

Abstract
CAR T cells have shown clinical efficacy for acute lymphoblastic leukemia, but this therapy has not been effective for acute myeloid leukemia (AML), and other treatment options are needed. Theoretically, CAR-NK cells have a more favorable toxicity profile compared to CAR T cells, especially in avoiding adverse effects such as cytokine release syndrome. However, the clinical evidence for this has not yet been reported. In the current study, we tested the safety of CD33-CAR NK cells in patients with relapsed and refractory AML. At doses up to 5 × 109 (5 billion) cells per patient, no significant adverse effects were observed. CAR NK-92 cells can be produced at much lower cost compared to CAR T cells, and we believe after being optimized, they will be widely accessible for the treatment of cancer.
Keywords: CAR NK-92 cells, CD33, safety test, relapsed, refractory, acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is a genetically and clinically heterogeneous myeloid cell malignancy. Although the rate of complete remission can be as high as 80% following initial induction chemotherapy, the majority of AML patients will eventually progress to relapsed or refractory (RR) disease [1]. RR-AML carries a poor prognosis and introduces new challenges for treating these patients. Recently, several new drugs and treatment strategies have been used for RR-AML including hypomethylating agents, histone deacetylase inhibitors, immunomodulatory drugs, targeted tyrosine kinase inhibitors, and cellular therapies. An ongoing cellular therapy approach to treating cancer involves modified chimeric antigen receptor (CAR) T cells. Given the clinical success of CD19-CAR T cell-based therapies against B cell malignancies, several studies have attempted to incorporate a similar approach toward treating AML. Unfortunately, CAR T cells have not shown much promise in treating AML. This is mainly because the antigens present in AML (e.g., CD123) that could be targeted by CAR T cells are also highly expressed in normal myeloid cells and hematopoietic stem cells. Thus, targeting these antigens by CAR T cells may cause severe myeloablative toxicity [2], as well as other adverse effects including off-target effects, cytokine release syndrome, and neurotoxicity. There is a growing interest in CAR NK cells for their therapeutic potential [3-12], specifically in using a modified CAR-engineered form of the NK-92 cell line, which can provide an unlimited number of cells and thus a more cost-effective approach to therapy.
We herein report a first-in-man phase I trial of CD33-CAR NK-92 cells in three patients with RR-AML. To our knowledge, this is the first report of a phase I clinical study using CAR NK-92 cells in the treatment of RR-AML. The objective of this study was to explore the safety of escalating infusion doses of CAR NK-92 cells in these RR-AML patients with high tumor burden.
Materials and methods
We used a third generation CAR lentiviral construct containing both CD28 and 4-1BB co-stimulatory molecules, with an Fc fragment inserted between the CD33 single chain variable fragment (scFv) and CD28 to detect transduced CAR cells. NK-92-MI cells, which express human IL-2 and were originally purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), were transduced with the CD33-CAR construct. The transduction efficiency was determined by flow cytometry. After confirming the transduction efficiency was over 90%, the transduced cells were expanded. All cell processing and transduction protocols were undertaken in a Good Manufacturing Practice (GMP) facility. The in vitro cytotoxicity of CD33-CAR NK-92 cells was determined by 4-hr standard 51Cr release assays, using the human HL-60 promyelocytic leukemia cell line as target cells. Three refractory and relapsed AML patients, treated with salvage chemotherapy, were infused with 60Co-irradiated (10 Gy) CD33-CAR NK-92 cells. The doses of the infused CD33-CAR NK-92 cells are detailed in the Results section. The Clinical Trials Identification Number for our study was NCT02944162. All human work was approved by the Soochow University Institutional Review Board.
Results
The third generation CAR lentiviral construct is shown in Figure 1A. The NK-92 cells were transduced with the above-mentioned CD33-CAR vector with an efficiency over 90% (Figure 1B). In vitro cytotoxicity assays demonstrated a moderate enhancement of cytotoxicity of CD33-CAR NK-92 cells against the human HL-60 promyelocytic leukemia cell line compared to parental NK-92 cells (Figure 1C). We then treated three AML patients with salvage chemotherapy, followed by infusion of 60Coirradiated (10 Gy) CD33-CAR NK-92 cells.
Figure 1.
