

Abstract
Background
Chitotriosidase (Chitnase 1, Chit1), a major true chitinase in humans, is induced in childhood asthma and has been implicated in the pathogenesis of a variety of inflammatory and tissue remodeling responses. We hypothesized that Chit1 plays a significant role in the pathogenesis of allergic asthma. To identify the role of Chit1, the mechanisms that underlie these contributions and the relevance of these murine findings to childhood asthma.
Methods
Wild type and Chit1-deficient mice and cells in culture were used to define the roles of Chit1 in models of allergic adaptive Th2 inflammation. In addition, the levels of sputum Chit1 were evaluated in pediatric asthma patients and compared to control.
Results
The levels of sputum Chit1 were significantly increased in the patients with childhood asthma. Mice with Chit1 null mutation demonstrated enhanced allergic Th2 inflammatory and cytokine and IgE responses to OVA or house dust mite allergen sensitization and challenge. However, the expression levels of TGF-β1 were significantly decreased with a diminished number of Foxp3+ regulatory T cells (Treg) in the lungs of Chit1−/− mice compared to WT controls. In vitro, the absence of Chit1 significantly reduced TGF-β-stimulated conversion of CD4+CD25- naïve T cells to CD4+Foxp3+ Treg cells, suggesting Chit1 is required for optimal effect of TGF-β1 in Treg cell differentiation.
Conclusion
Chit1 plays a protective role in the pathogenesis of allergic inflammation and asthmatic airway responses via regulation of TGF-β expression and Foxp3+ Treg cells.
Keywords: chitotriosidase, asthma, TGF-beta1, regulatory T cell, Th2 response
Graphical abstract
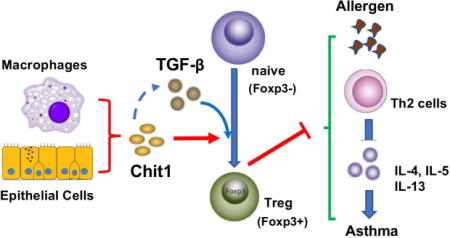
INTRODUCTION
Asthma is one of the most common chronic diseases in the world and is a major cause of morbidity in the USA (1, 2). In keeping with its public health impact, the immune and cellular events that are involved in asthma pathogenesis have been studied intently. These studies have demonstrated that exaggerated Type 2 immune responses play an important role in disease pathogenesis (3). However, the processes that control type 2 responses in normal airways and how these control mechanisms fail to function in asthma have not been adequately defined.
The 18 glycosyl hydrolase gene family contains true chitinases and chitinase-like proteins (CLP) that bind but do not cleave chitin. The former includes moieties such as acidic mammalian chitinase (AMCase) and chitotriosidase (chitinase 1; Chit1) which play important roles in the control of chitin containing pathogens (4). The latter includes the prototypic CLP, chitinase 3-like-1 (Chi3L1) (BRP-39 in mice and YKL-40 in man) which is an impressive regulator of cell survival, inflammation and remodeling (4). Recent studies have demonstrated that AMCase and Chit1 play important roles in the regulation and stimulation of Th2 responses (5, 6). These studies have demonstrated that the levels of Chit1 are increased in asthma and other diseases characterized by inflammation and remodeling and, in some cases, correlate with fungal sensitization (5, 7, 8). Moreover, studies from our laboratory and others have demonstrated that Chit1 is the major chitinase in the human lung and that genetic variations in the CHIT1 are associated with the frequency of atopy and atopic biomarkers, including blood eosinophils, serum IgE, and eosinophil cationic protein (ECP) (9, 10). However, the mechanism(s) by which Chit1 contributes to the pathogenesis of Th2 inflammation and the relationship between Chit1 and asthma have not been defined.
Immune responses in the lung and other organs are regulated by a variety of mechanisms including regulatory T cells (Tregs) (11). This can be readily appreciated in asthma and Th2 inflammation where Tregs deficiencies have been noted and Tregs inhibit type 2 inflammatory responses. In the mouse, CD4+CD25−Foxp3− cells can be induced to express Foxp3 and manifest the phenotypic and functional qualities of Tregs when activated in vitro by T-cell receptor stimulation in the presence of TGF-β (12). Recent studies from our laboratory have demonstrated that Chit1 augments TGF-β1 signaling (8). However, a relationship between Chit1 and Tregs has not been defined and the role of Chit1 in the generation of Tregs and their ability to control Th2 inflammation has not been defined.
We hypothesized that Chit1 is induced in childhood asthma and allergen-induced airway inflammation mice where it plays an important role in disease promotion by regulating Th2 inflammation. To test this hypothesis, we characterized the levels of Chit1 in sputum for children with asthma and controls. We also compared the adaptive Th2 responses induced after antigen sensitization and challenge in wild type and Chit1 null mice and the role of Tregs in the differences that were noted.
METHODS
Animals
All experiments used 7-13-wk-old, sex- and age-matched mice, housed under specific pathogen-free conditions. Chitotriosidase null mutant (Chit1−/−) and TGF-β1 transgenic (Tg) mice were generated on a C57BL/6 background and maintained as previously described (8, 13). All experiments were performed in compliance with Korea Research Institute of Bioscience and Biotechnology and approved by the institutional review boards of Yonsei University College of Medicine Council of Science and Technology.
Single cell isolation and flow cytometry
Lung, spleen, lymph node (LN) and thymus cells were isolated (14, 15). For intracellular staining, cells were fixed and permeabilized with Foxp3 Staining Kit (eBioscience, San Diego, CA) or Fix/Perm reagent kit (BD Biosciences). Samples were run on a FACS LSRII flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Human Subjects
A total of 140 children aged 6.6-12.2 years were enrolled in this study, divided into 2 groups: atopic asthma and controls. Subjects with asthma were classified on the basis of consistent respiratory symptoms verified by physicians, together with the presence of bronchial hyperresponsiveness in methacholine challenge (PC20 ≤16 mg/ml) or at least 12% reversibility of forced expiratory volume in 1s (FEV1) after inhalation of β2 agonist, in accordance with previous guidelines (16). Children treated with systemic corticosteroids due to asthma exacerbation in the preceding month were excluded from the study. The control group visited the hospital for general health workup or vaccination, and had no history of chronic diseases, recurrent wheezing, or respiratory infection for at least 4 weeks prior to evaluation.
Spirometry and subsequent sputum induction were performed at the initial visit. A methacholine challenge test and serum sampling for peripheral blood eosinophil count and IgE measurements were performed at the second visit. This study was approved by the Institutional Review Board of Severance Hospital (protocol no. 4-2004-0036). Written informed consent was obtained from the participants and their parents. Details on sputum induction, IgE measurement, spirometry, and methacholine challenge test are provided in the Method of Online Supplemental Information.
Statistical analyses
Normally distributed data were analyzed using the Student t-test for two groups or ANOVA followed by Tukey test for 3 or more groups. Chi-square test was used with categorical variables. Correlations between Chit1 levels and PC20 were evaluated using the Pearson test. All statistical analyses were performed using SPSS 20 (version 22.0, IBM Corp, Armonk, NY).
Details on the other methods employed in this study are included in the Methods of Online Supplemental data.
RESULTS
Chit1 accumulation in sputum from childhood asthma
Sputum samples were obtained from childhood asthma and compared with similar samples from age- and sex- matched controls. As shown in the table 1, the blood eosinophil counts, levels of total IgE and levels of serum ECP were increased in children with asthma compared to controls (p<0.001). Importantly, the levels of sputum Chit1 were significantly higher in the asthmatics compared to the controls (1262±69.97 pg/mL vs. 1758±129.2 pg/mL, P=0.001; fig. 1A).
Table 1.
