 5201–5206. 10.1002/anie.200351780. [DOI] [PubMed] [Google Scholar]; c Andrews L.; Wang X. J. Am. Chem. Soc. 2003, 125, 11751–11760. 10.1021/ja036307q. [DOI] [PubMed] [Google Scholar]; d Andrews L. Chem. Soc. Rev. 2004, 33, 123–132. 10.1039/b210547k. [DOI] [PubMed] [Google Scholar]
- a Tsui E. Y.; Müller P.; Sadighi J. P. Angew. Chem., Int. Ed. 2008, 47, 8937–8940. 10.1002/anie.200803842. [DOI] [PubMed] [Google Scholar]; Hydride-bridged binuclear gold(I) phosphine complexes have also been reported:; b Escalle A.; Mora G.; Gagosz F.; Mézailles N.; Le Goff X. F.; Jean Y.; Le Floch P. Inorg. Chem. 2009, 48, 8415–8422. 10.1021/ic901014r. [DOI] [PubMed] [Google Scholar]
- Roşca D.-A.; Smith D. A.; Hughes D. L.; Bochmann M. Angew. Chem., Int. Ed. 2012, 51, 10643–10646. 10.1002/anie.201206468. [DOI] [PubMed] [Google Scholar]
- Pintus A.; Rocchigiani L.; Fernandez-Cestau J.; Budzelaar P. H. M.; Bochmann M. Angew. Chem., Int. Ed. 2016, 55, 12321–12324. 10.1002/anie.201607522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinhans G.; Hansmann M. M.; Guisado-Barrios G.; Liles D. C.; Bertrand G.; Bezuidenhout D. I. J. Am. Chem. Soc. 2016, 138, 15873–15876. 10.1021/jacs.6b11359. [DOI] [PubMed] [Google Scholar]
- a Roşca D.–A.; Wright J. A.; Hughes D. L.; Bochmann M. Nat. Commun. 2013, 4, 2167. 10.1038/ncomms3167. [DOI] [PubMed] [Google Scholar]; b Roşca D.-A.; Fernandez-Cestau J.; Morris J.; Wright J. A.; Bochmann M. Science Adv. 2015, 1, e1500761. 10.1126/sciadv.1500761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews:; a Roşca D.-A.; Wright J. A.; Bochmann M. Dalton Trans. 2015, 44, 20785–20807. 10.1039/C5DT03930D. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kumar R.; Nevado C. Angew. Chem., Int. Ed. 2017, 56, 1994–2015. 10.1002/anie.201607225. [DOI] [PubMed] [Google Scholar]
- Pintus A.; Bochmann M. RSC Adv. 2018, 8, 2795–2803. 10.1039/C7RA13481A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Joost M.; Amgoune A.; Bourissou D. Angew. Chem., Int. Ed. 2015, 54, 15022–15045. 10.1002/anie.201506271. [DOI] [PubMed] [Google Scholar]; b Rekhroukh F.; Estevez L.; Mallet-Ladeira S.; Miqueu K.; Amgoune A.; Bourissou D. J. Am. Chem. Soc. 2016, 138, 11920–11929. 10.1021/jacs.6b07035. [DOI] [PubMed] [Google Scholar]
- a Comas-Vives A.; González-Arellano C.; Corma A.; Iglesias M.; Sánchez F.; Ujaque G. J. Am. Chem. Soc. 2006, 128, 4756–4765. 10.1021/ja057998o. [DOI] [PubMed] [Google Scholar]; b Casado R.; Contel M.; Laguna M.; Romero P.; Sanz S. J. Am. Chem. Soc. 2003, 125, 11925–11935. 10.1021/ja036049x. [DOI] [PubMed] [Google Scholar]; c Lv H.; Zhan J.-H.; Cai Y.-B.; Yu Y.; Wang B.; Zhang J.-L. J. Am. Chem. Soc. 2012, 134, 16216–16227. 10.1021/ja305204y. [DOI] [PubMed] [Google Scholar]
- a Klatt G.; Xu R.; Pernpointner M.; Molinari L.; Hung T. Q.; Rominger F.; Hashmi A. S. K.; Köppel H. Chem. - Eur. J. 2013, 19, 3954–3961. 10.1002/chem.201203043. [DOI] [PubMed] [Google Scholar]; b Ung G.; Bertrand G. Angew. Chem., Int. Ed. 2013, 52, 11388–11391. 10.1002/anie.201306550. [DOI] [PubMed] [Google Scholar]; c Reis M. C.; López C. S.; Kraka E.; Cremer D.; Faza O. N. Inorg. Chem. 2016, 55, 8636–8645. 10.1021/acs.inorgchem.6b01188. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Krieger J.-P.; Gómez-Bengoa E.; Fox T.; Linden A.; Nevado C. Angew. Chem., Int. Ed. 2017, 56, 12862–12865. 10.1002/anie.201705557. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Linden A.; Nevado C. Angew. Chem., Int. Ed. 2015, 54, 14287–14290. 10.1002/anie.201505533. [DOI] [PubMed] [Google Scholar]
- David B.; Monkowius U.; Rust J.; Lehmann C. W.; Hyzak L.; Mohr F. Dalton Trans. 2014, 43, 11059–11066. 10.1039/C4DT00778F. [DOI] [PubMed] [Google Scholar]
