

Abstract
Background
The increasing incidence of oropharyngeal squamous cell carcinoma (OPSCC) is mainly related to human papillomavirus (HPV) infection. As OPSCCs are often diagnosed at an advanced stage, mortality and morbidity remain high. There are no diagnostic biomarkers for early detection of OPSCC.
Methods
Serum from 25 patients with stage I–II OPSCC, and 12 healthy controls, was studied with quantitative label-free proteomics using ultra-definition MSE. Statistical analyses were performed to identify the proteins most reliably distinguishing early-stage OPSCCs from controls. P16 was used as a surrogate marker for HPV. P16-positive and P16-negative tumours were analysed separately.
Results
With two or more unique proteins per identification, 176 proteins were quantified. A clear separation between patients with early-stage tumours and controls was seen in principal component analysis. Latent structures discriminant analysis identified 96 proteins, most reliably differentiating OPSCC patients from controls, with 13 upregulated and 83 downregulated proteins in study cases. The set of proteins was studied further with network, pathway and protein–protein interaction analyses, and found to participate in lipid metabolism, for example.
Conclusions
We found a set of serum proteins distinguishing early-stage OPSCC from healthy individuals, and suggest a protein set for further evaluation as a diagnostic biomarker panel for OPSCC.
Subject terms: Tumour biomarkers, Diagnostic markers
Introduction
The worldwide annual incidence of head and neck cancers is almost 700,000, and 380,000 patients succumb to their disease annually.1 Oropharyngeal squamous cell carcinoma (OPSCC) accounts for ~20% of all new head and neck cancers, and the incidence is expected to rise over the following decades.1–3 This increase is mainly due to the cancers related to the human papillomavirus (HPV), and particularly due to its high-risk genotype HPV-16.2, 4
Traditionally, the main risk factors for OPSCC have been smoking and heavy alcohol consumption.5 Patients diagnosed with HPV-related OPSCC tend to be younger, and the consumption of alcohol and tobacco is often lower or even absent.6 HPV-related tumours have a better prognosis, a lower risk of secondary malignancies and the disease responds better to (chemo)radiotherapy.5, 7 It is also of note that HPV-associated OPSCCs in tobacco users behave like classical tobacco-associated OPSCCs.8 While the de-escalation of HPV-positive OPSCC patients’ treatment is under investigation,7 patients with HPV-negative OPSCC still require heavy treatment and the prognosis remains poor.6 At the moment, the only way to improve the prognosis of patients with HPV-negative tumours would be to diagnose them earlier.
Currently, there are no diagnostic biomarkers for OPSCC to enhance its detection at an earlier stage. Brush samples, used successfully for cervical cancer screening, have been shown to be ineffective in screening HPV-positive OPSCCs, and no diagnostic biomarkers from standard bio-fluids exist.9 HPV vaccinations could eventually decrease the epidemic of HPV-related OPSCC; however, even if effective vaccination programmes were launched, the decrease in incidence would only be seen after a couple of decades.10
Protein expression levels in both tumour tissue and serum samples of patients with OPSCC have been studied, showing some alterations, compared with those of healthy controls.11–16 However, these studies have often been targeted to recognised proteins, based on earlier studies on other cancers. Discovery-driven mass spectrometry proteomics offers the possibility to discover novel biomarkers and pathways, as well as to associate the findings with clinical aspects.
Our objective was to compare the serum protein profiles of patients with early-stage OPSCC and of healthy controls, to promote early cancer diagnostics. For early-stage tumours, we chose stage I and stage lI tumours (eighth edition of TNM classification of malignant tumours, 2016). Protein p16, i.e. cyclin-dependent kinase inhibitor 2A, is used as a surrogate marker for HPV status at our department and also in this study. The protein was first presented for OPSCC by Klussmann et al. and is now an established immunohistological marker, widely used instead of the arduous and expensive HPV detection and typing.17 We analysed the serum samples in ultra-definition MSE (UDMSE) mode. Of three data-independent data-acquisition methods available in the Synapt G2-S (MSE, high-definition MSE (HDMSE) and UDMSE), the last one was chosen as it gives the best protein coverage on the sample.18 Based on the proteomic changes revealed, we aimed to find a set of proteins that are possibly usable as a biomarker panel for early-stage OPSCC.
Materials and methods
Patients and serum samples
Serum samples from 25 patients diagnosed with stage I–II OPSCC were collected prior to treatment between the years 2012 and 2015 at the Department of Otorhinolaryngology—Head and Neck Surgery, Helsinki University Hospital, Helsinki, Finland. After collection, the samples were allowed to clot at room temperature (RT) before they were centrifuged at 4 °C (1000 × g) to separate serum. Sera were stored at –70 °C until all were assayed at the same time. The inclusion strategy by the TNM status was based on the eighth edition of TNM classification of malignant tumours, dividing HPV-positive and HPV-negative OPSCCs as separate entities,8 and protein p16 status was used as a surrogate marker for HPV. Twelve serum samples from age-matched and gender-matched control patients were received from the Finnish Red Cross Blood Service.
Written informed consent was obtained from all patients. The study plan was approved by the institutional Research Ethics Board at the Helsinki University Hospital (DNr. 51/13/03/02/2013).
Reagents
Reagents for serum pre-processing, Pierce Swell Gel Blue Albumin Removal Discs, Pierce Centrifuge columns and Pierce C18 Spin Columns, were acquired from Thermo Scientific (Rockform, IL, USA), solvents and high-purity HPLC reagents from Waters (Milford, MA, USA) and other reagents from Sigma-Aldrich (St Louis, MO, USA).
Serum treatment and protein digestion
The workflow has been described previously in detail.19 In brief, the samples were thawed, and after the depletion of the top 12 proteins with Pierce Top 12 protein depletion columns, the protein concentration was measured by a bicinchonic acid assay kit (Pierce, Thermo Scientific, Rockform, IL, USA) for the total protein concentration. Top 12 protein-depleted serum samples corresponding to 350 µg of total protein were dried in a speed vacuum (Savant, Thermofisher), and then dissolved in 6 M urea and 100 mM Tris-HCl (pH 7.4). Reduction of disulphide bonds was performed with 10 mM of dithiothreitol (DTT) for 60 min at RT, and thereafter 30 mM iodoacetamide was used for alkylating the proteins for 60 min in the dark at RT. Protein digestion was performed with trypsin (Promega, Madison, WI) for 18 h at +37 °C after the consumption of excess iodoacetamide by adding DTT again (30 mM DTT, 60 min at RT). Samples were diluted 1:10 with high-purity Milli-Q water (Millipore, Billerica, MA, USA) before addition of trypsin. Finally, the samples were purified in C18 spin columns, and dried in a speed vacuum and dissolved in 0.1% formic acid containing 12.5 fmol Hi3 peptide mixture (Waters) per µl. All of the procedures described were performed according to the manufacturer’s instructions, wherever applicable.
Liquid chromatography—ultra-definition MSE
Four-microlitre samples corresponding to 1.4 µg of total protein were injected to the ultra-performance liquid chromatography (UPLC) system (Waters Corporation, Billerica, MA, USA).18 TRIZAIC nanoTile 88-µm × 100-mm HSS-T3u wTRAP was applied as a separating device before mass spectrometry (MS). After loading and trapping, the samples were washed for 2 min at 8.0 µl/min with 1% buffer B. The analytical gradient was used as follows: 0–1 min 1% B; at 2 min 5% B; at 65 min 30% B; at 78 min 50% B; at 80 min 85% B; at 83 min 85% B; at 84 min 1% B and at 90 min 1% B with 450 nl/min. Buffer A consists of 0.1% formic acid in water and buffer B consists of 0.1% formic acid in acetonitrile (Sigma-Aldrich).
