

Abstract
Phosphatidylserine (PS) exposure during apoptosis leads to silent clearance of cells without adverse immune reactions to self-proteins. Given the biological functions of PS in cellular cleanup and global immunosuppression, we hypothesized that administration of PS-protein complexes would reduce immunogenicity. Here, we report that exposing Pompe disease mice to acid alpha glucosidase (rhGAA) with PS or immunosuppressant dexamethasone (Dex) resulted in lower anti-rhGAA-antibodies than in animals receiving rhGAA alone. However, upon re-challenge with rhGAA, only PS-rhGAA pre-exposed mice displayed a durable hypo-responsiveness even after PS administration ceased. Thus, pre-exposure of antigens administered together with PS were not silently cleared but the immune system acquired memory about the antigen that averted mounting of a response during re-challenge. In Hemophilia A mice, PS hypo-responsiveness towards Factor VIII was reversed by administration of function-blocking antibody against the PS receptor TIM-4, implicating this receptor in PS’s effect. Moreover, pre-exposure of myelin oligodendrocyte glycoprotein peptide with PS delayed the onset and reduced severity of experimental autoimmune encephalomyelitis. These observations suggest that PS’s function in apoptosis is not limited to silent antigen clearance without immune responses toward self-proteins, but shows that PS reduces immune response during re-challenge to several antigens that also involves initiation of antigen tolerance.
Keywords: Phospholipids, Liposomes, Apoptosis, Immune Response, Proteins, Biotechnology
INTRODUCTION
Immunogenicity, the unwanted immune response against therapeutic proteins, is a major clinical complication that impacts safety and efficacy of several life-saving protein-based therapies1. Clinical complications of replacement therapies for diseases such as Hemophilia A (HA), a bleeding disorder, and Pompe disease, a lysosomal disorder, are good examples of unwanted antibody responses to therapeutic proteins that negatively impact clinical efficacy2–4. HA is characterized by the deficiency and/or dysfunction of a coagulation protein Factor VIII (FVIII) and replacement therapy using recombinant FVIII is the first line of therapy. A major clinical complication is the development of anti-FVIII antibodies that abrogate the biological activity of the protein in about 15-30% of the patients2. This unwanted antibody response is more damaging in Pompe disease, which is caused by the lack of the lysosomal storage protein acid alpha glucosidase (GAA). GAA breaks down glycogen to glucose and the lack of this protein leads to accumulation of glycogen in muscles. Enzyme replacement therapy using recombinant GAA is complicated by the development of anti-GAA antibodies in about 90% of the patients leading to loss of efficacy3. Once high titer antibodies are established, very few effective clinical options are available and, therefore, reducing or eliminating immune response would address an important medical need.
Phosphatidylserine (PS) contains a phosphoserine head group, glycerol backbone and one or two saturated or unsaturated acyl chains and is generally present in the inner leaflet of the cell membrane. The phospholipid asymmetry and PS exposure during apoptosis has received considerable interest in investigating the effect of PS on the immune system. During apoptosis, PS flips from the inner leaflet of the lipid bilayer to the outer leaflet of the plasma membrane of the apoptotic cells. The externalized PS induces multiple signals that ultimately result in the immune system to clear the dead cells without any immunological reactions5–8. These signals have been referred to as “find me, eat me, and ignore/tolerate me.” The “eat me” signal is essential for recognition and engulfment by phagocytosis. PS binding by apoptotic cells triggers the release of anti-inflammatory cytokines, including IL-10, TGF-β, and PGE2, and simultaneously decreases the secretion of inflammatory cytokines TNF-α, IL-1β, and IL-12. These signals prevent the immune system from mounting an immune response against self-proteins and thus maintain tolerance towards self-proteins, generally referred as ignore/tolerate me signal. Overall, PS mediates the silent clearance of apoptotic cells and thus can evade or suppress immune surveillance. Based on the known biological functions of PS as a cleanup crew and immune suppressor, we hypothesized that administration of proteins in the presence of PS would reduce immunogenicity of therapeutic proteins.
METHODS
Materials
Phosphatidylcholine (PC) and brain phosphatidylserine (PS) were purchased from Avanti Polar Lipids (Alabaster, AL). Full length recombinant human factor VIII (FVIII) was a generous gift from the Hemophilia Center of Western New York (Buffalo, NY). Recombinant human acid alpha-glucosidase (rhGAA) was provided by Genzyme Corporation (Cambridge, Massachusetts). Mouse MOG35-55 was purchased from AnaSpec Inc. (Fremont, CA). All solvents and buffer salts were obtained from Fisher Scientific (Fairlawn, NJ). Murine TIM-4 antibody was purchased from BioLegend (San Diego, CA). Anti-FVIII monoclonal antibody ESH8 was obtained from American Diagnostica Inc. (Greenwich, CT). Alkaline phosphatase-conjugated goat anti-mouse Ig antibody was purchased from Southern Biotech (Birmingham, AL). p-nitrophenylphosphate (pNPP) substrate system was purchased from KPL Inc. (Gaithersburg, MD). Horseradish peroxidase-conjugated goat anti-mouse IgG antibody and 3,3′,5,5′-tetramethylbenzidine substrate (TMB) were purchased from Sigma Aldrich (St. Louis, Missouri). Endosafe Endochrome-K® Kit was purchased from Charles River Laboratories (Charleston, SC). NUNC MaxiSorp 96 well plates were purchased from Thermo Fisher Scientific (Waltham, MA).
