

Abstract
Tumor metastasis suppressor factor BRMS1 can regulate the metastasis of breast cancer and other tumors. Here we report scinderin (SCIN) as a novel transcriptional target of BRMS1. SCIN protein belongs to the cytoskeletal gelsolin protein superfamily and its involvement in tumorigenesis remains largely illusive. An inverse correlation between the expression levels of BRMS1 and SCIN was observed in hepatocellular carcinoma (HCC) cells and tissues. On the molecular level, BRMS1 binds to SCIN promoter and exerts a suppressive role in regulating SCIN transcription. FACS analysis and caspase 9 immunoblot reveal that knockdown of SCIN expression can sensitize HCC cells to chemotherapeutic drugs, leading to suppression of tumor growth in vivo. Consistently, overexpression of SCIN protects cells from apoptotic death, contributing to increased xenografted HCC cell growth. In summary, our study reveals SCIN as a functional apoptosis regulator as well as a novel target of BRMS1 during HCC tumorigenesis. Inhibition of SCIN might bring a potential cancer therapy approach.
Keywords: BRMS1, scinderin, cell apoptosis, hepatocellular carcinoma, transcriptional regulation
Introduction
Human breast cancer metastasis suppressor 1 (BRMS1) was first discovered and cloned in 2000 by Seraj et al. [1]. Later on, it has been demonstrated that BRMS1 plays a suppressive role in regulating tumor metastasis in breast cancer, melanoma, non-small cell lung cancer, hepatocellular carcinoma, among others [2-5]. In consistent with these findings, the expression level of BRMS1 has been found to be significantly down-regulated in multiple metastatic tumor tissues [6-9]. It has been shown that BRMS1 mainly acts as a component of Sin3a•HDAC complex, which regulates chromatin status and further affects gene transcription efficiency [10,11]. We have previously reported two well-defined tumor-metastasis related genes, osteopontin (OPN) and death-associated protein kinase 1 (DAPK1), which were under transcriptional control of BRMS1 in hepatocellular carcinoma (HCC) cell lines [9,12].
Herein, we further identified Scinderin (SCIN, also known as adseverin) as a novel transcriptional target of BRMS1. SCIN is located in 7p21.3 and encodes a Ca2+-dependent actin filament severing and capping protein. SCIN protein was discovered in chromaffin cells in 1990 but its biological functions remain largely illusive [13]. As a member of the gelsolin superfamily, SCIN has the core structure of gelsolin-like domains and is involved in vesicle transport, exocytosis via regulating the polymerization and disassembly of F-actin network [14-16]. In addition, SCIN also exhibits activities in regulating other cellular processes such as cell differentiation, osteoclastogenesis, among others [17-19]. The involvement of SCIN in tumor development was first investigated in the megakaryoblastic leukemia cells [20]. Miura et al. later found that SCIN expression is significantly increased in the cisplatin-resistant urothelial cancer patients by comparison with cisplatin-sensitive control group [21]. Knockdown of SCIN led to an increase of mitochondria-mediated cell apoptosis in vitro. Recently, the expression of SCIN has been shown to be up-regulated in human prostate cancer, lung carcinoma and gastric cancer, suggesting an oncogenic role of SCIN in regulating tumor growth [22-24]. However, the molecular mechanism accounting for abnormal SCIN expression in tumor remains unknown.
In our study, several lines of evidence demonstrated that SCIN expression is transcriptionally suppressed by BRMS1 in HCC cells. The association relationship between BRMS1 and SCIN expression in HCC tissues and cells was studied through western blot analysis and the transcriptional mechanism was illuminated through dual luciferase assay and ChIP experiment. To further investigate the function of SCIN, lentivirus carrying shRNA against SCIN or recombinant plasmid overexpressing SCIN were utilized. It is found that knockdown of endogenous SCIN sensitized HCC cells to apoptosis, whereas expression of SCIN protected cells from apoptotic cell death. Moreover, SCIN promoted HCC cell growth in xenografted tumor mice. Taken together, our findings characterized SCIN as another functional downstream effector of BRMS1, providing another molecular pathway accounting for BRMS1’s tumor suppressive role in HCC cells.
Materials and methods
Tumor specimens
Fresh surgical specimens of HCC, including tumor tissues and the neighboring pathologically nontumorous liver tissues, were obtained from liver cancer patients at Zhongshan Hospital, Shanghai, China. Written informed consent was obtained from all these patients. Tissue samples were immediately frozen in liquid nitrogen after surgery and later stored at -80°C.
Quantitative real-time PCR (qRT-PCR)
RNA extracted from tissues or cultured cells using TRIzol reagent (Life Technologies, USA) was used for reverse transcription using PrimeScirptTM RT reagent and gDNA Eraser kit (Takara, Japan). Quantitative PCR analysis was performed with the CFX Connection detection system (Bio-Rad, USA) using SYBR Green Supermix kit (Takara). Cycle parameters were 95°C for 5 min hot start and 40 cycles of 95°C for 5 sec, 58°C for 10 sec and 72°C for 20 sec. Samples with no cDNA templates were used as negative control to rule out contamination in each run. Melting curves were analyzed to confirm the specificity of the PCR product. Primers for BRMS1: forward, 5’-ACTGAGTCAGCTGCGGTTGCGG-3’; reverse, 5’-AAGACCTGGAGCTGCCTCTGGCGTGC-3’. Primers for SCIN: forward, 5’-TCTGCGTTCCTGACTGTTC-3’; reverse, 5’-GACCTCCTTTCTTTGATGTTCC-3’. 18S rRNA was used as the internal control. Primers for 18S rRNA: forward, 5’-GTAACCCGTTGAACCCCATT-3’; reverse, 5’-CCATCCAATCGGTAGTAGCG-3’.