The data were acquired with UDMSE with Synapt G2-S UDMS (Waters Corporation) including ion mobility spectroscopy (IMS). The data range was 100–2000m/z, scan time 1 s, IMS wave velocity 650 ms−1 and collision energy ramped in trap between 20 and 60 V. Calibration was performed by Glu1-fibrinopeptide B MS2 fragments and Glu1-fibrinopeptide B precursor ion, used during the acquisitions as a lock mass. In total, 10% of the samples were acquired as triplicates to validate the results, and further analysis was conducted with Progenesis QI for Proteomics software (Nonlinear Dynamics, Newcastle, UK) (Supplement S2—triplets).
The mass spectrometry proteomics data have been deposited into the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD008445.20
Data analysis
The data analysis was described previously in detail.21 Briefly, Progenesis QI for proteomics software (Version 3, Nonlinear Dynamics) was used for processing raw files. Peptide identification was run with Uniprot human FASTA sequences (UniprotKB Release 2015_09, 20205 sequence entries), and label-free protein quantification was performed with the Hi-N method (Protein Lynx Global Server).22 The samples were spiked with 12.5 fmol/µl of CLPB_ECOLI (P63285, ClpB protein) peptides (Hi3 Escherichia Coli Standard, Waters).
The peptide identification parameters were fixed modification of cysteine (carbamidomethyl) and variable modification of methionine (oxidation). The peptide error tolerance was set to a maximum of 10 ppm, the false-discovery rate was limited to less than 2% and default values (in Progenesis QI for Proteomics) were used for the rest of the parameters.
The quantified proteins in all comparisons were compared by ANOVA on a protein-by-protein basis and their expression levels were considered significantly different if the ANOVA p value was <0.05. Principal component analysis (PCA), offering the visualisation of the main axes of variation in the data groups, was performed by Progenesis QI for proteomics. Processing the Progenesis QI data with EZinfo 3.0 software (a statistical tool released in December 2014, Umetrics, Sweden), supervised OPLS–DA modelling was performed. With a p(corr) cut-off of ± 0.80, a variance versus correlation plot (S-plot) and a list of S-plot proteins was generated from OPLS–DA data.
Protein–protein interactions, pathways and networks
STRING 10.5 database illustrates known and predicted protein–protein interactions (PPI),23 and was used for PPI analyses, giving a sophisticated view of possible and known interactions between proteins. PPI analyses were conducted to filter the S-plot proteins and project them to connected pathways and/or co-expression. Medium stringency was used for inferring the networks from protein lists on the STRING DB and textmining was excluded as a setting.
The network and canonical pathway overrepresentation analyses were conducted through the use of Ingenuity pathway analysis (IPA; QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) with default parameters to identify which networks and pathways were most enriched in our protein list.24 IPA networks differ from PPIs in their way of connecting proteins. In addition to the proteins actually present, they combine the information about possible connector proteins (not present in the user-supplied list). This allows another way of finding the networks the proteins are enriched into. IPA analyses were conducted on the proteins with the ANOVA p value < 0.05 and S-plot proteins were then separately matched to the proteins in enriched networks.
Results
Metadata and workflow
Twenty-five serum samples from patients with stage I and stage II OPSCC, together with 12 samples from healthy controls were studied. Of the 25 patients with stage I–II tumours, 12 had p16-positive and 13 had p16-negative tumours.
The tumour localisation was tonsil in 15 (60%) of the 25 patients, base of the tongue in 8 (32%), the soft palate in 8 (32%) and posterior wall of the oropharynx in 1 (4%). Sixty percent of the patients were male and 40% were female. The age of the patients varied from 36 to 78 years with the median age being 60.85 (average 60.92). More detailed clinical parameters are provided in Supplementary Table 1. The data analysis workflow is presented in Fig. 1.
Fig. 1.
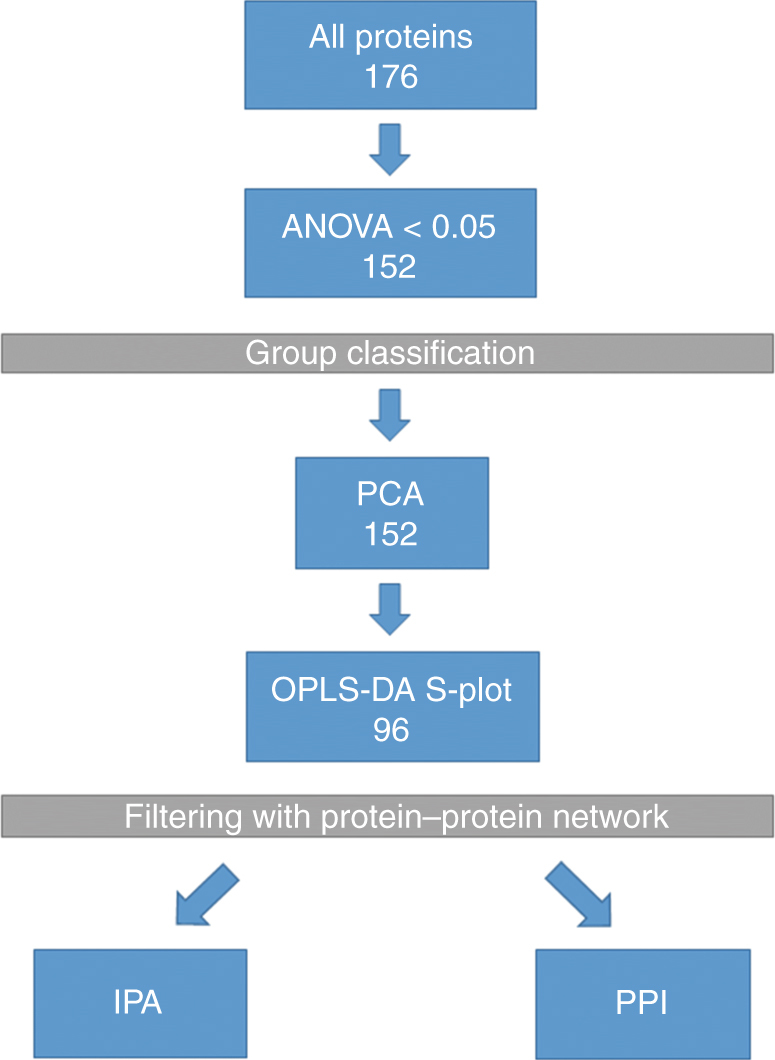
Data analysis workflow. Protein quantification data were from ultra-definition MSE, and proteins with two or more unique peptides were approved for identification. ANOVA cut-off of 0.05 was used. PCA: principal component analysis is used to visualise the variation between groups. OPLS–DA: latent structures discriminant analysis brings data for the S-plot for an efficient comparison of protein expression profiles. PPI: protein–protein interaction network gives the known and predicted functional and physical associations between single proteins in the S-plot. IPA: Ingenuity pathway analysis is an analysis tool revealing pathways and potential networks associated with the given data
All early-stage OPSCCs versus controls
Protein identification and PCA
With the criterion of two or more unique peptides per protein identification, 176 proteins were quantified from all cases and controls were analysed. The identified proteins were compared by ANOVA on a protein-to-protein basis. With the ANOVA cut-off of 0.05, 152 proteins with two or more unique peptides were quantified (Supplementary Table 2). Based on serum protein expression levels of patients with early-stage OPSCC and healthy controls, the two groups were found to be separated in PCA (Fig. 2).
Fig. 2.

