

Abstract
Unsupported non-bridged uranium–carbon double bonds have long been sought after in actinide chemistry as fundamental synthetic targets in the study of actinide-ligand multiple bonding. Here we report that, utilizing Ih(7)-C80 fullerenes as nanocontainers, a diuranium carbide cluster, U=C=U, has been encapsulated and stabilized in the form of UCU@Ih(7)-C80. This endohedral fullerene was prepared utilizing the Krätschmer–Huffman arc discharge method, and was then co-crystallized with nickel(II) octaethylporphyrin (NiII-OEP) to produce UCU@Ih(7)-C80·[NiII-OEP] as single crystals. X-ray diffraction analysis reveals a cage-stabilized, carbide-bridged, bent UCU cluster with unexpectedly short uranium–carbon distances (2.03 Å) indicative of covalent U=C double-bond character. The quantum-chemical results suggest that both U atoms in the UCU unit have formal oxidation state of +5. The structural features of UCU@Ih(7)-C80 and the covalent nature of the U(f1)=C double bonds were further affirmed through various spectroscopic and theoretical analyses.
While metal–carbon double bonds are common in transition metal chemistry and catalysis, unsupported uranium–carbon double bonds remain highly challenging to prepare. Here, the authors stabilize and characterize a U=C=U cluster containing unusually short, unsupported double bonds inside an Ih(7)-C80 fullerene cage.
Introduction
Understanding the nature of actinide-ligand multiple bonding remains a modern day challenge owing to the complex electronic structures of these elements, and as a consequence, our comprehension of their chemistries lags behind that of the more commonly studied transition metals1–5. Importantly, recent developments in the synthesis and study of molecular uranium complexes containing a variety of U=E(L) or U≡E(L) moieties (E = O, S, Se, Te, N, NR, P, As; L = other ligands) have provided valuable insights regarding the participation of the 5f and 6d valence orbitals in chemical bonding1,2,6–27. Conspicuously missing from this list are U=C bonds, specifically those which lack ancillary heteroatom or chelating support. While metal–carbon double bonds (i.e., Schrock carbenes) are common in transition metal chemistry and catalysis, unsupported U=C bonds have long remained a major and outstanding synthetic target. Some success towards the synthesis of An=C bonds has been realized through the use of heteroatom stabilizing chelating ligands such as U=[C(Ph2PNSiMe3)2](O)Cl2 or U=[C(Ph2PS)] (BH4)2 (THF)2, among others4,27–33. In a few rare instances, the isolation of non-chelated U=C bonds has been achieved in complexes such as (η5-C5H5)3U=CHPMe2Ph and [N(SiMe3)2]3U=CHPPh3, using formally dianionic ylidic ligands31,34. Electronically, the U=C units found in these methanediide ([κ3-CL2]2−) or ylidic ([C(H)(PR3)]2−) examples are best described as highly polarized, nucleophilic carbenes with σ and modest π bonding overlap between uranium and carbon. The α-carbon bonded heteroatom(s) (e.g., phosphorous) aids the delocalization of the carbon-centered charge accumulation. While compounds possessing unsupported U=C bonds are difficult to prepare under typical synthetic conditions, Andrews and co-workers have identified alkylidene and even alkylidyne species such as H2C=UX2 and HC≡UX3 (X= H, F, Cl or Br) in low-temperature noble-gas matrix isolation experiments, providing evidence for such bonding motifs35–37. However, these compounds are too reactive to be isolated under typical synthetic conditions. The knowledge gained from studying stable compounds featuring U=C multiple-bonds can be extended to uranium carbide ceramics, which offer improved thermal density and higher conductivity over current UO2 nuclear fuels38–40.
Based on our recent success with the isolation of new actinide endohedral fullerenes, we turned our attention towards the synthesis and isolation of carbon cages encapsulating clusters that possess U=C bonds. The internal hollow cavity of C2n-fullerenes can encapsulate and stabilize novel metallic clusters, especially some which are highly reactive and virtually impossible to prepare independently41–43. Recently, we reported the first crystallographically characterized examples of actinide endohedral metallofullerenes (Th@C82 and U@C2n, 2n = 74, 82) and described their unique electronic properties43,44. We therefore hypothesized that fullerene cages would provide an ideal architecture and electronic environment to trap and stabilize unique actinide clusters with novel bonding motifs.
Herein, using the Ih(7)-C80 fullerene cage as a molecular nanocontainer, we report, to the best of our knowledge, the first structurally characterized example of unsupported uranium U=C bonds found in UCU@Ih(7)-C80, possessing the unprecedented dimetallic-carbide cluster U=C=U. X-ray crystallographic analysis reveals two very short U=C bonds of 2.033(5)/2.028(5) Å, with an unexpected nonlinear U=C=U bond angle of 142.8(3)°.
Results
Synthesis of U2C@C80
U2C@C80 was synthesized by the Krätschmer–Huffman arc discharge method45. Graphite rods, packed with graphite and finely dispersed U3O8, were vaporized in an arcing chamber under a He atmosphere. The compound was isolated and purified using a multistage high-performance liquid-chromatography protocol (HPLC). The composition and purity of the isolated U2C@C80 was confirmed by high-resolution matrix-assisted laser desorption-ionization time-of-flight positive-ion-mode mass spectrometry (MALDI-TOF/MS), which presents a prominent molecular ion peak with a mass-to-charge ratio of m/z = 1448.103 (Supplementary Fig. 1), corresponding to the [U2C81]+ empirical formula. The mass spectral isotopic distribution pattern matches the theoretically predicted one, thus confirming the molecular composition. In addition, an energy dispersive spectroscopic analysis of the purified sample was employed to determine the elementary composition of the compound. The spectrum shows characteristic peaks of uranium and carbon (Supplementary Fig. 2), which confirmed the assignment of this molecule to U2C81. Moreover, the powder X-ray diffraction pattern of U2C@C80 proves that the purified sample of U2C@C80 used for the experimental characterizations below, exists as a pure phase (Supplementary Fig. 5).
Molecular structure of UCU@Ih(7)-C80·[NiII-OEP]
The molecular structure of UCU@C80 was determined by single-crystal X-ray diffraction analysis. Slow diffusion of nickel(II) octaethylporphyrin (NiII-OEP) in benzene into a CS2 solution of UCU@C80 yielded black UCU@Ih(7)-C80·[NiII-OEP] cocrystals (1). In cocrystal 1, UCU@C80 is observed to adopt a slightly distorted icosahedral Ih(7)-C80 cage structure (Fig. 1a) that is highly ordered. Similarly, inside the fullerene cage, the central carbon atom C0 of the endohedral UC0U unit is fully ordered, while the U atoms are slightly disordered. Two major U positions (U1 and U2) have common dominant occupancy of 0.853(3), while the residual occupancies are located on both sides within ca. ½ to 1 Å distance from the two main positions, possibly indicating some large amplitude motions of the UC0U cluster (Supplementary Fig. 6). The distances between the major U1,2 sites and the carbons of the adjacent aromatic rings of the cage all lie within a narrow range of around 2.50 Å (2.471(5) to 2.543(5) Å, Fig. 1b), corresponding to distances of the ring centers Cti to Ui of 2.042(1) Å and angles Cti−Ui−C0≈159(1)°. The U–C0 bonds are 2.03 Å (U1–C0 = 2.033(5) Å, U2–C0 = 2.028(5) Å), forming a nearly linear Ct1–U1—U2–Ct2 chain (Fig. 1b) bridged by a non-linear –C0– carbide anion.