Cell culture and transfection
HCC cell lines SK-Hep1, Huh7, QGY-7701, Hep3B, SMMC-7721 and human embryonic kidney cell line 293T (HEK293T) were cultured with Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (Gibco, USA) in 5% CO2 humidified atmosphere at 37°C. Cells of 70% confluency were transfected with indicated plasmids or siRNA using Lipofectin 2000 (Invitrogen, USA) according to the manufacturer’s instructions. Small interfering RNA against BRMS1 as previously described was used [9] and the target sequence of si-SCIN was: 5’-CGAGATGAGCTGACAACAT-3’. In experiments evaluating cells’ sensitivity to apoptosis stimulation, etoposide (Sigma, USA) and cisplatin (Abcam, USA) were directly added to complete medium with indicated dosages.
Western blot analysis
Protein samples were separated by different concentrations of SDS-PAGE and then transferred to PVDF membranes. Membranes were incubated in 5% fat-free milk to avoid non-specific binding. After blocking, membranes were incubated with specific primary antibodies against different proteins at 4°C overnight. Membranes were then washed in TBST for four times and incubated with HRP-conjugated secondary antibody for 1 h at room temperature. Later on, membranes were washed in TBST for four times before being visualized by enhanced chemiluminescence (Pierce, USA) on a molecular imager ChemiDoc XRS+ system (Bio-Rad). Antibodies used include anti-BRMS1 (Abcam), anti-SCIN (Abcam), anti-caspase 9 (cell signaling technology, USA), anti-β-actin (Sigma), anti-Myc (Sigma), anti-Flag (Sigma), peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG (Jackson, USA).
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed according to our previous work [12]. Briefly, HEK293T cells were seeded at 1×107/dish in 100 mm dishes and then transfected with Myc-tagged BRMS1 plasmid or empty vector. At 36 h after transfection, cells were treated with 4% formaldehyde (Amresco, USA) to make them crosslinked and fixed. Cell nuclei were first released by lysis buffer in pH 8.1 and then sonicated. Genomic DNA fragment was immuno-precipitated by anti-Myc antibody at 4°C overnight after treating with protein A-agarose beads. Samples containing mouse IgG were used as negative control to exclude genomic contamination. Treated genomic DNA fragments were collected, washed, de-crosslinked and then digested with Proteinase K. PCR was applied to examine the quantity of these DNA fragments recovered by phenol/chloroform extraction using the following primers: forward, 5’-TCGTCTCCTCTGTCAACT-3’; reverse, 5’-TTATTCGCCGCCACTTTA-3’.
Dual-luciferase assay
Cells were seeded into 24-well plates at a density of 1×105 per well and transfected with indicated SCIN promoter segments or pGL3-Basic empty vector. Internal control pRL-TK vector (20 ng/well) was used for normalization. Primers for SCIN promoter were: forward, CGGCTAGCTTGCCCAATGAAATGACAGA; reverse, CCAAGCTTAGTTTGCGGTCCCCTTTACT. SCIN-P1 primers were: forward, CGGCTAGCTTGCCCAATGAAATGACAGA; reverse, CCAAGCTTTCCTTTGTGCATACCCATCA. pGL3-SCIN promoter 2 primers were: forward, CGGCTAGCCCTGCTGCTCTCGGTTTAGT; reverse, CCAAGCTTAGTTTGCGGTCCCCTTTACT; pGL3-SCIN promoter 3 primers were: forward, CGGCTAGCACAAAGGAGTGCCAAGCAGT; reverse, CCAAGCTTGAGCAGCAGGAGGAACCTTA.
Flow cytometry analysis
For cell cycle phase analysis, cells were harvested and resuspended in 0°C 70% ethanol to fix overnight. DNA was stained with propidium iodide (50 µg/mL) and treated with RNase (100 µg/mL) and then analyzed by FACSCalibur (BD Biosciences, CA, USA). The apoptotic cell population corresponds to cells in sub-G1 phase. Each result was representative of three independent experiments with triplicate samples for each condition.
Cell proliferation assay
Cells were plated in 96-well plates at a density of 1500 cells/well. In the serum deprivation assay, medium was replaced by DMEM after 24 hours’ culture with complete medium. Cell proliferation was detected using CCK8 assay. Generally, culture medium was replaced by 0.5 mg/mL CCK8 (Dojindo, Japan) diluted in DMEM. Cells were then kept in 37°C for 4 hours and absorbance at a wavelength of 490 nm was read by a microtiter reader (Bio-RAD, USA). The final growth curve was constructed using the absorbance data.
Tumor formation in nude mice
Three-week-old BALB/c female nude mice were obtained from Shanghai Laboratory Animal Co. Ltd. (SLAC, China) and maintained on standard laboratory chow. Cells were harvested and resuspended in DMEM to a final concentration of 2×107 cells/ml. Cells were then subcutaneously injected into each mouse. Tumors were visible around five days after injection. Mice were sacrificed till the average size of the tumors was around 800 mm3. Photos were taken, and weight of tumors was measured (n = 5).
Histological studies
Xenografted tumor tissues were embedded in paraffin and cut into sections (4 μm thickness) for immunohistochemistry studies. The avidin-biotin complex method was used in Ki-67 immunostaining. Briefly, endogenous peroxidase was blocked with H2O2. After BSA blocking, sections were incubated with anti-Ki-67 antibody (Abcam, USA) at 4°C overnight, followed by biotinylated secondary antibody. The sections were further treated with diaminobenzidine and counterstained with hematoxylin. TUNEL (terminal deoxynucleotide transferase mediated dUTP nick end labeling) assay was performed using the in situ Cell Apoptosis Detection kit (Boster, China) according to the manufacturer’s instructions.
