

Abstract
Although strip films are a promising platform for delivery of poorly water-soluble drug particles via slurry casting, the effect of critical materials attributes (CMAs), e.g., super-disintegrants on critical quality attributes (CQAs), including film disintegration time (DT), remains under-explored. A two-level factorial design is considered to examine the impact of the super-disintegrant (SDI) type (sodium starch glycolate and croscarmellose sodium), their amount, and film thickness. SDIs were used with hydroxypropyl methylcellulose (HPMC-E15LV) and glycerin solutions along with viscosity matching. Fenofibrate, a model poorly water-soluble drug, was micronized and surface modified (MC-FNB) via fluid energy milling. Significant decreases in film DT, measured using three different methods, were observed due to the addition of SDIs. Percentage reduction in DT was a strong function of SDI amount, and thinner films disintegrated faster. Films with either higher SDI concentrations (>9%) or films under 80 μm, exhibited fast DT (<180s, Ph. Eur.). All thin films (50–60 μm) exhibited immediate release (>80% in 10 min). All films achieved good content uniformity, except for those with the lowest amount of SDI, attributed to insufficient viscosity and thickness non-uniformity due to the SDI. Finally, all films achieved adequate mechanical properties, notwithstanding minor negative impact of SDIs.
Keywords: polymeric drug delivery systems, content uniformity, microparticles, excipients, viscosity, dissolution, mechanical properties
Graphical abstract

1. Introduction
There is continued interest in strip films for oral drug delivery thanks to their notable advantages over traditional solid dosage forms, such as large surface area and dosing accuracy, as well as increased flexibility and portability as compared to most orodispersible tablets (ODTs).1 One of the most attractive aspects of films is their quick disintegration/dissolution after contact with saliva in the mouth, leading to improved acceptance and compliance among pediatric, geriatric, and dysphagic patients.1 More recently, various particle engineering techniques have been applied to develop films via slurry casting in contrast to solution casting, establishing polymeric strip films as a robust platform to embed relatively high drug loading (40 wt%) of poorly water-soluble drug nano or microparticles to achieve fast drug release, excellent drug content distribution and uniformity, and mechanical properties accompanied by fast dissolution.2-5 Unfortunately, those papers did not specifically examine the disintegration time and how it may be impacted by critical material attributes. Clearly, determination of film disintegration time is essential in the development and evaluation of this new dosage form. Thus, it is very essential to gain a systemic understanding of the disintegration behavior of films laden with poorly water-soluble drug particles via slurry casting.
Strip films benefit from access to a wide variety of usable excipients which can be used to control film properties without sacrificing the integrity of the format.1,6,7 Recent studies have begun to explore different film formers, film fillers or combinations used for the preparation of rapidly disintegrating oral films.8-10 Visser et al. observed that films with different film forming materials disintegrated in 20–206 s, such as hypromellose (HPMC), hydroxypropyl cellulose (HPC), sodium alginate (SA), and carbomer 974P. Garsuch et al. prepared very thin films (32–55 μm) with several types of sodium carboxymethyl cellulose (CMC), several types of hydroxypropyl methylcellulose (HPMC), a synthetic copolymer of polyethylene glycol-polyvinyl alcohol (Kollicoat IR) and SA, and these rather thin films achieved very fast disintegration time (<37 s). However, these papers focus on films composed of water-soluble drugs via solution casting.
Super-disintegrants (SDIs) are a class of compounds which primarily aid in the rapid disintegration of orally disintegrating tablets.11,12 This class of SDIs, e.g., sodium starch glycolate (SSG), croscarmellose sodium (CCS) and crospovidone (CP) has been shown to be effective at excipient concentrations as low as 2 to 10% in ODTs.13 However, it is not fully clear how these excipients function when used in other applications, and the exact advantages they render when employed.13 Azad et al. and Bilgili et al. showed that the SDIs serve as a novel class of stabilizers for nano-suspensions or dispersants of poorly water-soluble drugs in nanocomposites, and Gordon et al. showed that SDIs promote dissolution of poorly water-soluble drugs when incorporated intra-granularly.14-16 A few studies have incorporated SDIs to enhance dissolution rate of drugs from films. For example, Sagban and Ismail demonstrated that solution cast, water-soluble drug loaded films prepared with 5% CP have shorter disintegration time, dissolution time, as well as reduced mechanical properties.17 Susarla et al. incorporated SSG, CCS, and CP primarily as viscosity enhancers to improve drug content uniformity of films containing griseofulvin (GF) nanoparticles, and found that they accelerate dissolution times, compared to films formed using alternate methods of viscosity enhancements.18 Faster drug dissolution of donepezil hydrochloride from films containing SDIs was also reported in another work.19 Unfortunately, they did not examine disintegration time for such films.18 Ideally, mechanical properties such as a moderately high tensile strength and elongation at break, low Young's modulus, along with a short disintegration time are preferred for fast disintegrating films.20,21 To the best of our knowledge, none of the previous papers has investigated the effect of incorporating SDIs on reduction of disintegration time while maintaining proper mechanical properties, and the effect of SDI concentration on overall film properties.
Three major mechanisms of disintegration have been proposed for tablets, which include capillary action (wicking), swelling, and deformation (strain recovery).13 In principle, these mechanisms are not independent and are often synergistic, leading to the break-up of the tablet matrix and disruption of particle-particle bonds.13 Similarly, imbibition of water is also the first stage of the disintegration process for film matrix and thus capillary action is of primary importance, as it will affect all the other disintegration mechanisms. Thus, the thickness of films and its effect on the water penetration path could impact disintegration time significantly. The film thickness has been claimed to impact the dissolution time of films laden with poorly water-soluble drugs.22 As film thickness increased from 26.2 μm to 123.2 μm, drug dissolution slowdown was demonstrated in pullulan films with xanthan gum as a viscosity enhancer.22 Therefore, it would be beneficial to explore the possible effect of decreasing thickness on reducing disintegration time and mechanical properties.
So far neither the US Pharmacopoeia nor the European Pharmacopoeia has defined a specific disintegration test or criteria of disintegration time for films. Generally, the frame and Petri dish methods are the two most popular disintegration tests used in literature.8 Mazumder et al. promoted a modified Petri dish method by incorporating an orbital shaker to mimic oral movement.23 In this work, disintegration time of films was obtained by comparing within USP tablet method, frame, and Petri dish methods. Additionally, the effect of shaking speed on the disintegration time of films using the modified Petri dish method was also examined.
With the groundwork in place for films as fast disintegrating/dissolving drug delivery system, it is necessary to gain a systemic understanding of the effect of incorporating varying amounts of SDIs as viscosity enhancers, on the intermediate critical quality attributes (I-CQAs) (thickness) and the critical quality attributes (CQAs) of films, particularly film disintegration time (DT) and mechanical properties. Therefore, in the present study, a two-level factorial design was considered to examine the impact of the super-disintegrant (SDI) type (SSG and CCS), their amount, and film thickness on CQAs. Different amounts of SSG or CCS were incorporated in the low molecular weight hydroxypropyl methylcellulose (HPMC-E15LV) polymer solution containing surface modified, micronized fenofibrate (MC-FNB), a model poorly water-soluble drug. Two different viscosity ranges, low and high, were attained for each key formulation following the viscosity matching approach.24 The resulting film precursors were cast and dried to form films loaded with MC-FNB at two different thickness ranges: low (50–60 μm) and high (90–100 μm). A medium thickness range (70–80 μm) was also considered for assessing film DT. These films were then characterized for drug content and uniformity, film structure, disintegration time, moisture content, mechanical properties, and drug dissolution rate. It is expected that this study will allow testing the main hypothesis that SDIs may provide ability to achieve fast disintegration (<180s, Ph. Eur.) of films laden with poorly water-soluble drug microparticles while retaining good content uniformity and mechanical properties.