Fig. 1.
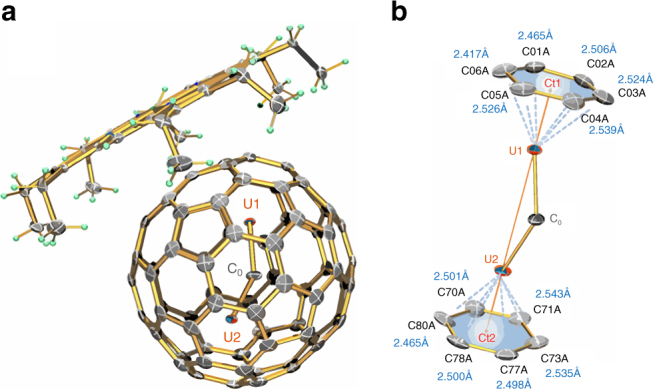
ORTEP drawing of UCU@Ih(7)-C80·[NiII-OEP] with 40% probability ellipsoids. a UCU@Ih(7)-C80·[NiII-OEP] structure showing the relationship between the fullerene cage and the [NiII-OEP] ligands. The two U1/U2 sites have common occupancy of 0.853(3). Four minor U sites (Supplementary Fig. 6) and the solvent molecules are omitted here for clarity. b Fragment view showing the interaction of the major U1–C0–U2 cluster with the closest aromatic ring fragments of the cage with centers Ct1 and Ct2. The orange line connects Ct1–U1–U2–Ct2
For hexa-coordinated uranium, Pyykkö found that two short and strong axial and four long and weak equatorial bonds are energetically near-equivalent to two long and weak axial and four short and strong equatorial bonds46,47. Therefore, particularly short axial bonds are expected when additional equatorial coordination is absent, as in the present case. Indeed, most U-Ct(arene) distances are reported between 2.5 and 2.9 Å, for instance 2.651(4) to 2.698(4) Å for the dinuclear U(V) inverse sandwich complex [{UV(Ts)}2(η6:η6-C6H5Me)]48. However, by far the most salient features of 1 are the unusually short U–C0 bond lengths, (U1–C0 = 2.033(5) Å, U2–C0 = 2.028(5) Å) and the unusual bond angle of 142.8(3)° for U1–C0–U2 (Table 1). In fact, the shortest U–C bond distance in poly-coordinated U-complexes to date is 2.184(3) Å, reported by Liddle et al. for the U(VI) carbene complex U=[C(Ph2PNSiMe3)2](O)Cl227. Interestingly, the U–C bond lengths of the UC0U unit are between the 2.067(7) Å and 1.948(7) Å, theoretically calculated distances for uranium alkylidene and alkylidyne complexes U=CH2(H)F and U≡CH(Cl)335–37, and are nearly identical to the 2.01 Å distance calculated from the sum of the covalent radii49. Furthermore, the U–C0 distances in 1 are longer than the triple bond of the U≡O unit (1.78 Å) in the uranyl dication UO22+, the U≡NR bonds (1.844(6) Å) of U(NtBu)2(I)2(THF)2), and the terminal U≡N bond (1.83(2) Å for the uranium nitride complex (TrenTIPS)UN), even when accounting for differences in the atomic radii of O, N, and C of ca. 0.05 Å each18,23,50.
Table 1.
Calculated and experimentally derived geometric parameters of UCU groups
| Geometric parameter | Calc.a UCU@(3I)2 (molecule) | Calc.a UCU@(C7H7)2 (molecule) | Calc.a UCU@Ih(7)-C80 (molecule) | Exptl.b UCU@Ih(7)-C80 •[NiII-OEP] (crystal) |
|---|---|---|---|---|
| U–C /Å | 2.024 | 2.074 | 2.022 | 2.033/2.028 |
| U∙∙∙U /Å | 3.918 | 4.118 | 3.917 | 3.849 |
| Ligand→U /Å | 2.33c | 2.52–2.55 | 2.47–2.51 | 2.47–2.54 |
| U–C–U / o | 150.9 | 166.1 | 151.3 | 142.8 |
| Fullerene C–C /Å | — | — | 100 × 1.43 ± 0.01 | 110 × 1.43 ± 0.03 |
| ‘π-Donating’ C–C /Å | — | 14 × 1.43 ± 0.01 | 20 × 1.46 ± 0.01 | 10 × 1.48 ± 0.01 |
aFrom scalar relativistic ZORA Kohn–Sham PBE VTZp approaches
bFrom crystal structure analysis by X-ray diffraction
cFor comparison of I−U with C−U, we have subtracted the I–C bond-radii difference of 0.55 Å from the actual I–U distance
Altogether, the bond lengths observed for 1 strongly support an Arene≡U=C=U≡Arene structure possessing two uranium–carbon double bonds formed by a bridging carbide atom, formally C4−. The bending of the U=C=U cluster of 1 was an unexpected exception to the common linear geometries of spσ1–pπ2 hybridized carbon atoms found for main-group molecules of type E=C=E (E=O, NR, CR2), though a few cases with angles between 180° and 90° have been theoretically proposed and observed in recent years51.
Computational studies of molecular structure and bonding in UCU@Ih(7)-C80
Quantum-chemical calculations were performed to further verify the unique structural parameters of UCU@Ih(7)-C80 and gain insight about the bonding of the encapsulated bent U=C=U cluster. The molecular structure was modeled for free UCU@Ih(7)-C80 and for LUCUL (L = C7H7, 3I) model molecules, using quasi-relativistic density-functional (DF) approximations (Supplementary Figs. 13, 14) and ab-initio CASSCF(10e,12o)-PT2 approaches (Table 1). Additional information of the structure, orbitals and electronic states of LUCUL is available in Supplementary Figs. 15–19 and Supplementary Tables 3–5. For the three calculated cases, the formal oxidation state52 is identified as 5+ for each U, derived from the formal [L3−≡>U5+<=C4−=>U5+<≡L3−] Lewis structure, where L3−≡>stands for the cage arene units that accept 3 electrons from each uranium atom and forms 3 dative pair bonds with the nearly empty U-5f6d valence shell. In the case of UCU@Ih(7)-C80, the two L3− units are the ligating arene units of the encapsulating C806– closed-shell cage. The C and I atoms of the model units (C7H7) and (3I) have similar electronegativity as C of (C80), and the model units also become closed-shell ligands upon accepting 3 electrons from each U. The effective physical partial charges in all three cases remain small, around −1 for formal C4−, around +½ to +1 for formal U5+, and small for the ligand atoms of C80, 2C7H7 or 6I (Supplementary Table 1).