Principal component analysis using serum protein expression data of early-stage OPSCC versus controls (two or more unique peptides, ANOVA p value < 0.05). Early-stage tumour samples are marked with red and controls are marked with blue
OPLS–DA
As another group classification method, OPLS–DA modelling was performed, and an S-plot was generated, presenting 96 proteins that most reliably distinguished patients from controls (Fig. 3). These proteins passed the p(corr) cut-off of ± 0.80 and were thus considered significantly different (Table 1). Of the 96 proteins, 13 were expressed in higher levels in early-stage OPSCCs when compared to controls, and the remaining 83 proteins had lower levels in cases compared with controls.
Fig. 3.

S-plot obtained from OPLS–DA regression analysis of the serum protein expressions in early-stage OPSCCs versus controls (p(corr) ± 0.80). The proteins were downregulated in tumour patients’ serum at the upper-right corner and upregulated on the lower left
Table 1.
S-plot proteins obtained from OPLS–DA regression analysis (p(corr) ± 0.80), of all stage I–II OPSCCs versus controls
| Primary accession | Peptide count | Unique peptides | Confidence score | ANOVA (p) | Max fold change | Highest mean | Lowest mean | Protein name | Gene name | p[1] | p(corr)[1] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P19105 | 2 | 2 | 10.19 | 0 | 184.10 | Stage I–II OPSCC | Control | Myosin regulatory light chain 12A | MYL12A | –0.04 | –0.95 |
| P01034 | 2 | 2 | 12.07 | 0 | Infinity | Stage I–II OPSCC | Control | Cystatin-C | CST3 | –0.02 | –0.95 |
| Q8N2Z9 | 3 | 3 | 15.95 | 0 | 14.92 | Stage I–II OPSCC | Control | Centromere protein S | APITD1 | –0.04 | –0.94 |
| Q09013;M0QX08 | 4 | 3 | 35.22 | 0 | 9.23 | Stage I–II OPSCC | Control | Myotonin-protein kinase | DMPK | –0.03 | –0.92 |
| P15428 | 2 | 2 | 11.07 | 0 | Infinity | Stage I–II OPSCC | Control | 15-hydroxyprostaglandin dehydrogenase [NAD(+)] | HPGD | –0.02 | –0.92 |
| Q8N3Y7 | 5 | 3 | 33.98 | 1.12E−10 | 10.36 | Stage I–II OPSCC | Control | Epidermal retinol dehydrogenase 2 | SDR16C5 | –0.03 | –0.91 |
| P36980 | 12 | 4 | 74.12 | 0 | 8.55 | Stage I–II OPSCC | Control | Complement factor H-related protein 2 | CFHR2 | –0.02 | –0.89 |
| A0A0B4J2B5 | 3 | 2 | 10.40 | 0 | 35.11 | Stage I–II OPSCC | Control | Protein IGHV3R16-9 (fragment) | IGHV3R16-9 | –0.01 | –0.87 |
| P48736 | 6 | 4 | 34.80 | 2.00E−14 | 8.29 | Stage I–II OPSCC | Control | Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform | PIK3CG | –0.04 | –0.86 |
| Q3MJ40 | 3 | 2 | 24.36 | 1.11E−16 | 10.49 | Stage I–II OPSCC | Control | Coiled-coil domain-containing protein 144B | CCDC144B | –0.02 | –0.85 |
| O94887 | 7 | 3 | 36.29 | 3.27E−07 | 3.80 | Stage I–II OPSCC | Control | FERM, RhoGEF and pleckstrin domain-containing protein 2 | FARP2 | –0.01 | –0.81 |
| Q9C091 | 8 | 6 | 43.13 | 0 | 9.39 | Stage I–II OPSCC | Control | GREB1-like protein | GREB1L | –0.03 | –0.81 |
| A0A1B0GUA9 | 2 | 2 | 11.10 | 0 | Infinity | Stage I–II OPSCC | Control | Uncharacterised protein | N/A | –0.02 | –0.80 |
| P07357 | 39 | 37 | 309.89 | 6.18E−07 | 1.69 | Control | Stage I–II OPSCC | Complement component C8 alpha chain | C8A | 0.06 | 0.80 |
| P05452 | 19 | 17 | 183.04 | 1.11E−07 | 1.52 | Control | Stage I–II OPSCC | Tetranectin | CLEC3B | 0.04 | 0.80 |
| P02753 | 48 | 42 | 241.90 | 2.55E−07 | 2.03 | Control | Stage I–II OPSCC | Retinol-binding protein 4 | RBP4 | 0.09 | 0.81 |
| P07996 | 26 | 24 | 196.30 | 1.55E−09 | 3.15 | Control | Stage I–II OPSCC | Thrombospondin-1 | THBS1 | 0.06 | 0.81 |
| P05543 | 20 | 18 | 142.13 | 3.40E−07 | 2.40 | Control | Stage I–II OPSCC | Thyroxine-binding globulin | SERPINA7 | 0.05 | 0.82 |
| P01019 | 81 | 76 | 374.07 | 7.15E−09 | 2.58 | Control | Stage I–II OPSCC | Angiotensinogen | AGT | 0.14 | 0.84 |
| Q6PI47 | 3 | 3 | 22.91 | 2.59E−08 | 2.44 | Control | Stage I–II OPSCC | BTB/PZ domain-containing protein KCTD18 | KCTD18 | 0.06 | 0.85 |
| Q7Z6K3 | 3 | 3 | 29.13 | 6.27E−09 | 6.48 | Control | Stage I–II OPSCC | Protein prenyltransferase alpha subunit repeat-containing protein 1 | PTAR1 | 0.03 | 0.85 |
| P00734 | 106 | 90 | 686.35 | 1.45E−09 | 1.82 | Control | Stage I–II OPSCC | Prothrombin | F2 | 0.13 | 0.85 |
| O14791 | 11 | 6 | 64.95 | 2.10E−08 | 3.10 | Control | Stage I–II OPSCC | Apolipoprotein L1 | APL1 | 0.01 | 0.85 |
| P03952 | 53 | 44 | 476.60 | 4.74E−10 | 1.63 | Control | Stage I–II OPSCC | Plasma kallikrein | KLKB1 | 0.07 | 0.85 |
| P42336 | 9 | 3 | 45.78 | 8.43E−09 | 2.46 | Control | Stage I–II OPSCC | Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform | PIK3CA | 0.01 | 0.86 |
| P43652 | 108 | 90 | 739.29 | 3.46E−09 | 1.88 | Control | Stage I–II OPSCC | Afamin | AFM | 0.12 | 0.86 |
| Q5VU65 | 5 | 3 | 22.62 | 1.67E−10 | 6.46 | Control | Stage I–II OPSCC | Nuclear pore membrane glycoprotein 210-like | NUP210L | 0.01 | 0.87 |
| P00740 | 16 | 14 | 113.09 | 7.90E−09 | 3.73 | Control | Stage I–II OPSCC | Coagulation factor IX | F9 | 0.04 | 0.87 |
| P04196 | 62 | 58 | 332.49 | 5.30E−09 | 2.62 | Control | Stage I–II OPSCC | Histidine-rich glycoprotein | HRG | 0.14 | 0.87 |
| P43251 | 7 | 6 | 49.04 | 1.04E−07 | 3.04 | Control | Stage I–II OPSCC | Biotinidase | BTD | 0.02 | 0.87 |
| P04004 | 83 | 65 | 503.49 | 1.96E−10 | 2.03 | Control | Stage I–II OPSCC | Vitronectin | VTN | 0.16 | 0.87 |
| Q8IVH4 | 3 | 3 | 26.12 | 9.95E−06 | 3.33 | Control | Stage I–II OPSCC | Methylmalonic aciduria type-A protein, mitochondrial | MMAA | 0.02 | 0.88 |