2. Materials and Methods
2.1 Materials
Fenofibrate (FNB; Jai Radhe Sales, Ahmedabad, India) was selected as a model BCS Class II poorly water-soluble drug. Pharmaceutical grade amorphous hydrophilic silica (M5P, Cabot Corporation, MA, USA) with a primary particle size of 16 nm was used as the coating material for milled coated FNB (MC-FNB) particles to enhance wettability and reduce agglomeration of micronized FNB particles. Low molecular weight hydroxypropyl methylcellulose (HPMC; Methocel E15 Premium LV, Mw∼40,000, The Dow Chemical Company, Midland, MI) and glycerin (Sigma–Aldrich, Saint Louis, MO, USA) were used as the film former and the film plasticizer, respectively. Sodium starch glycolate (SSG; Primojel, Princeton, NJ, USA) and croscarmellose sodium (CCS; Ac-Di-Sol, Newark, DE, USA) were used as a film filler and viscosity enhancer. Sodium dodecyl sulfate (SDS) (Sigma–Aldrich, Saint Louis, MO, USA) was used as the surfactant in the dissolution media. All materials were used as received, except for the MC-FNB, for which the procedure is described in the next section.
2.2 Preparation of milled coated FNB particles
Two-step dry milling of coated FNB powders was performed following protocols established in previous work, and utilized pre-mixing using Laboratory Resonant Acoustic Mixer (LabRAM, Resodyn Acoustic Mixers, Inc., Butte, MT, USA), followed by continuous milling in a fluid energy milling (FEM; qualification model, Sturtevant Inc., Hanover, MA, USA).25 For pre-mixing in LabRAM, as-received FNB powder (97 g) and M5P silica (3 g) were added to a plastic cylindrical jar that was shaken at a frequency of 61 Hz with an acceleration of 70 G for 5 min to ensure that the M5P silica particles were well attached and distributed among as-received FNB (AR-FNB) particles. Simultaneous micronization and surface modification of FNB (MC-FNB) particles was carried out using the FEM. Powder feeding rate was controlled by a volumetric feeder (Model 102M, Schenck Accurate, WI, USA) at 1 g/min. A constant feeding pressure (FP) of 45 psi and a constant grinding pressure (GP) of 40 psi were maintained throughout the milling process. The milled and coated FNB particles are referred to as MC-FNB. The particle size was reduced from 9.43 μm (AR-FNB) to 4.20 μm (MC-FNB) after the simultaneous micronization and surface modification process.
2.3 Preparation of film precursor and film laden with MC-FNB particles
The slurry casting method for films containing nano/micro drug particles has been established previously.2,18 Slurry casting involves the preparation of a polymer solution followed by addition of drug particles. The resulting film precursor is passed through a Doctor Blade (3700, Elcometer, MI, USA) for film casting, followed by drying.
In order to examine the impact of SDIs, their amounts, and film thickness, a two-level factorial design was employed. The response variable of interest is the film disintegration time (DT). However, in order to avoid the confounding effect on DT of the film precursor viscosity, which is an intermediate response variable that may have an impact on film mechanical strength and hence DT, viscosity matching had to be considered, as was suggested in previous literature.24 The experimental design followed is presented in Table S1 in the supplementary material.
Polymer solution was prepared by adding corresponding amounts of glycerin (3.3 g) and HPMC-E15LV (10 g) into 100 ml deionized water at 30–40 °C and 80–90 °C, respectively. A sufficient amount of time was allowed for the polymer to dissolve completely without any clumps, and the polymer solution was cooled down to room temperature while being stirred continuously. This polymer solution was referred to as F-baseline with a low shear viscosity of 1800–2000 cP. Next, a “viscosity matching” step was used in which two additional target viscosity ranges were identified for this investigation: 3500–4500 cP (low) and 9500–10500 cP (high). The careful selection of formulations without SDIs, and SDIs amounts for formulations with SDIs, is based on the F-baseline formulation, shown in Table 1 and explained below. For a fair comparison, the ratio of polymer/plasticizer/drug particles was maintained within each viscosity range.
Table 1. Composition and viscosity of polymer solutions and film precursors.
| Exp # | HPMC-E15 (g) | Glycerin (g) | DI water (ml) | Additive (g) | Viscosity of polymer solution (cP) | Viscosity of film precursor (cP) |
|---|---|---|---|---|---|---|
| F-baseline | 10 | 3.3 | 100 | 0.00 | 1,820 ± 275 | 3050 ± 234 |
| F-1 | 10 | 3.3 | 80 | 0.00 | 3,470 ± 149 | 5106 ± 209 |
| F-2 | 10 | 3.3 | 70 | 0.00 | 9,980 ± 541 | 11507 ± 427 |
| F-SSG-1 | 10 | 3.3 | 100 | 0.75 | 3,260 ± 389 | 5407 ± 507 |
| F-CCS-1 | 10 | 3.3 | 100 | 1.40 | 4,170 ± 365 | 5293 ± 197 |
| F-SSG-2 | 10 | 3.3 | 100 | 1.75 | 10,830 ± 550 | 12687 ± 437 |
| F-CCS-2 | 10 | 3.3 | 100 | 2.75 | 9,570 ± 70 | 11674 ± 354 |
Polymer/drug ratio was 4:1 for all formulations, drug amount is 2.5g for all formulations
In general, viscosity of polymer solutions may be varied through adjusting the amount of polymer or viscosity enhancers while fixing the amount of water. Here, a slightly different protocol was followed. According to the F-baseline, the formulations without SDIs, F-1 and F-2 were prepared by reducing water amount from 100ml to 80 ml and 70 ml to achieve low and high viscosity levels respectively, and adding the same amount of HPMC and glycerin. For formulations with SDIs, F-SSGs and F-CCSs, appropriate amounts of SSG or CCS were added to polymer solution (F-baseline) to match low and high viscosity levels. SDIs were added as the polymer solution cooled down to 50–60 °C, with continued stirring for additional 2 h. These HPMC-SDI mixtures are aqueous micro-particulate suspensions referred to as F-SSG-1, F-CCS-1 (low viscosity) and F-SSG-2, F-CCS-2 (high viscosity). Once again, the viscosity levels of the polymer solution before adding drug powder are provided in Table 1. Each of the resulting polymer solutions were mixed with MC-FNB (2.5 g) using a planetary mixer, called Thinky mixer (ARE-310, THINKY, CA, USA), for 10 min at 2100 rpm, followed by 3 min of de-aeration at 2200 rpm to form a homogeneous film precursor. To further ensure that no bubbles were present after deaeration, the precursor suspension was left 8–12 h at room temperature before casting. As a side note, initial investigation considered crospovidone (CP) also, but unfortunately it precipitated in the polymer solution and film precursor during the de-aeration process. As a result, CP was not considered for further investigations due to the miscibility issue of CP and HPMC polymer solution.
The investigation of the effect of film thickness on the film properties and disintegration time was also carried out. Hence, the wet casting thickness (provided in supplementary Table S2) was varied to achieve two desired thickness ranges of dry films (50–60 μm and 90–100 μm), for each formulation. In addition, films having thickness of 70–80 μm were also used for disintegration testing. The final deaerated film precursors were cast onto a plastic substrate (Scotchpak™ 9744, 3M, MN, USA) using a Doctor Blade (3700, Elcometer, MI, USA), and dried inside a laboratory-scale tape caster (TC-71LC, HED International, NJ, USA). The dry film was trimmed into dimensions of 8 cm × 15 cm pieces for further analysis.
2.4 Characterization methods
2.4.1 Viscosity
The apparent shear viscosities of polymer solutions and film precursors were measured with a rheometer (R/S-CC+, Brookfield Engineering, MA, USA) equipped with a shear rate-controlled coaxial cylinder (CC25), and a temperature-controlled water jacket (Lauda Eco, Lauda-Brinkmann LP, NJ, USA). Both were recorded at a low shear rate (2.2 s-1) and 25 ± 0.5 °C, representing the low-shear rate imparted during film casting at room temperature.26,27
2.4.2 Digital optical microscope
A digital optical microscope (Axio Lab.A1, Carl Zeiss, Germany) was used to evaluate the surface morphology of films. Sample films of F-1, F-SSG-1, and F-CCS-1 were cut into strips with dimensions 2 cm × 3 cm - a common commercial strip film size, and imaged at 5× magnification.
2.4.3 Scanning electron microscope (SEM)
A field emission scanning electron microscope (FESEM) (LEO1530VP GEMINI; Carl Zeiss Inc., MA, USA) was used to examine the cross-sectional structure of films (F-1, F-SSG-1, and F-CCS-1). A rectangular film sample with dimensions 0.3 cm × 0.5 cm was placed vertically on an aluminum stub with carbon tape and carbon coated via a sputter coater (EMS150T ES; Quorum Technologies Ltd, Laughton, UK) before imaging.