Animals
A colony of hemophilic mice with a targeted deletion in exon 16 of the FVIII gene (termed HA mice) was maintained onsite. Breeding pairs of 6neo/6neo GAA knockout mice (termed Pompe disease mice) were purchased from the Jackson Laboratories (Bar Harbor, ME), where a homozygous colony was bred in-house. In addition, female C57BL/6J mice were purchased from the Jackson Laboratories. All animal experiments were conducted under approval and following the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the University at Buffalo.
Preparation of Protein-Lipid Complexes
PS liposomes were prepared at a 30:70 molar ratio of PS to PC as previously described9. The size of the liposomes was confirmed using a NICOMP Model CW380 particle size analyzer from Particle Sizing Systems (Port Richey, FL) and lipid content was confirmed using a phosphate assay10. Zeta potential was analyzed for surface charge using Nanobrook Omni zeta potential analyzer from Brookhaven Instruments Corporation (Holtsville, NY) as previously described11. The protein to lipid ratio used was 1:10,000 for experiments using rhGAA and FVIII and 1:1,000 for MOG35-55. The proteins were associated with the liposomes by incubation at 37°C for 30 minutes. All formulations were tested for endotoxin level by using Endosafe Endochrome-K endotoxin assay kit (Charleston, SC) and endotoxin negative samples were used for in vivo studies.
Association Efficiency of PS-Protein Complexes
Encapsulation efficiency of the amount of protein associated with PS liposomes was accessed. For PS FVIII, encapsulation efficiency was previously determined using a discontinuous dextran gradient centrifugation and reported to be approximately 45.2 ± 16.8%12. For rhGAA and MOG35-55, encapsulation efficiency was determined using a size exclusion column prepared using G-150 and G-50 Sephadex beads, respectively, as previously described11. Briefly, one mole percent of rhodamine-phosphatidylethanolamine (PE) was added to the lipid mixture before evaporation and liposomes were subsequently prepared as previously described. The addition of rhodamine-PE served to fluorescently label the PS liposomes in order to detect liposomes as they eluted off the column. Column eluent was collected in fractions. Liposome content was quantified by rhodamine fluorescence by excitation at 560 nm and emission at 583 nm. Protein content was quantified by an activity assay for GAA activity and micro BCA protein assay kit from Thermo Fisher Scientific (Waltham, MA) for MOG35-55.
PS-rhGAA Immunogenicity Study
The relative immunogenicity of PS rhGAA and its ability to induce a durable tolerance was evaluated in Pompe disease mice. Pompe disease mice (n=8/group) were pre-treated with sub-therapeutic dose of 1 g rhGAA subcutaneously (SC) in the presence and absence of PS once a week for four weeks. Dexamethasone rhGAA (Dex-rhGAA) was used as a general immunosuppressive control. Throughout the duration of the study just prior to each weekly injection, mice were pre-dosed with 5 mg/kg of diphenhydramine via intraperitoneal (IP) injection to prevent hypersensitivity reactions. Two weeks after the last immunization, mice were re-challenged with 1 g of free rhGAA SC once a week for seven weeks. A blood sample was taken from the saphenous vein prior to each re-challenge injection to follow the course of anti-rhGAA antibody development. Two weeks after the last re-challenge, all mice were sacrificed and plasma collected via cardiac puncture in 10% v/v acid citrate dextrose solution. Plasma samples were stored at −80°C until further analysis.
Role of TIM-4 Immunogenicity Study
The involvement of TIM-4 in the ability of PS to induce tolerance was evaluated in HA mice. TIM-4 is expressed exclusively on APCs such as dendritic cells and macrophages13,14. To investigate the role of TIM-4 receptor in PS-mediated tolerance, we used a function blocking anti-TIM-4 antibody due to the exclusive expression of TIM-4 on antigen presenting cells (APCs) and the expected PS-mediated effects through dendritic cells by skewing the dendritic cells towards a tolerogenic phenotype13–15. Mice (n=12-14/group) were immunized with four weekly SC injections of 0.4 g FVIII in the presence and absence of PS. A third group of mice received a SC immunization of 15 g of anti-TIM-4 antibody. Thirty minutes after the administration of the anti-TIM-4 antibody, mice in this group received a SC injection of PS-FVIII (0.4 g) in close proximity to the administered anti-TIM-4 antibody. Following a two week washout period, half the animals from each group were sacrificed and plasma samples were collected as baseline samples for relative immunogenicity analysis of antibody titers. All remaining animals were re-challenged with two weekly SC injections of 0.4 g of free FVIII alone. Two weeks after the last re-challenge injection, all mice were sacrificed and plasma collected via cardiac puncture in 10% v/v acid citrate dextrose solution. Plasma samples were stored at −80°C until further analysis.