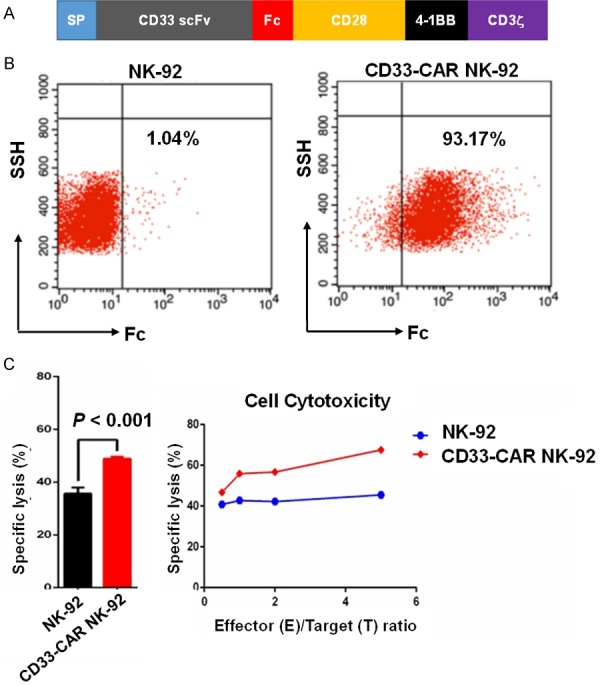
Preclinical characterization of CD33-CAR NK-92 cells. A. Diagram of the third generation CD33-CAR vector. B. A representative flow cytometric analysis showing NK-92 cells transduced with the CD33-CAR vector with efficiency greater than 90%. C. CD33-CAR NK-92 cells showed moderately enhanced killing capacity against the human promyelocytic leukemia cell line HL60 when compared to parental NK-92 cells. The cytotoxicity of CD33-CAR NK-92 cells was determined by 4-hr standard 51Cr release assays.
The first case was a 14-year-old girl diagnosed with AML1-ETO(+), C-KIT(+), M4 AML (intermediate risk). She attained complete remission (CR) after idarubicine (IDA) plus Ara-C (IDA 12 mg/m2/d × 3 d and Ara-C 100 mg/m2/d × 7 d). After 2 cycles of consolidation chemotherapy with an intermediate dose of Ara-C combined with fludarabine (FA: fludarabine 30 mg/m2/d × 5 d, d1-d5 and Ara-C 2 g/m2/d × 5 d, d1-d5), she received allo-HSCT from an unrelated 10/10 HLA-matched donor in July 2015. However, 15 months post-transplantation, the patient suffered from pain and swelling in the right elbow and right leg in October 2016. PET-CT scan imaging showed that several hypermetabolic lesions in the right elbow joint, left knee joint, and right knee joint. Skin biopsy of the right leg revealed extramedullary infiltration of AML with 99.8% CD33+ expression. Upon skin biopsy, analysis of AML1/ETO fusion gene (by transcript copy number) showed 20,588 copies per 10,000 ABL copies (all values normalized to ABL). In addition, bone marrow examination showed 4% blasts, AML1/ETO copy number of 1,574 per 10,000 ABL, WT1 mutation copy number of 247 per 10,000 ABL, and 94.2% donor chimerism determined by STR (short tandem repeats) analysis. Flow cytometry showed that minimal residual disease (MRD) was 1.4% with 99% CD33+ expression. These results collectively demonstrated molecular relapse in conjunction with bone marrow and extramedullary relapse. After chemotherapy with high dose cytarabine combined with mitoxantrone (MTZ) (Ara-C 2 g/m2/q12 h × 4 d, d1-d4; MTZ 8 mg/m2/d × 2 d, d5-d6), the patient received 3 doses of CD33-CAR NK-92 infusion in December 2017: 3 × 108, 6 × 108, and 1 × 109 cells on days 1, 3, and 5, respectively (Figure 2A). The number of CAR NK-92 cells in the peripheral blood were 4 × 102/ml, 3.6 × 103/ml and 2.9 × 103/ml on days 3, 5, and 8 post-infusion. The patient suffered a moderate fever (38.5°C) on the next day following the second infusion that abated one day later (Figure 2B). Analysis of cytokines showed that interleukin (IL)-6 and IL-10 were drastically increased on day 6 (i.e., following the third cell infusion on day 5), but quickly changed back to normal in 48 hr. However, IL-17A, IL-2, IL-4, TNF-α, and IFN-γ were not changed after cell infusion (Figure 2C), and a grade I cytokine release syndrome (CRS) was observed. The patient experienced relief of pain after the chemotherapy treatment and CAR NK cell infusion. One month following CAR NK cell treatment, BM evaluation showed 0% blasts. MRD was 1.7 × 10-3 with 88.2% CD33+ expression. AML1/ETO and WT1 copy numbers decreased to 308 and 12 per 10,000 ABL, respectively (Figure 2D). The patient presented with right leg pain again at 3 weeks after the CAR NK-92 infusion. She then received local radiotherapy (36 Gy total). Two months later, the patient’s right leg lesions progressed. Four months after CAR NK-92 infusion, the patient presented with hematological relapse with 76% blasts in the BM, and further treatment was not pursued.