2.4.4 Determination of drug content and uniformity in films
A previously established protocol for determining the drug content and uniformity of films was followed.28 Ten circular samples ∼0.72 cm2 in area were punched out randomly from film samples and dissolved in 100 ml of 7.2 mg/ml SDS solution with continuous stirring for a minimum of 3 h. A Thermo Scientific Evolution 300 UV-vi spectrophotometer (Thermo Fisher Scientific Inc., MA, USA) was used to measure the UV absorbance of each sample using the appropriate wavelength of 290 nm for each dissolved sample, and then the concentration was calculated according to an established calibration curve. The thickness was measured using a digital micrometer (Mitutoyo Corporation, Kanagawa, Japan) with an accuracy of 0.001 mm (1 μm) at 10 random points of an 8 cm × 15 cm size film to obtain a reliable estimate of the film thickness. The average and standard relative deviation (RSD) values of film thickness, drug dose per unit area (mg/cm2), and weight percentage of drug in the film (wt% FNB) of ten samples were recorded. Moreover, percentage of label claim (%LC) and the acceptance value (AV) were also calculated to determine the content uniformity of films.29,30
2.4.5 Disintegration time with three test methods
The in-vitro disintegration time was estimated by three methods: USP, frame, and Petri dish. In addition, the modified Petri dish method with orbital shaker was also used to evaluate the effect of shaking speed and the pH of disintegration media on the disintegration time of films. The summary of main features of the disintegration test methods are presented in Table 2. The average values of disintegration time and standard deviations were calculated over six samples.
Table 2. Summary of disintegration test methods.
| USP method | Frame method | Petri dish method | Modified petri dish method | |
|---|---|---|---|---|
| Number of samples | 6 | 6 | 6 | 6 |
| Amount of media | 600 ml | 0.15 ml | 4 ml | 4 ml |
| Type | dynamic | static | static | dynamic |
| End-point | visual | visual | visual | visual |
USP method
The disintegration time of the films was determined in 600 ml of deionized water (DI water) at 37 ± 0.5 °C, using a USP disintegration test apparatus (DT 2, Sotax, Switzerland).31
Petri dish method
One circular sample ∼0.72 cm2 in area was placed in 4 ml of DI water in a petri dish. The time taken for the film to disintegrate into tiny particles was measured.8 Figs. 1 (a)–(b) show the onset and complete disintegration points.
Fig. 1.

Disintegration of film: Petri dish method: (a) at onset and (b) after complete disintegration; frame method: (c) at onset and (d) after complete disintegration.
Frame method
A rectangular piece of film (1.5 cm × 3 cm) was held in a slide frame and 0.15 ml of DI water was placed on film surface using a pipette. The time taken for the water to penetrate through the film was recorded.8 Figs. 1 (c)–(d) show the onset and complete disintegration points.
Modified Petri dish method
In order to imitate the movement of the tongue, a modified Petri dish method involving an orbital shaker proposed by Mazumder et al.23, was used in this study. The petri dish was placed into an orbital shaker maintained at a temperature of 37 ± 0.5 °C, with three shaking speeds (0 rpm, 35 rpm and 65 rpm). In order to identify the influence of media pH, DI water (pH=5.7) and a phosphate buffer solution (PBS, pH=6.8) were used. The PBS was prepared according to the published protocols.32 The time taken for the film to disintegrate into tiny particles was recorded.
2.4.6 Mechanical properties of the films
Film mechanical properties were measured using a TA-XT Plus Texture Analyzer (Stable Microsystems, UK). 3–5 rectangular film strips with dimensions of 5 cm × 1.5 cm were held between two clamps positioned at an initial distance of 3 cm. The films were then subjected to an extension (at a constant speed of 1 mm/s) until the breaking point (i.e., tensile failure). Tensile strength (TS), Young's modulus (YM), and percentage elongation (E%) were calculated from the stress versus strain data. The average and standard deviation of TS, YM and E% were computed over three film strips.
2.4.7 Thermo-gravimetric analysis (TGA)
TGA was performed using a TGA/DSC1/SF STARe system (Mettler Toledo Inc., OH, USA). A ∼8 mg film sample was placed in a standard ceramic crucible and heated under nitrogen flow from 25 °C to 150 °C at a constant rate of 10 °C/min, maintained at 150 °C for 15 min, heated to 250 °C at a rate of 10 °C/min, and finally cooled back to 25 °C at a rate of −10 °C/min.28
2.4.8 Differential scanning calorimetry (DSC)
A differential scanning calorimeter (DSC, Mettler Toledo, Inc., OH, USA) was used to determine the melting point of AR-FNB and MC-FNB particles in the films. In a standard aluminum pan, a ∼8 mg film sample was heated under nitrogen flow from 25 °C to 150 °C at a constant rate of 10 °C/min, and then cooled back to 25 °C at a rate of −10 °C/min.
2.4.9 X-ray diffraction (XRD)
X-ray diffraction was performed to determine the crystallinity of AR-FNB, placebo films and drug particles in the films. Diffraction patterns were acquired to analyze the amorphous/crystalline behavior of these samples using Philips X'Pert (Almelo, Netherlands), scanning a 2θ angle in the range 5–35° (0.01° step).
2.4.10 Dissolution
Dissolution experiments of films containing MC-FNB particles were performed following previously established protocols using an automated flow-through cell dissolution apparatus (USP IV, Sotax, Switzerland), equipped with six flow-through cells (Ø 22.6 mm) and 0.2 μm HT Tuffryn® disc filters (Pall Corporation, NY, USA).33 A ruby ball (Ø 5 mm) and 3 g of glass beads (Ø 1 mm) were placed at the bottom of the cone to ensure uniform flow of dissolution media entering the cell. Punched circular samples from each film with an area of ∼0.72 cm2 were horizontally positioned in the cells with 2 g of glass beads on the top. The temperature of cells was maintained at 37 ± 0.5 °C and 100 ml dissolution media (7.2 mg/ml SDS aqueous solution) was circulated at a flow rate of 16 ml/min. Dissolution results are reported as percentage FNB released as a function of time for an average of six circular samples from each type of film.
2.4.11 Statistical analysis
Basic calculations were performed using Microsoft Excel (Microsoft Office 2010, USA). Results for disintegration time and mechanical properties are expressed as mean ± SD (standard deviation) while content uniformity results are expressed as mean with RSD% (relative standard deviation). In order to analyze the impact of the addition of SDIs and film thickness on the disintegration time and mechanical properties, Student's t-test and General linear model of ANOVA were used and the results (p value and main factor plot) were reported (Minitab® 18).
3. Results and discussion
3.1 Viscosity of polymer solutions and film precursors
As discussed in Section 2.3, the viscosity match approach was necessary in lieu of changing polymer amount to keep the ratio of polymer/plasticizer/drug constant. Appropriately selecting amounts of DI water and SDIs within each viscosity range (3500–4500 cP and 9500–10500 cP) was expected to minimize the impact of polymer solution viscosity on film properties.24 As discussed before and shown in Table 1, the water amount was reduced from 100 ml (F-baseline) to 80 ml (F-1) and 70 ml (F-2), leading to viscosity increases from 1820 cP (F-baseline) to 3470 cP (low) and 9980 cP (high) for F-1 and F-2, respectively. Adding selected amount of SSG or CCS to F-baseline according to their swelling capacities18 caused increases in viscosity, consequently matching low and high viscosity levels. As a result, viscosities of F-SSG-1 and F-CCS-1 are 3260 cP and 4170 cP (low), and the viscosities of F-SSG-2 and F-CCS-2 are 10830 cP and 9570 cP (high). The proposed viscosity matching approach generally worked well except for two cases where the achieved viscosity values were slightly outside the desired ranges. Thanks to relatively low drug loading (13–15%), the viscosity of film precursors only increased by 1000–2000 cP after adding drug microparticles compared to polymer solutions of all formulations; shown in Table 1.