| P08185 | 27 | 23 | 212.28 | 7.43E−10 | 2.33 | Control | Stage I–II OPSCC | Corticosteroid-binding globulin | SERPINA6 | 0.07 | 0.88 |
| P02749 | 124 | 109 | 472.25 | 1.30E−10 | 2.15 | Control | Stage I–II OPSCC | Beta-2-glycoprotein 1 | APH | 0.27 | 0.88 |
| Q14624 | 138 | 123 | 984.03 | 4.93E−10 | 1.91 | Control | Stage I–II OPSCC | Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 | 0.19 | 0.89 |
| P08697 | 38 | 36 | 326.03 | 4.46E−11 | 1.95 | Control | Stage I–II OPSCC | Alpha-2-antiplasmin | SERPINF2 | 0.08 | 0.89 |
| P00915 | 3 | 2 | 16.19 | 0 | Infinity | Control | Stage I–II OPSCC | Carbonic anhydrase 1 | CA1 | 0.01 | 0.89 |
| Q9UK55 | 8 | 7 | 43.36 | 0 | 8.09 | Control | Stage I–II OPSCC | Protein Z-dependent protease inhibitor | SERPINA10 | 0.08 | 0.89 |
| Q15102 | 2 | 2 | 10.00 | 0 | 1344.35 | Control | Stage I–II OPSCC | Platelet-activating factor acetylhydrolase IB subunit gamma | PAFAH1B3 | 0.01 | 0.89 |
| P20851 | 10 | 6 | 78.00 | 0 | 45.67 | Control | Stage I–II OPSCC | C4b-binding protein beta chain | C4BPB | 0.03 | 0.89 |
| P05090 | 24 | 23 | 200.64 | 2.42E−14 | 2.69 | Control | Stage I–II OPSCC | Apolipoprotein D | APD | 0.09 | 0.90 |
| P02774 | 206 | 181 | 1148.20 | 2.81E−11 | 1.79 | Control | Stage I–II OPSCC | Vitamin D-binding protein | GC | 0.31 | 0.90 |
| P19652 | 31 | 20 | 133.66 | 1.44E−15 | 6.38 | Control | Stage I–II OPSCC | Alpha-1-acid glycoprotein 2 | RM2 | 0.07 | 0.90 |
| Q52LA3 | 4 | 3 | 26.69 | 6.92E−09 | 19.54 | Control | Stage I–II OPSCC | Protein lin-52 homologue | LIN52 | 0.02 | 0.90 |
| P01591 | 3 | 2 | 21.76 | 0 | 20.46 | Control | Stage I–II OPSCC | Immunoglobulin J chain | JCHAIN | 0.01 | 0.91 |
| P02748 | 80 | 70 | 553.63 | 3.29E−11 | 2.21 | Control | Stage I–II OPSCC | Complement component C9 | C9 | 0.12 | 0.91 |
| P41222 | 2 | 2 | 11.91 | 0 | 13700.40 | Control | Stage I–II OPSCC | Prostaglandin-H2 D-isomerase | PTGDS | 0.02 | 0.91 |
| P00747;Q02325;Q15195 | 154 | 132 | 1214.37 | 2.25E−11 | 2.00 | Control | Stage I–II OPSCC | Plasminogen | PLG | 0.19 | 0.91 |
| Q9NZP8 | 3 | 2 | 19.85 | 2.79E−07 | 194.65 | Control | Stage I–II OPSCC | Complement C1r subcomponent-like protein | C1RL | 0.02 | 0.92 |
| P80108 | 7 | 7 | 57.61 | 4.83E−11 | 3.28 | Control | Stage I–II OPSCC | Phosphatidylinositol–glycan-specific phospholipase D | GPLD1 | 0.03 | 0.92 |
| P0C0L5 | 394 | 16 | 2558.66 | 2.67E−08 | 6.78 | Control | Stage I–II OPSCC | Complement C4-B | C4B | 0.11 | 0.93 |
| P01031 | 131 | 116 | 977.48 | 3.05E−13 | 2.28 | Control | Stage I–II OPSCC | Complement C5 | C5 | 0.13 | 0.93 |
| Q9Y2H2 | 7 | 5 | 31.34 | 1.11E−16 | 14.32 | Control | Stage I–II OPSCC | Phosphatidylinositide phosphatase SAC2 | INPP5F | 0.08 | 0.93 |
| P09871 | 50 | 42 | 381.14 | 1.18E−13 | 2.02 | Control | Stage I–II OPSCC | Complement C1s subcomponent | C1S | 0.08 | 0.93 |
| Q96KN2 | 18 | 16 | 138.56 | 0 | 3.87 | Control | Stage I–II OPSCC | Beta-Ala-His dipeptidase | CNDP1 | 0.05 | 0.94 |
| P0C0L4 | 393 | 14 | 2503.26 | 9.99E−16 | 49.09 | Control | Stage I–II OPSCC | Complement C4-A | C4A | 0.19 | 0.94 |
| P08294 | 4 | 3 | 29.94 | 0 | 36.98 | Control | Stage I–II OPSCC | Extracellular superoxide dismutase [Cu–Zn] | SD3 | 0.02 | 0.94 |
| Q7LC44 | 3 | 2 | 22.46 | 7.34E−13 | 316.16 | Control | Stage I–II OPSCC | Activity-regulated cytoskeleton-associated protein | ARC | 0.02 | 0.94 |
| O95445 | 12 | 11 | 85.78 | 1.55E−15 | 3.46 | Control | Stage I–II OPSCC | Apolipoprotein M | APM | 0.06 | 0.94 |
| P02649 | 70 | 62 | 492.63 | 3.32E−14 | 3.40 | Control | Stage I–II OPSCC | Apolipoprotein E | APE | 0.16 | 0.94 |
| P27918 | 10 | 8 | 82.75 | 0 | 3.61 | Control | Stage I–II OPSCC | Properdin | CFP | 0.04 | 0.94 |
| P22352 | 23 | 20 | 129.93 | 1.11E−16 | 3.73 | Control | Stage I–II OPSCC | Glutathione peroxidase 3 | GPX3 | 0.07 | 0.95 |
| P07358 | 60 | 51 | 461.28 | 2.22E−16 | 2.42 | Control | Stage I–II OPSCC | Complement component C8 beta chain | C8B | 0.07 | 0.95 |
| P22792 | 28 | 25 | 266.69 | 3.08E−14 | 2.57 | Control | Stage I–II OPSCC | Carboxypeptidase N subunit 2 | CPN2 | 0.09 | 0.95 |
| Q08629 | 4 | 2 | 34.99 | 1.01E−08 | 31.43 | Control | Stage I–II OPSCC | Testican-1 | SPCK1 | 0.03 | 0.95 |
| Q04756 | 8 | 7 | 54.20 | 0 | 228.12 | Control | Stage I–II OPSCC | Hepatocyte growth factor activator | HGFAC | 0.04 | 0.95 |
| P00736 | 54 | 43 | 423.82 | 2.89E−15 | 2.58 | Control | Stage I–II OPSCC | Complement C1r subcomponent | C1R | 0.10 | 0.95 |
| O75636 | 8 | 7 | 88.24 | 0 | 24.29 | Control | Stage I–II OPSCC | Ficolin-3 | FCN3 | 0.07 | 0.95 |
| A0A096LPE2;P35542 | 35 | 15 | 202.65 | 0 | 6.02 | Control | Stage I–II OPSCC | Protein SAA2–SAA4 | SAA2–SAA4 | 0.06 | 0.96 |
| P06681 | 31 | 8 | 254.03 | 6.01E−13 | 4.48 | Control | Stage I–II OPSCC | Complement C2 | C2 | 0.04 | 0.96 |
| P19827 | 126 | 107 | 848.22 | 3.33E−16 | 3.17 | Control | Stage I–II OPSCC | Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH1 | 0.24 | 0.96 |
| P15169 | 10 | 9 | 93.43 | 0 | 9.89 | Control | Stage I–II OPSCC | Carboxypeptidase N catalytic chain | CPN1 | 0.06 | 0.96 |
| Q13790 | 3 | 2 | 27.72 | 1.10E−13 | 22.60 | Control | Stage I–II OPSCC | Apolipoprotein F | APF | 0.02 | 0.96 |
| P07360 | 19 | 15 | 166.71 | 2.64E−14 | 2.87 | Control | Stage I–II OPSCC | Complement component C8 gamma chain | C8G | 0.09 | 0.96 |
| P02747 | 8 | 8 | 60.45 | 6.85E−14 | 4.08 | Control | Stage I–II OPSCC | Complement C1q subcomponent subunit C | C1QC | 0.05 | 0.96 |