Subject Characteristics
| Characteristics | Asthma (n=80) | Control (n=60) | P value |
|---|---|---|---|
| Age, y | 8.3 (6.6–10.9) | 8.9 (6.8–12.2) | n.s. |
| Sex, male (%) | 53 (66.2) | 35 (58.3) | n.s. |
| Blood eosinophil,/μL | 551.8 ± 407 | 165.8 ± 94.2 | < 0.001 |
| Total IgE, IU/mL | 421.5 (202–1636) | 47.9 (21.5–87.4) | < 0.001 |
| Serum ECP, μg/L | 14.4 (6.8–28.1) | 7.5 (4.9–17.3) | < 0.001 |
| FEV1, % predicted | 89.4 ± 18.3 | 105.2 ± 11.5 | < 0.001 |
| Change in FEV1 after BD, % | 10.2 (1.6–18) | 0.65 (0–5.8) | < 0.001 |
| FEV1/FVC, % | 87.0 ± 12.9 | 102.5 ± 8.0 | < 0.001 |
| FEF25-75%, % predicted | 62.1 ± 25.6 | 95.7 ± 20.8 | < 0.001 |
Values are presented as mean ± standard deviation, median (interquartile range) or number (%). ECP, eosinophil cationic protein; FEV1, forced expiration volume in one second; BD, bronchodilator; FVC, forced vital capacity; FEF25-75%, forced expiratory flow at 25–75% of vital capacity; n.s., not specified
Figure 1. Chit1 expression in childhood asthma and allergen-induced airway inflammation mice.
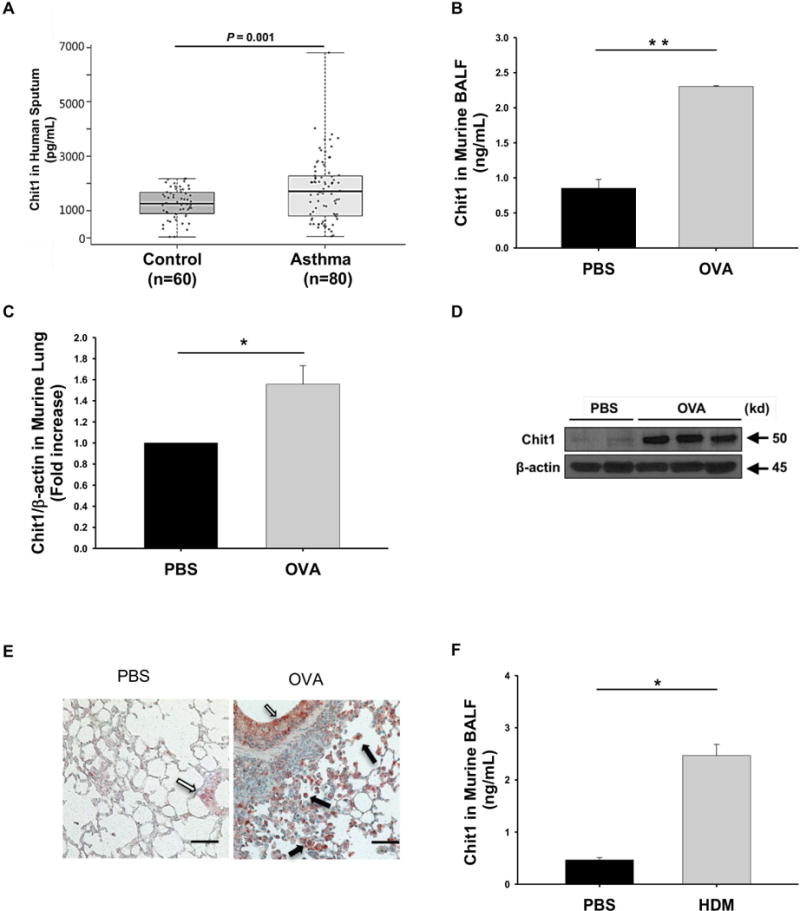
(A) Chit1 levels in supernatants of induced sputum from patients with normal control subjects (n = 60) and asthma (n = 80). (B-E) WT and Chit1−/− mice were sensitized with OVA/Alum and challenged with OVA. 24h After the last OVA challenge, the levels of Chit1 protein and mRNA expression were assessed via (B) ELISA and (C) real-time PCR and (D) Western blot. (E) Representative immunohistochemistry on lung sections with vehicle (PBS) or OVA challenge (solid arrows, alveolar macrophages; open arrow, airway epithelial cells). (F) WT and Chit1−/− mice were sensitized with house dust mite (HDM)/Alum and challenged with HDM. The level of Chit1 in bronchoalveolar lavage (BAL) fluids was assessed using ELISA. The values in panels B-D, F represent the triplicate evaluations in a minimum of 5 mice each group. Panels E is representative photograph of minimum 5 mice each group. Scale bars in E; 100 μm, respectively. *p<0.05, **p<0.01.by Pearson test and student’s t-test.
Regulation and accumulation of murine pulmonary Chit1 during aeroallergen challenge
To define the roles of Chit1, we evaluated the regulation and accumulation of pulmonary Chit1 in murine allergic inflammation. Models were utilized that employ the chitin-free aeroallergen, OVA, and HDM which contains chitin (17). The levels of Chit1 protein and mRNA were significantly increased in the lungs from OVA sensitized and challenged mice compared with controls (fig. 1B–1D). IHC evaluation demonstrated that airway epithelial and inflammatory cells, especially alveolar macrophages, are the major cells expressing Chit1 in the lungs of aeroallergen challenged mice (fig. 1E). These effects were not specific for chitin-free aeroallergens because similar increases in the levels and sites of Chit1 expression were noted in the lungs from the mice sensitized and challenged with HDM (fig. 1F).
Role(s) of Chit1 in allergen-induced airway inflammation and airway responsiveness
In these experiments, WT and Chit1−/− mice were sensitized and challenged with OVA and the resulting inflammatory responses were evaluated. In the absence of OVA exposure, BAL cell accumulation, IgE production and parameters of airway responsiveness were similar in WT and Chit1−/− mice. Interestingly, after OVA sensitization and challenge, Chit1−/− mice manifest significantly increased BAL total cells and eosinophil recovery (fig. 2A), increased total and OVA-specific IgE accumulation (fig. 2B), and heightened airway responsiveness to methacholine (fig. 2C) compared to WT mice. Histologic evaluations demonstrated enhanced perivascular and peribronchial inflammatory cell infiltration in comparisons of Chit1−/− and WT mice (fig. 2D). The noted effects of Chit1 in airway inflammation and responsiveness were not specific for chitin-free aeroallergens because similar changes were noted in comparisons of lungs from Chit1−/− and WT mice challenged with HDM (fig. S1A and S1D). However, heightened IgE response in the WT mice with HDM challenges compared to OVA challenge mice were noted (fig. S1B and fig 2B), and that blunted statistical difference in total IgE levels between Chit1−/− and WT mice challenged with HDM.
Figure 2. Role of Chit1 in allergen-induced airway inflammation and airway hyperresponsiveness.