The atomic free valence and the spin and orbital populations (Supplementary Table 1) indicate a single f-electron on each U atom. The experimental U–C0 bond lengths, though unprecedentedly short (2.03 Å), are well reproduced in all three computed models (2.02 Å for ligands C80 or (I3)2, 2.07 Å for (C7H7)2). Further, the observed unusual U–C0–U bond angle of 142.8° is comparable to those computed for UCU@C80 (151.3°) and (3I)UCU(3I) (150.9°), while the bulkier ligands of (C7H7)UCU(C7H7) led to 166.1°, which is still far from linear (Table 1). Neither the bond lengths nor the bond angle of the UCU unit appear to be significantly distorted by the encapsulation in C80.
We chose the localized equivalent molecular orbital picture of the two simpler model systems to sketch the correlated and spin–orbit coupled valence electronic structure of ligated UC0U. (C7H7)UCU(C7H7) (Fig. 2) and (3I)UCU(3I) (Supplementary Fig. 15) gave very similar results. Each U forms a strong σ-bond to the central C0 atom through covalent overlap of hybridized U(5 f6d) orbitals with C(2s2p) hybrids. The U–C bonds of dominant π-character can be described as three-center two-electron (3c2e) bonds that possess some σ-admixture. The large electronegativity difference of C and U (1.3 Pauling units) leads to bond-pair polarization toward C0, especially for the π-type pairs, which are strongly localized on C0 and lead to its effective negative charge. The U–C interactions are characterized by Mayer bond orders (BO) of 1.4 from DF calculations (comparable to 1.6 for the U=As double bond in [K(B15C5)2] {[N(CH2CH2NSiiPr3)3] U=AsH})26 and by a Roos effective bond order (EBO) of 1.9 (counting the formally bonding—antibonding NOs from our CASSCF calculation of (C7H7)UCU(C7H7), which substantiates the U=C double bond character).
Fig. 2.

Natural Localized Molecular Orbitals (NLMOs) of sandwiched UCU. The UCU@(C7H7)2 molecule formally consists of a (UCU)6+ unit between two aromatic (C7H7)3− rings. Spin–orbit coupled multi-reference all-electron relativistic wavefunction calculations (state-averaged CASSCF) were analyzed with the NBO program [S27]. The contour values of the NLMOs generated thereby are ±0.042√(e/Å3). a The two 1-center U-5f1-type NLMOs, each occupied by one electron. b The U–C (left) and C–U(right) 2-center 2-electron (2c2e) σ-pair NLMOs, each consisting of 67% C-2sp1, 31% U-5f½ 6d and 2% tail-contribution from the other U. c The vertical π-type and the in-plane dominantly π-type U–C–U 3-center 2-electron (3c2e) NLMOs of 65% C-2ps and 17% of each of the two U-5f½ 6d
The bonding motif is nicely illustrated by the electron localization function (ELF) map in Fig. 3, which shows the electron–density accumulation of the two two-center U–C σ-bonds and of the in-plane three-center U–C–U π-type bond, as well as the multiple maxima of the inner-valence non-bonding 5f electrons. Thus, the bending of the U=C=U unit can be interpreted in terms of the localized negative charge build-up on C0, which generates lone electron-pair density and gives rise to an sp1/sp2-hybridized type geometry at carbon with bond angles in the middle between 120° and 180°. This charge accumulation is partially offset by donation into the U(5f6d) orbitals. Thus the bonding model can be described as highly polarized U=C interactions with partial π-overlap strengthened by electrostatic attractive forces between the cationic U5+ atoms and the anionic C04− carbide bridge.
Fig. 3.

ELF-plot of ligated UCU in the molecular plane. The left and right ligands are two (I3) groups. ELF is the ‘electron localization function’ of Becke and Edgecombe, showing electronic accumulations in the inner atomic shells, in the atomic lone-pair regions, and in covalent and dative bond regions
Spectroscopic properties of UCU@Ih(7)-C80
Nuclear Magnetic Resonance (NMR) spectroscopy was used to characterize UCU@Ih(7)-C80. The 13C NMR spectrum of UCU@Ih(7)–C80 measured at 298 K shows only two sharp signals at 138.53 and 125.14 ppm with a 3:1 intensity ratio, corresponding to the sets of 60 and 20 equivalent carbon atoms of the unperturbed Ih–C80 fullerene cage (Supplementary Fig. 7). The apparent symmetry is likely due to fast and large-amplitude librations of the cage, similar to those proposed for M2@Ih-C80 (M = La, Ce) and M3N@C80 (M = Sc, Y)42,53,54. Unfortunately, no signal was detected for the single bridging C0 carbon atom, likely due to limited sample amount, as well as to paramagnetic broadening effects from the unpaired 5f1 electrons on each uranium. Noteworthy is that the 13C NMR spectrum measured at lower temperature (i.e., 283 K) shows slightly shifted signals at 138.35 and 124.65 ppm, respectively, with a slightly larger chemical shift difference (Δδ = 13.70 ppm) than those observed at 298 K (Δδ = 13.39 ppm). Such a trend is very similar to that observed for Ce2@Ih–C8055, consistent with the paramagnetic nature of UCU (see below) and with the presence of an unpaired f electron on the formal U5+ (5f16d07s0) ions (Supplementary Table 1)54. On the other hand, the slight temperature dependence of Δδ/ΔT = 0.02 ppm/K might also be accounted for by the libration of the UCU cluster that neutralizes the paramagnetic effects (mainly pseudocontact interactions) for all carbon atoms.
We also utilized Fourier transform infrared absorption (FTIR), Raman emission and photoluminescence (PL) spectroscopies together with quantum-chemical calculations to further characterize UCU@Ih(7)-C80. An experimental and theoretical spectral overview is presented in Fig. 4. The high-wavenumber range from 1600 to 1100 cm‒1 contains 104 carbon cage C–C stretching vibrations, resembling those of other Ih-C80 based endohedral fullerenes such as Ln3N@C8041,56. Typical characteristics are the major overlaid bands around 1380 cm−1 and the featureless gap between 1100 and 900 cm‒1. The remaining 130 collective deformations originating from bending and torsional motions of the carbon cage atoms show up between 900 and 200 cm−1.
Fig. 4.