PS-MOG35-55 EAE Study
Animal studies were conducted using six week old female C57BL/6J mice (n=10/group). Mice were immunized with four weekly SC injections of 1 g MOG35-55 in the presence and absence of PS. A third group of animals received SC injections of Tris buffer. On the fifth week, experimental autoimmune encephalomyelitis (EAE) was induced in all animals with commercially available MOG35-55 and Complete Freund’s Adjuvant (CFA) emulsion using Hooke kits according to manufacturer’s instructions (Hooke Laboratories, EK-2110, Lawrence, MA). Briefly, 100 L of MOG35-55/CFA emulsion were injected SC at two sites (200 L/mouse) followed by an intraperitoneal injection of 60 ng of pertussis toxin (PTX) at 2 and 24 hours after disease induction (Day 0). All mice were monitored daily for thirty days after the disease induction and assigned a clinical score ranging from 0 to 5 in a blinded fashion according to guidelines provided by Hooke Laboratories. Briefly, clinical scores were assigned as followed: 0, no sign of disease; 1, limp tail; 2, limp tail and hind limb weakness; 3, complete hind limb paralysis; 4, complete hind limb and forelimb paralysis; 5, moribund. Easy access to wet chow and water was provided for the mice for the duration of the disease. At the conclusion of the monitoring period, all animals were sacrificed. Splenocytes were collected from immunized animals and prepared for analysis using flow cytometry with BD LSRFortessa (Pittsburgh, PA). Cells were stained with FITC conjugated to anti-CD4 antibody and PE conjugated to anti-FoxP3 antibody according to manufacturer’s protocol (Miltenyi Biotec, Auburn, CA). Quadrant analysis of double positive cells were analyzed using FlowJo software (Ashland, OR). To account for the spectral overlap of FITC and PE, compensation using single label FITC and PE controls were acquired and compensation carried out using BD FACSDiva software provided by the manufacturer.
Determination of anti-drug antibodies
Total anti-rhGAA IgG and anti-FVIII antibody titers were determined by ELISA as previously described9,16.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism (La Jolla, CA). One-way ANOVA followed by Tukey’s post-hoc analysis was performed as indicated. Area under the curve (AUC) values were calculated as indicated and non-parametric Kruskal-Wallis test with Dunn’s post-hoc analyses were performed as indicated in the rhGAA and MOG studies. P values < 0.05 were considered statistically significant.
RESULTS
Biophysical Characterization of PS-Protein Complexes
The protein was associated with PS containing liposomes as described in the Methods section. The average size of PS liposomes was 208.8 ± 71.6 nm (mean ± SD), and zeta potential was measured as having an average surface charge of −28.88 ± 7.52 mV (mean ± SD). The association efficiency of PS associated with rhGAA, FVIII, and MOG35-55 were determined to be 41.5 ± 0.2%, 45.2 ± 16.8%12, and 45.6 ± 5.1%, respectively.
PS-rhGAA Immunogenicity Study
Pompe disease mice were pre-exposed with the different formulations according to the immunization protocol described in Figure 1A. Baseline plasma samples were taken at week 6 before re-challenge with free rhGAA in order to assess relative immunogenicity of rhGAA, PS-rhGAA, and Dex-rhGAA. As seen in Figure 1B, on week 6, the mean titers for animals in the PS-rhGAA treatment group (3060 ± 1038 [mean titers ± standard error of the mean (SEM)]) were similar to those for animals in the Dex-rhGAA treatment group (4182 ± 1236 [mean ± SEM]). Pre-treatment with all formulations were stopped on week 4 and all animals were then re-challenged with free rhGAA to determine whether the reduction in antibody responses is due to tolerance induction. Further, the aggressive, multi-week re-challenges of free rhGAA would help address if the hypo-responsiveness induced by PS pre-exposure is durable and long-lasting.
FIGURE 1.


PS induces hypo-responsiveness towards rhGAA. (A) Immunization schedule utilized for PS-rhGAA immunogenicity study (n=8/group) in Pompe disease mice. Open circles (○) represent immunizations during the pre-exposure period and the closed triangles (▼) represent re-challenge injections. The closed squares (■) represent the terminal samples. (B) Overall time course of anti-rhGAA antibody titer progression (mean ± SEM) in Pompe disease mice pre-treated with free rhGAA (black), PS-rhGAA (red), or Dex-rhGAA (blue). * denotes statistical significance with p < 0.05 by non-parametric Kruskal-Wallis with Dunn’s post-hoc analysis of mean AUC values.
If PS induces tolerance towards the antigen, then the titers should remain unchanged despite the aggressive re-challenge. From Figure 1B, Pompe disease mice pre-treated with PS-rhGAA continued to maintain lower antibody responses compared to those pre-treated with Dex-rhGAA and free rhGAA. Analyses of the overall area under the curve (AUC) values indicate significant differences between PS-rhGAA pre-treated animals compared to free rhGAA pre-treated animals. Further, PS-rhGAA pre-treated animals displayed a 70% and 63% reduction in AUC compared to free rhGAA pre-treated animals and Dex-rhGAA pre-treated animals, respectively. On week 14, two weeks after the last re-challenge, PS-rhGAA pre-treated animals showed significant reduction in antibody responses (5662 ± 1812 [mean ± SEM]) compared to free rhGAA pre-treated animals (29594 ± 6521 [mean ± SEM]). Although not statistically significant, PS-rhGAA pre-treated animals displayed a trend towards lower antibody responses compared to Dex-rhGAA pre-treated animals (16404 ± 6321 [mean ± SEM]). In fact, despite the similar antibody responses between Dex-rhGAA and PS-rhGAA treated animals on week 6, once Dex-rhGAA treatment stopped and animals were subsequently re-challenged with free rhGAA, animals displayed an increase in antibody development while PS-rhGAA treated mice maintained a hypo-responsive state, suggesting PS induces immunological tolerance towards rhGAA and that this tolerance is durable. The results showed that PS function is not limited to silent clearance but that it teaches the immune system to tolerate an antigenic protein. Further, the comparison between PS and Dex suggests a mechanistic difference between the two, implying that PS is not a global immune suppressor.