Figure 2.
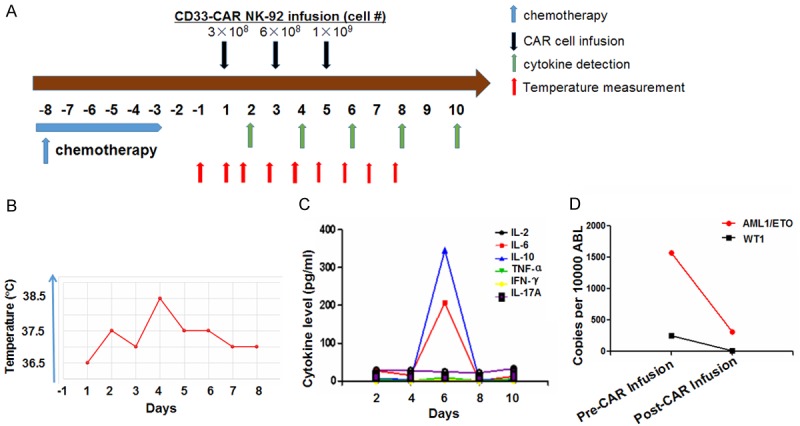
Safety test of CD33-CAR NK-92 cells for the treatment of relapsed and refractory AML. A. Patients receiving chemotherapy from day -8 to -3 were infused with CD33-CAR NK-92 cells on days 1 (first day of cell infusion), 3, and 5. B. Body temperature of patients was measured daily after first CD33-CAR NK-92 cell infusion. C. Cytokine levels in serum were detected by ELISA every other day after first CD33-CAR NK-92 cell infusion (as indicated). D. The copy number of the minimal residual disease biomarkers AML1-ETO and WT1 was assessed by PCR in patients prior to and post CD33-CAR NK-92 cell infusion.
The second case was a 24-year-old male who presented in 2009 with fever and fatigue. He was diagnosed with M4 AML containing chromosomal abnormality t(3;16). He received two courses of HAG chemotherapy (homoharringtonine +Ara-C+G-CSF) leading to complete remission, followed by 10 cycles of consolidation therapy with various regimens. In 2014, the patient relapsed with extramedullary infiltration of leukemia cells in his eyes. After local radiotherapy and high-dose cytarabine chemotherapy, the local lesion improved. In May 2015, his disease relapsed with 8% blasts in the BM, and WT1 was 8,000 per 10,000 ABL. He received multiple chemotherapy treatments, but complete remission was not achieved. In November 2016, the bone marrow examination showed 27% blasts with 20.4% CD33+ expression. After chemotherapy with FLAG regimen (fludarabine 30 mg/m2/d × 5 d, Ara-C 1 g/m2/d × 5 d and G-CSF 300 ug/d d0-d5), he received three doses of irradiated CAR NK-92 cells: 3 × 108, 6 × 108, and 1 × 109 cells on days 1, 3, and 5, respectively. CAR NK-92 cells in the peripheral blood were detected at 7.4 × 103/ml, 7.6 × 103/ml, and 2.9 × 103/ml at days 3, 5, and 8 post-first infusion. Interleukin (IL)-17A and IL-10 were substantially elevated on day 6 (following the third infusion), whereas IL-6, IL-2, IL-4, TNF-α, and IFN-γ were not changed. A moderate fever (38.5°C) was observed after infusion on day 1, but returned to normal by day 2. The patient presented with only grade I CRS. One month following CAR NK-92 infusion, bone marrow examination showed 75% blasts with 49% CD33+ and 45.8% CD123+ expression. In March 2017, two months after CAR NK-92 infusion, the patient received allo-HSCT from an unrelated donor and achieved CR. He unfortunately relapsed 4 months after allo-HSCT and died of grade IV severe GVHD after salvage chemotherapy combined with donor lymphocyte infusion (DLI).