Observed and theoretically predicted vibrational features of UCU@Ih(7)-C80. The left upper curve (black) presents the observed Infrared absorption (IR) spectrum vs. wavenumber from 1600 to 600 cm‒1, with quantum-chemical density-functional simulation below (in blue). On the right is the observed Raman emission (in red) vs. wavenumber from 600 to 100 cm‒1, with calculated wavenumbers (‘Theor. Freqs.’) in the 600–0 cm‒1 range (below in magenta). Observed local UCU vibrations above 100 cm‒1 are indicated by heavy dots. Observed and calculated spectral C80-cage features represent either a single mode, or an overlay of several near-degenerate ones. indicates a contamination by CS2
Quantum-chemical calculations suggest that an endohedral cluster of n atoms (here n = 3) shows 3 frustrated translational rocking modes against the cage (at 33, 59, and 115 cm‒1), 3 frustrated torsional wagging modes (at 16, 50, and 142 cm‒1), and 3n – 6 = 3 internal vibrations. The latter comprise the UC0U bending mode (at 97 cm‒1), the symmetric stretch (coupled to near-degenerate cage deformations from 250 to 300 cm‒1), and the asymmetric stretch (at 780 cm‒1). The observed weak features in the Raman spectrum above 100 cm‒1 at 126, 148, and 277 cm‒1 agree reasonably well with the predicted UCU modes given above (in bold italics, estimated as weakly Raman active). Notably, the asymmetric UCU stretch appears as a pronounced feature in the IR spectrum at 785 cm‒1, which is consistent with the high negative charge on the C0 atom of the endohedral UCU cluster, and with the crystallographic observation of very short axial U=C bonds.
Further, an experimental violet-blue photo-luminescence progression of U2C@Ih(7)-C80 starting at a wavelength of 430 nm (~2.95 eV) in steps of ca. 1425 cm‒1 (Supplementary Fig. 8) may originate from a ligand-to-metal charge-transfer excitation of the highest occupied molecular orbital, which has dominant density on the fullerene cage near the U atoms. There are also Cring-breathing vibrations around 1400 cm−1 localized in the spatial Cring–U–C–U–Cring region.
The redox properties of UCU@Ih(7)-C80, as investigated by means of cyclic voltammetry, show a surprisingly small electrochemical gap of only 0.83 eV (Table 2, Supplementary Fig. 9). Most reported transition metal cluster fullerenes of Ih(7)-C80 have gaps larger than 1 eV41. Typical M3N@Ih(7)-C80 fullerenes have gaps of 1.8–2.2 V. In particular, the first reduction potential of UCU@Ih(7)-C80 (−0.41 V) is much more positive than that of any other reported M3N@Ih(7)-C80 fullerene57, e.g., −1.26 V for Sc3N@Ih(7)-C8058 and −0.94 V for TiLu2C@Ih(7)-C8059. UCU@Ih(7)-C80 obviously exhibits a much better electron-accepting ability than other Ih(7)-C80 clusterfullerenes. This significant difference between UCU@Ih(7)-C80 and other reported M3N@C80 fullerenes indicates a major difference of their electronic structures, which may be caused by the encapsulation of the U=C0=U cluster with an open d–f shell electronically interacting with the cage. Interestingly, when compared to most other Ih(7)-C80 cluster fullerenes, the redox potentials of UCU@Ih(7)-C80 are close to those of dilanthanide-fullerene, such as La2@Ih(7)-C80, which exhibits an electrochemical gap of 0.87 eV60. In general, the electrochemical gaps are related to the HOMO-LUMO gaps of the closed-shell fullerene subsystems. However, in the present case, the encapsulated U=C0=U cluster has a low-lying open U-5f shell, causing an abnormally small f–f gap of the UCU@Ih(7)-C80 molecule. The reversible reduction of UCU@Ih(7)-C80 is also quite remarkable, since M3N@Ih(7)-C80 fullerenes with group 3 metals exhibit irreversible reductions57.
Table 2.
Redox potentials and electrochemical gaps of UCU@Ih(7)–C80, La2@Ih(7)–C80, and Sc3N@Ih(7)–C80
| Fullerenes | E 2+/+ a | E +/0 a | E 0/− a | E -/2− a | E 2−/3− a | E gap b | Ref. |
|---|---|---|---|---|---|---|---|
| UCU@Ih(7)-C80 | +1.05d | +0.42c | −0.41c | −1.34c | 0.83 | This work | |
| La2@Ih(7)-C80 | +0.95c | +0.56c | −0.31c | −1.72c | −2.13d | 0.87 | 60 |
| Sc3N@Ih(7)-C80 | +0.97c | −1.26d | −1.62d | −1.82d | 2.23 | 58 |
aRedox potentials in V vs. ferrocene couple
bElectrochemical gaps in eV
cHalf-wave potential (reversible redox process)
dPeak potential (irreversible redox process)
The XPS spectrum of UCU@Ih(7)-C80 was also recorded (Supplementary Fig. 10). The U-4f7/2 ionization appears at 378.6 eV. In general, ionization energies increase with the formal oxidation state, e.g., from the U-4f7/2 value of ca. 3771/3 eV for the neutral uranium metal to ca. 3813/4 eV for some uranyl(VI) salts. The present value of 378.6 eV is consistent with the very low effective charges on U as computed for UC0U@C80 (see below, U ~ +1.0e; Co ~ −1.0e, Cfullerene ~ ± 0.0e), caused by the strong electron pair donation of formal C806− and C04− to the formal U5+ ions.
To gain further insight into the unique electronic characteristics of the U=C=U cluster in UCU@C80, the magnetic susceptibility was measured (see Supplementary Fig.11 and Methods). The magnetic curves exhibit two regimes, (i) one at “high temperatures” from 300 K down to ca. 60 K, and (ii) another one at “low temperatures” from ca. 40 K down to 2 K. The “high temperature” region exhibits a huge, basically temperature-independent paramagnetism (TIP) of yet unknown origin. It is unknown for uranium complexes with common ligands21,22,48,61–65 and may possibly be due to the cooperative coupling of UCU and C80 in the solid and related to the low band-gap calculated for UCU@C80 (see Supplementary “Quantum Computational Methods”).
At “low temperatures”, we observed a Curie type paramagnetism ~μ2/T on top of the TIP, with net μ-values increasing with temperature from below to above a μBohr, which appears compatible with hardly coupled unique units of type η3L≡U(V)=C=U(V)≡Lη3, i.e., the endohedral di-uranium carbide fullerene molecules. The “low-temperature” literature values of μeff21,22,48,61–65 for mono- and bridged di-metallic U(V) and U(IV) complexes, typically increase, too, from low values up to 3 μBohr. It shows the expected temperature dependent weak coupling of two spins and with the two partially quenched U-f±3 orbital angular momenta, the experimental curves being reasonably reproduced by numerical simulations (see Supplementary Method “Quasi-relativistic correlated ab initio approaches” on the (C7H7)UCU(C7H7) model).