Statistical analysis
Categorical data were analyzed by Fisher’s exact test. Comparisons of quantitative data were analyzed by Student’s t-test. We considered two groups with a p value less than 0.05 to be different (*), and with a p value less than 0.01 to be significantly different (**).
Results
SCIN and BRMS1 levels were inversely correlated in HCC cells and tissues
Previously, we have applied microarray analysis of whole genome expression to screen for novel transcriptional targets of BRMS1 [12]. One rarely-studied gene, SCIN, was found to be suppressed in two stable SK-Hep1 cell clones overexpressing exogenous BRMS1 by comparison with control cell clones (Figure 1A). Western blot analysis result in consistent with this was shown in Figure 1B. To ensure that SCIN expression was specifically impaired by BRMS1, different dosages of recombinant BRMS1 plasmid were transiently introduced into QGY-7701 cells. As shown in Figure 1C, endogenous SCIN expression was gradually decreased upon Myc-BRMS1 overexpression in a dosage-dependent manner. Furthermore, Huh-7 cells with relative higher endogenous BRMS1 levels were subjected to RNA interference. As shown in Figure 1D and 1E, both mRNA and protein levels of endogenous SCIN were increased after endogenous BRMS1 was successfully knocked down in Huh-7.
Figure 1.
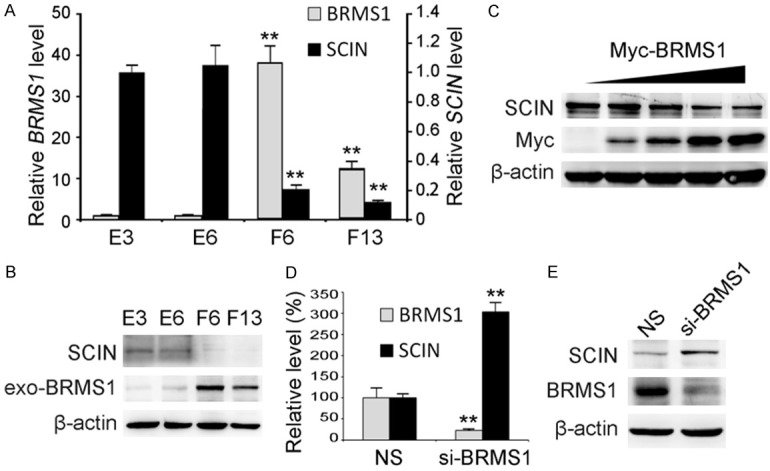
The expression level of SCIN was suppressed by BRMS1. (A) Relative BRMS1 and SCIN mRNA levels in SK-Hep1 cell clones stably expressing exogenous BRMS1 (F6 and F13) and control cell clones (E3 and E6) were analyzed through qRT-PCR. Values were normalized to internal control 18S rRNA and expressed as mean ± SD, n = 3. (B) The protein levels of exogenous BRMS1 (exo-BRMS1) and endogenous SCIN in SK-Hep1 cell clones were investigated through western blot. β-actin was used as a loading control. (C) Myc-tagged BRMS1 (Myc-BRMS1) and endogenous SCIN protein levels in QGY-7701 cells transiently expressing different dosages of Myc-BRMS1 were investigated through western blot. Relative longer exposure of endogenous SCIN is shown. Endogenous BRMS1 and SCIN expression levels in Huh-7 cells infected with lentivirus expressing siRNA targeting BRMS1 (siBRMS1) or non-silencing control siRNA (NS) were analyzed through qRT-PCR (D) and western blot (E).
In order to further evaluate the relationship between BRMS1 and SCIN, different cancer cell lines and HCC tissues were investigated using western blot. As shown in Figure 2A, the BRMS1 and SCIN levels in most cancer cell lines were inversely correlated. In addition, we found that SCIN protein was up-regulated while BRMS1 was mainly suppressed in HCC tissues (T) compared with the corresponding non-tumorous tissues (N). Of note, the expression pattern of SCIN was inversely correlated with BRMS1 in 7 paired HCC tissues such as H1, H2, H5, among others. This finding provides another piece of evidence that BRMS1 probably exerted a transcriptional suppression effect on SCIN expression during tumorigenesis.
Figure 2.
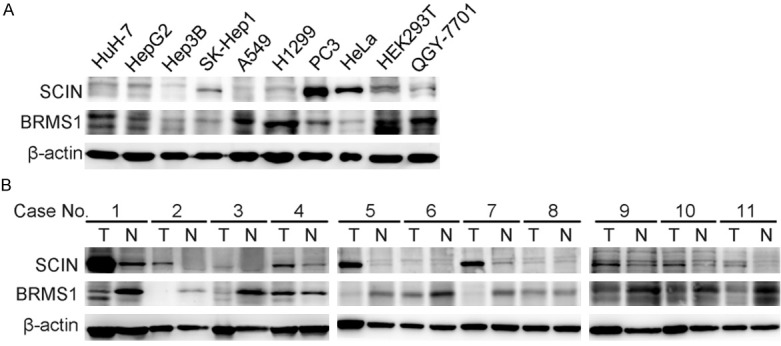
The protein level of SCIN was inversely correlated with BRMS1 level in HCC cells and tissues. A. Western blot analysis of BRMS1 and SCIN levels in HEK293T, five HCC cell lines (Huh-7, HepG2, Hep3B, SK-Hep1, QGY-7701), two lung carcinoma cell lines (A549, H1299), one prostate cancer cell line (PC3) and one cervical carcinoma cell line (HeLa). B. Western blot analysis of BRMS1 and SCIN levels in 11 paired HCC specimens. T, tumor tissues; N, corresponding non-tumorous tissues.