The third case was a 49-year-old woman who presented with fever and fatigue. She was diagnosed with CD33+ M4 AML with normal karyotype and multiple mutations including NRAS(+), NPM1(+), DNMT3A(+), and TET2(+). The patient achieved CR after induction chemotherapy of decitabine combined with HAG (homoharringtonine +Ara-C+G-CSF). One month later, the patient suffered extramedullary recurrence in the skin. The skin lesions disappeared after an intermediate dose of Ara-C+IDA chemotherapy (Ara-C 1.5 g/m2/q12 h × 4 d, d1-d4; IDA 8 mg/m2/d × 2 d, d5-d6). The patient then received Ara-C+etoposide (Ara-C 1.5 g/m2/q12 h × 4 d, d1-d4; etoposide 100 mg/m2/d × 2 d, d5-d6) for consolidation therapy after which the mutations in the above four genes were undetectable. In January 2017, 6 months after diagnosis, the patient relapsed with 37.5% blasts in the BM. Flow cytometry showed 99.9% CD33+ expression. The copy number of WT1 increased to 11,890 per 10,000 ABL. Further genetic analysis revealed that the gene mutations TET2, NPM1, and DNMT3A were present again. The patient was given salvage chemotherapy with Ara-C+MTZ (Ara-C 1.5 g/m2/q12 h × 4 d, d1-d4; MTZ 10 mg/m2/d × 2 d, d5-d6), followed by 3 doses of CAR NK-92 cell infusion: 1 × 109, 3 × 109, and 5 × 109 cells on days 1, 4, and 7, respectively. IL-17A was elevated in the serum, whereas the levels of IL-6, IL-2, IL-4, TNF-α, IL-10, and IFN-γ did not change after cell infusion. The patient suffered from a high fever (40°C) after cell infusion that returned to normal in two days. However, the patient had no response to the treatment. Ten days after CAR NK cell infusion, BM examination showed 79.5% blasts with 94.6% CD33+ expression.
Discussion
Here we demonstrated that CD33-CAR NK-92 cell infusions at doses up to 5 × 109 cells per patient can be safely applied with no substantial adverse effects. We performed infusions in three RR-AML patients, in a series of three increasing doses per patient. We were able to detect CAR NK-92 cells in the blood of patients following infusion. We plan to expand upon the results of this study to design future clinical trials. Our goal is to improve our study design so as to identify patients who can best respond to this type of therapy. One way to design a more robust study involves identifying patients whose tumor cells have high surface expression of a CAR-targeted gene. For example, the CAR NK cells used in this study were specifically engineered to target the CD33 antigen. Patients with higher surface CD33 expression may respond better to CD33-CAR-directed therapy. We also have a concern regarding the irradiation of CAR NK-92 cells. Irradiation of CAR NK-92 cells prior to infusion was required because the parental cell line was originally derived from a lymphoma patient [13]. In our study, we were able to detect the irradiated cells up to one week after cell infusion. However, the long-term survival of irradiated cells in a patient is unlikely. There are other effects of irradiating the CAR NK-92 cells, including decreased cytotoxic potential (data not shown). This presents a challenge when attempting to treat a rapidly progressing cancer such as AML. If irradiated CAR NK-92 cells have decreased cytotoxicity, the applications of this therapy may in fact be better optimized for a more slowly developing disease, such as multiple myeloma (MM). Of note, our recently reported preclinical studies showed that CS1-CAR NK-92 cells are effective against MM in vitro and in vivo [4]. They also appear to have more robust anti-tumor activity against MM compared to that of the CD33-CAR NK-92 cells that were used against AML [4]. In light of decreased cytotoxicity, we also plan to address the above concerns regarding irradiation, so as to maintain the potential use of CAR NK-92 cells for AML treatment. The purpose of irradiation is to prevent excessive expansion of the NK-92 cell line in the patient. However, if we can avoid this complication with an alternative method, irradiation of the cells will no longer be necessary. In lieu of irradiating the cells, we propose incorporating suicide genes (two different ones are preferred) into the NK-92 cell line, allowing control of the life span of the cells. This will allow us to better maintain the viability and cytotoxicity of the CAR NK cells, which will translate into extended anti-tumor activity in the patient.
The mechanism of CAR-mediated therapies (of both T and NK cells) depends on the binding affinity and specificity of a CAR with its associated antigen. This binding interaction facilitates the effector response designed to eliminate malignant cells. Because of this, changes in antigen binding can have a large impact on the effectiveness of CAR-mediated therapy. In our preclinical investigation, CD33-CAR NK-92 cells showed moderate efficacy in vitro. We expect that enhancing CD33-CAR binding will improve the efficacy of the CAR NK cells. For example, a modified CAR that is engineered with higher affinity for CD33 may achieve an improved clinical outcome. It has been reported that variations of the CD33 transcript exist in AML, and these differences may affect the binding affinities to a CAR [14]. In addition, there may be other AML-associated antigens that can be targeted by CAR NK cells. One example is FLT3, which is expressed on the surface of AML blasts, and was the subject of our recent report [15]. A combination CAR-mediated therapy, in which multiple antigens are simultaneously targeted in a patient by their corresponding CARs, may provide a synergistic effect and improved clinical response. Additional research to optimize the targets of CAR NK cells will certainly be valuable to improving future clinical trials.