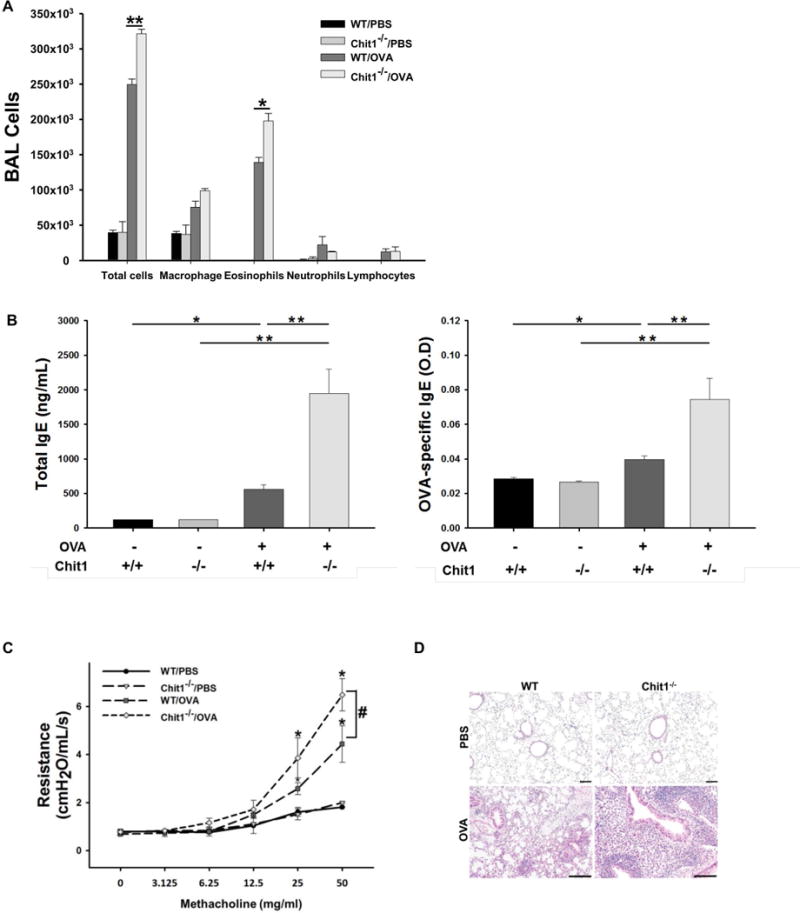
WT and Chit1−/− mice were sensitized with OVA/Alum and challenged with OVA. (A) Total and differential cell counts in BAL fluid. (B) Serum levels of total and OVA-specific IgE measured by ELISA. (C) Airway resistance in response to inhaled methacholine in WT and Chit1−/− with and without OVA challenge was measured by Flexivent system. (D) Representative lung sections with H&E staining. Panel A is representative of two separate analysis. The values in panels A-C represent the triplicate evaluations in a minimum of 5 mice each group. Panel D is representative photograph of minimum 5 mice each group. *p<0.05, **p<0.01, #p<0.05 (WT/OVA vs. Chit1−/−/OVA). Scale bars in D, 50 and 100 μm, respectively.
Role of Chit1 in aeroallergen regulation of Th2 cytokines, TGF-β1 and IL-10
To understand the mechanism(s) by which Chit1 regulates allergen-induced inflammation, we next characterized the expression and accumulation of cytokines in BAL fluids and lungs from WT and Chit1−/− mice. As previously reported (18), OVA sensitization and challenge stimulated, the expression of IL-4, IL-5, and IL-13 in lungs from WT mice. Importantly, these inductive events were significantly augmented in the lungs of Chit1−/− mice when compared to WT mice (fig. 3A, 3B). OVA sensitization and challenge also stimulated the expression of TGF-β1 and IL-10 in lungs from WT mice. In contrast to the Th2 cytokines noted above, these inductive events were markedly blunted in lungs from Chit1−/− mice compared to WT mice (fig. 3C, 3D). Enhanced Th2 cytokine and decreased anti-inflammatory cytokine responses were also noted in comparisons of WT and Chit1−/− mice challenged with HDM (fig. S1C). In combination, these results demonstrate that Chit1 inhibits allergen-stimulated Th2 cytokine production while enhancing the expression of the anti-inflammatory cytokines TGF-β1 and IL-10.
Figure 3. Role of Chit1 in allergen-induced expression of Th2 cytokines, TGF-β and IL-10 in the lung.
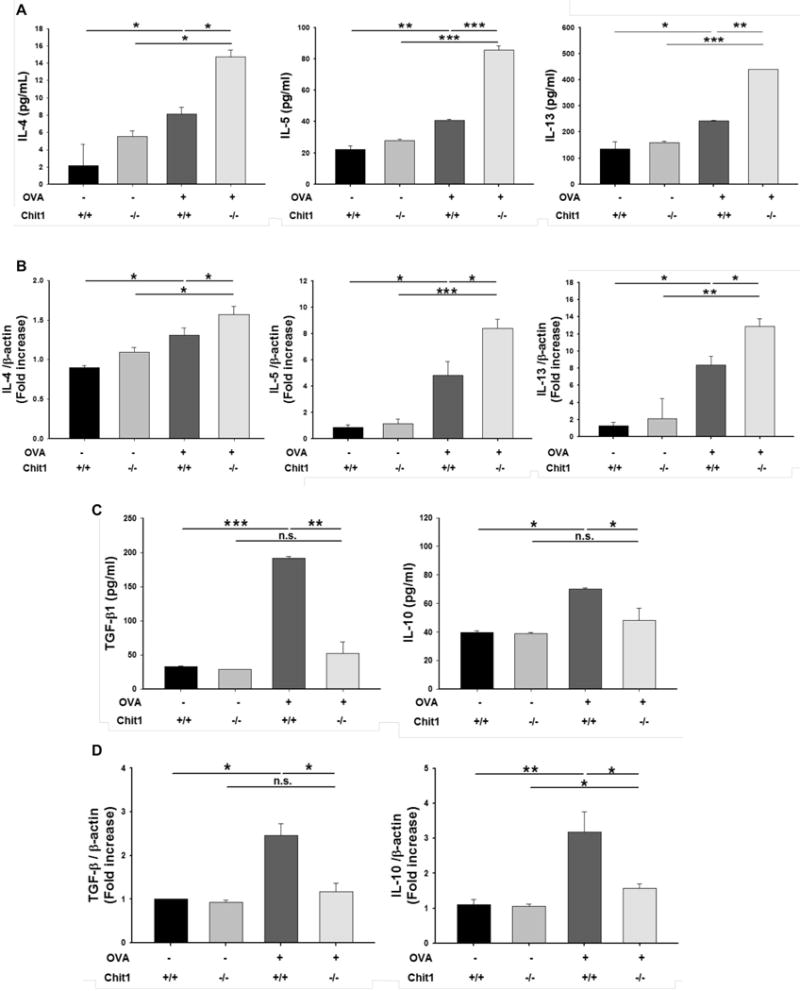
Levels of IL-4, IL-5, IL-13, TGF-β and IL-10 in the lungs from WT and Chit1−/− mice after PBS or OVA challenge were evaluated. (A and C) The protein levels of IL-4, IL-5, IL-13, TGF-β and IL-10 in BAL fluid measured by ELISA. (B and D) The mRNA expression levels of IL-4, IL-5, IL-13, TGF-β and IL-10 measured by real-time RT-PCR. The values represent the mean±SEM of duplicate evaluations in a minimum of 5 mice in each group. *p<0.05, **p<0.01, ***p<0.001, n.s.=not significant.
Role of Chit1 in CD4+ T cell population in the lung of allergen challenged mice
Studies were next undertaken to assess whether Chit1 plays a role in T cell population. After OVA challenge, the number of CD4+GATA3+Foxp3- Th2 cells and CD4+GATA3-Foxp3+ Tregs were evaluated. In keeping with the enhanced inflammation noted in Chit1−/− mice, these studies revealed a significant increase in the number of CD4+GATA3+Foxp3- cells in the lungs from Chit1−/− mice compared to WT mice (fig. 4A–4C). On the other hand, CD4+GATA3-Foxp3+ cells accumulation in the lung of Chit1−/− mice was significantly lower than in WT mice (fig. 4A–4C). These Chit1-induced effects on Th2 and Tregs were lung-specific, because similar differences were not noted in mediastinal lymph nodes (mLN) or spleens from WT and Chit1−/− animals (fig. 4A–4C). These results indicate that Chit1 inhibits allergen-induced Th2 cell differentiation while enhancing Foxp3+ Tregs development in the lung.
Figure 4. Role of Chit1 in CD4+ T cell population in murine model of allergic asthma.