3.2 Multi-faced characterization of films laden with MC-FNB particles
3.2.1 Optical digital images and SEM images of films
Figs. 2 (a)–(c) and Figs. 3 (a)–(c) show the surface and cross-sectional images of films made from F-1, F-SSG-1, and F-CCS-1. Optical images from Figs. 2 (a)–(c) showed distinguishable differences between the HPMC films and HPMC-SDI films. Film made from F-1 exhibited a homogenous and smooth surface (Fig. 2 (a)). On other hand, some aggregates and uneven structure in the films of F-SSG-1 and F-CCS-1 can be observed (Figs. 2 (b)–(c)), which could be caused by the intrinsically larger size and aggregation of insoluble SDIs particles (red arrows). SEM images (Figs. 3 (a)–(c)) also showed that these films present quite different morphologies as a result of embedded SDI particles. Films of F-1 show a more homogenous, uniform and denser cross-sectional structure (Fig. 3 (a)). Whereas, Figs. 3 (b)–(c) revealed embedded SDIs particles (red arrows) and small pores in the cross-sectional structures of films. SEM images of films with SDIs are in good agreement with Susarla et al..18 Furthermore, these images could shed light on the poor performance in mechanical properties of films with SDIs (Section 3.2.7).
Fig. 2.

Digital optical images of film surface: (a) F-1, (b) F-SSG-1, and (c) F-CCS-1. Scale bars are 200 μm each.
Fig. 3.

Cross-sectional SEM images of films: (a) F-1, (b) F-SSG-1, and (c) F-CCS-1.
3.2.2 Content uniformity
Recent papers demonstrate that films containing uniformly dispersed, poorly water-soluble drug nano or micro particles have very low relative standard deviation (RSD) values relevant to drug content uniformity.5,28 Susarla et al. have shown that films made from high viscosity polymer solution containing SDIs (10,000–20,000 cP) exhibited very low RSD values of drug content and uniformity, indicating uniformly distributed nanoparticles in dry films.18 The present results are designed to examine if such trends for drug content uniformity continue for polymer solutions with viscosities lower than 10,000 cP, specifically, at two viscosity ranges, 3500–4500 cP and 9500–10500 cP). Table 3 (a) presents the content uniformity of films at a thickness of 50–60 μm for all formulations including the average and relative standard deviation (RSD) values for thickness, drug amount per unit area, weight percentage of drug, mean percentages of label claim (mean %LC) and acceptance value (AV). Whereas the results of films at higher thickness of 90–100 μm are provided in Table 3 (b).
Table 3(a). ontent uniformity of films at the thickness of 50–60 μm.
| Exp # | Film thickness (μm) | RSD (%) | API dose 2 (mg/cm2) | RSD (%) | API wt% | RSD (%) | Mean %LC | Acceptance value (AV) |
|---|---|---|---|---|---|---|---|---|
| F-baseline | 55 | 5.2 | 1.0 | 2.6 | 15.3 | 5.9 | 101 | 14 |
| F-1 | 59 | 4.5 | 1.1 | 3.6 | 14.8 | 0.7 | 97 | 3 |
| F-2 | 58 | 4.9 | 1.1 | 4.6 | 15.3 | 1.4 | 101 | 3 |
| F-SSG-1 | 52 | 7.1 | 0.9 | 7.2 | 14.4 | 8.7 | 99 | 21 |
| F-CCS-1 | 53 | 5.1 | 0.8 | 5.9 | 13.4 | 5.1 | 95 | 14 |
| F-SSG-2 | 51 | 5.3 | 0.8 | 2.7 | 14.1 | 2.4 | 103 | 7 |
| F-CCS-2 | 50 | 4.7 | 0.6 | 2.8 | 13.4 | 3.2 | 103 | 10 |
Table 3(b). Content uniformity of films at the thickness of 90-100 μm.
| Exp # | Film thickness (μm) | RSD (%) | API dose 2 (mg/cm2) | RSD (%) | API wt% | RSD (%) | Mean %LC | Acceptance value (AV) |
|---|---|---|---|---|---|---|---|---|
| Exp # | thickness (μm) | RSD (%) | API dose 2 (mg/cm2) | RSD (%) | API wt% | RSD (%) | Mean %LC | Acceptance value (AV) |
| F-baseline | 94 | 4.3 | 1.6 | 5.2 | 15.8 | 2.9 | 104 | 10 |
| F-1 | 91 | 5.9 | 1.6 | 4.6 | 14.8 | 3.2 | 98 | 8 |
| F-2 | 95 | 5.0 | 1.7 | 3.9 | 13.8 | 2.0 | 90 | 12 |
| F-SSG-1 | 102 | 6.5 | 1.3 | 7.2 | 14.9 | 5.2 | 103 | 14 |
| F-CCS-1 | 101 | 5.6 | 1.2 | 5.7 | 13.6 | 3.6 | 97 | 13 |
| F-SSG-2 | 90 | 4.0 | 1.2 | 5.8 | 12.4 | 2.7 | 91 | 14 |
| F-CCS-2 | 101 | 4.0 | 1.3 | 4.0 | 13.7 | 2.4 | 105 | 10 |
All HPMC films exhibited good RSD values (<6%) in terms of thickness, drug dose per unit area, and FNB (drug) loading, as well as acceptable AV values (<15) and mean %LC close to 100%. As anticipated, films made from F-baseline, which has the lowest viscosity led to higher RSD value compared to F-1 and F-2. Nonetheless, those values still meet the content uniformity criteria of the United State Pharmacopeia (USP) and European Pharmacopeia (Ph. Eur.), demonstrating the capability of forming films containing uniformly dispersed surface modified micronized drug particles, even when a low viscosity HPMC polymer solution (<5000) was used.
HPMC-SDI films, on the other hand, have higher RSD values for all cases, as well as higher AV values compared to F-1 and F-2 at the same viscosity ranges. This could be mainly attributed to thickness non-uniformity due to the uneven distribution of the SDI particles, confirmed by digital optical and SEM images. This contradicts the claim that the addition of gums as viscosity enhancers promotes better thickness uniformity.18 Another surprising set of results, as compared with Susarla et al.,18 is that viscosity values lower than 10,000 cP led to very good content uniformity, except when the viscosity fell below 4,000 cP. In part, this unique behavior of SDIs may stem from the fact that the d90 particle sizes of the SDI particles (23.30 μm for CCS and 25.03 μm for SSG) may also play some role for films at lower viscosity formulations. Interestingly, thinner films made from F-SSG-1 have highest RSD values (>6%) and also a high AV number (>15). All other HPMC-SDI films still meet the content uniformity criteria of USP and Ph. Eur.. Therefore, these results suggest that the viscosity of formulations that include SDIs may need to be above ∼4000 cP to achieve good content uniformity of drug in films. In this respect, both polymer solution viscosity and the addition of insoluble SDIs, which are correlated with the drug particle dispersion in the films, have influence on the drug content uniformity.
Interestingly, notwithstanding some issues with the AV results, the films at a thickness of 50–60 μm did not have higher RSD values of thickness compared to 90–100 μm films. For thicker films of 90–100 μm, excellent drug content RSD values (<6%), good mean %LC (91–105%) levels, and AV results (<15) are observed. However, as was the case for thinner films, RSD values of thickness and API dose per unit area of F-SSG-1 are just above acceptable levels, mostly likely due to its lower polymer solution viscosity (Table 3 (b)). In all of these results, overall good content uniformity may be attributed to the good dispersion and mixing of surface modified FNB microparticles as well as the high shear rate provided by the Thinky mixer. As a result, the thickness variations did not have any significant impact on the drug content uniformity of films.
3.2.3 Moisture content of films
Presence of SDIs could have an effect on the moisture content after drying due to their tendency to absorb water. Therefore, TGA analysis was performed on films of all formulations and the results are presented in Fig. 4. The first mass loss for films can be ascribed to the free or bound water content. Film made from F-baseline exhibited greater weight loss (7.7%) compared to films made from F-1 and F-2 (6.5%), which may due to higher water content in the polymer solution after the same drying time. In contrast, the weight loss of films prepared from HPMC-SDI formulations had lower moisture content ranged in 5.4–6.6%, indicating the incorporation of insoluble/swelling SDIs does not significantly hinder the drying efficiency that could cause an increase in moisture content of dry films. The second mass loss was due to decomposition of glycerin, as reported in the literature.26 The high moisture content was most likely due to the much higher water content of polymer solution used in this study, compared to the previous work.5 Although the moisture content of all formulations in this study is somewhat higher (5.4–7.7%), films are still peelable and non-tacky.
Fig. 4.
