

Prions of lower eukaryotes are transmissible protein particles that propagate by converting homotypic soluble proteins into growing protein assemblies. Prion activity is conferred by so-called prion domains, regions of low complexity that are often enriched in glutamines and asparagines (Q/N).
KEYWORDS: amyloid, neurodegeneration, prion-like, prions, protein misfolding
ABSTRACT
Prions of lower eukaryotes are transmissible protein particles that propagate by converting homotypic soluble proteins into growing protein assemblies. Prion activity is conferred by so-called prion domains, regions of low complexity that are often enriched in glutamines and asparagines (Q/N). The compositional similarity of fungal prion domains with intrinsically disordered domains found in many mammalian proteins raises the question of whether similar sequence elements can drive prion-like phenomena in mammals. Here, we define sequence features of the prototype Saccharomyces cerevisiae Sup35 prion domain that govern prion activities in mammalian cells by testing the ability of deletion mutants to assemble into self-perpetuating particles. Interestingly, the amino-terminal Q/N-rich tract crucially important for prion induction in yeast was dispensable for the prion life cycle in mammalian cells. Spontaneous and template-assisted prion induction, growth, and maintenance were preferentially driven by the carboxy-terminal region of the prion domain that contains a putative soft amyloid stretch recently proposed to act as a nucleation site for prion assembly. Our data demonstrate that preferred prion nucleation domains can differ between lower and higher eukaryotes, resulting in the formation of prions with strikingly different amyloid cores.
INTRODUCTION
Prions are infectious protein aggregates that propagate by autocatalytically converting normal cellular prion proteins into ordered protein assemblies with cross-β structure, so-called amyloids. The only bona fide prions identified so far in mammals cause fatal transmissible spongiform encephalopathies (TSE) (1). The cellular isoform of the mammalian prion is PrPC, a protein tethered to the cell membrane by a glycosylphosphatidylinositol anchor. Its conformational conversion generates infectious entities capable of transmitting between cells, within species, and sometimes even between species (2). Amyloid formation is also a common feature of neurodegenerative diseases of noninfectious origin (3). While epidemiological evidence is lacking that classical neurodegenerative diseases are transmissible, the mechanism of intra- and intercellular molecular templating is shared by many protein aggregates (4).
In analogy to mammalian prions, several proteins of Saccharomyces cerevisiae can adopt self-replicating prion conformations that induce heritable phenotypic traits (5, 6). These fungal protein aggregates are also termed prions and replicate by a mechanism of seeded polymerization in which a seed of a misfolded protein templates the conversion of the soluble protein into a self-perpetuating amyloid state. Fragmentation of existing prion fibrils by the yeast chaperone machinery then leads to the formation of new seeds and exponential multiplication of heritable entities (7, 8). The prion domains (PrDs) of most identified yeast prions are inherently disordered and enriched for asparagine (N) and/or glutamine (Q) residues, with charged and hydrophobic residues being underrepresented (9–13). The compositional similarity of PrDs of known S. cerevisiae prions encouraged the development of computational tools that successfully identified similar domains in several yeast proteins with unknown prion propensity (10, 13–16). Surprisingly, scoring of entire proteomes with prion algorithms predicts that at least 1% of mammalian proteins contain similar prion-like domains (PrLDs) (17, 18). Several mammalian proteins with predicted PrLDs drive liquid-liquid phase transitions for the transient formation of membrane-less ribonucleoprotein complexes. Mutations in the respective domains of disease-associated human proteins have been linked to muscular and neurodegenerative pathomechanisms (19). In light of the many Q/N-rich proteins in higher eukaryotes, it is possible that prion-like self-replication underlies other unresolved epigenetic phenomena and diseases of unknown etiology. So far, however, evidence for bona fide prions derived from human proteins with predicted PrLDs is lacking. Indeed, a recent study demonstrates limitations of prion algorithms to accurately predict the prion propensity of prion-like proteins in higher organisms, likely because host-dependent intracellular factors influence aggregation or prion behavior of a given protein (20).
On a cellular level, prion characteristics include rare spontaneous or template-assisted conversion of the protein into its prion conformation, multiplication of seeds, and natural infection of bystander cells (21, 22). Proof of principle that a prototype yeast prion domain can behave as an infectious entity in mammalian cells comes from studies on the aggregation behavior of the best-studied S. cerevisiae prion, Sup35, in mouse cells (23–25). In yeast, Sup35 serves as a translation termination factor that rarely switches into an inactive prion conformation (26, 27). Its PrD N domain drives prion propagation and assembles into fibrils with cross-β structure in vitro (28–31). While the amino acid composition of the N domain is a major determinant of its activity, distinct subdomains within the N domain exert specific but somewhat overlapping functions in prion biogenesis in S. cerevisiae (12, 32, 33). The highly charged middle (M) domain (amino acids [aa] 124 to 250) stabilizes the prion conformer during yeast mitosis and meiosis (34) and increases solubility of the protein in its non-prion state (35). The carboxy-terminal C domain (residues 251 to 685) mediates translation termination function but is otherwise dispensable for prion formation (35, 36). Sup35 NM does not share sequence homology with mammalian proteins and is thus ideally suited to study prion behavior in the absence of a potential loss-of-function phenotype. In analogy to prion induction in S. cerevisiae (37, 38), cytosolically expressed NM adopts a prion state in mammalian cells upon exposure to exogenous in vitro-fibrillized recombinant NM (23). Induced NM aggregates behave as infectious entities that faithfully transmit vertically and horizontally (24, 39). Here, we set out to define the subdomains in the Sup35 PrD that mediate the sequential steps of prion formation in mouse neuroblastoma cells. We demonstrate that the differential steps of prion biogenesis are preferentially driven by a carboxy-terminal region of the N domain. Importantly, this domain contains a soft amyloid core proposed to mediate first intermolecular contacts during prion assembly (40). Initiation of prion assembly by the carboxy-terminal region leads to the formation of NM prions that differ strikingly from wild-type prion variants propagating in S. cerevisiae.
RESULTS
The Sup35 N domain drives aggregation in mammalian cells.
We have recently shown that the Sup35 NM domain exhibits prion properties in mammalian cells (23, 24). Here, we use the term “prion” to describe the self-templating and intercellular transmission properties of Sup35 NM in our cellular model in analogy to the well-established fungal prions (41). Myc epitope-tagged N or M domains and a mutant coding for NM Δ 138-250, shown to maintain the prion phenotype in S. cerevisiae (Fig. 1A) (42), were tested for their ability to aggregate upon induction by untagged recombinant NM fibrils or by endogenous green fluorescent protein (GFP)-tagged NM prions. Transiently transfected N2a cells showed diffuse expression of the Myc-tagged mutants in the cytoplasm (Fig. 1B). Proteins lacking parts of the M domain also localized to the nucleus. The reason for the presence of N derivatives in the nucleus is unknown, as the protein lacks a predicted classical or proline-tryosine nuclear localization signal (43). Exposure of cells to in vitro-fibrillized untagged recombinant NM revealed that the N domain was crucial for aggregate induction by exogenous NM seeds (Fig. 1C and D). Myc-tagged mutants with deletions in the M domain aggregated in cytoplasm and nuclei. In contrast, M alone was incapable of forming aggregates. A sedimentation assay of cell lysates from transfected cells confirmed that before (Fig. 1E) or after (Fig. 1F) exposure to preformed untagged recombinant NM fibrils, M-Myc expressed in N2a cells remained soluble. When N2a cell clone 2CG11 stably producing NM-GFP prions (24) was transfected with plasmids coding for the Myc epitope-tagged NM mutants (Fig. 1G), aggregation was only observed when the N domain was present, suggesting that, in analogy to the S. cerevisiae system, NM aggregation in mammalian cells is mediated by the same domain (Fig. 1H). Significantly more cells with double-stained NM-GFP aggregates were found for NM-Myc Δ 138-250 than for N-Myc (Fig. 1I). Thus, while the M domain alone is incapable of forming aggregates, a stretch of amino-terminal amino acid residues in M engages in the binding to preexisting NM-GFP prions (Fig. 1I). This is consistent with the finding that residues 124 to 137 are important for variant-specific maintenance of Sup35-derived prions in yeast (42).