BRMS1 was able to transcriptionally suppress SCIN expression
To investigate the regulatory mechanism between BRMS1 and SCIN, the promoter region (-508 to +512 bp) of human SCIN gene was cloned from human genomic DNA and a series of deletion mutants, including SCIN-P1 (-508 to -157 bp), SCIN-P2 (-164 to +151 bp), SCIN-P3 (+142 to +512 bp), were constructed subsequently. The above four DNA fragments were then inserted into pGL3-Basic vector to detect their transcriptional activity using luciferase assay. As shown in Figure 3A, SCIN-P2 exhibited the highest report gene’s activity among all the promoter constructs. Next, when cells were co-transfected with different dosages of BRMS1 plasmid, the luciferase activity of SCIN-P2 gradually decreased upon BRMS1 expression in a dosage-dependent manner (Figure 3B). To further examine whether BRMS1 could bind to this SCIN promoter region, chromatin immunoprecipitation was carried out with cells overexpressing myc-tagged BRMS1. As shown in Figure 3C, the +38 to +134 bp region of SCIN-P2 was specifically immunoprecipitated by anti-Myc antibody, but not IgG control, in both HEK293T and SK-Hep1 cells. Taken together, these data suggested a transcriptional suppression relationship between BRMS1 and SCIN.
Figure 3.

BRMS1 could transcriptionally suppress the activity of the promoter of SCIN. A. The full length SCIN promoter (SCIN-P) and three deletion mutants (SCIN-P1, P2 and P3) were constructed into luciferase reporter plasmid. Schematic representations of SCIN promoter constructs are presented in the left panel. Relative reporter activities in HEK293T cells were investigated through dual luciferase assay (right panel). Values are presented as mean ± SD, n = 3. B. Relative reporter activities of SCIN-P2 were examined after co-transfection with different dosages of Myc-tagged BRMS1. Values are presented as mean ± SD, n = 3. C. Gel electrophoresis photos of the ChIP assay to detect the binding ability between BRMS1 protein and SCIN promoter in HEK293T and SK-Hep1 cells. Soluble chromatin was prepared from cells transfected with Myc-tagged BRMS1 and immunoprecipitated by an anti-Myc antibody or IgG control. Specific primers were utilized in the PCR analysis. Blank controls (BC) with no DNA template were utilized to rule out contamination and soluble chromatin before immunoprecipitation was utilized as input control.
SCIN was involved in regulating HCC cells’ sensitivity to apoptotic stimulus
To elucidate the biological functions of SCIN during HCC development, we utilized lentivirus-mediated RNA interference in QGY-7701 cells. As shown in Figure 4A, the specific SCIN-targeting siRNA successfully knocked down endogenous SCIN expression (KD) by comparison with non-silencing siRNA control (NS). Next, we investigated several cellular behaviors in QGY-7701 cells under different conditions. No significant difference was found in the cell cycle distribution or cell growth under normal culture conditions (Figure 4B, 4C). However, when cells were deprived of serum, cell growth capacities were significantly impaired in SCIN knockdown cells compared with control cells (Figure 4D), indicating that SCIN knockdown might sensitize HCC cells to apoptotic stimulus. To confirm this hypothesis, the chemotherapeutic drug cisplatin was utilized in the apoptosis assay as reported previously [21]. Not surprisingly, flow cytometry analysis revealed a higher level of apoptosis in SCIN knockdown cells compared with control cells upon cisplatin treatment (Figure 4E). Moreover, cleavage of caspase 9 was specifically activated in SCIN knockdown cells, but not in the control cells, after cisplatin treatment in a dosage-dependent manner (Figure 4F).
Figure 4.
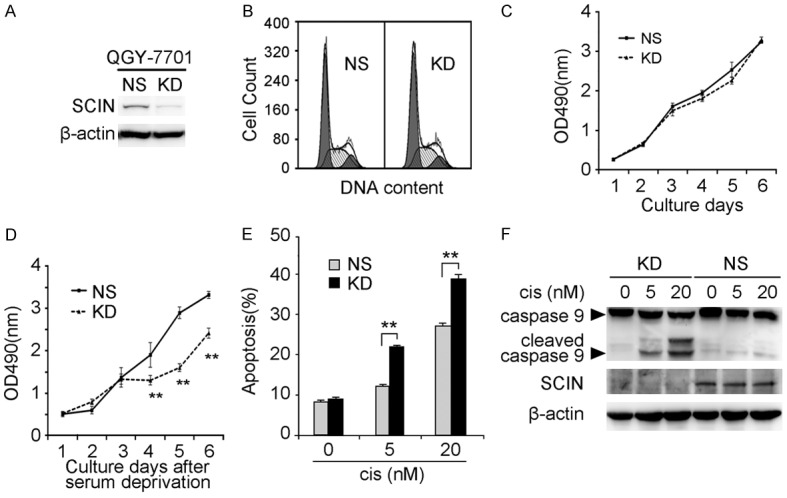
Effects of SCIN knockdown on QGY-7701 cells. (A) Endogenous SCIN level in QGY-7701 cells infected with lentivirus expressing shRNA targeting SCIN (KD) or non-silencing shRNA (NS) was investigated by western blot. β-actin was used as a loading control. (B) Representative flow cytometric images of lentivirus-infected QGY-7701 cells from cell cycle analysis. Cell growth curves of lentivirus-infected QGY-7701 cells under normal culture condition (C) and serum deprivation (D). Values are indicated as mean ± SD, n = 5. Lentivirus-infected QGY-7701 cells treated with indicated dosages of cisplatin (cis) were subjected to cell apoptotic analysis through sub-G1 analysis (E) and caspase 9 immunoblot (F). Values are indicated as mean ± SD, n = 3.