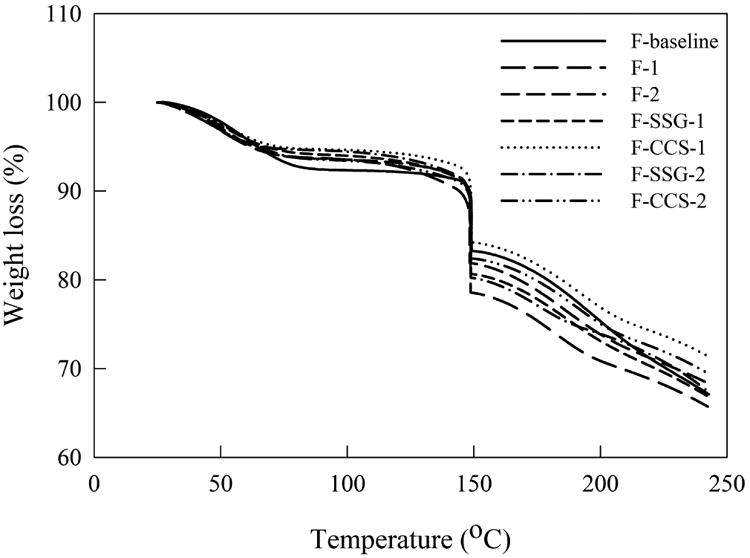
TGA curves for films of all formulations.
3.2.4 Crystallinity of films
Differential scanning calorimetry (DSC) analysis of AR-FNB particles as well as films laden with MC-FNB particles show (Fig. 5) a thermal event at 80–82 °C, which corresponds to the melting endotherm for FNB, and sharp endothermic peaks indicating the crystalline nature of FNB. As evident from the DSC profiles, the crystalline state of FNB particles has been preserved after milling drug particles and film manufacturing process.
Fig. 5.
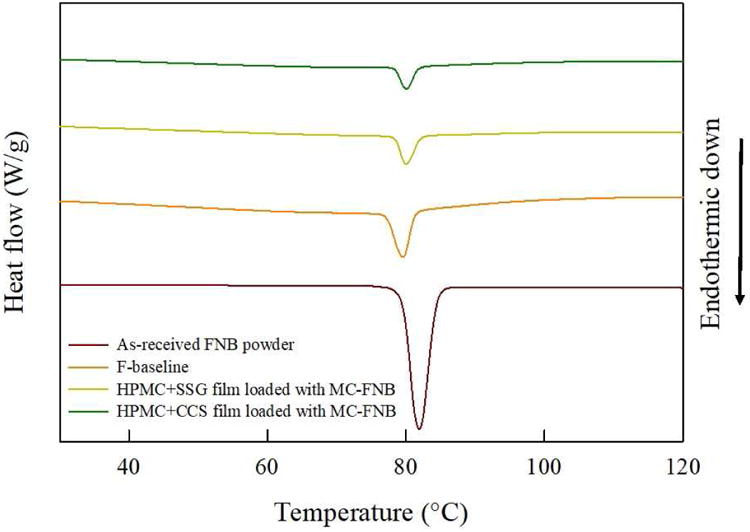
DSC curves of as-received FNB particles, placebo film, and films with SDIs laden with MC-FNB particles.
In addition, XRD analysis was performed to study the crystal structure of drug particles and the drug incorporated in films. Fig. 6 shows the XRD patterns of AR-FNB, placebo films and films laden with MC-FNB particles. AR-FNB particles presented sharp, high intensity peaks at the main diffraction angles (2θ) 10.0°–25.0°. Placebo films did not show any peak due to the amorphous nature of HPMC and SDIs. For the films laden with MC-FNB particles, the characteristic peaks in range 15.0°–25.0° were attributed to FNB, confirming that the crystalline structure of drug was preserved during the milling, mixing and drying processes. These results agree well with the DSC results that all processed dry powder and films consisted of crystalline FNB. This is significant in the context of maintaining the API form stability and release characteristics of the product over time.34
Fig. 6.
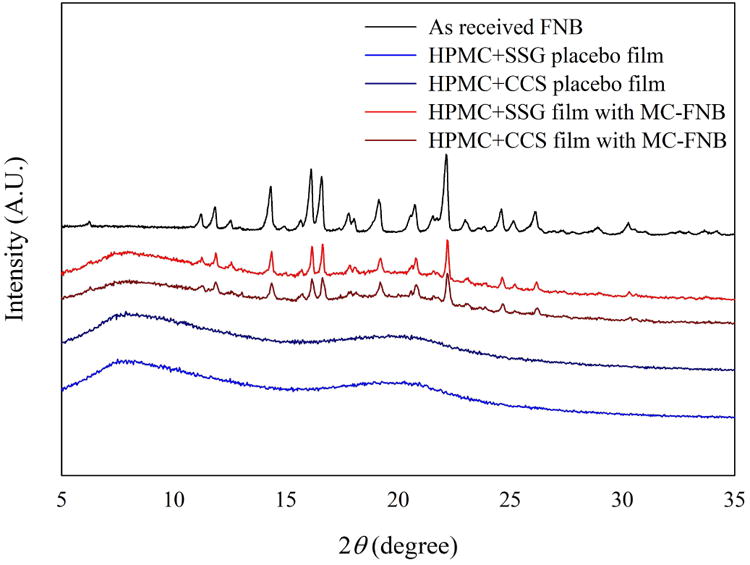
XRD diffractograms of as-received FNB particles, placebo films with SDIs, and films with SDIs laden with MC-FNB particles.
3.2.5 Comparison of disintegration time of USP, Petri dish and frame methods
The disintegration time of films laden with MC-FNB particles made from all formulations, as per the experimental design (Table S1), along with one additional thickness range tested by USP, Petri dish and frame methods are presented in Fig. 7. The USP method shown in Fig. 7 (a), has broader standard deviation (SD) and distribution of disintegration time compared to Petri dish and frame methods for all films. The prime reason for this large variation is due to difficulties in visual observation of films in the event they fold in the basket or attach to the plastic disk during the disintegration process. Disintegration time of most film samples showed good reproducibility with small standard deviations (SD) for frame (Fig. 7 (c)) and Petri dish (Fig. 7 (b)) methods, and there is no significant difference (p>0.05, details provided in supplementary Table S3) between both methods. The reader is reminded that for the Petri dish method, the time measured is for complete film disintegration as shown in Fig. 1. Therefore, Petri dish and frame methods were deemed suitable for determining disintegration times of films and this lends support to previous findings in the literature.8 Overall, all films under 80 μm and films with higher SDIs concentrations (>9%) at the thickness of 90–100 μm, exhibited fast disintegration time (<180s for dispersible tablets) according to Ph. Eur..35 Furthermore, films with SDIs under 60 μm disintegrated much faster (<50 s) than the required disintegration time for dispersible tablets. In particular, the film made from F-CCS-2 met the fast disintegration criteria of orodispersible tablets by FDA (<30s).31
Fig. 7.
Disintegration time of films of all formulations at three thickness ranges measured by (a) USP, (b) Petri dish, and (c) frame methods.
Regarding the effect of the addition of SDIs and the reduction of film thickness on the disintegration time, significant improvement (p<0.05) in terms of shorter disintegration time were obtained from both factors. HPMC films disintegrated in 70–380s compared to 30–220s of HPMC-SDIs films. The mean disintegration time for formulations including SDIs followed the trend of F-SSG > F-CCS, most likely caused by the higher concentration of CCS as compared to SSG in films. Regarding the effect of thickness, the observed trends were as expected; the disintegration time of thinner films was faster. Films 90–100 μm thick disintegrated in 150–380s, films 70–80 μm thick disintegrated in 70–180s, and films 50–60 μm thick disintegrated in 30–75s.
Results may be better interpreted through Figs. 8 (a)–(b), which show that the disintegration time of films has a nearly linear relationship with SDI concentration and film thickness (50–60 μm, 70–80 μm, 90–100 μm). As expected, for fixed thickness, the addition of SDI is clearly advantageous. Also, the main effects plot of thickness, SDI concentrations and viscosity generated using General Linear Model of Minitab® (supplementary Fig. S4) lends the same conclusion. Since it is difficult to visualize the extent of reduction in disintegration time due to addition of SDIs and their amounts in Figs. 8 (a) and (b), the percentage decrease of disintegration time is shown in Fig. 8 (c) as a function of SDI amount. Although the scatter in the linear trend is high, the R2 value of 0.84 indicates a nearly linear trend. In this figure, since all three thickness values are found to have a nearly similar trend, only a single trend line is shown (reader may refer to supplementary material Fig. S5 for a version with three separate trend lines). Accordingly, the percentage reduction in the disintegration time is largely controlled by the amount of SDI at any thickness. Also, the percentage reduction in disintegration time values for all films at thickness ranges of 90–100 μm, 70–80 μm, and 50–60 μm for a fixed type and amount of SDI are comparable to each other. Therefore, addition of SDI and in larger amounts greatly reduces the disintegration time for all films, regardless of their thickness in the ranges studied.