Role of TIM-4 Immunogenicity Study
To investigate the role of TIM-4 receptor in PS-mediated tolerance, we used a function blocking TIM-4 antibody (anti-TIM-4 antibody) due to the exclusive expression of TIM-4 on antigen presenting cells (APCs) and the expected PS-mediated effects through dendritic cells by skewing the dendritic cells towards a tolerogenic phenotype13–15. If the TIM-4 receptor is involved, then the administration of the function blocking TIM-4 antibody will prevent binding of PS to the TIM-4 receptor and we will observe a reversal of the PS-mediated effects. HA mice were immunized according to the schedule described in Figure 2A. According to Figure 2B, animals that received free FVIII alone showed robust total anti-FVIII antibody titers at baseline (4311 ± 1239 [mean ± SD]). Animals administered with PS-FVIII showed significantly lower total anti-FVIII antibody titers (1556 ± 212 [mean ± SD]). However, this reduction in antibody titers observed with PS was reversed upon administration of anti-TIM-4 antibody (5885 ± 1413 [mean ± SD]). In the presence of anti-TIM-4 antibody, the antibody responses are significantly higher than the PS-FVIII treatment and are statistically comparable to the free FVIII treatment group.
FIGURE 2.



PS-mediated hypo-responsiveness involves TIM-4 receptor. (A) Immunization schedule used for TIM-4 immunogenicity study (n=10-12/group) in HA mice. Open circles (○) represent immunizations during the pre-exposure period and the closed triangles (▼) represent re-challenge injections. The closed squares (■) represent the terminal samples. (B) Total anti-FVII antibody titers (mean ± SD) at baseline on week 6. (C) Total anti-FVIII antibody titers (mean ± SD) after re-challenge with free FVIII at week 9. * denotes statistical significance with p < 0.05 by one-way ANOVA with Tukey’s post-hoc analysis.
When treatments were stopped and animals were re-challenged with free FVIII, animals that were pre-treated with free FVIII continued to show increased anti-FVIII antibody titers (6647 ± 1228 [mean ± SD]) (Figure 2C). Animals that were pre-treated with PS-FVIII and subsequently re-challenged with free FVIII maintained lower anti-FVIII antibody titers (2152 ± 425 [mean ± SD]), significantly lower than the animals which received PS-FVIII along with the anti-TIM-4 antibody (6898 ± 1557 [mean ± SD]).
PS-MOG35-55 EAE Study
In order to investigate PS-mediated effects on a self-protein implicated in autoimmunity, animals were immunized with MOG35-55, a protein implicated in multiple sclerosis (MS), in the presence and absence of PS according to the schedule described in Figure 3A. On week 5, after pre-treatment with the different formulations stopped, all animals were induced with EAE, a murine model for human MS. Animals that developed EAE displayed the characteristic disease course, consistent with previously published results17–19. According to Figure 3B, mice that were pre-exposed to PS-MOG35-55 maintained statistically significant reduction in clinical scores compared to mice that were pre-treated with free MOG35-55 and buffer. Analyses of the overall AUC values of the clinical scores indicate a 40% and 44% reduction in AUC values of animals pre-treated with PS-MOG35-55 as compared to animals pre-exposed to buffer and free MOG35-55, respectively. Mice pre-treated with PS-MOG35-55 also displayed a statistically significant delay in disease onset and statistically significant reduction in cumulative disease score and peak disease score, as summarized in Table 1. Percentage of CD4+FoxP3+ cells that were isolated from splenocytes were compared between all treatment groups. In Figure 3C, an increase was observed for PS-MOG35-55 treatment group (1.72 ± 0.08% [mean ± SEM]) as compared to free MOG35-55 (1.34 ± 0.10% [mean ± SEM], *p < 0.05) and buffer (1.47 ± 0.05% [mean ± SEM]) treatment group, suggesting a role of Tregs in this PS-mediated function.
FIGURE 3.



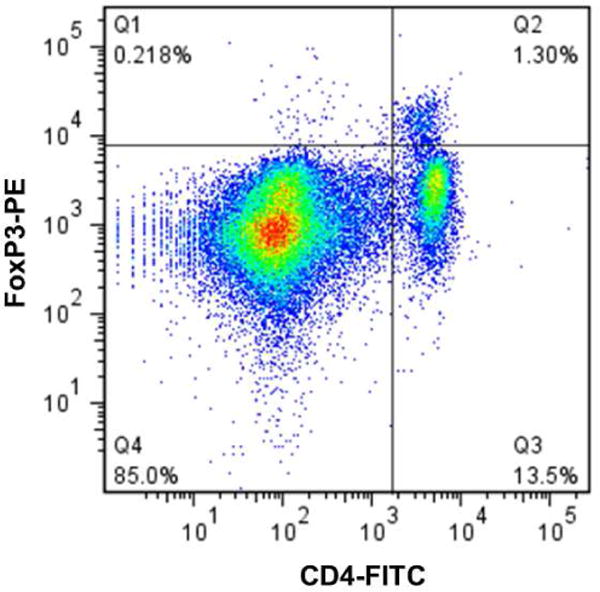


PS-MOG35-55 delays onset and reduces severity of EAE. (A) Immunization schedule used for PS-MOG35-55 study (n=10/group) in C57BL/6 mice. Open circles (○) represent immunizations during the pre-exposure period and the closed triangles (▼) represent the week of EAE induction. The closed squares (■) represent the terminal samples. (B) Clinical scores (mean ± SEM) of mice pre-treated with free MOG35-55 (black), PS-MOG35-55 (red), or buffer (blue) followed by EAE induction on week 5. ** denotes statistical significance with p < 0.01 and ¥ denotes statistical significance with p < 0.05 between PS-MOG35-55 pre-treatment and buffer and free MOG35-55, respectively, by non-parametric Kruskal-Wallis with Dunn’s post-hoc analysis of mean AUC values. (C) Percent expression of CD4+FoxP3+ Tregs isolated from splenocytes of EAE mice (mean ± SEM). * denotes statistical significance with p < 0.05 by one-way ANOVA with Tukey’s post-hoc analysis. (D) Representative dot plots of CD4+FoxP3+ Treg expression from splenocytes in EAE mice pre-treated with free MOG35-55 (left panel), buffer (middle panel), and PS MOG35-55 (right panel).