Attempts to further resolve the electronic structure using EPR spectroscopy were unsuccessful, as no clearly defined signal was observed at 4 K (Supplementary Fig. 12). Factors such as near degenerate electronic states and cage shielding effects as well as the huge TIP complicate the analysis. Thus, additional studies of the electronic properties of this unique and unprecedented system are warranted and will be communicated in due time.
Discussion
In summary, the overall agreement between the crystallographic, luminescence, Raman, IR, core-electronic, magnetic and voltammetric results, and the quantum-computational findings, conclusively show that a pure UCU@Ih(7)-C80 compound was synthesized, which consists of a weakly perturbed Ih-C80 cage and a sandwiched, bent and strongly bound polar endohedral near-symmetric U=C=U unit. The quantum-chemical results suggest that both U atoms in the UCU unit have a formal +5 oxidation state. The encapsulation causes little bond length or angle deformation of the UCU fragment. The encapsulation protects the axially ligated U atoms from binding with harder or more electronegative ligands like O or N (as compared to carbon in the forms of C04− and C806−) so that C0 can participate in primarily axial L≡>U=C0 bonding at unusually short U–C0 distances of 2.03 Å, and is not pushed into the weaker bonding equatorial plane of an E=U=E building block. The discovery of unsupported U=C bonding in a molecular compound confirms the distinction between “diagonal” and “poly-coordinated” uranium, and between “axial” covalent and “equatorial” dative bonding mechanisms. The work reported here offers a deeper understanding of the fundamentals of uranium bonding properties. This study also demonstrates that fullerene cages can be utilized as effective nanocontainers to stabilize and study rare and reactive clusters which contain actinide metal–ligand bonds.
Methods
Synthesis, separation and purification of U2C@Ih(7)-C80
A soot containing uranium fullerenes was synthesized by a direct-current arc discharge method. Graphite rods of U3O8 and graphite powder were annealed in a tube furnace at 1000 °C for 20 h under an Ar atmosphere and then burned in the arcing chamber under a 300 Torr He atmosphere. In total 1.87 g of graphite powder and 1.83 g of U3O8 (24:1 atom number ratio) were packed in each rod (6.7 g, without filling). The collected raw soot was refluxed in chlorobenzene under Ar atmosphere for 12 h and the solution was filtered. After solvent removal, the fullerenes were extracted by dissolving in toluene. On average ca. 140 mg of crude fullerene mixture per rod was obtained. In total, 60 carbon rods were burned in this work. Separation and purification of U2C@Ih(7)-C80 was achieved by a three-stage HPLC procedure (Supplementary Fig. 3, using columns including a Buckyprep M column (25 × 250 mm2, Cosmosil, Nacalai Tesque, Japan), a Buckprep D column (10 × 250 mm2, Cosmosil, Nacalai Tesque, Japan) and a Buckprep column (10 × 250 mm2, Cosmosil, Nacalai Tesque, Japan). Toluene was used as the mobile phase and the UV detector was adjusted to 310 nm for fullerene detection. The HPLC procedures and corresponding MALDI-TOF spectra for the isolated fractions are shown in Supplementary Figs. 3, 4. In total, ca. 2 mg of highly purified U2C@C80 was obtained for characterization.
Single crystal and powder X-ray diffraction analysis
Black co-crystals of UCU@Ih-C80·[NiII-OEP] were obtained by allowing the benzene solution of [NiII-OEP](1.2 mg/mL, 0.8 mL) and the CS2 solution of UCU@Ih-C80(1 mg/mL, 0.5 mL) to diffuse together. X-ray diffraction data were collected at 120 K using a diffractometer (APEX II; Bruker Analytik GmbH) equipped with a CCD collector. The Multiscan method was used for the absorption correction. The structure was resolved using direct methods (SIR2004) and refined on F2 with the full-matrix least-squares approach using SHELXL201466,67 within the WinGX package68.
Crystal Data for UCU@Ih-C80·[NiIIOEP]·1.5C6H6·CS2: C127H53N4NiS2U2, M = 2159.70, 0.05 × 0.05 × 0.03 mm3, monoclinic, P 21/c (No. 14), a = 17.678(4) Å, b = 16.970(3) Å, c = 26.695(5) Å, β = 106.65(3)°, V = 7673(3) Å3, Z = 4, ρcalcd = 1.934 g cm−3, μ = 4.578 mm−1, θ = 2.688–27.092°, T = 120 K, R1 = 0.0610, wR2 = 0.1045 for all data; R1 = 0.0388, wR2 = 0.0610 for 16,771 reflections (I > 2.0σ(I)) with 1278 parameters and 784 restraints. Goodness of fit indicator 1.007. Maximum residual electron density 2.096 e Å−3. For further details see the Supplementary Data 1, 2.
In addition, a powder X-ray diffraction analysis of UCU@C80 was conducted at the Shanghai Synchrotron Radiation Facility (SSRF) with wavelength λ = 0.6199 Å, and the intensity vs. 2θ pattern was recorded, see Supplementary Fig. 5.
Computational methods
Open-shell Kohn–Sham single-reference calculations were carried out with the ADF-2016, Gaussian-09 and ORCA-4.0 software, applying scalar and spin–orbit-coupled quasi-relativistic approaches (RECP or ZORA) with triple-ζ double-polarized basis sets. Bonding analyses were then performed using the NBO5.0 and Multiwfn codes. Multi-reference CASSCF, CASPT2, and RAS-SI/SO wave-function calculations were performed using the MOLCAS-V8.1 software applying the quasi-relativistic DKH Hamiltonian. Further details are given in the Supplementary “Quantum Computational Methods”.
Data availability
The coordinates for the X-ray structure of 1 are available free of charge from the Cambridge Crystallographic Data Centre under deposition no. CCDC 1572891. All other data supporting the findings of this study are available from the corresponding authors on request.
Electronic supplementary material
Description of Additional Supplementary Files
Acknowledgements
We cordially thank Prof. Xing Lu (Huazhong University of Science and Technology, China) for kind help with the crystallographic measurements and Prof. Marilyn Olmstead and Prof. Alan Balch (University of California at Davis, USA) for help with the data analysis. We also gratefully acknowledge the help with magnetic measurement by Prof. Taishan Wang and Mingzhe Nie (Institute of Chemistry, The Chinese Academy of Sciences) and helpful discussions of the magnetism by Prof. Stephen Liddle (University of Manchester, England), Prof, Thomas Albrecht-Schmitt and Dr. Ryan Baumbach (Florida State University, Tallahassee, USA) and Prof. Jörn Schmedt auf der Günne (University of Siegen, Germany). We thank Prof. Lin Wang and Dr. Cuiying Pei (Center for High Pressure Science &Technology Advanced Research) for the help with Powder XRD measurements. N.C. and L.F. thank the support from the NSFC (51772196, 51772195), the NSF of Jiangsu Province (BK20171211), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the project of scientific and technologic infrastructure of Suzhou (SZS201708). J.L. thanks the support from the NSFC (21433005, 91426302, 21521091, and 21590792), the Supercomputer Center of the Computer Network Information Center, the Chinese Academy of Sciences, and the Computational Chemistry Laboratory of the Department of Chemistry, Tsinghua University. L.E. thanks the US National Science Foundation (NSF) for generous support of this work under the NSF-PREM program (DMR 1205302) and the CHE-1408865. The Robert A. Welch Foundation is also gratefully acknowledged for an endowed chair to L.E. (Grant AH-0033). S.F. thanks the NSF-PREM program (DMR 1205302) and the Welch Foundation for support (AH-1922-20170325). J.A. thanks the U.S. Department of Energy, Office of Basic Energy Sciences, Heavy Element Chemistry program, grant DE-SC0001136 for support. G.V. and A.A.P. acknowledge the European Research Council under the European Union’s Horizon 2020 programme (Grant No. 648295 “GraM3”) for financial support; they also thank D. S. Krylov and A. U. B. Wolter (IFW Dresden) for their help with the magnetic measurements. W.H.E.S. thanks the support from the Theoretical Chemistry Center of Tsinghua University.