| P04070 | 4 | 2 | 23.64 | 1.11E−15 | 4.71 | Control | Stage I–II OPSCC | Vitamin K-dependent protein C | PRC | 0.01 | 0.96 |
| P13645 | 16 | 15 | 115.79 | 0 | 6.25 | Control | Stage I–II OPSCC | Keratin, type-I cytoskeletal 10 | KRT10 | 0.06 | 0.96 |
| P00748 | 3 | 3 | 18.59 | 0 | 679.60 | Control | Stage I–II OPSCC | Coagulation factor XII | F12 | 0.03 | 0.97 |
| P04003 | 82 | 74 | 628.10 | 1.11E−16 | 2.84 | Control | Stage I–II OPSCC | C4b-binding protein alpha chain | C4BPA | 0.18 | 0.97 |
| Q7Z794 | 6 | 3 | 44.34 | 0 | 48569.15 | Control | Stage I–II OPSCC | Keratin, type-II cytoskeletal 1b | KRT77 | 0.05 | 0.97 |
| P07225 | 32 | 27 | 244.61 | 0 | 3.08 | Control | Stage I–II OPSCC | Vitamin K-dependent protein S | PRS1 | 0.09 | 0.97 |
| P06276 | 7 | 6 | 56.00 | 0 | 11.44 | Control | Stage I–II OPSCC | Cholinesterase | BCHE | 0.04 | 0.97 |
| P27482 | 2 | 2 | 11.92 | 1.34E−07 | 25.89 | Control | Stage I–II OPSCC | Calmodulin-like protein 3 | CALML3 | 0.02 | 0.97 |
| P49908 | 9 | 7 | 46.07 | 0 | 41.43 | Control | Stage I–II OPSCC | Selenoprotein P | SELENP | 0.04 | 0.98 |
| O00750 | 11 | 4 | 59.49 | 0 | 4.02 | Control | Stage I–II OPSCC | Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit beta | PIK3C2B | 0.05 | 0.98 |
| P08571 | 6 | 6 | 51.89 | 0 | 6.40 | Control | Stage I–II OPSCC | Monocyte differentiation antigen CD14 | CD14 | 0.03 | 0.98 |
| Q9UGM5 | 8 | 8 | 54.43 | 1.08E−14 | 13.76 | Control | Stage I–II OPSCC | Fetuin-B | FETUB | 0.03 | 0.98 |
| P35858 | 32 | 29 | 277.24 | 0 | 11.46 | Control | Stage I–II OPSCC | Insulin-like growth factor-binding protein complex acid-labile subunit | IGFALS | 0.11 | 0.98 |
| O75882 | 22 | 20 | 150.98 | 0 | 15.94 | Control | Stage I–II OPSCC | Attractin | ATRN | 0.06 | 0.98 |
| P60709;P63261;P62736;P63267;P68032;P68133 | 6 | 3 | 45.19 | 0 | Infinity | Control | Stage I–II OPSCC | Actin, cytoplasmic 1 | ACTB | 0.05 | 0.99 |
| Q96IY4 | 5 | 5 | 35.72 | 0 | 18.33 | Control | Stage I–II OPSCC | Carboxypeptidase B2 | CPB2 | 0.04 | 0.99 |
| P17936 | 8 | 6 | 58.46 | 0 | 21.95 | Control | Stage I–II OPSCC | Insulin-like growth factor-binding protein 3 | IGFBP3 | 0.03 | 0.99 |
| P06396 | 76 | 3 | 631.02 | 1.11E−16 | 407.24 | Control | Stage I–II OPSCC | Gelsolin | GSN | 0.06 | 0.99 |
| P02746 | 12 | 11 | 106.18 | 0 | 8.16 | Control | Stage I–II OPSCC | Complement C1q subcomponent subunit B | C1QB | 0.07 | 0.99 |
| Q14520 | 19 | 17 | 146.18 | 0 | 7.40 | Control | Stage I–II OPSCC | Hyaluronan-binding protein 2 | HABP2 | 0.07 | 0.99 |
| Q5T6V5 | 5 | 4 | 38.73 | 0 | 64.40 | Control | Stage I–II OPSCC | UPF0553 protein C9orf64 | C9orf64 | 0.09 | 0.99 |
Protein–protein interactions
To further study our set of S-plot proteins and to try to identify the most relevant proteins, protein–protein interaction (PPI) webs were created using the STRING 10.5 database. Proteins with the most interactions, with connections to other proteins ranging from 9 to 16, were prothrombin (F2), plasminogen (PLG), alpha-2-antiplasmin (SERPINF2), histidine-rich glycoprotein (HRG), beta-2-glycoprotein 1 (APOH), carboxypeptidase B2 (CPB2), inter-alpha-trypsin inhibitor heavy chain H4 (ITIH4) and complement C2, C5, C4-A and C4-B (C2, C5, C4A and C4B).
According to the UNIPROT database,25 these proteins seemed to be associated with complement activation (early and late), extracellular matrix remodelling and lipid metabolism, for example. PPIs of the S-plot proteins are shown in Fig. 4.
Fig. 4.

PPI network of S-plot proteins (p(corr) ± 0.80) manifesting in the stage I–II OPSCC. The five serum proteins discussed in the article as possible biomarkers for early-stage OPSCC and suggested for further screening are circled: CFHR2 and MYL12A, upregulated in the tumour patients’ serum are circled with red, and the downregulated C9, FCN3 and C4BPA are circled with green
Pathways and networks
The top six IPA networks where the identified proteins were most enriched were 1. developmental disorder, hereditary disorder and immunological disease; 2. lipid metabolism, molecular transport and small-molecule biochemistry; 3. humoral immune response, inflammatory response, haematological system development and function; 4. cardiovascular disease, organismal injury and abnormalities and tissue morphology; 5. hereditary disorder, ophthalmic disease, organismal injury and abnormalities and 6. cell morphology, cellular development, cellular assembly and organisation. The score of the top six IPA networks ranged from 21 to 45. There were 13–23 proteins with the ANOVA p value < 0.05 participating in each of the networks and the total amount of focus molecules was 108. Of these, 46 were S-plot proteins (p(corr) ± 0.80). The network linked with lipid metabolism, containing 14 S-plot proteins, is illustrated in Fig. 5. The other five networks are presented in Supplementary Table 3. Altogether, among the S-plot proteins present in the top six IPA networks, four were upregulated in cases versus controls: complement factor H-related protein 2 (CFHR2), GREB1-like protein (GREB1L), myosin regulatory light chain 12A (MYL12A) and myotonin-protein kinase (DMPK). CFHR2 and MYL12A were also found to be binding in the PPI clusters. The remaining 42 S-plot proteins presented in the top three IPA networks were downregulated in cases versus controls, and the majority of these were also present in the PPI clusters.