- Rocchigiani L.; Fernandez-Cestau J.; Budzelaar P. H. M.; Bochmann M. Chem. Commun. 2017, 53, 4358–4361. 10.1039/C7CC01628J. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Bronner C.; Wenger O. S. Dalton Trans. 2011, 40, 12409–12420. 10.1039/c1dt10636h. [DOI] [PubMed] [Google Scholar]; b Wong K. M.-C.; Chan M. M.Y.; Yam V. W.-W. Adv. Mater. 2014, 26, 5558–5568. 10.1002/adma.201306327. [DOI] [PubMed] [Google Scholar]
- Complex 4 has been independently identified as one of the products in the reaction of 1 with an excess of l-selectride: C. Nevado, personal communication.
- a Hrobárik P.; Hrobáriková V.; Meier F.; Repiský M.; Komorovský S.; Kaupp M. J. Phys. Chem. A 2011, 115, 5654–5659. 10.1021/jp202327z. [DOI] [PubMed] [Google Scholar]; b Hrobárik P.; Hrobáriková V.; Greif A.; Kaupp M. Angew. Chem., Int. Ed. 2012, 51, 10884–10888. 10.1002/anie.201204634. [DOI] [PubMed] [Google Scholar]; c Greif A. H.; Hrobárik P.; Hrobáriková V.; Arbuznikov A. V.; Autschbach J.; Kaupp M. Inorg. Chem. 2015, 54, 7199–7208. 10.1021/acs.inorgchem.5b00446. [DOI] [PubMed] [Google Scholar]
- Greif A. H.; Hrobárik P.; Kaupp M. Chem. - Eur. J. 2017, 23, 9790–9803. 10.1002/chem.201700844. [DOI] [PubMed] [Google Scholar]
- For SO-induced shielding/deshielding effects on 13C shifts in gold complexes, see also:; a Vícha J.; Foroutan-Nejad C.; Pawlak T.; Munzarová M.; Straka M.; Marek R. J. Chem. Theory Comput. 2015, 11, 1509–1517. 10.1021/ct501089z. [DOI] [PubMed] [Google Scholar]; b Novotný J.; Vícha J.; Bora P. L.; Repiský M.; Straka M.; Komorovský S.; Marek R. J. Chem. Theory Comput. 2017, 13, 3586–3601. 10.1021/acs.jctc.7b00444. [DOI] [PubMed] [Google Scholar]
- Roşca D.-A.; Fernandez-Cestau J.; Hughes D. L.; Bochmann M. Organometallics 2015, 34, 2098–2101. 10.1021/om501165z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijamudheen A.; Karmakar S.; Datta A. Chem. - Eur. J. 2014, 20, 14650–14658. 10.1002/chem.201403867. [DOI] [PubMed] [Google Scholar]
- Estes D. P.; Norton J. R.; Jockusch S.; Wesley Sattler W. J. Am. Chem. Soc. 2012, 134, 15512–15518. 10.1021/ja306120n. [DOI] [PubMed] [Google Scholar]
- a Matsuura K.; Muto H. J. Phys. Chem. 1993, 97, 8842–8844. 10.1021/j100137a003. [DOI] [Google Scholar]; b Huang M.-B.; Liu Y. J. Phys. Chem. A 2001, 105, 923–929. 10.1021/jp0033418. [DOI] [Google Scholar]
- Clark H. C.; Ferguson G.; Goel A. B.; Janzen E. G.; Ruegger H.; Siew P. Y.; Wong C. S. J. Am. Chem. Soc. 1986, 108, 6961–6972. 10.1021/ja00282a021. [DOI] [Google Scholar]
- Lancaster S. J.; Rodriguez A.; Lara-Sanchez A.; Hannant M. D.; Walker D. A.; Hughes D. L.; Bochmann M. Organometallics 2002, 21, 451–453. 10.1021/om010942s. [DOI] [Google Scholar]
- a Currie L.; Rocchigiani L.; Hughes D. L.; Bochmann M. Dalton Trans. 2018, 47, 6333–6343. 10.1039/C8DT00906F. [DOI] [PubMed] [Google Scholar]; b Rocchigiani L.; Fernandez-Cestau J.; Budzelaar P. H. M.; Bochmann M. Chem. - Eur. J. 2018, 10.1002/chem.201801277. [DOI] [PubMed] [Google Scholar]
- Rocchigiani L.; Fernandez-Cestau J.; Agonigi G.; Chambrier I.; Budzelaar P. H. M.; Bochmann M. Angew. Chem., Int. Ed. 2017, 56, 13861–13865. 10.1002/anie.201708640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Note that 1H hydride shifts computed at the two-component quasi-relativistic 2c-ZORA(SO)/PBE0-XC level, including the previously neglected terms from the exchange-correlation (XC) response kernel (see refs (20c) and (32)), agree very well with those obtained using the four-component fully relativistic (mDKS) method along with the same hybrid functional (cf. Table 1).