Author contributions
N.C., L.E., and X.Z. conceived and designed the experiments. X.Z. and Y.W. synthesized and isolated all the compounds. W.L., J.L., W.H.E.S., X.C., A.H., S.G., D.C.S., T.J.D., and J.A. performed the computations and theoretical analyses. L.F. performed the crystallographic analysis. N.A. and S.D. provided the XPS analysis. X.L. carried out the NMR test. G.V. and A.A.P. measured and analyzed the magnetic data. N.C., L.E., W.H.E.S., J.L., L.F., S.F., X.Z, W.L., S.G., J.A., and S.W. co-wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Xingxing Zhang, Wan-lu Li, Lai Feng.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-05210-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jun Li, Email: junli@tsinghua.edu.cn.
Luis Echegoyen, Email: echegoyen@utep.edu.
Ning Chen, Email: chenning@suda.edu.cn.
References
- 1.Hayton TW. Metal-ligand multiple bonding in uranium: structure and reactivity. Dalton Trans. 2010;39:1145–1158. doi: 10.1039/B909238B. [DOI] [PubMed] [Google Scholar]
- 2.Hayton TW. Recent developments in actinide-ligand multiple bonding. Chem. Commun. 2013;49:2956–2973. doi: 10.1039/c3cc39053e. [DOI] [PubMed] [Google Scholar]
- 3.Liddle ST. The renaissance of non-aqueous uranium chemistry. Angew. Chem. Int. Ed. 2015;54:8604–8641. doi: 10.1002/anie.201412168. [DOI] [PubMed] [Google Scholar]
- 4.Gregson M, Wooles AJ, Cooper OJ, Liddle ST. Covalent uranium carbene chemistry. Inorg. Chem. 2015;35:262–294. [Google Scholar]
- 5.Patel D, Liddle ST. f-Element-metal bond chemistry. Rev. Inorg. Chem. 2012;32:1. doi: 10.1515/revic.2012.0001. [DOI] [Google Scholar]
- 6.Fortier S, Brown JL, Kaltsoyannis N, Wu G, Hayton TW. Synthesis, molecular and electronic structure of UV(O)[N(SiMe3)2]3. Inorg. Chem. 2012;51:1625–1633. doi: 10.1021/ic201936j. [DOI] [PubMed] [Google Scholar]
- 7.Lewis AJ, Mullane KC, Nakamaru-Ogiso E, Carroll PJ, Schelter EJ. The inverse trans influence in a family of pentavalent uranium complexes. Inorg. Chem. 2014;53:6944–6953. doi: 10.1021/ic500833s. [DOI] [PubMed] [Google Scholar]
- 8.Lewis AJ, Carroll PJ, Schelter EJ. Stable uranium(VI) methyl and acetylide complexes and the elucidation of an inverse trans influence ligand series. J. Am. Chem. Soc. 2013;135:13185–13192. doi: 10.1021/ja406610r. [DOI] [PubMed] [Google Scholar]
- 9.Kosog B, La Pierre HS, Heinemann FW, Liddle ST, Meyer K. Synthesis of uranium(VI) terminal oxo complexes: molecular geometry driven by the inverse trans-influence. J. Am. Chem. Soc. 2012;134:5284–5289. doi: 10.1021/ja211618v. [DOI] [PubMed] [Google Scholar]
- 10.Fortier S, Wu G, Hayton TW. Synthesis of a nitrido-substituted analogue of the uranyl ion, [N═U═O]+ J. Am. Chem. Soc. 2010;132:6888–6889. doi: 10.1021/ja101567h. [DOI] [PubMed] [Google Scholar]
- 11.Hayton TW, Boncella JM, Scott BL, Batista ER. Exchange of an imido ligand in bis(imido) complexes of uranium. J. Am. Chem. Soc. 2006;128:12622–12623. doi: 10.1021/ja064400j. [DOI] [PubMed] [Google Scholar]
- 12.Brown JL, Fortier S, Lewis RA, Wu G, Hayton TW. A complete family of terminal uranium chalcogenides, [U(E)(N{SiMe3}2)3]− (E=O, S, Se, Te) J. Am. Chem. Soc. 2012;134:15468–15475. doi: 10.1021/ja305712m. [DOI] [PubMed] [Google Scholar]
- 13.Brown JL, Fortier S, Wu G, Kaltsoyannis N, Hayton TW. Synthesis and spectroscopic and computational characterization of the chalcogenido-substituted analogues of the uranyl ion, [OUE]2+ (E=S, Se) J. Am. Chem. Soc. 2013;135:5352–5355. doi: 10.1021/ja402068j. [DOI] [PubMed] [Google Scholar]
- 14.Fortier S, Kaltsoyannis N, Wu G, Hayton TW. Probing the reactivity and electronic structure of a uranium(V) terminal oxo complex. J. Am. Chem. Soc. 2011;133:14224–14227. doi: 10.1021/ja206083p. [DOI] [PubMed] [Google Scholar]
- 15.Smiles DE, Wu G, Hayton TW. Synthesis of uranium–ligand multiple bonds by cleavage of a trityl protecting group. J. Am. Chem. Soc. 2014;136:96–99. doi: 10.1021/ja411423a. [DOI] [PubMed] [Google Scholar]
- 16.Smiles DE, Wu G, Hrobárik P, Hayton TW. Use of 77Se and 125Te NMR spectroscopy to probe covalency of the actinide-chalcogen bonding in [Th(En){N(SiMe3)2}3]− (E=Se, Te; n=1, 2) and their oxo-uranium(VI) congeners. J. Am. Chem. Soc. 2016;138:814–825. doi: 10.1021/jacs.5b07767. [DOI] [PubMed] [Google Scholar]
- 17.Rosenzweig MW, et al. Uranium(IV) terminal hydrosulfido and sulfido complexes: insights into the nature of the uranium-sulfur bond. Chem. Sci. 2016;7:5857–5866. doi: 10.1039/C6SC00677A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayton TW, et al. Synthesis of imido analogs of the uranyl ion. Science. 2005;310:1941. doi: 10.1126/science.1120069. [DOI] [PubMed] [Google Scholar]