FIG 1.

N domain is sufficient for aggregate formation. (A) Schematic diagram of NM constructs. (B) N2a cells 48 h posttransfection of NM constructs. Ectopically expressed proteins were detected using pAb anti-Myc tag (red). Nuclei were stained with Hoechst (blue). Scale bar, 5 μm. (C) Experimental setup to study NM aggregate induction by untagged recombinant (rec.) NM fibrils. Wild-type N2a cells were transiently transfected with constructs N-Myc, NM-Myc Δ 138-250, and M-Myc. Twenty-four hours later, cells were exposed to untagged recombinant NM fibrils. (D) N2a cells transiently transfected with NM mutants and 24 h later exposed to 10 μM (monomer concentration) in vitro-fibrillized untagged NM. Ectopically expressed proteins were detected using pAb anti-Myc tag (red). Nuclei were stained with Hoechst (blue). (E) Sedimentation assay of cell lysates from N2a cells 24 h posttransfection with constructs. S, supernatant; P, pellet. Ectopically expressed protein was detected using MAb anti-Myc. (F) Sedimentation assay following exposure to 10 μM fibrillized untagged recombinant NM (monomer concentration). Cells were harvested 48 h posttransfection. The M domain alone exhibits an abnormal migration behavior, potentially due to net charge and/or charge distribution. (G) N2a cell clone 2CG11 stably producing NM-GFP aggregates was transiently transfected with NM mutants for 48 h. NM encoded by plasmids was detected using pAb anti-Myc tag (red). GFP is shown in green. Nuclei were stained with Hoechst (blue). Arrows indicate nuclear NM. Scale bar, 5 μm. (H) N2a NM-GFPagg cell clone 2CG11 was transiently transfected with constructs and fixed 48 h posttransfection. Shown is the percentage of NM-GFPagg cells with Myc-tagged aggregates. Bars represent mean values ± SEM (three independent transfection experiments; n = 3). At least 300 cells were analyzed per cell population. The Cochran-Mantel-Haenszel test was used (****, P ≤ 0.0001; ns, not significant). (I) Percentage of cells that contain Myc-tagged and GFP-tagged NM aggregates (shown in panel H) that show colocalization of NM-Myc and NM-GFP aggregates. Bars represent mean values ± SEM (n = 3). Statistical analysis was done using the Cochran-Mantel-Haenszel test (****, P ≤ 0.0001).
Deletion of the Q/N-rich region increases the rate of spontaneous aggregation.
We constructed a set of truncated NM mutants previously tested in S. cerevisiae for prion activities (Fig. 2A) (32). The N domain contains a region particularly enriched for Q/N (generally termed QNR; residues 1 to 39) that is crucially required for aggregate induction, polymerization, and amyloid core formation in yeast (11, 44). The five and a half degenerate octapeptide repeats (OPR) promote inheritance over multiple cell divisions by stabilizing intermolecular interactions (45, 46). Importantly the OPR also act as a recognition site for the chaperone Hsp104 that mediates fibril fragmentation in yeast (32, 47, 48). A single-amino-acid substitution (G58D) in the OPR has been shown to modulate Sup35 prion heritability by affecting either fragmentation or bidirectional sorting to progeny (49–52). Aggregate persistence in mitotically active cells is taken as a proxy for effective seed multiplication (22). To restrict expression of the NM derivatives to the cytosol, all deletion mutants contained the M domain. In N2a cell populations stably transduced with virus coding for hemagglutinin (HA) epitope-tagged proteins, NM derivatives generally remained soluble (Fig. 2B). Automated confocal microscopy analysis demonstrated stable transgene expression in at least 38% of the cells over a period of 13 cell culture passages (Fig. 2C and data not shown). In rare cases, we noticed cells expressing NM-HA Δ 1-39 that contained visible NM aggregates (Fig. 3A). Sedimentation assays confirmed that NM-HA proteins almost exclusively resided in a soluble state, with the exception of NM-HA Δ 1-39, where a fraction of the protein became insoluble (Fig. 3B). This is in line with our quantitative confocal microscopy analyses of transduced cell populations, demonstrating that up to 2.5% of cells expressing NM-HA Δ 1-39 exhibited NM aggregates, while spontaneous aggregates were found in less than 0.3% of cell populations expressing full-length NM-HA or other mutants (Fig. 3C). The highest numbers of NM-expressing cells with aggregates were also found in N2a NM-HA Δ 1-39 upon continuous passage (Fig. 3D). Lowest spontaneous aggregation was observed when the last 2.5 repeats and/or the carboxy-terminal N region (CTN) was deleted (Δ 39-123, Δ 75-123, and Δ 98-123). When cells were grown for several days, we noticed clusters of N2a cells with NM-HA Δ 1-39agg but not Δ 75-123agg, suggesting that spontaneously forming aggregates lacking the QNR were self-sustained (data not shown).
FIG 2.
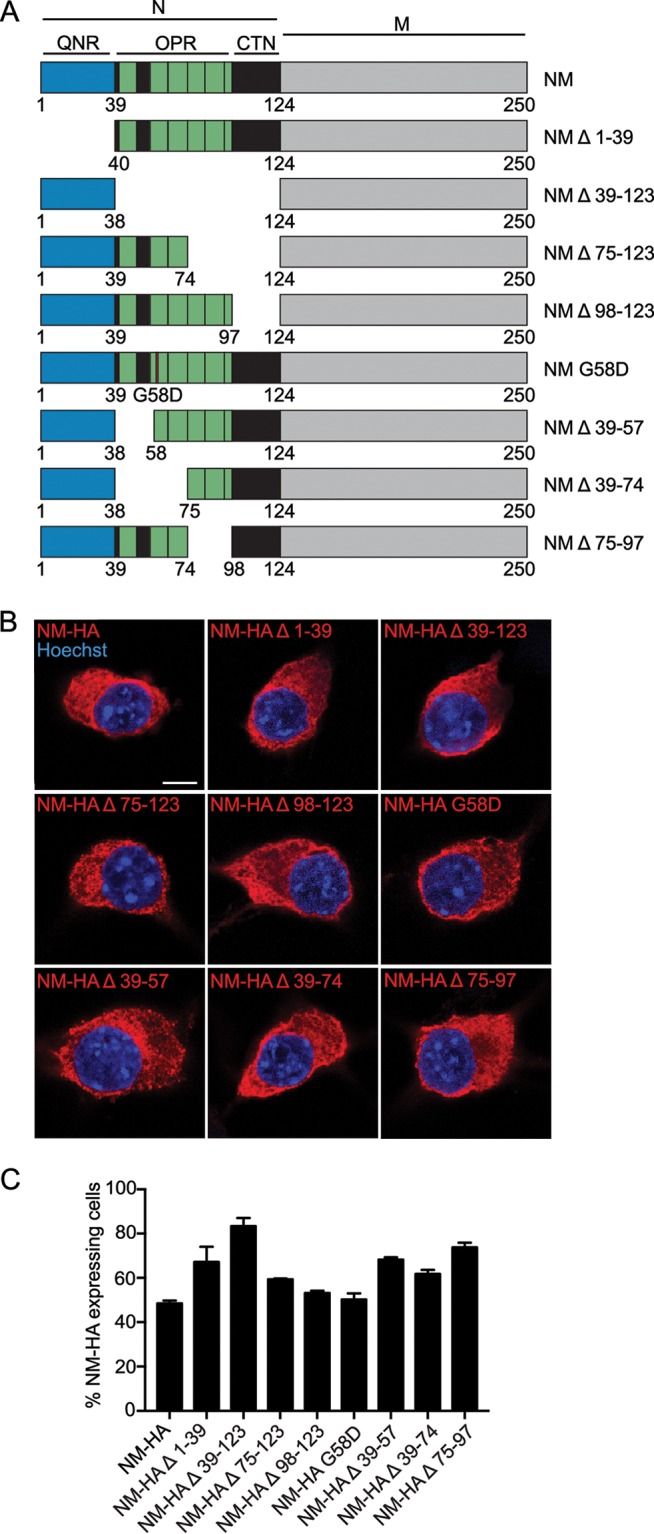
N2a cells stably express NM wild type and NM mutants. (A) Eight NM constructs were generated based on NM mutants already analyzed in S. cerevisiae for their prion propensity (32). QNR, residues 1 to 39; OPR, residues 40 to 97, containing repeats R1 to R5; R1, residues 41 to 49; R2, residues 56 to 64; R3, residues 65 to 74; R4, residues 75 to 83; R5, residues 84 to 93; R5.5, residues 94 to 97; CTN, residues 98 to 123. Note that the terms Q/N-rich region and Q/N-rich tract are used in the field to refer to this region of N (32, 50, 84). The carboxy-terminal region of N also is enriched for Q/N, albeit to a lesser degree. All constructs were tagged with an HA antibody epitope tag at the carboxy termini. (B) Bulk populations of N2a cells stably expressing wild-type and mutant NM-HA. NM was detected using MAb anti-HA (red). Nuclei were stained with Hoechst (blue). Scale bar, 5 μm. (C) Percentage of cells in the bulk population (passage one postthawing) expressing wild-type or mutant NM. Bars represent mean values ± SEM (n = 3). At least 14,000 cells were analyzed per cell population.