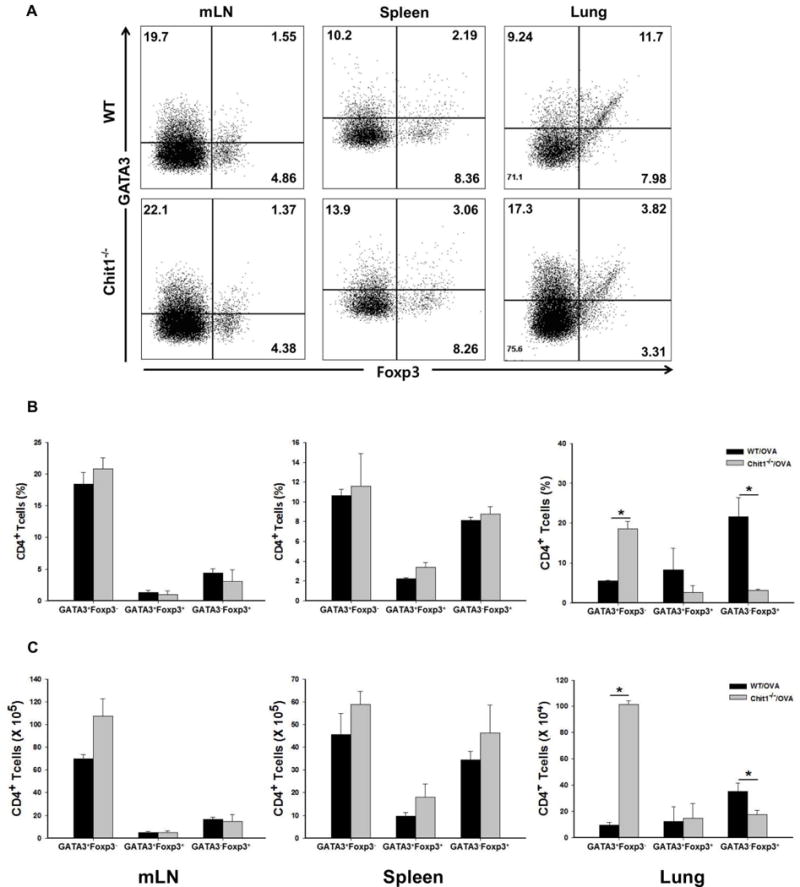
WT and Chit1−/− mice were sensitized with OVA/Alum and challenged with OVA. (A) 24 h after the last OVA challenge, cells were isolated from mediastinal lymph node (mLN), spleen and lung from WT and Chit1−/− mice, stained with anti-CD4, Foxp3, and GATA3, and analyzed by flow cytometry. (B) Bar graph represents percentage of CD4+GATA3+Foxp3-, CD4+GATA3+Foxp3+ and CD4+GATA3-Foxp3+ cells in the mLN, spleen and lungs from WT and Chit1−/− mice. (C) Graphs represent absolute numbers of CD4+GATA3+Foxp3-, CD4+GATA3+Foxp3+ and CD4+GATA3-Foxp3+ cells in the mLN, spleen and lungs from WT and Chit1−/− mice. Panel A is a representative FACS evaluations in a minimum of 5 mice each group. The values in panels B and C represent the mean±SEM of triplicate evaluations in a minimum of 5 mice each group. *p<0.05.
Role of Chit1 effect on the regulatory T cells in the absence of allergen challenge
To further understand the mechanisms that Chit1 uses to regulate Tregs, studies were undertaken to determine if Chit1 alters Foxp3+ Tregs development, proliferation or the ability of Tregs to suppress T cell proliferation. First, we compared the accumulation of CD4+Foxp3+ T cells in thymus, spleen, and mLN tissues from WT and Chit1−/− mice at baseline. Significant difference in CD4+Foxp3+ T cells accumulation in tissues from WT and Chit1−/− mice were not noted regardless of the source of the cells (fig. 5A, 5B). This suggests that Chit1 does not play a major role in the regulation of Foxp3+ Treg cell development in vivo under steady-state conditions. To determine if Chit1 regulates Treg cell proliferation, peripheral CD4+CD25+ T cells were purified from WT and Chit1−/− mice, and their ability to proliferate following in vitro TCR stimulation was evaluated. In this assay, both WT and Chit1−/−CD4+CD25+ T cells manifest strong proliferative responses and the frequency of dividing Chit1−/−CD4+CD25+ T cells was comparable in cells from WT and Chit1−/− mice (fig. 5C). And the suppressive ability of Tregs was assessed in vitro by evaluating the proliferation of naïve T cells (CD4+CD25-) co-cultured with WT or Chit1−/−CD4+CD25+ T cells. We found that Chit1−/− Tregs were as effective at suppressing proliferation of naïve T cells as WT Tregs (fig. 5D). Lastly, to examined the direct effects of Chit1 on Tregs proliferation, freshly isolated cells were stimulated in CD3/CD28 with or without Chit1. We found that Chit1 have no direct effect on the proliferation of Treg cells (fig. 5E). Therefore, Chit1-deficient Tregs have proliferative and suppressive abilities comparable to WT Tregs, highlighting the limited effect of Chit1 in Tregs development and function in the absence of allergen stimulation.
Figure 5. Role of Chit1 in the development, proliferation and functionality of regulatory T cells.
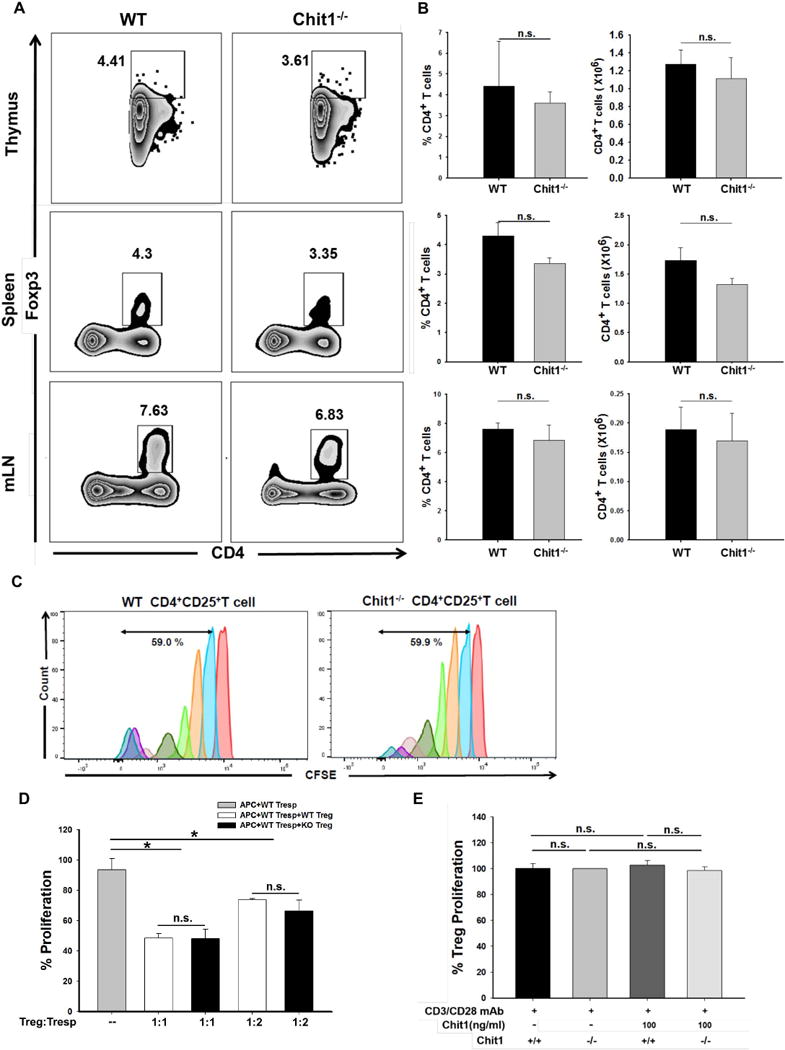
(A) Cells were isolated from thymus, spleen and mediastinal lymph node (mLN) from WT and Chit1−/− mice, stained with CD4 and Foxp3, and analyzed by flow cytometry (B) Bar graphs representing percentage and absolute numbers of CD4+Foxp3+ cells in the lungs from age- and sex-matched WT and Chit1−/− mice. (C) Regulatory T cell proliferation assay using carboxylfluorescein succinimidyl ester (CFSE) staining and FACS analysis. (D) Suppression of T cell-proliferative response by CD4+CD25+ T cells sorted from WT and Chit1−/− mice following co-culture with WT CD4+CD25- (Tresp) cells. (E) The proliferation of CD4+CD25+ Treg cell was measured by CCK-8. Isolated cells were stimulated with plate bound-anti CD3/CD28 in the presence of IL-2 with or without Chit1 following culture for 3 days. Panel A is a representative FACS evaluations in a minimum of 5 mice each group. The values in panel B represent the mean±SEM of FACS evaluations in a minimum of 5 mice each group. Panel C is a representative of CFSE evaluations in a minimum of 3 separate experiments. Panel D and E represent the mean±SEM of triplicate evaluations in a minimum of 2 separate experiments. *p<0.05, n.s.=not significant.