To further confirm the function of SCIN in regulating cell apoptosis, we established SMMC-7721 stable clone overexpressing flag-tagged SCIN (OE) and the control clone (Ctl) in parallel (Figure 5A). In consistent with the above findings, while no difference was observed in cell cycle distribution or cell growth under normal conditions between two clones (Figure 5B, 5C), cells overexpressing SCIN are less sensitive to serum deprivation compared with control cells (Figure 5D). Furthermore, SCIN overexpression protected SMMC-7721 clone from cisplatin-induced cell apoptosis and caspase 9 cleavage (Figure 5E, 5F). Collectively, these data strongly implied that SCIN level could modify HCC cells’ sensitivity to apoptotic stimulus.
Figure 5.
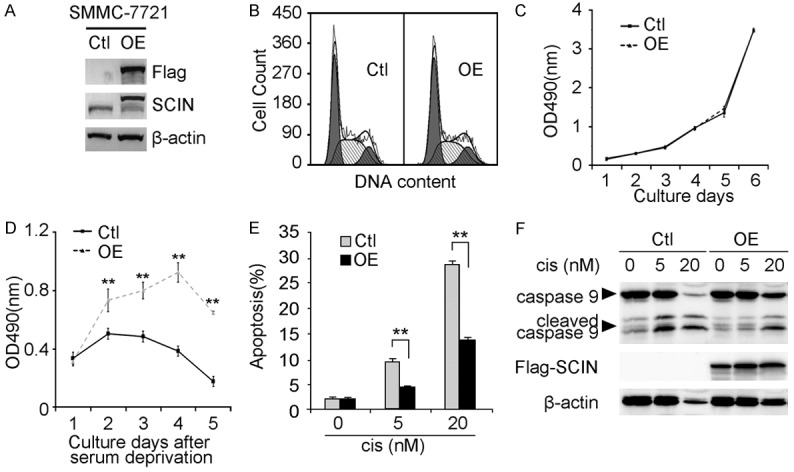
Effects of SCIN overexpression on SMMC-7721 cells. (A) Exogenous SCIN level in SMMC-7721 cells infected with lentivirus overexpressing flag-tagged SCIN (OE) or empty vector (Ctl) was investigated by western blot. β-actin was used as a loading control. (B) Representative flow cytometric images of lentivirus-infected SMMC-7721 cells determined from cell cycle analysis. Cell growth curves of lentivirus-infected SMMC-7721 cells under normal culture conditions (C) and serum deprivation (D). Values are indicated as mean ± SD, n = 5. Lentivirus-infected SMMC-7721 cells treated with indicated dosages of cisplatin (cis) were subjected to cell apoptotic analysis through sub-G1 analysis (E) and caspase 9 immunoblot (F). Values are indicated as mean ± SD, n = 3.
SCIN was able to regulate HCC cell growth in vivo
To explore the role of SCIN on HCC cell growth in vivo, tumorigenicity assay in BALB/c nude mice was performed. We first subcutaneously injected equal amounts of QGY-7701 KD cells and NS cells into nude mice separately. Tumors were visible in NS group from eight days after injection and they increase in size stably. However, no visible tumor was observed in all mice injected with KD cells throughout the whole experiment (22 days in total) (Figure 6A). Next, we investigated the effect of SCIN overexpression on xenografted tumor growth. As shown in Figure 6B, SMMC-7721 OE cells display dramatically increased tumor growth in nude mice compared with Ctl cells. The average tumor weight of the OE cells was 3.47-fold heavier than that of Ctl cells.
Figure 6.
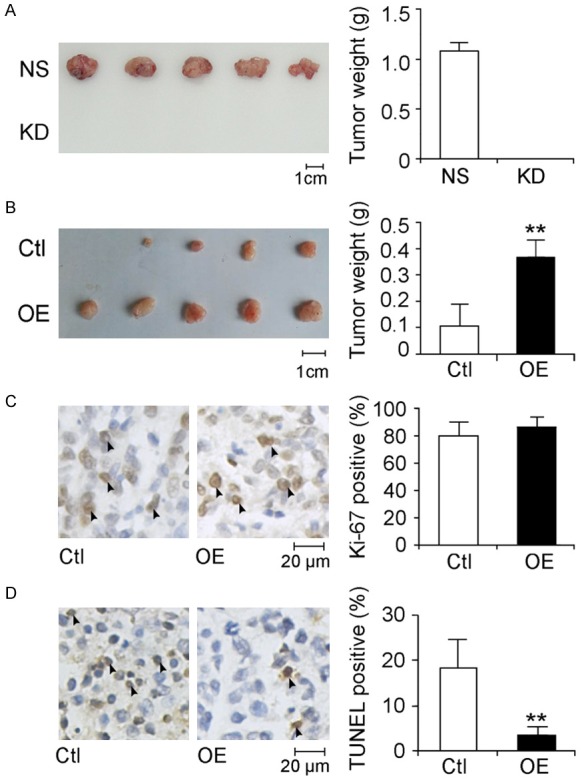
Evaluation of the function of SCIN on HCC cell growth in vivo. Photographs (left panels) and tumor weight measurements (right panels) of tumors dissected from nude mice engrafted with lentivirus-infected QGY-7701 cells (A) and SMMC-7721 cells (B). Scale bars, 1 cm. Values are indicated as mean ± SD, n = 5. (C) Immunohistochemistry staining against Ki-67 was performed in tumor tissues derived from lentivirus-infected SMMC-7721. Representative images of tumor sections are given in the left panel and typical Ki-67-positive cells are indicated with arrowheads. Scale bar, 20 μm. Ki-67 positive cell populations were statistically analyzed in the right panel. Values are indicated as mean ± SD, n = 8. (D) TUNEL assays were performed on tumor sections. Representative images of tumor sections are given in the left panel, and typical apoptotic cells are indicated with arrowheads. Scale bar, 20 μm. Apoptotic cell populations are statistically analyzed in the right panel. Values are indicated as mean ± SD, n = 8.