FIG 3.

Spontaneous aggregation propensities of full-length and mutant NM in the mammalian cytosol. (A) N2a NM-HA Δ 1-39 cell with spontaneously formed NM-HA aggregates. NM was detected using MAb anti-HA. The nucleus was visualized with Hoechst (blue). The arrow indicates NM-HA aggregates. Scale bar, 5 μm. (B) Sedimentation assay of cell lysates from N2a bulk populations expressing wild-type or mutant NM-HA. NM was detected using MAb anti-HA. S, supernatant; P, pellet. (C) Percentage of cells in the bulk population (one passage postthawing) that express the transgene and also spontaneously form NM aggregates. Bars represent mean values ± SEM from three independent experiments (n = 3), with at least 14,000 cells imaged per cell population. Statistical analysis was done using the Cochran-Mantel-Haenszel test (*, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001; ns, not significant). (D) Percentage of N2a NM-HA cells that express wild-type or mutant NM-HA and that spontaneously form NM aggregates over continuous culture (passage 1 to 13 postthawing). Bars represent mean values ± SEM (n = 3). At least 9,800 cells were imaged per cell population per passage. Please note variations in scaling. P, passage. (E) Expression of wild-type and mutant NM-HA in stable cell populations. NM was detected by Western blotting using MAb anti-HA. Actin served as a loading control. These data can be compared to the graph shown in Fig. 2C. (F) Stable cell populations expressing full-length NM-HA or NM-HA Δ 75-123 were transiently transfected with constructs coding for the homotypic proteins to increase the expression level of the transgene. N2a NM-HA Δ 1-39 cells were mock transfected. Overexpressed NM-HA Δ 75-123 was partially amino-terminally deleted, as has been observed previously for NM (85). NM was detected using MAb anti-HA. Actin served as a loading control. Additional lanes were excised for presentation purposes. (G) Quantitative analysis of expression shown in panel F (three independent transfections, n = 3). Shown is the fold difference in expression level compared to that of mutant NM-HA Δ 1-39. For NM-HA Δ 75-123, only the signal of the upper band was used for quantification. Bars represent mean values ± SEM. Statistical analysis was performed using one-way analysis of variance (*, P ≤ 0.05; ***, P ≤ 0.001). (H) Spontaneous aggregation of NM-HA and NM-HA mutants in cells shown in panels F and G. Aggregation was assessed 48 h posttransfection. At least 6,000 cells were analyzed per cell population. Bars represent mean values ± SEM. Statistical analysis was done using the Cochran-Mantel-Haenszel test (****, P ≤ 0.0001).
Prion induction in yeast increases with NM expression levels (31). To test if increased expression resulted in higher aggregate induction efficiencies, cell populations stably expressing full-length NM-HA or NM-HA deletion mutant Δ 75-123 (Fig. 3E) were transiently transfected with expression plasmids coding for homotypic NM proteins to achieve higher expression levels (Fig. 3F). Western blot analysis revealed approximately 2- to 4-fold higher NM levels in lysates of N2a NM-HA or NM-HA Δ 75-123 cells than in lysates of N2a NM-HA Δ 1-39 cells (Fig. 3G). Still, NM-HA Δ 1-39 spontaneously formed aggregates at significantly higher rates than NM-HA or NM-HA Δ 75-123 (Fig. 3H). Increasing transgene expression 3- to 4-fold by transient transfection of N2a NM-HA, N2a NM-HA Δ 75-123, or N2a NM-HA Δ 98-123 cell populations with corresponding plasmids only insignificantly increased NM puncta formation, resulting in less than 0.25% positive cells (data not shown). Collectively, these findings suggest that mutant NM-HA Δ1-39 has an increased ability to self-aggregate into prions.
The last 2.5 repeats and the carboxy-terminal N region are essential for prion induction by preexisting seeds.
To study prion induction by exogenous seeds, N2a cell populations were exposed to untagged recombinant NM fibrils (Fig. 4A and B), and cells were analyzed for NM-HA aggregate induction in passage one (Fig. 4C). Independent of the NM-HA mutant expressed, untagged fibrils seeded protein polymerization in all cell populations (Fig. 4C), albeit at different frequencies (Fig. 4D). Deletion of the QNR (Δ 1-39) or the point mutation G58D only slightly affected numbers of aggregate-bearing cells compared to those of cells that express full-length NM-HA (Fig. 4D). Deletion of the first three repeats (Δ 39-74) had some effect on aggregate induction. In contrast, deletion of the last 2.5 repeats and/or the CTN (Δ 39-123, Δ 75-123, Δ 98-123, and Δ 75-97) drastically impaired aggregation. Sedimentation assays confirmed smaller amounts of insoluble NM-HA when the last 2.5 repeats and/or the CTN were deleted (Fig. 4E).
FIG 4.

Template-assisted aggregate induction by fibrillized untagged recombinant NM or endogenous NM-GFP prions. (A) Experimental setup to study the propensity of the NM mutants to form aggregates upon exposure to in vitro-fibrillized recombinant NM. (B) Electron microscopy image of untagged recombinant NM fibrils. Scale bar, 100 nm. (C) N2a cell populations exposed to 10 μM untagged recombinant NM fibrils (monomer concentration) for 24 h. Ectopically expressed wild-type and mutant NM were detected using MAb anti-HA (red), and nuclei were stained with Hoechst (blue). Scale bar, 5 μm. (D) Percentage of cells harboring NM-HA aggregates upon induction with 10 μM untagged recombinant NM fibrils (monomer concentration). Bars represent mean values ± SEM (three independent induction experiments; n = 3). At least 12,500 cells per cell population were imaged. Statistical analysis was done using the Cochran-Mantel-Haenszel test (****, P ≤ 0.0001). (E) Sedimentation assay of cell lysates from N2a bulk populations exposed to 10 μM fibrillized untagged recombinant NM (monomer concentration). Cells were harvested 24 h after aggregate induction. Ectopically expressed NM was detected using MAb anti-HA. S, supernatant; P, pellet. Additional lanes were excised for presentation purposes.
To determine if preexisting endogenous NM-GFP prions could sequester NM-HA mutants, N2a NM-GFPagg cells were transiently transfected with empty vector or constructs coding for NM-HA wild type or NM-HA mutants (Fig. 5A). Note that NM-GFPagg cells produce both long fibril-like and punctate NM-GFP aggregates (Fig. 5B). When transfected with constructs coding for NM-HA derivatives, HA-tagged NM aggregates were found in all cell populations (Fig. 5C). At least 36% of NM-GFPagg-bearing cells in all NM-HA derivative-expressing cells contained NM-HA aggregates independent of the mutant (Fig. 5D). Lowest NM-HA aggregate induction was found when parts of the OPR and the CTN (Δ 39-123 and Δ 75-123) were deleted. In cells exhibiting both NM-GFP and NM-HA derivative aggregates, NM-HA Δ 39-123 hardly ever decorated NM-GFP aggregates (Fig. 5C and E). Deletion of either the last 2.5 OPR or the CTN (Δ 75-97, Δ 75-123, or Δ 98-123) drastically decreased coaggregation with full-length NM. Instead, aggregated mutants often appeared as individual puncta (Fig. 5C and E). The presence of independent NM-HA mutant aggregates in cells producing NM-GFP prions suggests that prions lacking the last 2.5 repeats and the CTN or the CTN alone are structurally less compatible with full-length NM prions. Collectively, these data suggest that parts of the OPR and the CTN mediate intermolecular contacts between soluble NM with preexisting exogenous or endogenous NM seeds.