- 19.Andrez J, et al. Synthesis and reactivity of a terminal uranium(IV) sulfide supported by siloxide ligands. Chem. Sci. 2016;7:5846–5856. doi: 10.1039/C6SC00675B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King DM, Liddle ST. Progress in molecular uranium-nitride chemistry. Coord. Chem. Rev. 2014;266-267:2–153. doi: 10.1016/j.ccr.2013.06.013. [DOI] [Google Scholar]
- 21.Tsoureas N, Kilpatrick AFR, Inman CJ, Cloke FGN. Steric control of redox events in organo-uranium chemistry: synthesis and characterisation of U(V) oxo and nitrido complexes. Chem. Sci. 2016;7:4624–4632. doi: 10.1039/C6SC00632A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King DM, et al. Synthesis and structure of a terminal uranium nitride complex. Science. 2012;337:717–720. doi: 10.1126/science.1223488. [DOI] [PubMed] [Google Scholar]
- 23.King DM, et al. Isolation and characterization of a uranium(VI)–nitride triple bond. Nat. Chem. 2013;5:482–488. doi: 10.1038/nchem.1642. [DOI] [PubMed] [Google Scholar]
- 24.Anderson NH, et al. Elucidating bonding preferences in tetrakis(imido)uranate(VI) dianions. Nat. Chem. 2017;9:850–855. doi: 10.1038/nchem.2767. [DOI] [PubMed] [Google Scholar]
- 25.Gardner BM, et al. Triamidoamine-uranium(IV)-stabilized terminal parent phosphide and phosphinidene complexes. Angew. Chem. Int. Ed. 2014;53:4484–4488. doi: 10.1002/anie.201400798. [DOI] [PubMed] [Google Scholar]
- 26.Gardner BM, et al. Triamidoamine uranium(IV)–arsenic complexes containing one-, two- and threefold U–As bonding interactions. Nat. Chem. 2015;7:582–590. doi: 10.1038/nchem.2279. [DOI] [PubMed] [Google Scholar]
- 27.Mills DP, et al. Synthesis of a uranium(VI)-carbene: reductive formation of uranyl(V)-methanides, oxidative preparation of a [R2C=U=O]2+ Analogue of the [O=U=O]2+ uranylion (R=Ph2PNSiMe3), and comparison of the nature of UIV=C, UV=C, and UVI=C double bonds. J. Am. Chem. Soc. 2012;134:10047–10054. doi: 10.1021/ja301333f. [DOI] [PubMed] [Google Scholar]
- 28.Cooper OJ, et al. The nature of the UC double bond: pushing the stability of high-oxidation-state uranium carbenes to the limit. Chem. Eur. J. 2013;19:7071–7083. doi: 10.1002/chem.201300071. [DOI] [PubMed] [Google Scholar]
- 29.Lu EL, et al. Synthesis, characterization and reactivity of a uranium(VI) carbene imido oxo complex. Angew. Chem. Int. Ed. 2014;53:6696–6700. doi: 10.1002/anie.201403892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold PL. Organometallic actinides: Now U=C it. Nat. Chem. 2009;1:29–30. doi: 10.1038/nchem.153. [DOI] [PubMed] [Google Scholar]
- 31.Cramer RE, Maynard RB, Paw JC, Gilje JW. A uranium-carbon multiple bond. crystal and molecular structure of (η5-C5H5)3UCHP(CH3)2(C6H5) J. Am. Chem. Soc. 1981;103:3589–3590. doi: 10.1021/ja00402a065. [DOI] [Google Scholar]
- 32.Tourneux JC, Berthet JC, Mézailles N, Ephritikhine M. Exploring the uranyl organometallic chemistry: from single to double uranium-carbon bonds. J. Am. Chem. Soc. 2011;133:6162–6165. doi: 10.1021/ja201276h. [DOI] [PubMed] [Google Scholar]
- 33.Cantat T, et al. The U=C double bond: synthesis and study of uranium nucleophilic carbene complexes. J. Am. Chem. Soc. 2009;131:963–972. doi: 10.1021/ja807282s. [DOI] [PubMed] [Google Scholar]
- 34.Fortier S, Walensky JR, Wu G, Hayton TW. Synthesis of a phosphorano-stabilized U(IV)-carbene via one-electron oxidation of a U(III)-ylide adduct. J. Am. Chem. Soc. 2011;133:6894–6897. doi: 10.1021/ja2001133. [DOI] [PubMed] [Google Scholar]
- 35.Lyon JT, Andrews L. Formation and characterization of the uranium methylidene complexes CH2=UHX (X=F, Cl, and Br) Inorg. Chem. 2006;45:1847–1852. doi: 10.1021/ic051785i. [DOI] [PubMed] [Google Scholar]
- 36.Lyon JT, Hu HS, Andrews L, Li J. Formation of unprecedented actinide carbon triple bonds in uranium methylidyne molecules. Proc. Natl Acad. Sci. USA. 2007;104:18919–18924. doi: 10.1073/pnas.0707035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho HG, Andrews L. Matrix preparation and spectroscopic and theoretical investigation of small high oxidation-state complexes of groups 3-12, 14, lanthanide and actinide metal atoms: carbon-metal single, double and triple bonds. Coord. Chem. Rev. 2017;335:76–102. doi: 10.1016/j.ccr.2016.12.012. [DOI] [Google Scholar]
- 38.Du J, Jiang G. A systematic theoretical study of UC6: structure, bonding nature, and spectroscopy. Inorg. Chem. 2017;56:13794–13800. doi: 10.1021/acs.inorgchem.7b01871. [DOI] [PubMed] [Google Scholar]
- 39.Utton CA, et al. Laser melting of uranium carbides. J. Nucl. Mater. 2009;385:443–448. doi: 10.1016/j.jnucmat.2008.12.031. [DOI] [Google Scholar]
- 40.Frad, W. A. Advances in Inorganic Chemistry and Radiochemistry Vol. 11, 153–247 (Academic Press, New York and London, 1968).