Furthermore, xenografted tumor tissues were embedded in paraffin and subjected to immunohistochemistry studies. As shown in Figure 6C, there is no obvious difference in the fraction of Ki-67-positive cells, suggesting SCIN has less effect on regulating cell proliferation in vivo. By contrast, the TUNEL assay revealed a lower apoptotic cell population in OE group than in Ctl group (Figure 6D), indicating SCIN overexpression protected cells from apoptosis in vivo.
Discussion
Our study identified SCIN as a novel transcriptional target of BRMS1 in HCC cells and SCIN was able to promote tumor cell growth through modulating the apoptotic levels in cells. Accumulating evidence demonstrated that BRMS1 has an essential role in regulating gene transcription mainly through interacting with Sin3•HDAC chromatin remodeling complexes or NF-κB transcription factor [10,25,26]. Although BRMS1 suppresses several metastasis-related genes’ expression through deacetylation of RelA/p65 subunit of NF-κB, such as OPN, uPA and CXCR4 [3,26,27], we ruled out the possibility that NF-κB was involved in the transcriptional regulation of SCIN mainly because there is no NF-κB binding site within the promoter of SCIN. On the other hand, Sin3•HDAC is a complex associated with a number of DNA-binding proteins and mediates transcriptional regulation of a variety of protein-coding genes and miRNAs implicated in diverse biological functions [28]. After the ChIP assay, which revealed a binding relationship between BRMS1 protein and the +38 to +134 bp region of SCIN promoter, we performed in silico prediction and found a putative GC binding factor (GCF) binding site (GCGCCGCCT) within the binding region. GCF is able to bind to GC-rich sequence and suppress the expression of several tumor-related genes [29,30]. It is interesting to note that the expression of the growth factor receptor (EGFR) gene is under the control of both GCF and Sin3•HDAC [11,30]. Together with our data, we raised the hypothesis that Sin3•HDAC might act in concert with GCF on SCIN expression. However, this still remains to be experimentally investigated in our future work.
Although the association of SCIN with cancer has been revealed for a long time, multiple reports addressing the role of SCIN in tumor development were controversial among different investigators. Expression of SCIN in megakaryoblastic leukemia cells induced cell apoptosis, inhibited cell proliferation and tumoriogenesis [20]. By contrast, silencing of SCIN in gastric cancer cells suppressed cell invasion and tumor metastasis [24]. In addition, different changes of cell cycle distribution and different cyclins affected by SCIN were reported in 3 different studies [22,23,31]. Although heterogenity of tumor cell lines might partially explain these disparities, more comprehensive studies are required to uncover the relationship between SCIN and cancer. In our present work, an anti-apoptotic role of SCIN has been revealed in HCC cells by different approaches both in vitro and in vivo, while no obvious changes were shown in cell cycle distribution or cell proliferation. Our finding was in consistent with the report in human bladder cancer cell, where SCIN was identified as the cisplatin-resistant marker via interacting with VDAC and further inhibiting cell apoptosis [21]. In addition, SCIN has 63% homology with gelsolin, and gelsolin superfamily has been demonstrated to play an important role in cell apoptosis by modulating dynamic actin assembly [32,33]. Overexpression of gelsolin in Jurkat cells strongly inhibited caspase-3 activation and cell apoptosis level triggered by different reagents [34]. Consistently, gelsolin or villin knockout mice exhibited higher apoptotic cell population, shorter survival and enhanced caspase activation in liver after exposure to stimulatory Fas antibody [35-37]. Taken together, the results presented here highlight that SCIN might also be another important gelsolin superfamily member mediating the crosstalk between apoptosis pathway and cytoskeleton reorganization.
Furthermore, abnormal apoptosis has been demonstrated to affect HCC growth and metastasis [38,39]. As we previously reported, BRMS1 also exhibits a strong activity in regulating HCC cell apoptosis, which further contributes to its suppressive role in tumorigenesis and tumor metastasis [5,9]. A few proteins, including OPN, Bim and Bcl-2, have been demonstrated to partially account for the pro-apoptotic effect of BRMS1 in breast cancer cells [40,41]. Given that overexpression of SCIN has a similar effect as knockdown of BRMS1 in regulating HCC cells’ sensitivity to apoptotic stimulus, we raised the hypothesis that SCIN might be another downstream molecule employed by BRMS1 in modulating cancer cell apoptosis pathway. Investigations of how these different transcriptional targets of BRMS1 collaborate to regulate cell apoptosis are therefore of great importance and interest in the future.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31000558) and 2012 “Chen Guang” project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation.
Disclosure of conflict of interest
None.