FIG 5.

Template-assisted aggregate induction by endogenous NM-GFP prions. (A) Illustration of the experiment. N2a NM-GFPagg cell clone 2CG11 was transiently transfected with empty vector or HA-tagged NM full-length or NM mutant constructs. Cells were fixed 48 h posttransfection. (B) N2a NM-GFP cell clone 2CG11 stably producing NM-GFP aggregates was mock transfected. Nuclei were stained with Hoechst (blue). Note that aggregates can exhibit a fibrillar morphology (left) or exist as puncta (right). Scale bars, 5 μm. (C) 2CG11 cells transiently transfected with HA-tagged NM full-length or NM mutant constructs. NM-HA was detected using MAb anti-HA (red). Some NM-GFP aggregates also stained with Hoechst, most likely due to nucleic acids incorporated in the aggregates. Mutant NM-HA Δ 39-123 hardly ever coalesced with NM-GFP aggregates. Scale bars, 5 μm. (D) Shown is the percentage of NM-GFPagg cells with NM-HA aggregates compared to NM-GFPagg cells expressing NM-HA (soluble or aggregated). Bars represent mean values ± SEM (four independent transfection experiments; n = 4). At least 630 cells were analyzed per cell population. Statistical analysis was done using the Cochran-Mantel-Haenszel test (*, P ≤ 0.05; ****, P ≤ 0.0001; ns, not significant). (E) Percentage of NM-GFP/NM-HA aggregate-bearing cells (D) that show colocalization of NM-HA and NM-GFP aggregates. Bars represent mean values ± SEM. Statistical analysis was done using the Cochran-Mantel-Haenszel test (****, P ≤ 0.0001; ns, not significant).
Vertical transmission of NM aggregates to progeny.
A hallmark of mammalian and yeast prions is their stable maintenance in dividing cell populations (21, 53). The persistence of aggregates over multiple cell divisions is taken as a proxy for aggregate multiplication (22, 53). We compared the number of cells bearing NM-HA aggregates to the number of cells expressing NM-HA over continuous passages after fibril exposure (Fig. 6A and B). Independent of the NM-HA construct, the number of aggregate-bearing cells decreased over time, but loss appeared to slow down between passages six and nine for full-length NM-HA, mutant G58D, and NM-HA aggregates lacking the QNR (Fig. 6B). The initial presence of unstable prion variants has been observed in yeast (31, 54). Less than 13% of cells with aggregates were observed for mutants lacking the last 2.5 repeats and/or the CTN (Δ 39-123, Δ 75-97, Δ 75-123, and Δ 98-132). As spontaneous aggregate induction was less than 0.3% except for the NM-HA mutant without the QNR (Fig. 3A to D), the persistence of aggregates suggests that at least a subpopulation of induced aggregates was able to propagate. We used the Cox proportional hazard model to assess the fold change in the loss of aggregate-bearing cells over multiple cell divisions. A higher fold change in cells with mutant NM aggregates compared to cells with wild-type NM aggregates (set to zero) was taken as a measure for lower mitotic stability (Fig. 6C). Generally, deletions in NM decreased the mitotic stability of aggregates. NM-HA Δ 75-97 and Δ 98-123 aggregates were lost most frequently, suggesting that the last 2.5 repeats and the CTN are most important for stable inheritance. NM-HA G58D showed increased mitotic stability compared to that of full-length NM-HA (Fig. 6C). This mutation has been shown to modulate prion activity dependent on prion variant as well as genetic background of the yeast host by affecting either seed fragmentation or transmission to progeny (26, 49, 55). To monitor the fate of induced NM aggregates upon cell division, N2a cell lines were generated that stably express full-length NM or NM mutant Δ 1-39 or Δ 75-123 fused to GFP (Fig. 6D). Aggregation was induced by exposure to untagged recombinant NM fibrils. Distribution of NM-GFP aggregates to both daughter cells also confirmed that mutant NM aggregates that lack either the QNR or the last 2.5 repeats and the CTN are bidirectionally transmitted to daughter cells. We conclude that deletion of the last 2.5 repeats and the CTN had strongest effects on mitotic prion stability.
FIG 6.

Maintenance of NM-HA aggregates over multiple cell divisions. (A) Experimental setup to study aggregate maintenance over several cell passages. In a given population, the number of cells stably expressing the transgene was determined for each passage. These cells were then analyzed for NM-HA aggregates. (B) Percentage of cells expressing wild-type or mutant NM-HA that harbor aggregates over multiple cell divisions. Cell populations were exposed to 10 μM untagged recombinant NM fibrils (monomer concentration) and subsequently passaged every 2 to 3 days. Cells with aggregates were determined by automated confocal microscopy analysis (n = 3 biological replicates). Bars represent mean values ± SEM. P, passage. At least 7,900 cells were analyzed per cell population per passage. Proliferation doubling time for N2a NM-HA cells is approximately 13 h (data not shown). This corresponds to approximately 51 doublings within 13 passages. (C) Fold change of the probability for NM-HA mutant cell populations to lose aggregate-harboring cells over time compared to cells expressing full-length NM-HA. >0 denotes lower mitotic stability than NM-HA, and <0 denotes higher mitotic stability than NM-HA. Bars represent mean values, and error bars represent the upper limit for the confidence interval (n = 3). The Cox proportional hazard model was used for statistics (cell count, >12,500; ****, P ≤ 0.0001). (D) Time-lapse analysis of N2a cell lines stably expressing NM-GFP full-length, Δ 1-39, or Δ 75-123. Cells were exposed to 10 μM untagged recombinant NM fibrils for 24 h (monomer concentration). Two passages postinduction, cells were subjected to live cell imaging for a total of 15 h. Scale bar, 10 μm.
NM mutants are infectious.
Contrary to known behavior in S. cerevisiae, NM prions in mammalian cells infect naive bystander cells (24). To explore if NM deletion mutants exhibit infectious properties, we first generated donor populations with a higher number of aggregate-bearing cells by transfecting untagged recombinant NM fibrils into N2a NM-HAsol, Δ 1-39sol, and Δ 75-123sol cell populations using Effectene (Fig. 7A). Cloning was subsequently performed (Fig. 7B) to isolate cell populations with high prion replication ability (56). A total of 24 N2a NM-HA clones was isolated, 14 of which produced aggregates in 80 to 100% of total cells. For NM-HA Δ 1-39, 16 out of 20 clones stably propagated aggregates. For NM-HA Δ 75-123, the initial round of cloning generated only 2 out of 144 clones with aggregates in only 40 to 70% of cells, suggesting mitotic instability. Subcloning generated 86 clones, with 21 clones harboring visible aggregates in at least 50% of the cells. Four clones/subclones were chosen for the NM-HA wild type and the two NM-HA mutants that exhibited a high percentage (≥80%) of cells harboring aggregates (Fig. 7B). Cell clones displayed punctate aggregates that were evenly distributed throughout the cytosol or clustered in certain areas, and occasionally large, fibrillar aggregates were observed (Fig. 7C). SDD-AGE analysis (23) confirmed high-molecular-weight NM aggregates (Fig. 7D), demonstrating that NM deletion mutants also were able to form amyloid fibrils (57).
FIG 7.