TABLE 1.
Clinical parameters of EAE in treated and control mice.
| EAE Onset (Day) | Peak Disease Score | Cumulative Disease Score | |
|---|---|---|---|
| Free MOG35–55 | 10.4 ± 0.64 | 4.15 ± 0.18 | 59.05 ± 3.47 |
| PS MOG 35–55 | 17.2 ± 2.38**, ¥¥ | 2.60 ± 0.45*, ¥¥ | 35.65 ± 6.73**, ¥ |
| Buffer | 10.3 ± 0.42 | 3.90 ± 0.18 | 63.90 ± 5.34 |
Data are expressed as mean ± SEM. Comparisons of PS MOG35-55 pre-treatment versus free MOG35-55 and buffer pre-treatment relating to EAE onset, peak disease score, and cumulative disease score following pre-exposure period and EAE induction on week 5.
denotes statistical significance at p < 0.05 between PS MOG35-55 and buffer, while
denotes statistical significance at p < 0.05 between PS MOG35-55 and free MOG35-55 by non-parametric Kruskal-Wallis with Dunn’s post-hoc analysis.
DISCUSSION
Based on the known biological functions of PS as a cleanup crew and a global immune suppressor, we hypothesized that administration of proteins in the presence of PS would reduce immunogenicity of therapeutic proteins. In support of this hypothesis, we previously established that the administration of four weekly injections of FVIII associated with PS reduced the development of anti-FVIII antibodies in HA mice compared to animals treated with free FVIII alone9,15,20. Similarly, PS associated with GAA reduced the anti-rhGAA antibody response in Pompe disease mice16. Following the pre-exposure of the mice to proteins associated with PS, the animals were re-challenged with protein alone, expecting a normal immunological response to the protein as the cleanup function of PS is no longer available. However, the animals that were pre-exposed to the protein in the presence of PS (but not other control lipids such as charge-matched phosphatidylglycerol (PG) or neutral PC) continue to show significantly lower anti-protein antibody development compared to mice treated with protein only15. Of particular interest is that the suppression of the antibody response to the protein persisted even after PS exposure has stopped. In order to gain further insight into the mechanism by which PS induced this immune suppression in Pompe disease mice, a comparison was made of the immunosuppression that occurs with Dex (Figure 1B). In sharp contrast to the Dex immune-suppressed mice, animals that were pre-exposed to protein in the presence of PS were found to be hypo-responsive to a challenge with free protein and that the hypo-responsiveness was durable (Figure 1B). Similar effects in the antibody responses were observed in HA mice that were given FVIII in the presence of Dex and PS, with the brain-derived PS15, soy-derived PS, and a more homogenous PS preparation (unpublished results). These data demonstrate that, unlike the transient Dex-induced immune-suppression, the pre-exposure of the proteins with PS induces a durable hypo-responsiveness. Further, the comparison between PS and Dex suggests a mechanistic difference between the two, implying that PS is not a global immune suppressor and can induce immunological tolerance. We conclude that immunological tolerance was induced towards the protein that occurred after its initial exposure in the presence of PS. Although PS is able to induce hypo- responsiveness in antibody development, the lack of complete abrogation of antibody responses may be due to the presence of unassociated protein in the PS formulation, as the free fraction was not separated in order to maintain a comparable protein dose in all treatment groups. Further, removal of unassociated protein was not attempted due to potential contaminants during separation procedures that could in turn confound the results.
Further, it was determined that the animals that were pre-treated with protein in the presence of PS responded normally when re-challenged with an unrelated antigen ovalbumin (Ova), indicating that the persistent hypo-responsiveness to the original PS associated protein was antigen specific21. Thus, the biological function of PS is not simply limited to its cleanup and immunosuppressive activity as previously thought, but it includes a very important functional and durable immune modulation or teaching/tolerance possibly by the ability of PS to convert an immunogen to a tolerogen15,16,21,22. The molecular and cellular level studies indicate that the teaching and tolerizing function of PS is mediated by multiple mechanisms including phenotypic changes in antigen presenting cells and also likely direct effect on B and T-cells. PS has been shown to induce phenotypic changes in dendritic cells and macrophages23–26, and increase in secretion of regulatory cytokines27. In addition, PS has been shown to inhibit T-cell activation and generation of Tregs that are consistent with immunological tolerance15,24. Further, PS-mediated effects involve reduction in memory B-cells and direct effect on T-cells and its signaling15,21. Based on the ability of PS to induce tolerance, it is reasonable to hypothesize that the immune system uses the apoptotic process not only to avoid immune responses to self-antigens during cell death (i.e. cleanup-mediated immune evasion and suppression), but also to learn about self-proteins and its mutations to maintain immune homeostasis. Thus, apoptotic events may not be a silent event but instead active learning process that occurs due to PS exposure and the teaching and tolerizing potential of PS.