Effect of TGF-β1 expression on Chit1 inhibition of Th2 inflammation
Since TGF-β1 expression and Treg differentiation were decreased in the lung under allergen sensitization and challenge in the absence of Chit1, studies were undertaken to see whether TGF-β1 is responsible for Chit1 regulation of allergic inflammation in the lung. In this evaluation, TGF-β1 Tg mice were used to express TGF-β1 specifically in the lung. After WT and Chit1−/−, TGF-β1 Tg (+), and double mutant mice of Chit1 and TGF- β1 Tg (TGF-β1 Tg(+)/Chit1−/−) were exposed to PBS (-) or OVA (+), then BAL and lung inflammation and Th2 cytokine expression were evaluated. As shown in figure 6A–6D, OVA challenge significantly increased of eosinophil-dominant BAL and tissue inflammation, serum levels of total IgE and BAL IL-4, IL-5, and IL-13 in WT animals. In the absence of Chit1, all these parameters of Th2 inflammation were significantly increased compared to WT animals. However, transgenic expression of TGF-β1 abrogated these increases seen in Chit1−/− mice to the levels of WT mice, suggesting that TGF-β1 is mediating Chit1 inhibition of Th2 inflammatory responses in the lung.
Figure 6. Effect of TGF-β1 expression on Th2 inflammation and Foxp3+Treg conversion.
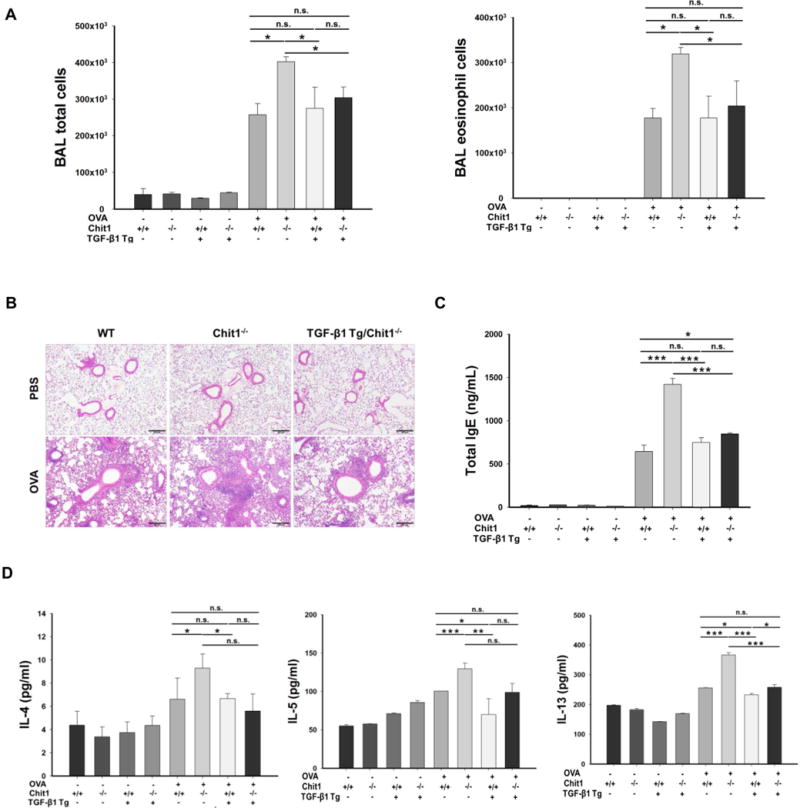
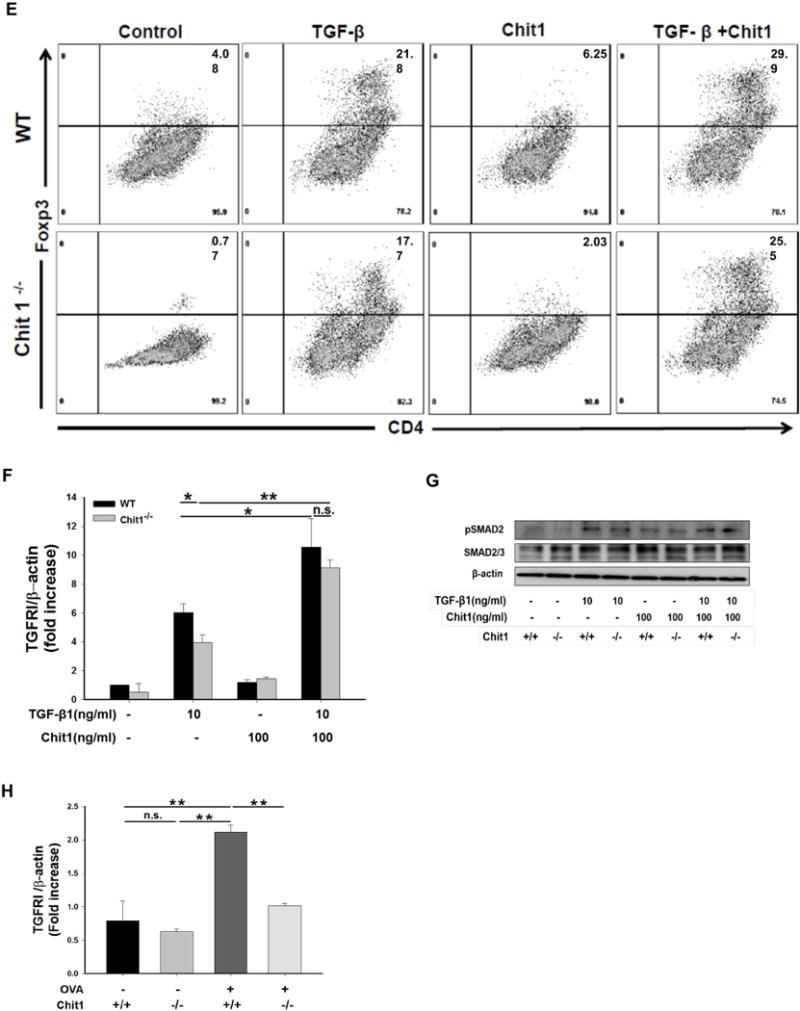
(A–D) WT, Chit1−/−, TGF-β1 Tg(+), and double mutant mice of Chit1 and TGF-β1 Tg (TGF-β1 Tg(+)/Chit1−/−) mice were sensitized with OVA/Alum and challenged with OVA then evaluated infalmmatoy and cytokine responses in the lung. (A) Total cell and eosinophil recovery in the BAL at 24 h after the last OVA challenge, (B) H&E stains for the evaluation of lung tissue inflammation. (C-D) total serum IgE levels, and BAL Th2 cyokine levels. (E-F) Naïve CD4+CD25- cells were isolated from spleen from WT and Chit1−/− mice. Cells were then stimulated with plate bound-anti CD3/CD28 in the presence of IL-2 and TGF-β1 with or without Chit1 following culture for 3 days. (E) A representative flow cytometry analysis on the cell that were harvested and stained with anti-CD4/Foxp3. (F) TGF-β receptor I mRNA expression determined by real time PCR. (G) Smad2 phosphorylation was assessed by Western blot in sorted naïve T cells stimulated by reombiniant TGF-β1 and Chit1, alone and in combination. (H) CD4+CD25+ cells were isolated from lung from WT and Chit1−/− mice. TGF-β receptor I mRNA expression determined by real time PCR. The values in panels A, C, D, F, H represent the mean±SEM and panel E is a representative FACS evaluations in a minimum of 5 mice each group. Panel B is a representative tissue section with H&E stains. G is representative of a minimum of three separate experiments. *p<0.05, **p<0.01, ***p<0.001, n.s.=not significant. Scale bar in panel B; 100 μm.