Induction of the prion state in bystander cells. (A) Untagged recombinant NM fibrils (10 μM monomer concentration) were either added directly or mixed with Effectene transfection reagent before addition to full-length and mutant NM-HA cell populations. Induction efficiency was determined at passage three postexposure. At least 400 cells per treatment were examined. Bars represent mean values ± SEM. (B) Cloning strategy to isolate N2a clones with stable NM aggregates. Cell populations were exposed to 10 μM untagged recombinant NM fibrils (monomer concentration) with Effectene for 24 h. Cell clones with NM-HA aggregates were subsequently isolated by limiting dilution cloning. For mutant NM-HA Δ 75-123, subcloning was required to isolate four subclones with stable aggregate propagation. (C) Confocal microscopy analysis of cell clones isolated by limiting dilution cloning. For NM-HA wild type and both NM-HA mutants, four clones/subclones were analyzed. NM was detected using MAb anti-HA (red). Nuclei were stained with Hoechst (blue). Scale bar, 5 μm. (D) SDD-AGE assay of cell lysates from N2a NM-HAagg clone 7A, Δ 1-39 clone 5D, Δ 75-123 subclone 8H-23, and the corresponding soluble bulk cell lines. NM-HA was detected using MAb anti-HA. (E) Clones with NM-HA aggregates were cocultured with recipient cell line N2a NM-GFPsol for 42 h. Cells were analyzed for recipient cells with NM-GFP aggregates (arrows). NM-HA was detected using MAb anti-HA (red). Nuclei were stained with Hoechst (blue). GFP is shown in green. Note that large NM-GFP aggregates also stained brightly with Hoechst dye. Scale bar, 5 μm. (F) Percentage of recipient cells harboring NM-GFP aggregates after 42 h of coculture with donor clones. Bars represent mean values of the combined data from the four NM-HA clones/subclones ± SEM (n = 4). Cell count, >79,000; ****, P ≤ 0.0001; ns, not significant. Statistical analysis was done using the Cochran-Mantel-Haenszel test.
All clones, except one subclone producing large aggregates (NM-HA Δ 75-123; clone 64H-3), induced aggregation of NM-GFP in recipient cells upon direct coculture (Fig. 7E). Consistent with our previous findings, intercellular NM prion induction was a rare event (24). Generally, intercellular induction appeared to be higher when donor cells produced small, punctate aggregates. We were unable to detect donor NM-HA aggregates in the recipient cells, as transmitted, seeding-competent NM polymers are usually too small to be visualized by confocal microscopy (25). A significantly increased induction rate was found for aggregates lacking the QNR compared to full-length NM; however, clonal differences cannot be excluded (Fig. 7F) (24). In summary, neither the QNR nor the last 2.5 repeats and the CTN are essential for intercellular aggregate transmission.
Efficiently propagating NM prions contain a carboxy-terminal protease-resistant core.
The amyloid core of Sup35 prions propagating in S. cerevisiae has been experimentally mapped to the QNR (58). We used limited proteolysis (Fig. 8A) to characterize differences in prion cores of NM-HA wild type and Δ 1-39 and Δ 75-123 mutants (59). Chymotrypsin preferentially cleaves at the carboxyl side of amide bonds of tyrosine, tryptophan, and phenylalanine. These residues are abundant in the N domain, but except for one residue (aa 129), they are not present in the M domain. Three tyrosine residues are also part of the HA antibody epitope tag. As expected, chymotrypsin cleavage sites became inaccessible to partial proteolysis in the tightly packed untagged recombinant NM fibril (Fig. 8B). When cell lysates of N2a cells with soluble or aggregated (Fig. 8C) full-length or mutant NM-HA derivatives were incubated with chymotrypsin, polymers exhibited higher resistance to proteolysis. Proteolysis of full-length NM-HAagg resulted in a sequential decrease in protein size, suggesting that amino-terminally located cleavage sites were accessible to proteases (Fig. 8C). Increasing chymotrypsin concentrations resulted in the appearance of a wild-type NM-HAagg band at approximately 38 kDa, a size approximately similar to that of the NM deletion mutants. Both NM-HAagg mutants Δ 1-39 and Δ 75-123 sustained proteolysis, generating approximately similarly sized fragments (Fig. 8C). Very weak low-molecular-weight bands of NM-HA Δ 75-123 most likely represent proteolytic fragments of residual soluble protein (compare to Fig. 8C, top panel). These findings argue that the entire carboxy termini of wild-type and mutant NM, including the HA tag, were protected (Fig. 8D). The involvement of the M domain in the amyloid core has recently been reported for some Sup35 prions in S. cerevisiae (60, 61). The fact that we observed similarly sized fragments for the chymotrypsin-treated NM-HAagg wild type and the undigested NM-HAagg deletion mutants that still harbor the carboxy-terminal HA tag strongly suggests that (i) the QNR in full-length NM-HAagg does not engage in the tightly packed core, (ii) aggregated NM-HA full-length and mutant NM-HA Δ 1-39 likely share a similar core, and (iii) NM-HAagg Δ 75-123 has a different amyloid core that also includes the QNR.
FIG 8.

Partial protease resistance of wild-type and mutant NM-HA. (A) Experimental setup to define the protected aggregate core of wild-type and mutant NM-HA by enzymatic proteolysis. (B) Limited proteolysis of in vitro-fibrillized untagged recombinant NM. Fibrils were treated with chymotrypsin at the indicated protease/total protein ratio for 1 h on ice or were left untreated. Proteins were analyzed by Western blotting. NM was detected by 4A5 anti-M domain antibody (epitope, aa 229 to 247). (C) Limited proteolysis of N2a NM-HAsol/agg wild type, Δ 1-39, and Δ 75-123 cells. Lysates of N2a NM-HAagg wild-type clone 7A, Δ 1-39 clone 5D, Δ 75-123 subclone 8H-23, and the corresponding bulk cell lines expressing soluble NM-HA were subjected to chymotrypsin at the indicated protease/total protein ratio or left untreated. Proteins were analyzed by SDS-PAGE and Western blotting. NM was detected by anti-HA antibody or 4A5 anti-M domain antibody. Differences in exposure times are indicated by separate boxes. (D) Putative fragments of full-length and mutant NM after limited proteolysis with chymotrypsin. Arrowheads indicate putative cleavage sites of chymotrypsin. Blue, QNR; green or black, OPR and CTN; gray, M domain; red, HA tag. Numbers refer to amino acid residues.
DISCUSSION
Q/N-rich PrLD reminiscent of prion domains of lower eukaryotes are abundant in the proteomes of higher eukaryotes (17, 18). Experimental evidence is lacking that their putative PrLDs can turn the protein into a bona fide prion. To understand in more detail the sequence features in Q/N-rich domains that can drive prion formation in mammalian cells, we made use of the prototype yeast prion domain Sup35 NM that also amplifies as a prion in the mammalian cytosol (23–25). Strikingly, subdomains that preferentially drive the sequential steps of prion biogenesis differ between S. cerevisiae and our murine cell line (Fig. 9A). In S. cerevisiae, the QNR mediates de novo prion induction (11, 32). This region is crucially required for propagation of all so-far tested wild-type Sup35 prion variants in S. cerevisiae, and its deletion abrogates prion activity (11, 32, 37, 47, 48, 62). In N2a cells, the QNR was not required for prion induction. Instead, de novo NM aggregate induction, exogenous and endogenous NM seed-induced prion formation, and maintenance were preferentially driven by the last 2.5 repeats of the OPR and the CTN. Depending on the prion variant and/or genetic background of the host, the minimal region required for stable prion maintenance in S. cerevisiae varies but consistently includes the amino-terminal OPR (32, 37, 42, 45–48) proposed to enhance fibril fragmentation by disaggregase Hsp104 (32, 50, 51, 63, 64). As Hsp104 has no homologue in the mammalian cytosol (65), NM seed multiplication in N2a cells must proceed by other means. Potentially, the metazoan Hsp70 machinery in conjunction with Hsp110 (66) or autophagy-related processes contribute to prion partitioning (67). Alternatively, NM prions might also form through a fibril-catalyzed secondary nucleation of oligomers, a process reported for Aβ-42 seed proliferation (68).
FIG 9.