References
- 1.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–2769. [PubMed] [Google Scholar]
- 2.Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, Mauger DT, Welch DR. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273:229–239. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 3.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res. 2005;65:3586–3595. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- 4.Smith PW, Liu Y, Siefert SA, Moskaluk CA, Petroni GR, Jones DR. Breast cancer metastasis suppressor 1 (BRMS1) suppresses metastasis and correlates with improved patient survival in non-small cell lung cancer. Cancer Lett. 2009;276:196–203. doi: 10.1016/j.canlet.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J, Wang Y, Qiao X, Saiyin H, Zhao S, Qiao S, Wu Y. Cloning and characterization of a novel human BRMS1 transcript variant in hepatocellular carcinoma cells. Cancer Lett. 2013;337:266–275. doi: 10.1016/j.canlet.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 6.Metge BJ, Frost AR, King JA, Dyess DL, Welch DR, Samant RS, Shevde LA. Epigenetic silencing contributes to the loss of BRMS1 expression in breast cancer. Clin Exp Metastasis. 2008;25:753–763. doi: 10.1007/s10585-008-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagji AS, Liu Y, Stelow EB, Stukenborg GJ, Jones DR. BRMS1 transcriptional repression correlates with CpG island methylation and advanced pathological stage in non-small cell lung cancer. J Pathol. 2010;221:229–237. doi: 10.1002/path.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Cheng Y, Tai D, Martinka M, Welch DR, Li G. Prognostic significance of BRMS1 expression in human melanoma and its role in tumor angiogenesis. Oncogene. 2011;30:896–906. doi: 10.1038/onc.2010.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y, Jiang W, Wang Y, Wu J, Saiyin H, Qiao X, Mei X, Guo B, Fang X, Zhang L, Lou H, Wu C, Qiao S. Breast cancer metastasis suppressor 1 regulates hepatocellular carcinoma cell apoptosis via suppressing osteopontin expression. PLoS One. 2012;7:e42976. doi: 10.1371/journal.pone.0042976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meehan WJ, Samant RS, Hopper JE, Carrozza MJ, Shevde LA, Workman JL, Eckert KA, Verderame MF, Welch DR. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279:1562–1569. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- 11.Hurst DR, Xie Y, Vaidya KS, Mehta A, Moore BP, Accavitti-Loper MA, Samant RS, Saxena R, Silveira AC, Welch DR. Alterations of BRMS1-ARID4A interaction modify gene expression but still suppress metastasis in human breast cancer cells. J Biol Chem. 2008;283:7438–7444. doi: 10.1074/jbc.M709446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao X, Yang X, Zhou Y, Mei X, Dou J, Xie W, Li G, Wang Y, Qiao S, Hu J, Wu Y. Characterization of DAPK1 as a novel transcriptional target of BRMS1. Int J Oncol. 2017;50:1760–1766. doi: 10.3892/ijo.2017.3930. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez Del Castillo A, Lemaire S, Tchakarov L, Jeyapragasan M, Doucet JP, Vitale ML, Trifaro JM. Chromaffin cell scinderin, a novel calcium-dependent actin filament-severing protein. EMBO J. 1990;9:43–52. doi: 10.1002/j.1460-2075.1990.tb08078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitale ML, Rodriguez Del Castillo A, Tchakarov L, Trifaro JM. Cortical filamentous actin disassembly and scinderin redistribution during chromaffin cell stimulation precede exocytosis, a phenomenon not exhibited by gelsolin. J Cell Biol. 1991;113:1057–1067. doi: 10.1083/jcb.113.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dumitrescu Pene T, Rose SD, Lejen T, Marcu MG, Trifaro JM. Expression of various scinderin domains in chromaffin cells indicates that this protein acts as a molecular switch in the control of actin filament dynamics and exocytosis. J Neurochem. 2005;92:780–789. doi: 10.1111/j.1471-4159.2004.02907.x. [DOI] [PubMed] [Google Scholar]
- 16.Trifaro JM, Gasman S, Gutierrez LM. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol (Oxf) 2008;192:165–172. doi: 10.1111/j.1748-1716.2007.01808.x. [DOI] [PubMed] [Google Scholar]
- 17.Nurminsky D, Magee C, Faverman L, Nurminskaya M. Regulation of chondrocyte differentiation by actin-severing protein adseverin. Dev Biol. 2007;302:427–437. doi: 10.1016/j.ydbio.2006.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassanpour S, Jiang H, Wang Y, Kuiper JW, Glogauer M. The actin binding protein adseverin regulates osteoclastogenesis. PLoS One. 2014;9:e109078. doi: 10.1371/journal.pone.0109078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Jiang H, Huang Y, Gong Q, Wang J, Ling J. Expression and function of the actin-severing protein adseverin in the proliferation, migration, and differentiation of dental pulp cells. J Endod. 2015;41:493–500. doi: 10.1016/j.joen.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 20.Zunino R, Li Q, Rose SD, Romero-Benitez MM, Lejen T, Brandan NC, Trifaro JM. Expression of scinderin in megakaryoblastic leukemia cells induces differentiation, maturation, and apoptosis with release of plateletlike particles and inhibits proliferation and tumorigenesis. Blood. 2001;98:2210–2219. doi: 10.1182/blood.v98.7.2210. [DOI] [PubMed] [Google Scholar]