Role of Chit1 in TGF-β1-stimulated Foxp3+ Treg conversion
TGF-β1 is an important regulator of T cell tolerance, in part through the conversion of CD4+CD25- T cells to inducible Treg cells (iTregs) in the periphery. Therefore, studies were undertaken to investigate the role(s) of Chit1 in the conversion of naïve T cells to iTregs. We stimulated naïve T cells (CD4+CD25-) in the presence of TGF-β1 and/or Chit1. The addition of TGF-β1 alone led to an increase in Foxp3 conversion. However, Chit1-deficient T cells showed less effective Foxp3 conversion compared with cells from WT mice (fig. 6E). In these evaluations, the TGF-β1-induced Foxp3 induction was significantly enhanced with the co-treatment with recombinant Chit1 (fig. 6E). These data demonstrate that Chit1 effect are notable only in the presence of TGF-β1 for further development of peripheral CD4+Foxp3+ iTreg cells from naïve T cells.
Role of Chit1 in the regulation of TGF-β-stimulated TGF-β receptor expression and signaling in T cells
Previous studies from our laboratory demonstrated that Chit1 interacts in a synergistic fashion with TGF-β1 to enhance TGF-β receptor expression and Smad phosphorlytion and the development of pulmonary fibrosis (8). Thus, to further understand the mechanism by which Chit1 participates in the genesis of Tregs, studies were undertaken to determine if Chit1 regulated TGF-β receptor I (TGFRI) expression and signaling during Treg development. These studies demonstrated that Chit1 significantly enhanced the ability of TGF-β1 to stimulate the expression of TGFRI in TGF-β1 and Chit1 stimulated T cells (fig. 6F). As TGF-β1 and/or Chit1 treatment in T cells showed increased TGFRI expression and therefore an increased sensitivity to TGF-β1 signaling, we next evaluated the activation of Smad2 in sorted naïve T cells treated with recombinant TGF-β1 and recombinant Chit1, alone and in combination. As expected, treatment of exogenous of TGF-β1 increased p-Smad2 in both WT and Chit1−/− T cells compared with stimulation with TCR alone, but the level of p-Smad2 in WT T cells was higher than that in Chit1−/− (fig. 6G). In these evaluations, the TGF-β1-induced TGFRI expression and signaling was significantly enhanced with the co-treatment with recombinant Chit1(fig. 6G). In addition, CD4+CD25+ T cells with OVA challenge, Chit1−/− mice showed significantly lower expression of TGFRI expression compared to WT CD4+CD25+ T cells (fig. 6H). When viewed in combination, these findings demosntrate that Chit1 enhances TGF-β-dependent Treg conversion, at least in part, via its ability to increase T cell TGFRI expression and signaling.
DISCUSSION
Although mammals do not have chitin or chitin synthase, they contain chitinases and CLP that can be detected in the circulation as well as in local tissues (4, 18, 19). Chit1, a major enzymatically active true chitinase, is produced, stored, and secreted by macrophages and neutrophils and plays important roles in innate immune homeostasis (20). This can be appreciated in the pivotal roles it plays in host defenses against chitin-containing pathogens such as fungi, protozoa and insects (20). Its importance as a sensitive biomarker of macrophage activation, can also be seen in its dysregulated expression in a variety of human diseases including Gaucher’s disease, diabetes, sarcoidosis, inflammatory bowel disease, atherosclerosis, Alzheimer’s disease and prostate cancer (21, 22). Recent studies from our laboratory and others have also demonstrated that Chit1 is dysregulated in lung diseases characterized by inflammation and remodeling such as asthma, COPD and pulmonary fibrosis (8, 19, 23). However, the roles of Chit1 in these diseases and the mechanisms it uses to contribute to disease pathogenesis have not been defined. To begin to address these deficiencies, we focused on the regulation of Chit1 and its roles in the pathogenesis of allergic asthma. This study demonstrates that Chit1 is expressed in an exaggerated manner in pediatric asthma. Increased expression and activity of Chit1 in patients with asthma was already reported by other studies (23, 24). Consistent with sputum data from children with asthma, the lung of allergen-induced mice showed a significant increase in the level of Chit1 compared with those of control mice. Chit1 is the most readily active chitinase in the lung from mammal and man (25) and increased in the patients with inflammatory disorder disease also in the animal models (26, 27). The reasons for the elevation of Chit1 might be associated with the protective role in the immune response against allergens. And our murine models of asthma-like adaptive Th2 inflammation also demonstrated that Chit1 inhibits aeroallergen induced Th2 inflammation, IgE production, type 2 cytokine elaboration and airways hyperresponsiveness while stimulating the anti-inflammatory cytokines TGF-β1 and IL-10. We also defined the novel mechanisms that underlie these responses by demonstrating that Chit1 plays a critical role in TGF-β1 stimulation of Tregs accumulation and that this response is mediated, at least in part, by the ability of Chit1 to augment the ability of TGF-β1 to stimulate T cell TGF-β1 receptor expression. When viewed in combination these studies suggest that Chit1 plays an important protective role in aeroallergen-induced Th2 pulmonary inflammation.
CD4+Foxp3+ Tregs can be generated in the thymus; natural Tregs (nTregs). Conventional CD4+ T cells can also be converted to Tregs in peripherial tissues via TGF-β-dependent mechanisms; induced Tregs (iTregs) (28). Importantly, iTreg cells potently inhibit TCR-driven T cell proliferation in vitro and when adaptively transferred in vivo (29). In many reports, TGF-β, in the presence of TCR stimulation, could induce Foxp3 expression in naïve peripheral CD4+CD25-Foxp3- T cells and converted them into Foxp3+ Treg cells, not only in murine (29–31), but also in human CD4+ T cells (29, 32). TGF-β1 is a pleiotropic cytokine that regulates the many cellular functions, such as proliferation, migration, differentiation and survival (33). TGF-β1 is not only known for its anti-inflammatory, but also for its proinflammatory effects in the inflammation, as demonstrated by its fibrotic effects on airway remodeling. Although, airway TGF-β1 expression have not been well quantified in children with asthma (34), increased TGF-β1 expression has been shown in the airways of asthmatic adults after allergen challenge (35, 36). On the other hand, TGF-β1 acts as a negative feedback mechanism to suppress of immune response, inducing T cell tolerance. Neutralization of TGF-β reduced the Treg cell induction in transplantation model (37) and systemic increases in TGF-β induced Foxp3+ Treg cell numbers in mice (38). Therefore, the conversion to Foxp3+ Treg cells were shown to be dependent on TGF-β in vivo. Moreover, we recently demonstrated that Chit1 interacts with TGF-β1 in the pathogenesis of tissue fibrosis by augmenting TGF-β1-induced expression of TGF-β receptors 1 and 2, Smad2 phosphorylation and the activation of MAPK ERK1/2 in fibroblasts (8). The present studies add to our understanding of the interactions of Chit1 and TGF-β1 by demonstrating that Chit1 is required for optimal TGF-β1 stimulation of Foxp3+ T cells in allergic inflammation and that TGF-β1-induced expression of TGF-β receptors on T cells was significantly impaired in the absence of Chit1. This raises the interesting possibility that the failure to induce Foxp3+ Tregs in Chit1−/− mice is due, at least in part, to this loss of TGF-β receptor expression and downstream signaling of TGF-β. The specific contribution of TGF-β in this Chit1-regulated response was further confirmed by the experiment using Chit1−/− and TGF-β1 Tg(+) mice that demonstrated transgenic expression of TGF-β1 abrogated exaggerated IgE and Th2 cytokine responses in the the absence of Chit1. In combination, the current studies highlight novel Chit1-TGF-β interactions that control Tregs accumulation, airway inflammation and AHR in aeroallergen-induced Th2 pulmonary responses.