Carboxy-terminal OPR and the CTN are involved in nucleation, template-assisted aggregation, and prion maintenance. (A) Summary of results. Spontaneous aggregation, template-assisted aggregation, maintenance, and transmission to bystander cells by bulk cell populations or cell clones stably expressing the respective NM-HA proteins. Coaggregation with endogenous NM-GFP aggregates was determined following transient overexpression of wild-type and mutant NM-HA in N2a NM-GFPagg cells. Shown is the percentage of cells showing NM-GFP/NM-HA aggregate colocalization per NM-GFP/NM-HA aggregate-bearing cell. Aggregate maintenance was assessed as a proxy for seed multiplication: ++++, slightly increased mitotic stability compared to NM-HAagg control cells (fold change, >−0.5 and <0); +++, only slightly altered mitotic stability compared to NM-HAagg control (fold change, >0 and <0.5); ++, lower mitotic stability compared to control (fold change, >0.5 and <1); +, lowest mitotic stability (fold change, >1 and <2). For transmission to bystander cells, a plus sign indicates that at least 1 in 1,000 recipient cells contained NM-GFPagg. n.d., not done. (B) Amino acid composition of different subdomains of the N domain. The position of the region within NM and its length are indicated at the top. The abundances of specific residues within the region are given as percentages. (C) Prediction of disorder (FoldIndex [86]) and prion domains according to PAPA (16), PLAAC (15), or pWaltz/PrionW (80). Images were designed using PLAAC (http://plaac.wi.mit.edu/). The region identified as the prion nucleating domain by PrionW is shown at the bottom. This region falls within the CTN (aa 98 to 123). (D) Putative model of NM prion nucleation and seeding in N2a cells. NM contains two putative nucleation sites. Within the mammalian cytosol, the carboxy-terminal site comprising aa residues 98 to 123 preferentially drives nucleation and template-assisted seeding (left). The amino-terminal nucleation site (residues 1 to 38) likely mediates self-recognition and assembly when the carboxy-terminal site is deleted (right).
The amino-terminal QNR (approximately aa 9 to 39) and the CTN (approximately aa 90 to 120) have been identified recently as short recognition elements that mediate intermolecular contacts during de novo amyloid assembly and in mature fibrils generated in vitro (69, 70). Accumulating evidence suggests that Sup35 prion variants are composed of distinct parallel and in-register superpleated β-structures that differ in the extent of their β-sheets (58, 60–62). Recombinant full-length NM fibril preparations contain mixtures of conformers (35, 71) with QNR-containing amyloid cores (58, 60, 72) that induce prion variants with similar core structures in S. cerevisiae (37, 38). However, amyloid fibrils generated from a synthetic NM peptide (aa 98 to 118) can induce a Sup35 prion phenotype when transformed into S. cerevisiae. It is unclear if the induced prion variant exhibits a tightly packed core extending to the QNR as other variants in yeast do (73). The emergence of the new wild-type NM prion variant in N2a cells that lacks the QNR in its tightly packed core could be due to the selective amplification of few NM conformers with a carboxy-terminal amyloid core already present in the recombinant fibril mixture. Alternatively, initially induced NM prions with amino-terminal amyloid cores could interconvert into conformers with greater fitness in mammalian cells (74, 75). The reason for increased fitness of NM prions with tightly packed cores lacking the QNR is unknown but could relate to the cellular machinery involved in fibril assembly and/or maintenance or inhibitory factors. NM prions propagating in N2a cells exhibit reduced infectivity in yeast, potentially because this prion variant is not adapted to the yeast disaggregating machinery (23). In line with this, a recent study showed that yeast Hsp104 inefficiently fragments NM prions whose amyloid core extends to the carboxy-terminal N region (76). So far, however, molecular mechanisms or cofactors required for prion biogenesis in mammals are ill defined (77).
Bioinformatic predictions of proteins with prion-like behavior have been notoriously difficult (10, 12–14, 78). Importantly, prion prediction algorithms have been developed and trained on Q/N-rich yeast prion domains active in S. cerevisiae. While even for lower eukaryotes developed prion predictors suffer from significant limitations (10), predictions can be even more challenging when it comes to predicting the aggregation propensity of human PrLD-containing proteins in more complex organisms (20). PLAAC and PAPA algorithms support the compositional model that predicts that weak intermolecular interactions over a long Q/N-rich disordered domain confer prion propensity (15, 16). pWaltz (webserver PrionW) postulates that a short sequence in the disordered Q/N-rich context acts as a soft amyloid core that mediates prion assembly (79, 80). Importantly, the putative amyloid core element identified by pWaltz/PrionW (aa 98 to 118) (Fig. 9B and C) is basically identical to the CTN that, together with the last 2.5 repeats, is involved in spontaneous and fibril-assisted prion induction in our N2a cell model. The CTN could therefore act as the nucleation site for initial assembly, while the adjacent repeats stabilize intermolecular contacts within the aggregate (Fig. 9D) (45). Collectively, our results are in line with the hypothesis that short amyloidogenic stretches embedded in Q/N-rich disordered domains drive prion assembly.
MATERIALS AND METHODS
Cell culture and reagents.
N2a cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (PAA). HEK 293T/17 cells were grown in Iscove's modified Dulbecco's medium (Sigma) supplemented with 1% glutamine, 10% FCS, and 3.024 g/ml NaHCO3. Chemicals were purchased from Sigma or Carl Roth, unless stated otherwise. Monoclonal antibody (MAb) rat anti-HA clone 3F10 (1:1,000) (Roche Diagnostics), MAb 4A5 rat anti-NM (M domain) (1:10) (81), MAb mouse anti-Myc epitope (Miltenyi Biotec), MAb antiactin clone C4 (1:5,000) (MP Biomedicals), MAb mouse anti-HA (1:200) (Santa Cruz), MAb mouse anti-HA–Alexa Fluor 647 (1:500) (Biozol), or polyclonal antibody (pAb) rabbit anti-Myc tag (Abcam) was used. Horseradish peroxidase-conjugated antibodies were from Dianova, and fluorescently labeled antibodies were from Life Technologies. Cytoplasm was stained with HCS CellMask (Life Technologies). Effectene (Qiagen) or Lipofectamine 2000 (Thermo Scientific) was used for transfection.
Generation of NM-HA- and NM-GFP-expressing cell lines.
Constructs for N, M, NM Δ 138-250, and NM Δ 39-123 were ordered from Thermo Scientific or DNA 2.0. All other NM constructs were generated by PCRs with mutation-specific primers using the previously described NM-HA construct (33) as the template. Carboxy-terminally Myc- or HA-tagged constructs were cloned into the mammalian expression vector pJ602 and the lentiviral vector pRRL.sin.PPT.hCMV.Wpre (82). Gateway technology was used to generate lentiviral plasmids coding for GFP-tagged full-length NM and NM mutants (24). Viral particles were produced as described previously and used for transduction of N2a cells (24).
Sedimentation assay.
Cells were incubated with 10 μm in vitro-fibrilized NM (monomer concentration) for 24 h or left untreated. Cleared cell lysates (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.5% Triton X-100, 0.5% sodium deoxycholate, protease inhibitors) were centrifuged at 20,000 × g for 20 min at 4°C (the pellet was the insoluble fraction). Supernatant fractions were precipitated with 4× methanol at −20°C, and protein was pelleted at 2,120 × g for 25 min at 4°C (soluble fraction). The insoluble fraction and 1/6 of the soluble fraction dissolved in TNE buffer (50 mM Tris-HCl [pH 7.4], 100 mM NaCl, 0.1 mM EDTA) were loaded. Protein was detected using Pierce ECL (Life Technologies) or ECL Prime (GE Healthcare).
SDD-AGE.
The semidenaturing detergent-agarose gel electrophoresis (SDD-AGE) assay was performed as described previously (83). Cells were lysed (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, complete protease inhibitor) for 30 min on ice. Cleared supernatant was mixed with 4× sample buffer (2× TAE, 20% glycerol, 8% SDS, bromphenol blue), incubated at room temperature for 5 min, and separated on a 1.5% agarose gel supplemented with 0.1% SDS. Proteins were transferred onto a nitrocellulose membrane using capillary transfer.
Immunofluorescence staining and confocal microscopy analysis of N2a cells.