The molecular mimicry of apoptotic machinery to induce tolerance towards antigenic proteins is known28,29. For example, the administration of foreign antigen Ova loaded in apoptotic cells induced tolerance towards Ova28. In addition, dendritic cells pulsed with self-antigen containing apoptotic bodies prevent autoimmune conditions29. In addition, the anti-inflammatory effects of PS were further shown in experimental models of disease such as murine paw edema, myocardial infarction, EAE, and type I diabetes30–33. However, a direct link between the tolerizing potential of apoptotic process and externalization of PS could not be made as the known function of cleanup is insufficient to completely explain the tolerogenic potential of apoptotic events. The ability of PS to selectively and durably downregulate the immune responses to immunogenic molecules, as seen in Figures 1–3,15,16 provides a necessary link between the tolerogenic potential of apoptotic events and PS exposure. Because not all PS exposure is representative of apoptosis6 and, in addition to several other signals that accompany apoptosis, it is reasonable to assume that the use of PS liposomes as apoptotic mimicry may be an oversimplification of a complex process. Nonetheless, antigen in the presence of PS liposomes (with no other biological molecules) modulates the immune response via the teaching/tolerizing function that could involve effects on T-cells such as T-cell inhibition and Treg induction.
It is likely that apoptotic events may be characterized by signature PS signals such as biophysical characteristics (PS clustering, surface charge density, size of secreted vesicles)34 and structural changes (oxidation, unsaturation)35. It is possible a unique structural characteristic of PS is evolutionarily chosen to exert teaching activity over cleanup function. In addition, the uptake and receptor that PS engages could determine the function of cleanup/immunosuppression versus teaching/tolerance, as there are at least thirteen known receptors and bridging molecules for PS8,13,36. One such molecule is TIM receptors13. TIM-4 is expressed exclusively on APCs that plays an important role in tolerance. HA animals that were administered with TIM-4 function blocking antibody reversed PS-mediated reduction in antibody development after re-challenge (Figures 2B and 2C), suggesting a role for PS receptors in its teaching function. Overall, even though other factors including the addition of a ligand with PS to elicit “PS + ligand” recognition as well as the involvement of other PS receptors and bridging molecules cannot be ruled out for the tolerogenic potential of apoptotic events,36 the teaching/tolerance inducing function of PS is at least partly responsible.
The ability of PS to teach immune system to learn about an antigenic protein could be a useful therapeutic approach to treat unwanted immune responses. For example, this function of PS is clinically useful in preventing and treating immunogenicity of therapeutic proteins and antibodies, autoimmune conditions such as MS32, type I diabetes33 and allergies as observed with gluten, and in gene therapy. The pre-exposure of a therapeutic protein in the presence of PS modulates the immune response to an antigen and, hence, could effectively desensitize the immune system of the host, and prevent the establishment of unwanted responses when patients are on protein therapy. Such desensitization therapy significantly reduced antibody development in preclinical murine models of HA and Pompe Disease (Figures 1 and 2)15,16. This desensitization approach could also be therapeutically used to prevent or slow down progression of autoimmune conditions or allergies by re-teaching the immune system. In order to further investigate whether PS can teach immune system to be tolerant of an antigen and desensitize that animal to prevent EAE development in an animal model of human MS, we desensitized the animal with MOG peptide in the presence and in the absence of PS liposomes and induced the disease using established procedures (Figure 3B). As is shown in Figure 3B and 3C, the animals that were immunized with MOG peptide in the presence of PS displayed a delay in disease onset and reduced disease severity and mice pre-treated with PS observed an increased expression of CD4+FoxP3+ Tregs. Despite the small increase in Treg expression observed, this difference could be immunologically significant as Tregs have suppressive functions that mediate the tolerogenic environment. However, the clinical potential of the teaching of PS could be broadly realized if one can reverse an established response after the disease or unwanted immune response sets in. In HA mice with pre-existing anti-FVIII antibodies, we previously demonstrated that PS administration could be used to slow the progression of further antibody development21. It is possible that the treatment protocol, such as duration and route of desensitization, and PS structural properties regarding the design of the particles could effectively be used to reverse or substantially slow down the progress of a disease condition.
Apoptotic mimicry and PS exposure has been shown to be exploited by several parasites and viral particles to evade host immune responses by silent clearance6,37. For example, Listeria monocytogenes expose high levels of PS to promote parasite survival and replication6,38. Viruses have also been shown to use apoptotic mimicry and PS externalization to gain entry to cells and replication. This has led to several treatment approaches to block PS by anti-PS antibodies or by annexin V, so that these invasive agents do not use apoptotic mimicry to gain cell entry and evade and suppress host immune system6,39. PS exposure on tumor-associated microvesicles such as exosomes has been causally linked to their inhibition of T-cell function that may contribute to tumors’ immune escape40. Some promising preclinical and clinical studies suggest the use of PS blocking is a strategy to treat these conditions. For example, a monoclonal anti-PS antibody is currently being tested in clinical trials with cancer patients. However, the efficacy of these therapies, particularly in later stages of the disease, would depend on extend of teaching/tolerance inducing effect of PS. In such situations, therapeutic approaches should focus on not only inhibiting cell entry but also reverse any already tolerance that is established towards antigenic components of parasites, tumor or virus particles.