Fig. 8.
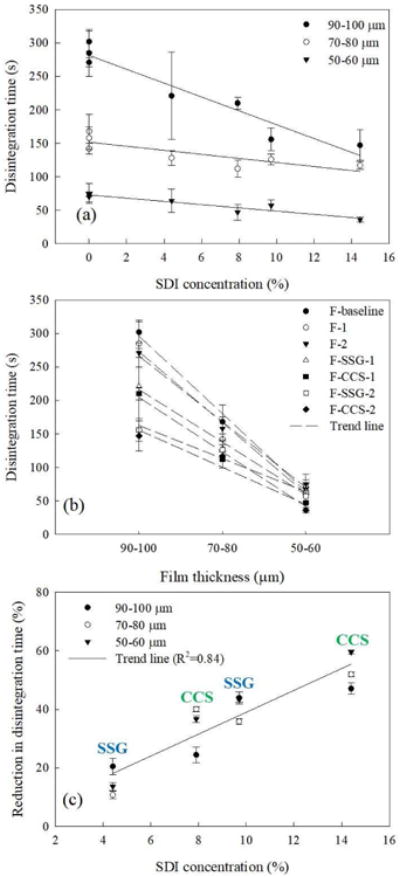
Disintegration time analysis of films: (a) disintegration time as functions of SDI concentration, (b) disintegration time as function of film thickness, (c) percentage reduction in disintegration time as a function of SDI concentration.
3.2.6 The effect of shaking speed and disintegration medium
The 50–60 μm films made from F-CCS-2 was used to investigate the effect of shaking speed (0 rpm, 35 rpm, and 65 rpm) on disintegration time (Fig. 9 (a)). Interestingly, the results show that the shaking speed did not affect the disintegration time at 0 rpm, 35 rpm or 65 rpm, indicating the shaking motion did not facilitate water penetration rate into the film matrix, or the water uptake rate of HPMC or SDIs. Fig. 9 (b) shows the effect of disintegration medium on the disintegration time of films (50–60 μm) of F-baseline, F-SSGs and F-CCSs, including DI water (pH=5.7) and PBS buffer (pH=6.8). Disintegration time of all films was shown to be slightly longer in higher pH medium (PBS buffer) compared to lower pH medium (DI water). Our results are in line with Preis et al.'s finding that HEC films have increasing disintegration time in media which contains more additives (water<buffer<SSF).7
Fig. 9.
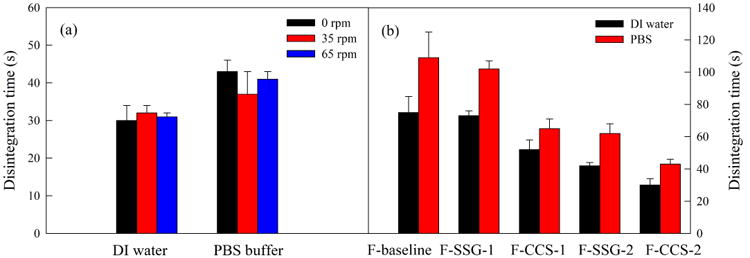
Disintegration time of selected thin films tested by modified Petri dish method: (a) shaking speeds of orbital shaker; (b) disintegration media (DI water vs PBS).
3.2.7 Mechanical properties of films
An ideal rapidly disintegrating film should possess good mechanical properties, such as moderate tensile strength (TS) and high elongation at break (E%), required for films to maintain their integrity, as well as being soft enough (low Young's modulus (YM)) to have a pleasant sensation in the buccal cavity.22,36 It has been reported that the type and amount of polymer, plasticizer, surfactant and drug all have profound effects on the mechanical properties of films.6,9,23,24,28,37,38 The results so far indicate that the addition of SDIs reduce the disintegration time of films. However, the extent of the effect of SDIs along with film thickness on mechanical properties of films loaded with poorly water-soluble drug microparticles is unknown. Therefore, the mechanical properties; tensile strength (TS), Young's modulus (YM) and percentage of elongation (E%), of 50–60 μm and 90–100 μm films of all formulations were measured and are shown in Figs. 10 (a)–(c). The TS, YM, and E% were determined from the stress-strain curves obtained from experiments using the texture analyzer.
Fig. 10.
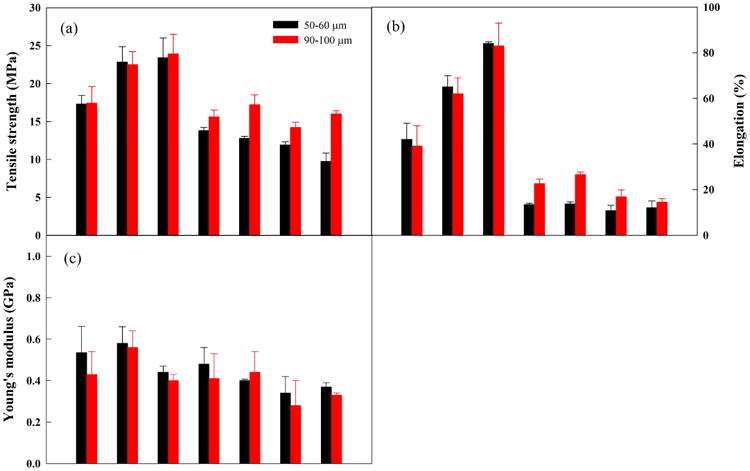
Mechanical properties of films made from all formulations at 50-60 μm and 90-100 μm: (a) tensile strength, (b) elongation (%), and (c) Young's modulus.
HPMC films (F-baseline, F-1 and F-2) resulted in the higher TS (17–23 MPa), E% (32–84%), and YM (0.4–0.58 GPa) compared to HPMC-SDI films which have TS in 9–17 MPa, E% in 10–22%, and YM in 0.28–0.48 GPa. Evidently, the incorporation of SDIs significantly influences the mechanical properties of films (p<0.05), and increasing the SDIs concentration in films exhibited decreases in TS, E%, and YM. This may be attributed to the embedded SDI particles inserting themselves between the polymer strands, thereby breaking the polymer-polymer interactions.23 In principle, the decreases in TS and E% lead to weaker and less ductile films, and are expected to be contributing factors toward enhancement of film disintegration and subsequent dissolution of drug microparticles. Hence, the mechanical properties results are in agreement with the disintegration time results of films. Regarding the effect of film thickness on mechanical properties (50–60 μm vs 90–100 μm), there are no significant changes (p>0.05) in TS, E% and YM of HPMC films. On the other hand, an obvious increasing trend of TS and E% was observed in HPMC-SDI films with minor decreases in YM with respect to increase in film thickness. The significant increases (p<0.05) in TS and E% with increasing thickness of HPMC-SDI films suggests that thinner HPMC-SDI films were more brittle and rigid. Remarkably, the values of TS (9.7–13.8 MPa), E% (10.7–13.7%), and YM (0.34–0.48 GPa) of HPMC-SDI films at the thickness of 50–60 μm are still in the proposed desirable ranges of ODFs, which are TS > 2 MPa, E% > 10%, and YM < 0.55 GPa as per the literature.20 These results offer sufficient evidence that films with SDIs can offer faster disintegration time while maintaining desired mechanical properties with respect to manufacturing and patient compliance.
Finally, rather interesting outcomes were observed in terms of the effect of increasing viscosity, via reducing water amount, on mechanical properties of HPMC films. It was found that increases in viscosity had positive effect on TS and E% however there was no drastic effect on YM of the films. One potential reason is that mechanical properties of films are related to distribution and density of intermolecular and intramolecular interactions in networks formed in films.39 Hence, high viscosity of polymer solution may result in forming a more compact structure or network in the film,40,41 which may be contributing to higher TS and E% but not YM of films.