Previous studies demonstrated that GATA3, a key regulator of Th2 polarization, inhibits the induction of T cell Foxp3 by binding to the Foxp3 promoter (39). In keeping with these findings, IL-4 has been shown to inhibit TGF-β-induced Foxp3 induction and prevent T cell conversion to a regulatory phenotype (40). Similar effects have been described for IL-6 which induces the differentiation into Th17 cells while inhibiting the generation of iTreg cells (40). Our studies demonstrate that aeroallergen sensitized and challenged Chit1 null mice manifest augmented Th2 responses and decreased numbers of Tregs. This led to the intriguing possibility that the Chit1 that is induced at sites of Th2 inflammation counteracts the effects of GATA3, Th2 cytokines, and/or IL-6 on Foxp3+ Tregs differentiation and that these effects are mediated by its ability to enhance T cell TGF-β receptor expression. Additional investigation will be required to assess the veracity of these speculations and the mechanisms that are involved.
Our studies demonstrate that the levels of TGF-β1 were significantly lower in lungs from allergen sensitized and challenged Chit1−/− mice than in lungs from WT controls. Interestingly, the lungs from the Chit1−/− mice also manifest impressive increases in inflammation including macrophages and eosinophils. These findings are in accord with the appreciation that macrophages are major sources of TGF-β1 and Chit1 (19, 41) and that allergen-stimulated alveolar macrophages act as antigen presenting cells (APCs) that promote the generation of Foxp3+ iTreg cells, that suppress the induction of lung inflammation, mucus production, and airway hyperreactivity (41, 42). They are also in accord with studies from our laboratory that demonstrated that the CLP chitinase 3-like-1 also plays a critical role in the stimulation of TGF-β1 elaboration. When viewed in combination, these studies demonstrate that Chit1 plays a critical role in the production of TGF-β1 by macrophages and other inflammatory cells at sites of inflammation and that this property may be a critical feature of 18 GH regulatory moieties.
The immune regulatory function of Chit1 acquired immune responses is still largely elusive compared to its function to innate immune response (for recent review, see (21)). In the prior study using lung-specific IL-13 transgenic mice, we have shown that IL-13 strongly induced the expression of Chit1 together with TGF-β (43), suggested that Th2 cytokine could be a major driver of Chit1 as well as TGF-β expression in asthmatic airways. In that setting, we have identified a significant role of Chit1 in fibrotic tissue repair response interacting with TGF-β. Up to date only a few studies are directed to address a specific role of Chit1 in adaptive allergic responses. Recent studies of Cryptococcus neoformans pulmonary infection demonstrated enhanced inflammation in lungs from Chit1 null versus WT mice (5). On superficial analysis this is the opposite of the results that were seen in aeroallergen sensitized and challenged Chit1−/− animals. Interestingly, the Th2 inflammation in the fungal model was dependent on the enzymatic activity of Chit1 and its ability to digest the chitin in the Cryptococcus to release free chitin which can be a potent macrophage activator (5). In contrast, in the current studies, enhanced inflammation was seen in Chit1−/− mice regardless of whether they were sensitized and challenged with ovalbumin which does not contain chitin or HDM which does. These observations suggest that these responses and the mechanisms that induce them are different. Support for this can be seen in the fact that the aeroallergen induced responses are manifestations of adaptive Th2 immunity while chitin induction of inflammation is a macrophage-induced innate immune response and that reflect a significant role of chitin as an adjuvant (17, 44–46). These studies also suggest that Chit1 and possibly Tregs play different roles in these responses. It is important to point out that studies in our laboratory and others have demonstrated that the immunological activities of chitin depend on its chemical modification, dose, route of exposure and size with proinflammatory and anti-inflammatory responses being induced with intermediate sized chitin (40–70μm) and small chitin (<40μm), respectively (47–50).
In summary, we have shown augmented expression of Chit1 in sputum from patients with asthma compared to healthy subjects. In murine models of asthma-like adaptive Th2 inflammation, these studies demonstrated that Chit1 inhibits aeroallergen induced Th2 inflammation, IgE production, type 2 cytokine elaboration and AHR while stimulating the anti-inflammatory cytokines TGF-β1 and IL-10. They also defined the novel mechanisms that underlie these responses by demonstrating that Chit1 plays a critical role in TGF-β1 induction and TGF-β1 stimulation of Tregs accumulation and that this response is mediated, at least in part, by the ability of Chit1 to augment the ability of TGF-β1 to stimulate T cell TGF-β1 receptor expression. When viewed in combination, these studies highlight a novel protective role of Chit1 in allergic inflammation and airway responses via regulation of TGF-β expression and its effector function on Tregs.
Supplementary Material
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2009-0075056 and 2016 R1D1A1B03931794) and funded by the Ministry of Science, ICT & Future Planning (NRF-2017R1A2B2004043), and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI17C0104). This work was also in part supported by grant U01 HL108638, PO1 HL114501, and R01 HL115813 from National Institute of Health (NIH), USA.
Abbreviations
- Chit1
chitotriosidase
- OVA
ovalbumin
- HDM
house dust mite
- Alum
aluminium hydroxide
- Foxp3
forkhead box protein 3
- GATA3
GATA binding protein 3
- Treg
regulatory T cell
- TGF-β
transforming factor-β
- BALF
bronchoalveolar lavage fluidAHR : airway hyperresponsiveness
Footnotes
DR. MYUNG HYUN SOHN (Orcid ID : 0000-0002-2478-487X)
PROF. CHUN GEUN LEE (Orcid ID : 0000-0002-9514-3658)
Author Contributions:
Conception and design: JYH, KEM, JAE, MHS, CGL; Data collection: JYH, ISS, KEL, MK, KWK, CML, MHS; Analysis and interpretation: JYH, KEL, MK, KWK, JAE, MHS, CGL; Drafting the manuscript for important intellectual content: JYH, KEM, JAE, MHS, CGL
Conflicts of Interest:
Authors declare that they have no conflicts of interest.