In conclusion, PS is not just a cleanup crew but also a well-meaning teacher that helps to maintain immune homeostasis. Further understanding of the biological function of this important bioactive lipid could lead to efficacious novel therapies.
Acknowledgments
This work was financially supported by National Institutes of Health grant R01 HL-70227 to Dr. Sathy V. Balu-Iyer. We would like to thank the assistance of the Confocal Microscope and Flow Cytometry Core Facility at the School of Medicine and Biomedical Sciences at the University at Buffalo. We would like to thank Dr. Wojciech Krzyzanski for use of the Auto Hematology analyzer. We are grateful to Dr. Robert Bies for independently reviewing the statistical analyses. We would like to thank Dr. Hongbiao Liu for his advice regarding the EAE studies. We are grateful to Dr. David Scott of USUHS and to the Hemophilia Center of Western New York for providing the gifts of the HA mice and recombinant FVIII, respectively.
Abbreviations
- HA
Hemophilia A
- FVIII
Factor VIII
- GAA
acid alpha glucosidase
- rhGAA
recombinant human GAA
- PS
phosphatidylserine
- SEM
standard error of the mean
- TIM-4
T-cell immunoglobulin and mucin 4
- MOG
myelin oligodendrocyte glycoprotein
- EAE
experimental autoimmune encephalomyelitis
- MS
multiple sclerosis
- Tregs
regulatory T cells
- PG
phosphatidylglycerol
- PC
phosphatidylcholine
- Dex
dexamethasone
- Ova
ovalbumin
- SC
subcutaneous
- Ab
antibody
- APC
antigen presenting cell
- CFA
Complete Freund’s Adjuvant
- AUC
area under the curve
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parenky A, Myler H, Amaravadi L, Bechtold-Peters K, Rosenberg A, Kirshner S, Quarmby V. New FDA draft guidance on immunogenicity. The AAPS journal. 2014;16(3):499–503. doi: 10.1208/s12248-014-9587-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lollar P, Healey JF, Barrow RT, Parker ET. Factor VIII inhibitors. Adv Exp Med Biol. 2001;489:65–73. doi: 10.1007/978-1-4615-1277-6_6. [DOI] [PubMed] [Google Scholar]
- 3.2006. Myozyme [package insert]. Genzyme Corporation
- 4.Banugaria SG, Patel TT, Kishnani PS. Immune modulation in Pompe disease treated with enzyme replacement therapy. Expert Review of Clinical Immunology. 2012;8(6):497–499. doi: 10.1586/eci.12.40. [DOI] [PubMed] [Google Scholar]
- 5.Chaurio RA, Janko C, Munoz LE, Frey B, Herrmann M, Gaipl US. Phospholipids: key players in apoptosis and immune regulation. Molecules. 2009;14(12):4892–4914. doi: 10.3390/molecules14124892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, Freimark B, Empig C, Mercer J, Schroit AJ, Schett G, Herrmann M. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell death and differentiation. 2016;23(6):962–978. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell death and differentiation. 1998;5(7):551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 8.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 9.Ramani K, Miclea RD, Purohit VS, Mager DE, Straubinger RM, Balu-Iyer SV. Phosphatidylserine containing liposomes reduce immunogenicity of recombinant human factor VIII (rFVIII) in a murine model of hemophilia A. Journal of pharmaceutical sciences. 2008;97(4):1386–1398. doi: 10.1002/jps.21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234(3):466–468. [PubMed] [Google Scholar]
- 11.Schneider JL, Dingman RK, Balu-Iyer SV. Lipidic Nanoparticles Comprising Phosphatidylinositol Mitigate Immunogenicity and Improve Efficacy of Recombinant Human Acid Alpha-Glucosidase in a Murine Model of Pompe Disease. Journal of pharmaceutical sciences. 2018;107(3):831–837. doi: 10.1016/j.xphs.2017.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purohit VS, Ramani K, Kashi RS, Durrani MJ, Kreiger TJ, Balasubramanian SV. Topology of factor VIII bound to phosphatidylserine-containing model membranes. Biochimica et biophysica acta. 2003;1617(1–2):31–38. doi: 10.1016/j.bbamem.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 13.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaitonde P, Ramakrishnan R, Chin J, Kelleher RJ, Bankert RB, Balu-Iyer SV. Exposure to Factor VIII Protein in the Presence of Phosphatidylserine Induces Hypo-responsiveness toward Factor VIII Challenge in Hemophilia A Mice. J Biol Chem. 2013;288(24):17051–17056. doi: 10.1074/jbc.C112.396325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider JL, Balu-Iyer SV. Phosphatidylserine Converts Immunogenic Recombinant Human Acid Alpha-Glucosidase to a Tolerogenic Form in a Mouse Model of Pompe Disease. Journal of pharmaceutical sciences. 2016;105(10):3097–3104. doi: 10.1016/j.xphs.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handbook of clinical neurology. 2014;122:173–189. doi: 10.1016/B978-0-444-52001-2.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nature protocols. 2006;1(4):1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- 19.Rangachari M, Kuchroo VK. Using EAE to better understand principles of immune function and autoimmune pathology. Journal of autoimmunity. 2013;45:31–39. doi: 10.1016/j.jaut.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purohit VS, Ramani K, Sarkar R, Kazazian HH, Jr, Balasubramanian SV. Lower inhibitor development in hemophilia A mice following administration of recombinant factor VIII-O-phospho-L-serine complex. J Biol Chem. 2005;280(18):17593–17600. doi: 10.1074/jbc.M500163200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramakrishnan R, Davidowitz A, Balu-Iyer SV. Exposure of FVIII in the Presence of Phosphatidyl Serine Reduces Generation of Memory B-Cells and Induces Regulatory T-Cell-Mediated Hyporesponsiveness in Hemophilia A Mice. Journal of pharmaceutical sciences. 2015;104(8):2451–2456. doi: 10.1002/jps.24513. [DOI] [PubMed] [Google Scholar]