Fig. 5.

IPA network 2. Lipid metabolism, molecular transport and small-molecule biochemistry with a score of 45. See text for details
In the canonical pathway analyses conducted with IPA, acute phase response signalling, LXR/RXR activation, FXR/RXR activation and the complement system were among the highest enriched pathways. The top canonical pathways are shown in Supplementary Figure 1.
Comparison between P16-negative and P16-positive tumours
Protein identification and PCA
In a comparison between p16-negative early-stage tumours and controls, 148 proteins were found with different expression levels in the serum samples. In the case of p16-positive early-stage tumours, the number was 152. When comparing the p16-negative and p16-positive groups with each other, 24 proteins were differently expressed. The protein identification tables are presented in Supplementary Table 2 and PCAs are presented in Supplementary Figures 4–6.
OPLS–DA
In the comparison between patients with early-stage p16-negative OPSCC and healthy controls, 103 proteins were presented in the S-plot (p(corr) ± 0.80), and 104 proteins were presented in the comparison of p16-positive tumours versus controls, respectively (Supplementary Figures 7 and 8). Of these, 96 were common between the two groups, shown in a Venn diagram (Supplementary Figure 9). It is of note, though, that the fold changes of the S-plot proteins were not identical in the two groups. The lists of S-plot proteins in the two comparisons are presented in Supplementary Table 4.
Protein–protein interactions
PPI networks of p16-positive and p16-negative groups were also studied separately, and the results showed great consistency with those from all early-stage OPSCC samples combined. Minor differences between p16-positive and p16-negative groups were detected, for example, there were slight differences in the protein interactions in the coagulation pathway. The PPI networks are shown in Supplementary Figures 10 and 11.
Pathways and networks
In the network analysis conducted with IPA, most networks were represented in both p16-positive and p16-negative patients’ data. Some differences were found, for example, haematological disease, haematological system development and function and organismal functions were a network solely enriched in the p16-positive group. The results are shown in Supplementary Table 3. In canonical pathway analyses, the top enriched pathways were also almost identical between p16-positive and p16-negative groups with some differences in their order (Supplementary Figures 2 and 3).
Discussion
The mortality of OPSCC ranges from 19 to 86%, the main predictive markers being tumour stage and HPV status.8, 26 At the moment, most tumours are diagnosed at an advanced stage, and thus the best way to improve their prognosis would be to diagnose them at an earlier stage.27 Currently, there are no known biomarkers to detect OPSCCs before clinical signs exist. When diagnosed, tumours are either visible or cause clinical symptoms, for example, dysphagia, pain etc.27 Discovering serum proteins that distinguish patients with early-stage cancer from healthy controls would be of great value from a diagnostic point of view. In order to identify possible proteins to be used as such biomarkers, we analysed serum samples of 25 patients diagnosed with stage I–II OPSCC and 12 healthy controls. Altogether, 176 serum proteins were reliably quantified, and the expression profiles of OPSCC patients differed clearly from those of healthy controls.
The discovery-driven nature of mass spectrometry-based analysis offers a unique chance to discover proteins and pathways that have not previously been studied in OPSCC. In previous serological studies, an association between serum antibodies towards HPV-16 early (E) antigens and HPV-positive OPSCC has been described, and these E antibodies have been studied as potential diagnostic biomarkers for HPV-related OPSCC. Recently, seropositivity for E6 antibodies was described as a highly sensitive (96%) and specific (98%) marker for HPV-positive OPSCC.12 However, in this study lacking a control group, the majority of patients had advanced stage tumours: there were 134 patients with stage lV tumours and 80 patients with stage I–III tumours. In addition, another study presenting an algorithm incorporating information about multiple E antibodies with a high sensitivity (83%) and specificity (99%) has been conducted for the detection of HPV-related OPSCC.11 In that study, age-matched and sex-matched healthy individuals served as healthy controls. However, analyses were made on patients with tumours of all stages and only a few represented early-stage tumours. Thus, the clinical use of the E antigens still remains an open question, as there is no information about their usability in early diagnostics, for example. In addition to E antigens, serum levels of matrix metalloproteinases (MMP1, 2 and 9) have been studied in oropharyngeal, laryngeal and hypopharyngeal carcinoma by Kalfert et al. and found not to serve as suitable prognostic tumour markers in these cancers.14 MMP1 expression was described as being significantly influenced by smoking and p16 expression. There was no control group in the study. Also, serum levels of IL-10, TNF-α, TGF-β, VEGF, Cyfra21-1, SCCAg, ferritin, CEA, CA19-9 and AFP have been studied in oral and oropharyngeal carcinoma patients.15, 16 In summary, until now, serological studies have not elucidated any biomarkers that will allow detection of oropharyngeal tumours at an early stage.
The OPLS–DA modelling generates a list of the most significant proteins in terms of group separation (S-plot proteins). This level of discrimination is difficult to obtain using other statistical methods. Statistically significant differences in expression of serum proteins between patients with early-stage OPSCC, when compared with controls, included 13 upregulated and 83 downregulated proteins. Of these, IPA networks and PPI analyses revealed interesting clusters of these proteins acting together. In the PPI network of the S-plot proteins of early-stage OPSCCs versus controls, examples of the pathways and biological processes visualised were complement activation (early and late), extracellular matrix remodelling, angiogenesis and possible tumour growth. Among the proteins with most interactions were complement C5, C4-A and C4-B (C5, C4A and C4B), prothrombin (F2), plasminogen (PLG), carboxypeptidase B2 (CPB2), alpha-2-antiplasmin (SERPINF2), histidine-rich glycoprotein (HRG) and insulin-like growth factor-associated proteins (IGFBP3, IGFALS). The complement cascade is one of the most studied biological processes in cancers.28 Dysregulated complement activation in the tumour microenvironment has been recently linked with increased inflammation and thus suppression of antitumour immune responses, leading to tumour cell proliferation, migration and invasive potential.29 The decrease of the plasmic complement C4-A has previously been described by Koifman et al. and Ornellas et al. in HPV-positive squamous cell carcinoma of the penis.30, 31 Also, genetic deficiency of the complement isoforms C4A or C4B may predict improved survival of metastatic renal cell carcinoma.32 In our study, serum levels of complement C4-A were lower in comparison with controls.