Cells were fixed with 4% paraformaldehyde and permeabilized for 10 min (0.5% Triton X-100). Antibodies were added in 2% blocking solution overnight at 4°C. Nuclei were visualized with Hoechst 33342 (1:10,000). Confocal microscopy was performed using an LSM 700 or LSM 710 (Zeiss, Jena, Germany). Cells on 96-well plates were stained with HCS CellMask blue stain (1:5,000), imaged with an automatic Cell Voyager 6000 confocal microscope (Yokogawa), and analyzed with Cell Profiler cell image analysis software. Time-lapse images of N2a NM-GFP cells were taken two passages after fibril exposure over a period of 15 h with an ApoTome (Zeiss) at 37°C and 5% CO2.
Aggregate induction by recombinant NM fibrils.
Untagged recombinant NM protein was prepared as described previously (24). To generate fibrils, NM was rotated head over tail for 24 h at 4°C at a concentration of 10 μM to 100 μM (monomer concentration). NM fibrils were sonicated for 3 min and added to cells (end concentration, 10 μM; monomer concentration). After 24 h the medium was exchanged.
Generation of N2a full-length NM-HAagg and mutant cell clones and coculture.
To enhance the fibril induction rate, untagged recombinant fibrils were mixed with the appropriate amount of Effectene transfection reagents (Qiagen) used for transfections in 24-well plates prior to addition to N2a cells. Cells were cultured for three passages, and the percentage of aggregate-bearing cells was determined. Aggregate-bearing cell clones were isolated. For NM-HAagg Δ 75-123, a second cloning step was necessary, as the first round yielded clones with a lack of mitotic stability. N2a NM-HAagg donor clones were cocultured with N2a NM-GFPsol cells (ratio, 1.5:1). After 42 h, cells were fixed and stained as described above. Confocal images were taken with an LSM700 (Zeiss) or Cell Voyager 6000.
EM analysis of fibril morphology.
Ten μM untagged recombinant NM fibrils (monomer concentration) was transferred to glow-discharged Formvar/carbon-coated electron microscopy (EM) grids (Plano GmbH). Samples were contrasted in 2% uranyl acetate and examined using a JEM-2200FS transmission electron microscope (JEOL) at 200 kV.
Image data analysis.
Confocal images were obtained using a Cell Voyager CV6000 with ×40 magnification. For NM aggregate colocalization, the LSM 700 with ×63 magnification was used. Images were analyzed with Cell Profiler. Nuclei and the associated cytoplasm were identified based on morphology parameters and intensities of Hoechst 33342 and HCS CellMask. NM-expressing cells were identified using intensity levels of immunofluorescently labeled NM. Aggregates were classified using the Ilastik segmentation tool kit. The numbers of wild-type or mutant NM-expressing cells and aggregate-bearing cells were determined.
Statistical analysis.
Individual experiments were performed at least three times, and for statistically analyzed data at least 550 cells per cell population were imaged. All data displayed in bar graphs are depicted as the means. Error bars represent the standard errors of means (SEM) or the upper limit of the confidence interval. P values of ≤0.05 were considered significant.
ACKNOWLEDGMENTS
We thank Meike Brömer and Sybille Krauss for helpful comments on this work. Ireen König, Philip Denner, and Birgit Kurkowsky helped with microscopy, automation, and image analysis.
Financial support was provided by the Helmholtz Wirkstoffforschung and the Centers of Excellence in Neurodegeneration (CoEN).
REFERENCES
- 1.Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Moore RA, Vorberg I, Priola SA. 2005. Species barriers in prion diseases–brief review. Arch Virol Suppl 2005:187–202. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. 2006. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Walker LC, Jucker M. 2015. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci 38:87–103. doi: 10.1146/annurev-neuro-071714-033828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crow ET, Li L. 2011. Newly identified prions in budding yeast, and their possible functions. Semin Cell Dev Biol 22:452–459. doi: 10.1016/j.semcdb.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liebman SW, Chernoff YO. 2012. Prions in yeast. Genetics 191:1041–1072. doi: 10.1534/genetics.111.137760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 8.Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. 2001. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol 21:4656–4669. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Z. 2011. The complexity and implications of yeast prion domains. Prion 5:311–316. doi: 10.4161/pri.5.4.18304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DePace AH, Santoso A, Hillner P, Weissman JS. 1998. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93:1241–1252. doi: 10.1016/S0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 12.Toombs JA, McCarty BR, Ross ED. 2010. Compositional determinants of prion formation in yeast. Mol Cell Biol 30:319–332. doi: 10.1128/MCB.01140-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison PM, Gerstein M. 2003. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol 4:R40. doi: 10.1186/gb-2003-4-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michelitsch MD, Weissman JS. 2000. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A 97:11910–11915. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lancaster AK, Nutter-Upham A, Lindquist S, King OD. 2014. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30:2501–2502. doi: 10.1093/bioinformatics/btu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toombs JA, Petri M, Paul KR, Kan GY, Ben-Hur A, Ross ED. 2012. De novo design of synthetic prion domains. Proc Natl Acad Sci U S A 109:6519–6524. doi: 10.1073/pnas.1119366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King OD, Gitler AD, Shorter J. 2012. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An L, Harrison PM. 2016. The evolutionary scope and neurological disease linkage of yeast-prion-like proteins in humans. Biol Direct 11:32. doi: 10.1186/s13062-016-0134-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. 2013. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul KR, Molliex A, Cascarina S, Boncella AE, Taylor JP, Ross ED. 2017. Effects of mutations on the aggregation propensity of the human prion-like protein hnRNPA2B1. Mol Cell Biol 37:e00652-. doi: 10.1128/MCB.00652-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE, Prusiner SB. 2007. Cell division modulates prion accumulation in cultured cells. Proc Natl Acad Sci U S A 104:17971–17976. doi: 10.1073/pnas.0708372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krauss S, Vorberg I. 2013. Prions ex vivo: what cell culture models tell us about infectious proteins. Int J Cell Biol 2013:704546. doi: 10.1155/2013/704546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krammer C, Kryndushkin D, Suhre MH, Kremmer E, Hofmann A, Pfeifer A, Scheibel T, Wickner RB, Schatzl HM, Vorberg I. 2009. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc Natl Acad Sci U S A 106:462–467. doi: 10.1073/pnas.0811571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofmann JP, Denner P, Nussbaum-Krammer C, Kuhn PH, Suhre MH, Scheibel T, Lichtenthaler SF, Schatzl HM, Bano D, Vorberg IM. 2013. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc Natl Acad Sci U S A 110:5951–5956. doi: 10.1073/pnas.1217321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S, Hossinger A, Hofmann JP, Denner P, Vorberg IM. 2016. Horizontal transmission of cytosolic Sup35 prions by extracellular vesicles. mBio 7:e00915-. doi: 10.1128/mBio.00915-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liebman SW, Derkatch IL. 1999. The yeast [PSI+] prion: making sense of nonsense. J Biol Chem 274:1181–1184. doi: 10.1074/jbc.274.3.1181. [DOI] [PubMed] [Google Scholar]
- 27.Uptain SM, Lindquist S. 2002. Prions as protein-based genetic elements. Annu Rev Microbiol 56:703–741. doi: 10.1146/annurev.micro.56.013002.100603. [DOI] [PubMed] [Google Scholar]
- 28.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. 2000. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 29.Santoso A, Chien P, Osherovich LZ, Weissman JS. 2000. Molecular basis of a yeast prion species barrier. Cell 100:277–288. doi: 10.1016/S0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 30.Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. 1994. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137:671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. 2004. Dissection and design of yeast prions. PLoS Biol 2:E86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohhashi Y, Ito K, Toyama BH, Weissman JS, Tanaka M. 2010. Differences in prion strain conformations result from non-native interactions in a nucleus. Nat Chem Biol 6:225–230. doi: 10.1038/nchembio.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JJ, Sondheimer N, Lindquist SL. 2002. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+]. Proc Natl Acad Sci U S A 99(Suppl 4):S16446–S16453. doi: 10.1073/pnas.252652099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. 1997. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89:811–819. doi: 10.1016/S0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 36.Chernoff YO. 2004. Amyloidogenic domains, prions and structural inheritance: rudiments of early life or recent acquisition? Curr Opin Chem Biol 8:665–671. doi: 10.1016/j.cbpa.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 37.King CY, Diaz-Avalos R. 2004. Protein-only transmission of three yeast prion strains. Nature 428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature 428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann J, Vorberg I. 2013. Life cycle of cytosolic prions. Prion 7:369–377. doi: 10.4161/pri.26414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batlle C, de Groot NS, Iglesias V, Navarro S, Ventura S. 2017. Characterization of soft amyloid cores in human prion-like proteins. Sci Rep 7:12134. doi: 10.1038/s41598-017-09714-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickner RB. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 42.Bradley ME, Liebman SW. 2004. The Sup35 domains required for maintenance of weak, strong or undifferentiated yeast [PSI+] prions. Mol Microbiol 51:1649–1659. doi: 10.1111/j.1365-2958.2003.03955.x. [DOI] [PubMed] [Google Scholar]