3.2.8 Dissolution
It is has been shown that thicker films lead to slower dissolution rate for films containing poorly water-soluble drugs.6 Here, dissolution profiles of both thin (50–60 μm) and thick films (90–100 μm) are presented in Fig. 11 for all formulations, using 100 ml 7.2 mg/ml SDS as the dissolution media. It is important to note that all films exhibited immediate release (>80% in 20 min). Films made from F-CCS-2 exhibited the fastest release rate of MC-FNB microparticles due to the high SDI concentration (14 wt% CCS), followed by similar release rates for films made from F-CCS-1 and F-SSG-2, owing to their similar concentrations of SDIs (8 wt% SSG and 9.6 wt% CCS). All HPMC films and films made with F-SSG-1 exhibited similar dissolution profiles. The FNB release rate of films made with F-SSG-1 was lowest among HPMC-SDI films, and similar to HPMC films, maybe due to its lower SDI concentration (4 wt% SSG). These results suggest that dissolution rate of films could be more dependent on SDI amount rather than the SDI type. The trend of dissolution rates correlate fairly well with disintegration times of films, and HPMC-SDI films disintegrate/dissolve faster than HPMC films. The faster release of drug from films is most likely attributed to faster breakdown/disintegration, which creates more pores and channels for the drug to diffuse out of the films.42
Fig. 11.
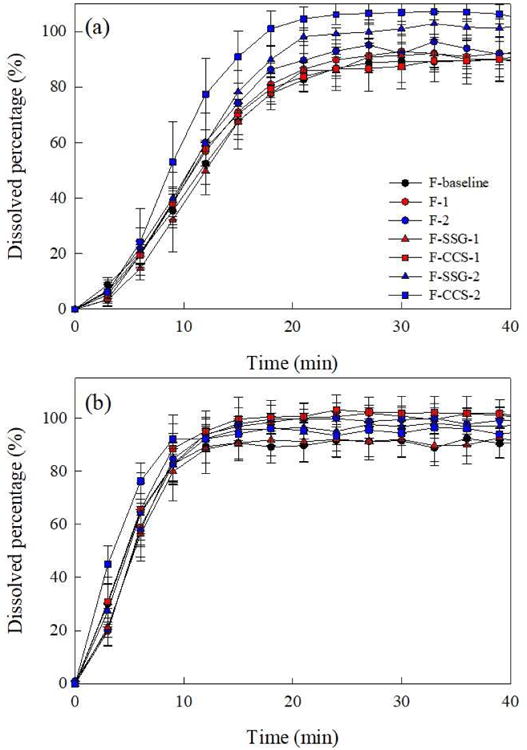
Dissolution profiles of (a) 90-100 μm and (b) 50-60 μm films from all formulations.
4. Conclusions
The impact of super-disintegrants (SDIs), their concentrations, and film thickness on various properties of films loaded with poorly water-soluble drug (fenofibrate) microparticles were investigated. A film precursor viscosity matching method was utilized to minimize the confounding effect of viscosity on the resulting film properties. Amongst three different disintegration testing methods, the Petri dish and frame methods were found to provide reliable measure of disintegration time, and the shaking speed of the Petri dish did not have an appreciable impact. The most important outcome of this study was that the addition of SDIs (sodium starch glycolate (SSG) and croscarmellose sodium (CCS)) led to reductions in disintegration time, which was found to be largely controlled by the amount of SDI at any thickness. All films under 80 μm thickness as well as thicker films (90–100 μm) with highest SDI (concentration > 9%) exhibited fast disintegration times (< 180 s) according to Ph. Eur. Furthermore, thinnest (under 60 μm) HPMC-SDI films disintegrated within 50 s and exhibited immediate release (< 80% in 10 min). All the films with SDIs achieved desired mechanical properties even though SDIs had somewhat negative impact on the film mechanical properties (tensile strength, elongation percentage and Young's modulus). All films with or without SDIs, except for films with the lowest level of SDIs, had very good drug content uniformity (RSD < 6%, AV < 15). Although the presence of SDIs in films led to slightly higher moisture retention upon drying, the final moisture content was still low, and resulted in peelable, non-tacky films with crystalline FNB microparticles. Overall, these results demonstrate that SDIs impart faster disintegration (<180 s, Ph. Eur.) of films laden with poorly water-soluble drug microparticles while achieving very good content uniformity and acceptable mechanical properties.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Science Foundation (NSF) in part through the ERC (EEC-0540855) award and from the National Institute of Health (NIH) NIH-U01 in part through award U01FD005521. We also thank Dr. Sonal Mazumder and FDA team for their valuable input on disintegration test experimental design.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borges AF, Silva C, Coelho JFJ, Simões S. Oral films: Current status and future perspectives: I-Galenical development and quality attributes. J Control Release. 2015;206:1–19. doi: 10.1016/j.jconrel.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Sievens-Figueroa L, Bhakay A, Jerez-Rozo JI, Pandya N, Romañach RJ, Michniak-Kohn B, Iqbal Z, Bilgili E, Davé RN. Preparation and characterization of hydroxypropyl methyl cellulose films containing stable BCS Class II drug nanoparticles for pharmaceutical applications. Int J Pharm. 2012;423(2):496–508. doi: 10.1016/j.ijpharm.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Beck C, Sievens-Figueroa L, Gärtner K, Jerez-Rozo JI, Romañach RJ, Bilgili E, Davé RN. Effects of stabilizers on particle redispersion and dissolution from polymer strip films containing liquid antisolvent precipitated griseofulvin particles. Powder Technol. 2013;236(Supplement C):37–51. [Google Scholar]
- 4.Bhakay A, Vizzotti E, Li M, Davé R, Bilgili E. Incorporation of fenofibrate nanoparticles prepared by melt emulsification into polymeric films. J Pharm Innov. 2016;11(1):53–63. [Google Scholar]
- 5.Zhang L, Li Y, Abed M, Davé RN. Incorporation of surface-modified dry micronized poorly water-soluble drug powders into polymer strip films. Int J Pharm. 2018;535(1–2):462–472. doi: 10.1016/j.ijpharm.2017.11.040. [DOI] [PubMed] [Google Scholar]
- 6.Krull SM, Patel HV, Li M, Bilgili E, Davé RN. Critical material attributes (CMAs) of strip films loaded with poorly water-soluble drug nanoparticles: I. Impact of plasticizer on film properties and dissolution. Eur J Pharm Sci. 2016;92:146–155. doi: 10.1016/j.ejps.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preis M, Gronkowsky D, Grytzan D, Breitkreutz J. Comparative study on novel test systems to determine disintegration time of orodispersible films. J Pharm Pharmacol. 2014;66(8):1102–1111. doi: 10.1111/jphp.12246. [DOI] [PubMed] [Google Scholar]
- 8.Garsuch V, Breitkreutz J. Comparative investigations on different polymers for the preparation of fast-dissolving oral films. J Pharm Pharmacol. 2010;62(4):539–545. doi: 10.1211/jpp.62.04.0018. [DOI] [PubMed] [Google Scholar]
- 9.Cilurzo F, Cupone IE, Minghetti P, Selmin F, Montanari L. Fast dissolving films made of maltodextrins. Eur J Pharm Biopharm. 2008;70(3):895–900. doi: 10.1016/j.ejpb.2008.06.032. [DOI] [PubMed] [Google Scholar]
- 10.Visser JC, Weggemans OAF, Boosman RJ, Loos KU, Frijlink HW, Woerdenbag HJ. Increased drug load and polymer compatibility of bilayered orodispersible films. Eur J Pharm Sci. 2017;107(Supplement C):183–190. doi: 10.1016/j.ejps.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Srinarong P, Faber JH, Visser MR, Hinrichs WLJ, Frijlink HW. Strongly enhanced dissolution rate of fenofibrate solid dispersion tablets by incorporation of superdisintegrants. Eur J Pharm Biopharm. 2009;73(1):154–161. doi: 10.1016/j.ejpb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Augsburger L, Brzecko A, Shah U, Hahm H. Characterization and functionality of super disintegrants. In: Swarbrick J, Boylan JC, editors. Encyclopedia of Pharmaceutical Technology. New York, NY: Marcel Dekker Inc; 2000. pp. 269–291. [Google Scholar]