A common approach to biomarker signature discovery for any given group of patient samples is to perform a classification analysis such as the one we have done (OPLS–DA). However, by using different approaches to discovery, different molecules that differentiate the disease can be found. Moreover, the biological interpretation is often difficult due to the complicated nature of how gene/protein signatures are found, including the lack of causal relationships between protein expression and disease. The two aforementioned shortcomings are currently preventing biomarkers from becoming standard clinical tools. To circumvent these problems, network-based approaches have been proposed to be integrated with feature-selection algorithms.33 These network-based approaches include protein–protein interactions, canonical pathways and Gene Ontology annotations, which can help interpret the feature selection for various purposes including biomarker discovery.34 However, different approaches to these network-based methods lead to slightly different results, such as those employed by STRING DB or IPA.35 The methodology in the present work was chosen according to what has been suggested by deep analysis of common network-building software modules, i.e. that at least two different methods should be used for the purpose of network inference.35 To be able to filter our protein set, and to further identify a potential panel of proteins to serve as a diagnostic panel, IPA network analysis was conducted. There were six networks considered significant, having a score of 21 or more and at least 13 focus proteins.36 The first and third of the top six IPA networks with the best scores and focus molecules were developmental disorder, hereditary disorder and immunological disease and humoral immune response, inflammatory response, haematological system development and function. These networks were associated with complement activation, thus being consistent with the data received from PPI analyses. Proteins found in the second network, lipid metabolism, molecular transport and small-molecule biochemistry, were associated with lipoprotein metabolism and lipid digestion, mobilisation and transport. Most solid tumours tend to get hypoxic and are thus acidic.37 This causes tumour cells to increase their uptake of apolipoproteins, handle fatty acids more rapidly and enhance their cholesterol biosynthesis.37 These functions have been shown to have a big influence on tumour cell growth.38 Alterations of serum levels of apolipoproteins have previously been reported to be associated with breast, lung and colorectal cancers.39 In our material, most of the apolipoproteins participating in the networks were downregulated in the OPSCC serum compared to controls, except for apolipoprotein C-IV (APOC4) that was upregulated. This seems logical considering the increased uptake of apolipoproteins by tumour cells.
Two S-plot proteins, CFHR2 and MYL12A, upregulated in early OPSCC when compared with controls, were found in both PPI clusters and among the top six IPA networks. Out of the 42 downregulated S-plot proteins presented in the top six IPA networks, complement component C9 (C9), ficolin-3 (FCN3) and C4b-binding protein alpha chain (C4BPA) had the best p(corr), fold change and intensity values, and were also present in the PPI clusters (Fig. 5). In our opinion, together, these five proteins should be further studied as a potential future panel for early OPSCC diagnostics. Being all among S-plot proteins and present in both IPA and PPI networks, they had the best ability to identify cases from controls. CFHR2 is a complement factor found to regulate alternative complement pathway activation.40 MYL12A is a myosin regulatory subunit that regulates muscle cell contraction.25 This protein has been thought to potentially participate in DNA damage repair,41 and upregulation of MYL12A mRNA has been associated with non-small-cell lung carcinoma previously.42 C9 is a member of the membrane attack complex, participating in the final component of the complement system.41 FCN3 has a role in the activation of the complement pathway through the activation of the lectin pathway.41 Downregulation of C9, FCN3 and C4BPA mRNAs has previously been associated with liver cancer.43, 44 C4BPA, together with C4BPB, forms a multimeric protein participating in complement activation in the classical pathway.41 It is of note that, owing to very small abundances of C4BPB, there is little or no utility for this protein, as it will be hard to detect it reliably with classical clinical chemistry settings. However, C4BPA has all the characteristics of being clinically useful due to good abundance in serum samples, high confidence of identification, good fold change and statistical significance (Table 1 and Supplementary Table 2).
The ratio between upregulated and downregulated proteins and the networks in which these proteins were participating made us hypothesise that in the case of early-stage OPSCCs, the main reason for the change in serum proteome could be a tumour-specific response in the host system, not necessarily proteins originating from the actual tumour. When comparing our results with earlier serum proteomics studies on cancer patients, we discovered that 11 proteins out of the 152 quantified proteins in OPSCC serum were also expressed in the serum of patients with pancreatic cancer and 47 proteins were expressed in the serum of oral cavity squamous cell carcinoma (OSCC).45, 46 This finding indicates that changes in the levels of some serum proteins most likely reflect a general response to cancer, with still the largest part being specific to the disease. Even though the networks and functions of the proteins with altered expression levels in OPSCC were quite generalised to cancer, the protein combinations seem to be unique. Interestingly, the differences between OSCC and OPSCC, although smaller than in comparison to pancreatic cancer, were significant. Although cases in the current study represented early tumours, whereas tumours in the OSCC study were of all TNM stages,46 it is likely that this significant difference in the protein expression profiles is also due to tumour-specific changes in serum. In addition to these possible changes due to histological and anatomical differences between OPSCC and OSCC, another possible reason for the OSCC/OPSCC difference is the viral origin in half of the OPSCC tumours studied.8 The role of HPV in tongue cancers is not established.
When serum samples of patients with p16-positive and p16-negative tumours were compared with each other, 24 proteins were differently expressed in the two groups. S-plot proteins resulting from comparing each group with healthy controls were almost exclusively shared between the two groups, although the fold changes of the proteins’ expressions varied. IPA canonical pathways and networks and PPI network analyses were created separately for p16-negative and p16-positive early-stage OPSCCs versus control data. The majority of the interacting proteins were shared by both groups, as expected, as all the cases represent early-stage OPSCC. Some minor differences in protein interactions segregating the two groups were discovered. For example, a network haematological disease, haematological system development and function and organismal functions were only present in the IPA networks of the p16-positive group. All in all, based on serum proteomics, p16-positive and p16-negative early-stage OPSCCs seemed to be mostly similar, although some specific proteins, networks and PPIs were found.
These results strengthen the current knowledge of OPSCC being a disease with versatile altering events in protein expression levels, and further the knowledge in associating networks and interactions. Most probably, the changes seen in serum protein levels reflect the general host response, tumour-specific host response and leaking of tumour-specific proteins into the bloodstream. The expression levels of 96 S-plot proteins were able to reliably distinguish early-stage OPSCCs from healthy controls. Network and PPI analyses provided some additional information of the proteins, with the ability to filter out a smaller set of proteins—putatively representing a potential panel of biomarkers. This is important, as instead of seeking a single protein, the opportunity to form a panel of proteins with both upregulated and downregulated abundancies could serve as a more dependable composition for decision making in future diagnostics. We suggest that the panel of five serum proteins; CFHR2, MYL12A, C9, FCN3 and C4BPA, identified with these methods, might serve as a diagnostic biomarker for early-stage OPSCC.
To conclude, we have demonstrated how serum proteomics is capable of differentiating patients with early-stage OPSCC from healthy controls. This finding has a great potential to improve the early diagnostics of OPSCC. More importantly, the present study and our earlier work will allow us to further delineate differences between different head and neck cancers in terms of their characteristic serum-biomarker profiles. Further screening of the five above-mentioned proteins in a larger cohort of patients would be necessary to establish their value for clinical use.
Electronic supplementary material
Author contributions
AT: writing of the manuscript; MS: preparing of the samples, mass spectrometry, data analysis and participating in writing the manuscript; AM: participating in writing the paper and study design planning; PM: collection of serum samples; RS: data processing and statistics; AD: participating in manuscript writing and English-language revision and figure preparation; TC: collection of clinical data; TT: preparing of the samples; SJ: mass spectrometry and data analysis and SR: design of the study and writing of the manuscript.
Ethics approval
Written informed consent was obtained from all patients. The study plan was approved by the institutional Research Ethics Board at the Helsinki University Hospital (DNr. 51/13/03/02/2013). The current study was performed in accordance with the Declaration of Helsinki.
Availability of data and materials
The mass spectrometry proteomics is available from the ProteomeXchange Consortium via the PRIDE partner repositorywith the data set identifier PXD008445.
Funding
This study was supported by Korvatautien tutkimussäätiö, Jane and Aatos Erkko Foundation and the University of Helsinki, Medical Faculty.
Competing interests
The authors declare that they have no competing interests.
Footnotes
These authors contributed equally: Anna Tuhkuri and Mayank Saraswat.
Electronic supplementary material
Supplementary information is available for this paper at 10.1038/s41416-018-0162-2.