- a Autschbach J. Mol. Phys. 2013, 111, 2544–2554. 10.1080/00268976.2013.796415. [DOI] [Google Scholar]; b Greif A. H.; Hrobárik P.; Autschbach J.; Kaupp M. Phys. Chem. Chem. Phys. 2016, 18, 30462–30474. 10.1039/C6CP06129J. [DOI] [PubMed] [Google Scholar]
- In organic compounds 1H NMR shifts are often considered to shift downfield (upfield) upon decrease (increase) of the atomic charge at the hydrogen atom.
- This is in line with the very recent predictions and detailed analysis of 1H NMR shifts for hypothetical linear HAuIL hydrides, where the strong trans ligands were shown to destabilize the σ(Au–H)-type bonding MOs.21 These have a deshielding δSO contribution and compete with the shielding effect of Au(dπ)-based MOs. Once the σ(Au–H)-type bonding MO has moved substantially above Au(dπ)-based MOs, mixing of these two types of scalar relativistic MOs into spinors becomes less favorable (the more so, the larger the energy gap between them) and results in an overall δSO deshielding, and thus positive 1H hydride shifts.
- The remarkable deviations are found for complexes with π-acceptor (CO and PH3) ligands. This can be attributed to the more pronounced π-back-donation (Au → L) in the electron-richer Au(I) hydrides as compared to Au(III) congeners.
- Despite the tendency of increasing negative charge at the hydride ligand upon increasing the σ-donor strength of the trans ligand, no reasonable correlation between atomic charges at the hydride ligand and 1H NMR shifts for a wide set of ligands can be established.
- Useful correlations between ligand NMR shifts and M–L bond covalency, assessed by QTAIM delocalization indices, have been recently demonstrated also for actinide-chalcogen and organothorium complexes:; a Smiles D. E.; Wu G.; Hrobárik P.; Hayton T. W. J. Am. Chem. Soc. 2016, 138, 814–825. 10.1021/jacs.5b07767. [DOI] [PubMed] [Google Scholar]; b Smiles D. E.; Wu G.; Hrobárik P.; Hayton T. W. Organometallics 2017, 36, 4519–4524. 10.1021/acs.organomet.7b00202. [DOI] [Google Scholar]; In these multiple-bonded f0 systems, the ligand 77Se/125Te/13C shifts are, however, dominated by paramagnetic shielding contribution and the opposite trend was observed, i.e., the ligand NMR shifts became more deshielded along with increasing M–L bond covalency.
- Miller W. T. Jr; Sun K. K. J. Am. Chem. Soc. 1970, 92, 6985–6987. 10.1021/ja00726a054. [DOI] [Google Scholar]
- a Smith D. A.; Roşca D.-A.; Bochmann M. Organometallics 2012, 31, 5998–6000. 10.1021/om300666j. [DOI] [Google Scholar]; b Chambrier I.; Rosca D.-A.; Fernandez-Cestau J.; Hughes D. L.; Budzelaar P. H. M.; Bochmann M. Organometallics 2017, 36, 1358–1364. 10.1021/acs.organomet.7b00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca D.-A.; Smith D. A.; Bochmann M. Chem. Commun. 2012, 48, 7247–7249. 10.1039/c2cc33104g. [DOI] [PubMed] [Google Scholar]
- Coles S. J.; Gale P. A. Chem. Sci. 2012, 3, 683–689. 10.1039/C2SC00955B. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.