- 13.Desai PM, Liew CV, Heng PWS. Review of disintegrants and the disintegration phenomena. J Pharm Sci. 2016;105(9):2545–2555. doi: 10.1016/j.xphs.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 14.Azad M, Afolabi A, Bhakay A, Leonardi J, Davé R, Bilgili E. Enhanced physical stabilization of fenofibrate nanosuspensions via wet co-milling with a superdisintegrant and an adsorbing polymer. Eur J Pharm Biopharm. 2015;94(Supplement C):372–385. doi: 10.1016/j.ejpb.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 15.Gordon MS, Rudraraju VS, Rhie JK, Chowhan ZT. The effect of aging on the dissolution of wet granulated tablets containing super disintegrants. Int J Pharm. 1993;97(1):119–131. doi: 10.1002/jps.2600820222. [DOI] [PubMed] [Google Scholar]
- 16.Bilgili E, Dave R, Bhakay A, Azad M. Systems and methods for superdisintegrant-based composite particles for dispersion and dissolution of agents. ed. US Patents 2013
- 17.Sagban TH, Ismail KY. Formulation and evaluation of orodispersible film of sildenafil citrate. Int J Pharm Pharm Sci. 2014;6(2):81–86. [Google Scholar]
- 18.Susarla R, Afolabi A, Patel D, Bilgili E, Davé RN. Novel use of superdisintegrants as viscosity enhancing agents in biocompatible polymer films containing griseofulvin nanoparticles. Powder Technol. 2015;285(Supplement C):25–33. [Google Scholar]
- 19.Lakshmi PK, Lavanya D, Ali MMH. Effect of synthetic super disintegrants and natural polymers in the preparation of donepezil hydrochloride fast disintegration films. International Current Pharmaceutical Journal. 2014;3(3):243–246. [Google Scholar]
- 20.Visser JC, Dohmen WMC, Hinrichs WLJ, Breitkreutz J, Frijlink HW, Woerdenbag HJ. Quality by design approach for optimizing the formulation and physical properties of extemporaneously prepared orodispersible films. Int J Pharm. 2015;485(1):70–76. doi: 10.1016/j.ijpharm.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann EM, Breitenbach A, Breitkreutz J. Advances in orodispersible films for drug delivery. Expert Opin Drug Deliv. 2011;8(3):299–316. doi: 10.1517/17425247.2011.553217. [DOI] [PubMed] [Google Scholar]
- 22.Krull SM, Ma Z, Li M, Dave RN, Bilgili E. Preparation and characterization of fast dissolving pullulan films containing BCS class II drug nanoparticles for bioavailability enhancement. Drug Dev Ind Pharm. 2016;42(7):1073–1085. doi: 10.3109/03639045.2015.1107094. [DOI] [PubMed] [Google Scholar]
- 23.Mazumder S, Pavurala N, Manda P, Xu X, Cruz CN, Krishnaiah YSR. Quality by Design approach for studying the impact of formulation and process variables on product quality of oral disintegrating films. Int J Pharm. 2017;527(1–2):151–160. doi: 10.1016/j.ijpharm.2017.05.048. [DOI] [PubMed] [Google Scholar]
- 24.Krull SM, Ammirata J, Bawa S, Li M, Bilgili E, Davé RN. Critical material attributes of strip films loaded with poorly water-soluble drug nanoparticles: II. Impact of polymer molecular weight. J Pharm Sci. 2017;106(2):619–628. doi: 10.1016/j.xphs.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Han X, Ghoroi C, To D, Chen Y, Davé RN. Simultaneous micronization and surface modification for improvement of flow and dissolution of drug particles. Int J Pharm. 2011;415(1–2):185–195. doi: 10.1016/j.ijpharm.2011.05.070. [DOI] [PubMed] [Google Scholar]
- 26.Susarla R, Sievens-Figueroa L, Bhakay A, Shen Y, Jerez-Rozo JI, Engen W, Khusid B, Bilgili E, Romañach RJ, Morris KR, Michniak-Kohn B, Davé RN. Fast drying of biocompatible polymer films loaded with poorly water-soluble drug nano-particles via low temperature forced convection. Int J Pharm. 2013;455(1-2):93–103. doi: 10.1016/j.ijpharm.2013.07.051. [DOI] [PubMed] [Google Scholar]
- 27.Dave RN, Susarla R, Khusid B, Bhakay AA, Bilgili EA, Muzzio F. System and method for fabrication of uniform polymer films containing nano and micro particles via continuous drying process. ed. US Patents 2014 [Google Scholar]
- 28.Krull SM, Susarla R, Afolabi A, Li M, Ying Y, Iqbal Z, Bilgili E, Davé RN. Polymer strip films as a robust, surfactant-free platform for delivery of BCS Class II drug nanoparticles. Int J Pharm. 2015;489(1-2):45–57. doi: 10.1016/j.ijpharm.2015.04.034. [DOI] [PubMed] [Google Scholar]
- 29.European Pharmacopoeia. Monographs, 82 Ibuprofen ed. Strasbourg, France: European Directorate for the Quality of Medicines (EDQM); 2014. [Google Scholar]
- 30.U.S. Pharmacopoeia. <905> Uniformity of Dosage Units ed. Rockville, MD: United States Pharmacopeial Convention, Inc; 2006. [Google Scholar]
- 31.U.S. Department of Health and Human Services, editor. Food and Drug Administration. Guidance for industry – Orally disintegrating tablets. Center for Drug Evaluation and Research (CDER); 2008. [Google Scholar]
- 32.Mashru RC, Sutariya VB, Sankalia MG, Parikh PP. Development and evaluation of fast-dissolving film of salbutamol sulphate. Drug Dev Ind Pharm. 2005;31(1):25–34. doi: 10.1081/ddc-43947. [DOI] [PubMed] [Google Scholar]
- 33.Sievens-Figueroa L, Pandya N, Bhakay A, Keyvan G, Michniak-Kohn B, Bilgili E, Davé RN. Using USP I and USP IV for discriminating dissolution rates of nano- and microparticle-loaded pharmaceutical strip-films. AAPS PharmSciTech. 2012;13(4):1473–1482. doi: 10.1208/s12249-012-9875-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogt M, Kunath K, Dressman JB. Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: Comparison with commercial preparations. Eur J Pharm Biopharm. 2008;68(2):283–288. doi: 10.1016/j.ejpb.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 35.European Pharmacopoeia, editor. European Pharmacopoeia. Oromucosal preparations, uniformity of content (method 296), uniformity of mass (method 295) and disintegration time (monograph 0478), ed. Strasbourg, France: European Directorate for the Quality of Medicines (EDQM); 2014. [Google Scholar]
- 36.Mohammadi R, Mohammadifar MA, Rouhi M, Kariminejad M, Mortazavian AM, Sadeghi E, Hasanvand S. Physico-mechanical and structural properties of eggshell membrane gelatin- chitosan blend edible films. Int J Biol Macromol. 2017;107:406–412. doi: 10.1016/j.ijbiomac.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Krull SM, Moreno J, Li M, Bilgili E, Davé RN. Critical material attributes (CMAs) of strip films loaded with poorly water-soluble drug nanoparticles: III. Impact of drug nanoparticle loading. Int J Pharm. 2017;523:33–41. doi: 10.1016/j.ijpharm.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brniak W, Maślak E, Jachowicz R. Orodispersible films and tablets with prednisolone microparticles. Eur J Pharm Sci. 2015;75:81–90. doi: 10.1016/j.ejps.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Leceta I, Guerrero P, de la Caba K. Functional properties of chitosan-based films. Carbohydr Polym. 2013;93(1):339–346. doi: 10.1016/j.carbpol.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 40.de Moura MR, Aouada FA, Avena-Bustillos RJ, McHugh TH, Krochta JM, Mattoso LHC. Improved barrier and mechanical properties of novel hydroxypropyl methylcellulose edible films with chitosan/tripolyphosphate nanoparticles. J Food Eng. 2009;92(4):448–453. [Google Scholar]
- 41.Prakash Maran J, Sivakumar V, Thirugnanasambandham K, Kandasamy S. Modeling and analysis of film composition on mechanical properties of maize starch based edible films. Int J Biol Macromol. 2013;62:565–573. doi: 10.1016/j.ijbiomac.2013.09.027. [DOI] [PubMed] [Google Scholar]
- 42.Satyanarayana DA, Keshavarao KP. Fast disintegrating films containing anastrozole as a dosage form for dysphagia patients. Arch Pharm Res. 2012;35(12):2171–2182. doi: 10.1007/s12272-012-1215-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.