- 43.Marfori M, Mynott A, Ellis JJ, Mehdi AM, Saunders NF, Curmi PM, Forwood JK, Boden M, Kobe B. 2011. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim Biophys Acta 1813:1562–1577. doi: 10.1016/j.bbamcr.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 44.Chien P, Weissman JS. 2001. Conformational diversity in a yeast prion dictates its seeding specificity. Nature 410:223–227. doi: 10.1038/35065632. [DOI] [PubMed] [Google Scholar]
- 45.Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, Smirnov VN. 1993. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol Microbiol 7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 46.Liu JJ, Lindquist S. 1999. Oligopeptide-repeat expansions modulate “protein-only” inheritance in yeast. Nature 400:573–576. doi: 10.1038/23048. [DOI] [PubMed] [Google Scholar]
- 47.Parham SN, Resende CG, Tuite MF. 2001. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J 20:2111–2119. doi: 10.1093/emboj/20.9.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shkundina IS, Kushnirov VV, Tuite MF, Ter-Avanesyan MD. 2006. The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 172:827–835. doi: 10.1534/genetics.105.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kochneva-Pervukhova NV, Paushkin SV, Kushnirov VV, Cox BS, Tuite MF, Ter-Avanesyan MD. 1998. Mechanism of inhibition of Psi+ prion determinant propagation by a mutation of the N-terminus of the yeast Sup35 protein. EMBO J 17:5805–5810. doi: 10.1093/emboj/17.19.5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiSalvo S, Derdowski A, Pezza JA, Serio TR. 2011. Dominant prion mutants induce curing through pathways that promote chaperone-mediated disaggregation. Nat Struct Mol Biol 18:486–492. doi: 10.1038/nsmb.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doel SM, McCready SJ, Nierras CR, Cox BS. 1994. The dominant PNM2- mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 137:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pei F, DiSalvo S, Sindi SS, Serio TR. 2017. A dominant-negative mutant inhibits multiple prion variants through a common mechanism. PLoS Genet 13:e1007085. doi: 10.1371/journal.pgen.1007085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. 2000. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI(+)] of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. 2005. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J Biol Chem 280:8808–8812. doi: 10.1074/jbc.M410150200. [DOI] [PubMed] [Google Scholar]
- 55.Verges KJ, Smith MH, Toyama BH, Weissman JS. 2011. Strain conformation, primary structure and the propagation of the yeast prion [PSI+]. Nat Struct Mol Biol 18:493–499. doi: 10.1038/nsmb.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klohn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. 2003. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci U S A 100:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. 2003. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 58.Toyama BH, Kelly MJ, Gross JD, Weissman JS. 2007. The structural basis of yeast prion strain variants. Nature 449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 59.Foo CK, Ohhashi Y, Kelly MJ, Tanaka M, Weissman JS. 2011. Radically different amyloid conformations dictate the seeding specificity of a chimeric Sup35 prion. J Mol Biol 408:1–8. doi: 10.1016/j.jmb.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shewmaker F, Wickner RB, Tycko R. 2006. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc Natl Acad Sci U S A 103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shewmaker F, Kryndushkin D, Chen B, Tycko R, Wickner RB. 2009. Two prion variants of Sup35p have in-register parallel beta-sheet structures, independent of hydration. Biochemistry 48:5074–5082. doi: 10.1021/bi900345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang HY, Lin JY, Lee HC, Wang HL, King CY. 2008. Strain-specific sequences required for yeast [PSI+] prion propagation. Proc Natl Acad Sci U S A 105:13345–13350. doi: 10.1073/pnas.0802215105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cox B, Tuite M. 2018. The life of [PSI]. Curr Genet 64:1–8. doi: 10.1007/s00294-017-0714-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langlois CR, Pei F, Sindi SS, Serio TR. 2016. Distinct prion domain sequences ensure efficient amyloid propagation by promoting chaperone binding or processing in vivo. PLoS Genet 12:e1006417. doi: 10.1371/journal.pgen.1006417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 66.Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. 2012. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J 31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heiseke A, Aguib Y, Schatzl HM. 2010. Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol 12:87–97. [PubMed] [Google Scholar]
- 68.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. 2013. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krishnan R, Lindquist SL. 2005. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tessier PM, Lindquist S. 2007. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature 447:556–561. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gorkovskiy A, Thurber KR, Tycko R, Wickner RB. 2014. Locating folds of the in-register parallel beta-sheet of the Sup35p prion domain infectious amyloid. Proc Natl Acad Sci U S A 111:E4615–E4622. doi: 10.1073/pnas.1417974111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luckgei N, Schutz AK, Bousset L, Habenstein B, Sourigues Y, Gardiennet C, Meier BH, Melki R, Bockmann A. 2013. The conformation of the prion domain of Sup35p in isolation and in the full-length protein. Angew Chem Int Ed Engl 52:12741–12744. doi: 10.1002/anie.201304699. [DOI] [PubMed] [Google Scholar]
- 73.Sant'Anna R, Fernandez MR, Batlle C, Navarro S, de Groot NS, Serpell L, Ventura S. 2016. Characterization of amyloid cores in prion domains. Sci Rep 6:34274. doi: 10.1038/srep34274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 75.Bateman DA, Wickner RB. 2013. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet 9:e1003257. doi: 10.1371/journal.pgen.1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DeSantis ME, Shorter J. 2012. Hsp104 drives “protein-only” positive selection of Sup35 prion strains encoding strong [PSI(+)]. Chem Biol 19:1400–1410. doi: 10.1016/j.chembiol.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caughey B, Baron GS. 2006. Prions and their partners in crime. Nature 443:803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 78.Ross ED, Toombs JA. 2010. The effects of amino acid composition on yeast prion formation and prion domain interactions. Prion 4:60–65. doi: 10.4161/pri.4.2.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sabate R, Rousseau F, Schymkowitz J, Ventura S. 2015. What makes a protein sequence a prion? PLoS Comput Biol 11:e1004013. doi: 10.1371/journal.pcbi.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zambrano R, Conchillo-Sole O, Iglesias V, Illa R, Rousseau F, Schymkowitz J, Sabate R, Daura X, Ventura S. 2015. PrionW: a server to identify proteins containing glutamine/asparagine rich prion-like domains and their amyloid cores. Nucleic Acids Res 43:W331–W337. doi: 10.1093/nar/gkv490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krammer C, Kremmer E, Schatzl HM, Vorberg I. 2008. Dynamic interactions of Sup35p and PrP prion protein domains modulate aggregate nucleation and seeding. Prion 2:99–106. doi: 10.4161/pri.2.3.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Follenzi A, Naldini L. 2002. HIV-based vectors. Preparation and use. Methods Mol Med 69:259–274. [PubMed] [Google Scholar]
- 83.Halfmann R, Lindquist S. 2008. Screening for amyloid aggregation by semi-denaturing detergent-agarose gel electrophoresis. J Vis Exp 2008:838. doi: 10.3791/838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tuite MF, Cox BS. 2003. Propagation of yeast prions. Nat Rev Mol Cell Biol 4:878–890. doi: 10.1038/nrm1247. [DOI] [PubMed] [Google Scholar]
- 85.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. 1996. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 86.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. 2005. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]