- 21.Miura N, Takemori N, Kikugawa T, Tanji N, Higashiyama S, Yokoyama M. Adseverin: a novel cisplatin-resistant marker in the human bladder cancer cell line HT1376 identified by quantitative proteomic analysis. Mol Oncol. 2012;6:311–322. doi: 10.1016/j.molonc.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, Sun SQ, Yu YH, Wu WZ, Yang SL, Tan JM. Suppression of SCIN inhibits human prostate cancer cell proliferation and induces G0/G1 phase arrest. Int J Oncol. 2014;44:161–166. doi: 10.3892/ijo.2013.2170. [DOI] [PubMed] [Google Scholar]
- 23.Liu H, Shi D, Liu T, Yu Z, Zhou C. Lentivirus-mediated silencing of SCIN inhibits proliferation of human lung carcinoma cells. Gene. 2015;554:32–39. doi: 10.1016/j.gene.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Liu JJ, Liu JY, Chen J, Wu YX, Yan P, Ji CD, Wang YX, Xiang DF, Zhang X, Zhang P, Cui YH, Wang JM, Bian XW, Qian F. Scinderin promotes the invasion and metastasis of gastric cancer cells and predicts the outcome of patients. Cancer Lett. 2016;376:110–117. doi: 10.1016/j.canlet.2016.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samant RS, Clark DW, Fillmore RA, Cicek M, Metge BJ, Chandramouli KH, Chambers AF, Casey G, Welch DR, Shevde LA. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-kappaB activation. Mol Cancer. 2007;6:6. doi: 10.1186/1476-4598-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cicek M, Fukuyama R, Cicek MS, Sizemore S, Welch DR, Sizemore N, Casey G. BRMS1 contributes to the negative regulation of uPA gene expression through recruitment of HDAC1 to the NF-kappaB binding site of the uPA promoter. Clin Exp Metastasis. 2009;26:229–237. doi: 10.1007/s10585-009-9235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Zhang B, Lin Y, Yang Y, Liu X, Lu F. Breast cancer metastasis suppressor 1 inhibits SDF-1alpha-induced migration of non-small cell lung cancer by decreasing CXCR4 expression. Cancer Lett. 2008;269:46–56. doi: 10.1016/j.canlet.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 28.Kadamb R, Mittal S, Bansal N, Batra H, Saluja D. Sin3: insight into its transcription regulatory functions. Eur J Cell Biol. 2013;92:237–246. doi: 10.1016/j.ejcb.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Yang C, Yin L, Zhou P, Liu X, Yang M, Yang F, Jiang H, Ding K. Transcriptional regulation of IER5 in response to radiation in HepG2. Cancer Gene Ther. 2016;23:61–65. doi: 10.1038/cgt.2016.1. [DOI] [PubMed] [Google Scholar]
- 30.Kageyama R, Pastan I. Molecular cloning and characterization of a human DNA binding factor that represses transcription. Cell. 1989;59:815–825. doi: 10.1016/0092-8674(89)90605-3. [DOI] [PubMed] [Google Scholar]
- 31.Chen XM, Guo JM, Chen P, Mao LG, Feng WY, Le DH, Li KQ. Suppression of scinderin modulates epithelialmesenchymal transition markers in highly metastatic gastric cancer cell line SGC7901. Mol Med Rep. 2014;10:2327–2333. doi: 10.3892/mmr.2014.2523. [DOI] [PubMed] [Google Scholar]
- 32.Khurana S, George SP. Regulation of cell structure and function by actin-binding proteins: villin’s perspective. FEBS Lett. 2008;582:2128–2139. doi: 10.1016/j.febslet.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol. 2005;6:583–589. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- 34.Ohtsu M, Sakai N, Fujita H, Kashiwagi M, Gasa S, Shimizu S, Eguchi Y, Tsujimoto Y, Sakiyama Y, Kobayashi K, Kuzumaki N. Inhibition of apoptosis by the actin-regulatory protein gelsolin. EMBO J. 1997;16:4650–4656. doi: 10.1093/emboj/16.15.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harms C, Bosel J, Lautenschlager M, Harms U, Braun JS, Hortnagl H, Dirnagl U, Kwiatkowski DJ, Fink K, Endres M. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol Cell Neurosci. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Leifeld L, Fink K, Debska G, Fielenbach M, Schmitz V, Sauerbruch T, Spengler U. Anti-apoptotic function of gelsolin in fas antibody-induced liver failure in vivo. Am J Pathol. 2006;168:778–785. doi: 10.2353/ajpath.2006.050323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy S, Esmaeilniakooshkghazi A, Patnaik S, Wang Y, George SP, Ahrorov A, Hou JK, Herron AJ, Sesaki H, Khurana S. Villin-1 and gelsolin regulate changes in actin dynamics that affect cell survival signaling pathways and intestinal inflammation. Gastroenterology. 2018;154:1405–1420. e2. doi: 10.1053/j.gastro.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strand S, Hofmann WJ, Hug H, Muller M, Otto G, Strand D, Mariani SM, Stremmel W, Krammer PH, Galle PR. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells--a mechanism of immune evasion? Nat Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 39.Hu H, Li Z, Chen J, Wang D, Ma J, Wang W, Li J, Wu H, Li L, Wu M, Qian Q, Chen J, Su C. P16 reactivation induces anoikis and exhibits antitumour potency by downregulating Akt/survivin signalling in hepatocellular carcinoma cells. Gut. 2011;60:710–721. doi: 10.1136/gut.2010.220020. [DOI] [PubMed] [Google Scholar]
- 40.Phadke PA, Vaidya KS, Nash KT, Hurst DR, Welch DR. BRMS1 suppresses breast cancer experimental metastasis to multiple organs by inhibiting several steps of the metastatic process. Am J Pathol. 2008;172:809–817. doi: 10.2353/ajpath.2008.070772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26:8683–8696. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]