- 22.Ramakrishnan R, Balu-Iyer SV. Effect of Biophysical Properties of Phosphatidylserine Particle on Immune Tolerance Induction Toward Factor VIII in a Hemophilia A Mouse Model. Journal of pharmaceutical sciences. 2016;105(10):3039–3045. doi: 10.1016/j.xphs.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Doffek K, Sugg SL, Shilyansky J. Phosphatidylserine regulates the maturation of human dendritic cells. Journal of immunology. 2004;173(5):2985–2994. doi: 10.4049/jimmunol.173.5.2985. [DOI] [PubMed] [Google Scholar]
- 24.Gaitonde P, Peng A, Straubinger RM, Bankert RB, Balu-Iyer SV. Phosphatidylserine reduces immune response against human recombinant Factor VIII in Hemophilia A mice by regulation of dendritic cell function. Clinical immunology. 2011;138(2):135–145. doi: 10.1016/j.clim.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. The Journal of clinical investigation. 1998;101(4):890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. The Journal of cell biology. 2001;155(4):649–659. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Ma HM, Kukita T, Nakanishi Y, Nakanishi H. Phosphatidylserine-containing liposomes inhibit the differentiation of osteoclasts and trabecular bone loss. Journal of immunology. 2010;184(6):3191–3201. doi: 10.4049/jimmunol.0803609. [DOI] [PubMed] [Google Scholar]
- 28.Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. The Journal of experimental medicine. 2002;196(8):1091–1097. doi: 10.1084/jem.20021215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marin-Gallen S, Clemente-Casares X, Planas R, Pujol-Autonell I, Carrascal J, Carrillo J, Ampudia R, Verdaguer J, Pujol-Borrell R, Borras FE, Vives-Pi M. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes. Clinical and experimental immunology. 2010;160(2):207–214. doi: 10.1111/j.1365-2249.2009.04082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos GC, Fernandes D, Charao CT, Souza DG, Teixeira MM, Assreuy J. Apoptotic mimicry: phosphatidylserine liposomes reduce inflammation through activation of peroxisome proliferator-activated receptors (PPARs) in vivo. British journal of pharmacology. 2007;151(6):844–850. doi: 10.1038/sj.bjp.0707302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(5):1827–1832. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pujol-Autonell I, Mansilla MJ, Rodriguez-Fernandez S, Cano-Sarabia M, Navarro-Barriuso J, Ampudia RM, Rius A, Garcia-Jimeno S, Perna-Barrull D, Martinez-Caceres E, Maspoch D, Vives-Pi M. Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis. Nanomedicine. 2017;12(11):1231–1242. doi: 10.2217/nnm-2016-0410. [DOI] [PubMed] [Google Scholar]
- 33.Pujol-Autonell I, Serracant-Prat A, Cano-Sarabia M, Ampudia RM, Rodriguez-Fernandez S, Sanchez A, Izquierdo C, Stratmann T, Puig-Domingo M, Maspoch D, Verdaguer J, Vives-Pi M. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PloS one. 2015;10(6):e0127057. doi: 10.1371/journal.pone.0127057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD, Heffern CT, Henderson JM, Meron M, Lin B, Roux B, Schlossman ML, Steck TL, Lee KY, Adams EJ. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(15):E1463–1472. doi: 10.1073/pnas.1320174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsura T, Togawa A, Kai M, Nishida T, Nakada J, Ishibe Y, Kojo S, Yamamoto Y, Yamada K. The presence of oxidized phosphatidylserine on Fas-mediated apoptotic cell surface. Biochimica et biophysica acta. 2005;1736(3):181–188. doi: 10.1016/j.bbalip.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. The Journal of experimental medicine. 2010;207(9):1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendes Wanderley JL, Costa JF, Borges VM, Barcinski M. Subversion of Immunity by Leishmania amazonensis Parasites: Possible Role of Phosphatidylserine as a Main Regulator. J Parasitol Res. 2012;2012:981686. doi: 10.1155/2012/981686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Czuczman MA, Fattouh R, van Rijn JM, Canadien V, Osborne S, Muise AM, Kuchroo VK, Higgins DE, Brumell JH. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature. 2014;509(7499):230–234. doi: 10.1038/nature13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1(4):256–268. doi: 10.1158/2326-6066.CIR-13-0073. [DOI] [PubMed] [Google Scholar]
- 40.Kelleher RJ, Jr, Balu-Iyer S, Loyall J, Sacca AJ, Shenoy GN, Peng P, Iyer V, Fathallah AM, Berenson CS, Wallace PK, Tario J, Odunsi K, Bankert RB. Extracellular Vesicles Present in Human Ovarian Tumor Microenvironments Induce a Phosphatidylserine-Dependent Arrest in the T-cell Signaling Cascade. Cancer Immunol Res. 2015;3(11):1269–1278. doi: 10.1158/2326-6066.CIR-15-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]