References
- 1.Ferlay J, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Chaturvedi AK, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011;29:4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta B, Johnson NW, Kumar N. Global epidemiology of head and neck cancers: a continuing challenge. Oncology. 2016;91:13–23. doi: 10.1159/000446117. [DOI] [PubMed] [Google Scholar]
- 4.Syrjanen K, Syrjanen S, Lamberg M, Pyrhonen S, Nuutinen J. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. Int. J. Oral Surg. 1983;12:418–424. doi: 10.1016/S0300-9785(83)80033-7. [DOI] [PubMed] [Google Scholar]
- 5.Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol. 2010;11:781–789. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ang KK, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masterson L, et al. De-escalation treatment protocols for human papillomavirus-associated oropharyngeal squamous cell carcinoma: a systematic review and meta-analysis of current clinical trials. Eur. J. Cancer. 2014;50:2636–2648. doi: 10.1016/j.ejca.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Lydiatt WM, et al. Head and Neck cancers-major changes in the American Joint Committee on cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017;67:122–137. doi: 10.3322/caac.21389. [DOI] [PubMed] [Google Scholar]
- 9.Fakhry C, Rosenthal BT, Clark DP, Gillison ML. Associations between oral HPV16 infection and cytopathology: evaluation of an oropharyngeal “pap-test equivalent” in high-risk populations. Cancer Prev. Res. 2011;4:1378–1384. doi: 10.1158/1940-6207.CAPR-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015;33:3235–3242. doi: 10.1200/JCO.2015.61.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahlstrom KR, et al. Diagnostic accuracy of serum antibodies to human papillomavirus type 16 early antigens in the detection of human papillomavirus-related oropharyngeal cancer. Cancer. 2017;123:4886–4894. doi: 10.1002/cncr.30955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holzinger D, et al. Sensitivity and specificity of antibodies against HPV16 E6 and other early proteins for the detection of HPV16-driven oropharyngeal squamous cell carcinoma. Int. J. Cancer. 2017;140:2748–2757. doi: 10.1002/ijc.30697. [DOI] [PubMed] [Google Scholar]
- 13.Horn D, et al. Upregulation of pAKT(Ser473) expression in progression of HPV-positive oropharyngeal squamous cell carcinoma. Head. Neck. 2017;39:2397–2405. doi: 10.1002/hed.24910. [DOI] [PubMed] [Google Scholar]
- 14.Kalfert D, et al. Analysis of preoperative serum levels of MMP1, -2, and -9 in patients with site-specific head and neck squamous cell cancer. Anticancer Res. 2014;34:7431–7441. [PubMed] [Google Scholar]
- 15.Polz-Dacewicz M, Strycharz-Dudziak M, Dworzanski J, Stec A, Kocot J. Salivary and serum IL-10, TNF-alpha, TGF-beta, VEGF levels in oropharyngeal squamous cell carcinoma and correlation with HPV and EBV infections. Infect. Agent Cancer. 2016;11:45. doi: 10.1186/s13027-016-0093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan C, Yang K, Tang H, Chen D. Diagnostic values of serum tumor markers Cyfra21-1, SCCAg, ferritin, CEA, CA19-9, and AFP in oral/oropharyngeal squamous cell carcinoma. Onco. Targets Ther. 2016;9:3381–3386. doi: 10.2147/OTT.S105672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klussmann JP, et al. Expression of p16 protein identifies a distinct entity of tonsillar carcinomas associated with human papillomavirus. Am. J. Pathol. 2003;162:747–753. doi: 10.1016/S0002-9440(10)63871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Distler U, et al. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods. 2014;11:167–170. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 19.Kuusela P, et al. Changes in plasma protein levels as an early indication of a bloodstream infection. PLoS ONE. 2017;12:e0172987. doi: 10.1371/journal.pone.0172987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vizcaino JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(D1):D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saraswat M, et al. Human spermatozoa quantitative proteomic signature classifies normo- and asthenozoospermia. Mol. Cell Proteom. 2017;16:57–72. doi: 10.1074/mcp.M116.061028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell Proteom. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Szklarczyk D, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–d8. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pundir S, Martin MJ, O’Donovan C. UniProt protein knowledgebase. Methods Mol. Biol. 2017;1558:41–55. doi: 10.1007/978-1-4939-6783-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner S, et al. Human papillomavirus association is the most important predictor for surgically treated patients with oropharyngeal cancer. Br. J. Cancer. 2017;116:1604–1611. doi: 10.1038/bjc.2017.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371:1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol. Cancer Res. 2010;8:1453–1465. doi: 10.1158/1541-7786.MCR-10-0225. [DOI] [PubMed] [Google Scholar]
- 29.Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat. Rev. Immunol. 2017;18:5–18. doi: 10.1038/nri.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koifman L, et al. Proteomics analysis of tissue samples from patients with squamous cell carcinoma of the penis and positive to human papillomavirus. Int. Braz. J. Urol. 2015;41:642–654. doi: 10.1590/S1677-5538.IBJU.2014.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ornellas P, et al. Downregulation of C3 and C4A/B complement factor fragments in plasma from patients with squamous cell carcinoma of the penis. Int. Braz. J. Urol. 2012;38:739–749. doi: 10.1590/1677-553820133806739. [DOI] [PubMed] [Google Scholar]
- 32.Zafar GI, Grimm EA, Wei W, Johnson MM, Ellerhorst JA. Genetic deficiency of complement isoforms C4A or C4B predicts improved survival of metastatic renal cell carcinoma. J. Urol. 2009;181:1028–1034. doi: 10.1016/j.juro.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo Z, et al. Towards precise classification of cancers based on robust gene functional expression profiles. BMC Bioinformatics. 2005;6:58. doi: 10.1186/1471-2105-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cun Y, Frohlich H. Biomarker gene signature discovery integrating network knowledge. Biology. 2012;1:5–17. doi: 10.3390/biology1010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Human Genom. 2010;4:353–360. doi: 10.1186/1479-7364-4-5-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gopurappilly R, Bhonde R. Transcriptional profiling and functional network analyses of islet-like clusters (ILCs) generated from pancreatic stem cells in vitro. Genomics. 2015;105:211–219. doi: 10.1016/j.ygeno.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat. Rev. Cancer. 2017;17:577–593. doi: 10.1038/nrc.2017.77. [DOI] [PubMed] [Google Scholar]
- 38.Kondo A, et al. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 2017;18:2228–2242. doi: 10.1016/j.celrep.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Borgquist S, et al. Apolipoproteins, lipids and risk of cancer. Int J. Cancer. 2016;138:2648–2656. doi: 10.1002/ijc.30013. [DOI] [PubMed] [Google Scholar]
- 40.Eberhardt HU, et al. Human factor H-related protein 2 (CFHR2) regulates complement activation. PLoS ONE. 2013;8:e78617. doi: 10.1371/journal.pone.0078617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Leary NA, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji P, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 43.Chang Q, et al. JNK1 activation predicts the prognostic outcome of the human hepatocellular carcinoma. Mol. Cancer. 2009;8:64. doi: 10.1186/1476-4598-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamada S, et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene. 2004;23:5901–5911. doi: 10.1038/sj.onc.1207782. [DOI] [PubMed] [Google Scholar]
- 45.Saraswat M, et al. Comparative proteomic profiling of the serum differentiates pancreatic cancer from chronic pancreatitis. Cancer Med. 2017;6:1738–1751. doi: 10.1002/cam4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saraswat M, Makitie A, Agarwal R, Joenvaara S, Renkonen S. Oral squamous cell carcinoma patients can be differentiated from healthy individuals with label-free serum proteomics. Br. J. Cancer. 2017;117:376–384. doi: 10.1038/bjc.2017.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics is available from the ProteomeXchange Consortium via the PRIDE partner repositorywith the data set identifier PXD008445.