References
- 1.Trends in Asthma Prevalence, Health Care, and Mortality in the United States, 2001–2010. CDC. 2012 [PubMed] [Google Scholar]
- 2.Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach. World Health Organization; 2007. [Google Scholar]
- 3.Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol. 2015;15(1):57–65. doi: 10.1038/nri3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol. 2011;73:479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiesner DL, Specht CA, Lee CK, Smith KD, Mukaremera L, Elias JA, et al. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. PLoS Pathog. 2015;11(3):e1004701. doi: 10.1371/journal.ppat.1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Z, Zheng T, Homer RJ, Kim Y, Chen NY, Cohn L, et al. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304(5677):1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 7.Vicencio AG, Chupp GL, Tsirilakis K, He X, Kessel A, Nandalike K, et al. CHIT1 mutations: genetic risk factor for severe asthma with fungal sensitization? Pediatrics. 2010;126(4):e982–985. doi: 10.1542/peds.2010-0321. [DOI] [PubMed] [Google Scholar]
- 8.Lee CG, Herzog EL, Ahangari F, Zhou Y, Gulati M, Peng X, et al. Chitinase 1 is a biomarker for and therapeutic target in scleroderma-associated interstitial lung disease that augments TGF-β1 signaling. J immunol. 2012;189(5):2635–2644. doi: 10.4049/jimmunol.1201115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim KWPJ, Lee JH, et al. Association of genetic variation in chitotriosidase with atopy in Korean children. Annals of Allergy, Asthma & Immunology. 2013;110(6):444–449. doi: 10.1016/j.anai.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Seibold MA, Donnelly S, Solon M, Innes A, Woodruff PG, Boot RG, et al. Chitotriosidase is the primary active chitinase in the human lung and is modulated by genotype and smoking habit. Journal of Allergy and Clinical Immunology. 2008;122(5):944–950.e943. doi: 10.1016/j.jaci.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Selvaraj RK, Geiger TL. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J Immunol. 2007;178(12):7667–7677. doi: 10.4049/jimmunol.178.12.7667. [DOI] [PubMed] [Google Scholar]
- 13.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. The Journal of experimental medicine. 2004;200(3):377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knosp CA, Schiering C, Spence S, Carroll HP, Nel HJ, Osbourn M, et al. Regulation of Foxp3+ inducible regulatory T cell stability by SOCS2. J Immunol. 2013;190(7):3235–3245. doi: 10.4049/jimmunol.1201396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172(9):5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 16.Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. Journal of allergy and clinical immunology. 2007;120(5 Suppl):S94–138. doi: 10.1016/j.jaci.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 17.Kim LK, Morita R, Kobayashi Y, Eisenbarth SC, Lee CG, Elias J, et al. AMCase is a crucial regulator of type 2 immune responses to inhaled house dust mites. Proc Natl Acad Sci U S A. 2015;112(22):E2891–2899. doi: 10.1073/pnas.1507393112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med. 2009;206(5):1149–1166. doi: 10.1084/jem.20081271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho SJ, Weiden MD, Lee CG. Chitotriosidase in the Pathogenesis of Inflammation, Interstitial Lung Diseases and COPD. Allergy Asthma Immunol Res. 2015;7(1):14–21. doi: 10.4168/aair.2015.7.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Eijk M, van Roomen CP, Renkema GH, Bussink AP, Andrews L, Blommaart EF, et al. Characterization of human phagocyte-derived chitotriosidase, a component of innate immunity. Int Immunol. 2005;17(11):1505–1512. doi: 10.1093/intimm/dxh328. [DOI] [PubMed] [Google Scholar]
- 21.Elmonem MA, van den Heuvel LP, Levtchenko EN. Immunomodulatory Effects of Chitotriosidase Enzyme. Enzyme Res. 2016;2016:2682680. doi: 10.1155/2016/2682680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanneganti M, Kamba A, Mizoguchi E. Role of chitotriosidase (chitinase 1) under normal and disease conditions. J Epithel Biol Pharmacol. 2012;5:1–9. doi: 10.2174/1875044301205010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.James AJ, Reinius LE, Verhoek M, Gomes A, Kupczyk M, Hammar U, et al. Increased YKL-40 and Chitotriosidase in Asthma and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2016;193(2):131–142. doi: 10.1164/rccm.201504-0760OC. [DOI] [PubMed] [Google Scholar]
- 24.Bargagli E, Olivieri C, Margollicci M, Bennett D, Luddi A, Perrone M, et al. Serum chitotriosidase levels in patients with allergic and non-allergic asthma. Respiration. 2010;79(5):437–438. doi: 10.1159/000277664. [DOI] [PubMed] [Google Scholar]
- 25.Kim KW, Park J, Lee JH, Lee HS, Lee KE, Hong JY, et al. Association of genetic variation in chitotriosidase with atopy in Korean children. Annals of allergy, asthma, & immunology. 2013;110(6):444–449.e441. doi: 10.1016/j.anai.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF, Cheever AW, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. Journal of Clinical Investigation. 2006;116(7):2044–2055. doi: 10.1172/JCI27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Migliaccio CT, Buford MC, Jessop F, Holian A. The IL-4Ralpha pathway in macrophages and its potential role in silica-induced pulmonary fibrosis. Journal of leukocyte biology. 2008;83(3):630–639. doi: 10.1189/jlb.0807533. [DOI] [PubMed] [Google Scholar]
- 28.Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, et al. TGF-β-dependent induction of CD4⁺CD25⁺Foxp3⁺ Tregs by liver sinusoidal endothelial cells. J Hepatol. 2014;61(3):594–599. doi: 10.1016/j.jhep.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 29.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. The journal of immunology. 2004;172(9):5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 30.Chen W, Jin W, Hardegen N, Lei K, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. The Journal of experimental medicine. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 32.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110(8):2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan Y, Flavell RA. Regulatory T cells, transforming growth factor-beta, and immune suppression. Proceedings of the American Thoracic Society. 2007;4(3):271–276. doi: 10.1513/pats.200701-020AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown SD, Baxter KM, Stephenson ST, Esper AM, Brown LA, Fitzpatrick AM. Airway TGF-β1 and oxidant stress in children with severe asthma: association with airflow limitation. Journal of Allergy and Clinical Immunology. 2012;129(2):388–396, 396.e381. doi: 10.1016/j.jaci.2011.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minshall EM, Leung DY, Martin RJ, Song YL, Cameron L, Ernst P, et al. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. American Journal of Respiratory Cell and Molecular Biology. 1997;17(3):326–333. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- 36.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. Journal of Allergy and Clinical Immunology. 2003;111(6):1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 37.Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, et al. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. The journal of immunology. 2004;172(10):6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 38.Luo X, Yang H, Kim IS, Saint Hilaire F, Thomas DA, De BP, et al. Systemic transforming growth factor-beta1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta cell function in overtly diabetic nonobese diabetic mice. Transplantation. 2005;79(9):1091–1096. doi: 10.1097/01.tp.0000161223.54452.a2. [DOI] [PubMed] [Google Scholar]
- 39.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008;9(12):1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kastner L, Dwyer D, Qin FX. Synergistic effect of IL-6 and IL-4 in driving fate revision of natural Foxp3+ regulatory T cells. J Immunol. 2010;185(10):5778–5786. doi: 10.4049/jimmunol.0901948. [DOI] [PubMed] [Google Scholar]
- 41.Coleman M, Ruane D, Moran B, Dunne PJ, Keane J, Mills KH. Alveolar macrophages contribute to respiratory tolerance by inducing FoxP3 expression in naive T cells. Am J Respir Cell Mol Biol. 2013;48(6):773–780. doi: 10.1165/rcmb.2012-0263OC. [DOI] [PubMed] [Google Scholar]
- 42.Soroosh P, Doherty TA, Duan W, Mehta AK, Choi H, Adams YF, et al. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med. 2013;210(4):775–788. doi: 10.1084/jem.20121849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200(3):377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Da Silva CA, Chalouni C, Williams A, Hartl D, Lee CG, Elias JA. Chitin is a size-dependent regulator of macrophage TNF and IL-10 production. J Immunol. 2009;182(6):3573–3582. doi: 10.4049/jimmunol.0802113. [DOI] [PubMed] [Google Scholar]
- 45.Choi JP, Lee SM, Choi HI, Kim MH, Jeon SG, Jang MH, et al. House Dust Mite-Derived Chitin Enhances Th2 Cell Response to Inhaled Allergens, Mainly via a TNF-alpha-Dependent Pathway. Allergy Asthma Immunol Res. 2016;8(4):362–374. doi: 10.4168/aair.2016.8.4.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dubey LK, Moeller JB, Schlosser A, Sorensen GL, Holmskov U. Chitin enhances serum IgE in Aspergillus fumigatus induced allergy in mice. Immunobiology. 2015;220(6):714–721. doi: 10.1016/j.imbio.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 47.Lee CG, Da Silva CA, Lee J, Hartl D, Elias JA. Chitin regulation of immune responses: an old molecule with new roles. Curr Opin Immunol. 2008;20(6):684–689. doi: 10.1016/j.coi.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagener J, Malireddi RK, Lenardon MD, Koberle M, Vautier S, MacCallum DM, et al. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog. 2014;10(4):e1004050. doi: 10.1371/journal.ppat.1004050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, et al. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447(7140):92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shibata Y, Foster LA, Bradfield JF, Myrvik QN. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J Immunol. 2000;164(3):1314–1321. doi: 10.4049/jimmunol.164.3.1314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.