- 41.Popov AA, Yang S, Dunsch L. Endohedral fullerenes. Chem. Rev. 2013;113:5989–6113. doi: 10.1021/cr300297r. [DOI] [PubMed] [Google Scholar]
- 42.Stevenson S, et al. Small-bandgap endohedral metallofullerenes in high yield and purity. Nature. 1999;401:55–57. doi: 10.1038/43415. [DOI] [Google Scholar]
- 43.Wang Y, et al. Unique four-electron metal-to-cage charge transfer of Th to a C82 fullerene cage: complete structural characterization of Th@C3v(8)-C82. J. Am. Chem. Soc. 2017;139:5110–5116. doi: 10.1021/jacs.6b13383. [DOI] [PubMed] [Google Scholar]
- 44.Cai W, et al. Single crystal structures and theoretical calculations of uranium endohedral metallofullerenes (U@C2n, 2n = 74, 82) show cage isomer dependent oxidation states for U. Chem. Sci. 2017;8:5282–5290. doi: 10.1039/C7SC01711A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krätschmer W, Lamb LD, Fostiropoulos K, Huffman DR. Solid C60: a new form of carbon. Nature. 1990;347:354–358. doi: 10.1038/347354a0. [DOI] [Google Scholar]
- 46.Pyykkö P, Zhao Y. The large range of uranyl bond lengths: ab initio calculations on simple uranium-oxygen clusters. Inorg. Chem. 1991;30:3787–3788. doi: 10.1021/ic00019a046. [DOI] [Google Scholar]
- 47.Pyykkö P, Li J, Runeberg N. Quasirelativistic pseudopotential study of species isoelectronic to uranyl and the equatorial coordination of uranyl. J. Phys. Chem. 1994;98:4809–4813. doi: 10.1021/j100069a007. [DOI] [Google Scholar]
- 48.Patel D, et al. A formal high oxidation state inverse-sandwich diuranium complex: a new route to f-block-metal bonds. Angew. Chem. Int. Ed. 2011;50:10388–10392. doi: 10.1002/anie.201104110. [DOI] [PubMed] [Google Scholar]
- 49.Pyykkö P, Atsumi M. Molecular double-bond covalent radii for elements Li–E112. Chem. Eur. J. 2009;15:12770–12779. doi: 10.1002/chem.200901472. [DOI] [PubMed] [Google Scholar]
- 50.Fortier S, Hayton TW. Oxo ligand functionalization in the uranyl ion (UO22+) Coord. Chem. Rev. 2010;254:197–214. doi: 10.1016/j.ccr.2009.06.003. [DOI] [Google Scholar]
- 51.Frenking G. Dative bonds in main-group compounds: a case for more arrows! Angew. Chem. Int. Ed. 2014;53:6040–6046. doi: 10.1002/anie.201311022. [DOI] [PubMed] [Google Scholar]
- 52.Karen P. Oxidation state, a long-standing issue! Angew. Chem. Int. Ed. 2015;54:4716–4726. doi: 10.1002/anie.201407561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamada M, Mizorogi N, Tsuchiya T, Akasaka T, Nagase S. Metal atoms collinear with the spiro carbon of 6,6-open adducts, M2@C80(Ad) (M=La and Ce, Ad=adamantylidene) J. Am. Chem. Soc. 2008;130:1171–1176. doi: 10.1021/ja073924w. [DOI] [PubMed] [Google Scholar]
- 54.Yamada M, Mizorogi N, Tsuchiya T, Akasaka T, Nagase S. Synthesis and characterization of the D5h isomer of the endohedral dimetallofullerene Ce2@C80: two-dimensional circulation of encapsulated metal atoms inside a fullerene Cage. Chem. Eur. J. 2009;15:9486–9493. doi: 10.1002/chem.200900713. [DOI] [PubMed] [Google Scholar]
- 55.Yamada M, et al. Positional control of encapsulated atoms inside a fullerene cage by exohedral addition. J. Am. Chem. Soc. 2005;127:14570–14571. doi: 10.1021/ja054346r. [DOI] [PubMed] [Google Scholar]
- 56.Krause M, et al. Structure and sta.bility of endohedral fullerene Sc3N@C80: a Raman, infrared and theoretical analysis. J. Chem. Phys. 2001;115:6596–6605. doi: 10.1063/1.1399298. [DOI] [Google Scholar]
- 57.Chaur ManuelN, Melin F, Ortiz, Angy L, Echegoyen L. Chemical, electrochemical, and structural properties of endohedral metallofullerenes. Angew. Chem. Int. Ed. 2009;48:7514–7538. doi: 10.1002/anie.200901746. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L, Chen N, Fan L, Wang C, Yang S. Electrochemistry of Sc3N@C78 and Sc3N@C80 (Ih): On achieving reversible redox waves of the trimetal nitride endohedral fullerenes. J. Electroanal. Chem. 2007;608:15–21. doi: 10.1016/j.jelechem.2007.04.015. [DOI] [Google Scholar]
- 59.Svitova AL, et al. Endohedral fullerene with µ3-carbido ligand and titanium-carbon double bond stabilized inside a carbon cage. Nat. Commun. 2014;5:3568. doi: 10.1038/ncomms4568. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki T, et al. Electrochemistry and ab initio study of the dimetallofullerene La2@C80. Angew. Chem. Int. Ed. 1995;34:1094–1096. doi: 10.1002/anie.199510941. [DOI] [Google Scholar]
- 61.Rosen RK, Andersen RA, Edelstein NM. [(MeC5H4)3U]2[μ-1,4-N2C6H4]: a bimetallic molecule with antiferromagnetic coupling between the uranium centers. J. Am. Chem. Soc. 1990;112:4588–4590. doi: 10.1021/ja00167a092. [DOI] [Google Scholar]
- 62.Spencer LP, et al. Cation–cation interactions, magnetic communication, and reactivity of the pentavalent uranium ion [U(N tBu)2]+ Angew. Chem. Int. Ed. 2009;48:3795–3798. doi: 10.1002/anie.200806190. [DOI] [PubMed] [Google Scholar]
- 63.Chen L, et al. A mixed-valent uranium phosphonate framework containing UIV, UV, and UVI. Chem. Eur. J. 2016;22:11954–11957. doi: 10.1002/chem.201602863. [DOI] [PubMed] [Google Scholar]
- 64.Schmidt AC, Heinemann FW, Lukens WW, Meyer K. Molecular and electronic structure of dinuclear uranium Bis-μ-Oxo complexes with diamond core structural motifs. J. Am. Chem. Soc. 2014;136:11980–11993. doi: 10.1021/ja504528n. [DOI] [PubMed] [Google Scholar]
- 65.Graves CR, Kiplinger JL. Pentavalent uranium chemistry−synthetic pursuit of a rare oxidation state. Chem. Commun. 2009;2009:3831–3853. doi: 10.1039/b902969a. [DOI] [PubMed] [Google Scholar]
- 66.Burla MC, et al. SIR2004: an improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005;38:381–388. doi: 10.1107/S002188980403225X. [DOI] [Google Scholar]
- 67.Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sec. C. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farrugia L. WinGX and ORTEP for Windows: an update. J. Appl. Crystallogr. 2012;45:849–854. doi: 10.1107/S0021889812029111. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The coordinates for the X-ray structure of 1 are available free of charge from the Cambridge Crystallographic Data Centre under deposition no. CCDC 1572891. All other data supporting the findings of this study are available from the corresponding authors on request.