Another way to improve CAR NK cell clinical trials will involve considering the ideal patient population for this type of therapy. The three patients in this study all had relapsed and refractory AML. Due to the heterogeneous clinical presentation of AML, it is unclear which subsets of patients will best respond to CAR NK cell therapy. For example, CAR NK cells may contribute to a favorable clinical outcome when used in conjunction with chemotherapy to prevent relapse, as opposed to treating relapsed and refractory AML patients. There may also be certain patients who respond to CAR NK cells as a first-line treatment (before chemotherapy). More investigation is required to determine which specific patient factors may influence the response to CAR NK cell therapy.
In summary, here we report a phase I clinical trial of CD33-CAR NK cells for relapsed and refractory AML patients. Before the current work, the safety of CAR NK cell therapy for AML had not been demonstrated. We believe that optimized off-the-shelf CAR NK-92 cells will provide a novel treatment option that is clinically advantageous and much more cost-effective compared to CAR T cells. Our phase I study did not demonstrate obvious clinical efficacy, yet this first-in-man clinical trial showed that this therapy can be safely used in RR-AML patients with high tumor burden. We believe that with future modifications (e.g., optimizing antigen affinity and preclinical efficacy, identifying patients with less aggressive disease, and avoiding the need for irradiation of the cell line), CAR NK-92 therapy may provide a safe, economic, off-the-shelf product for the treatment of AML and/or other cancers.
Acknowledgements
This work was supported in part by research grants from the National Key R&D Program of China (2016YFC0902800, 2016YFC1303403), NSFC Major Project of International Cooperation and Exchanges (81320108023), National Natural Science Foundation of China (81270645, 81000222), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Innovation Capability Development Project of Jiangsu Province (BM2015004), Frontier Clinical Technical Project of the Science and Technology Department of Jiangsu Province (BE2017655), Jiangsu Provincial Medical Talent (ZDRCA2016045), Six Talent Peaks Project in Jiangsu Province (No. SWYY-CXTD-010). This study was also supported by grants from the American Cancer Society (RSG-14-243-01-LIB), the Leukemia & Lymphoma Society (TRP), the NIH (AI129582, NS106170, and CA185301), a grant from the Gabrielle’s Angel Foundation for Cancer Research, and a gift from Greif, Inc (Delaware, Ohio).
Disclosure of conflict of interest
None.
References
- 1.Walter RB, Othus M, Burnett AK, Lowenberg B, Kantarjian HM, Ossenkoppele GJ, Hills RK, Ravandi F, Pabst T, Evans A, Pierce SR, Vekemans MC, Appelbaum FR, Estey EH. Resistance prediction in AML: analysis of 4601 patients from MRC/NCRI, HOVON/SAKK, SWOG and MD Anderson Cancer Center. Leukemia. 2015;29:312–20. doi: 10.1038/leu.2014.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, Danet-Desnoyers G, Scholler J, Grupp SA, June CH, Kalos M. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–54. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, Chen L, Wang Y, Wang H, Yi L, Elder JB, Wang QE, He X, Kaur B, Chiocca EA, Yu J. A combination therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7:27764–77. doi: 10.18632/oncotarget.8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu J, Deng Y, Benson D, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, He X, Devine S, Zhang X, Caligiuri M, Hofmeister C, Yu J. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–27. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. 2017;31:37–54. doi: 10.1016/j.smim.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Han J, Chu J, Chan WK, Zhang J, Wang Y, Cohen JB, Victor A, Meisen WH, Kim SH, Grandi P, Wang QE, He X, Nakano I, Chiocca EA, Glorioso JC 3rd, Kaur B, Caligiuri MA, Yu J. CAR-engineer NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patientderived glioblastoma stem cells. Sci Rep. 2015;5:11483. doi: 10.1038/srep11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147. doi: 10.4161/onci.28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lui E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, Gagea M, Banerjee P, Cai R, Bdaiwi M, Basar R, Muftuoglu M, Li L, Marin D, Wierda W, Keating M, Champlin R, Shpall E, Rezvani K. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–31. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, Ottmann OG, Tonn T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J Cell Mol Med. 2016;20:1287–94. doi: 10.1111/jcmm.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bonig H, Kohl U, Kloess S, Kohler S, Holtgreve-Grez H, Jauch A, Schmidt M, Schubert R, Kuhlcke K, Seifried E, Klingemann HG, Rieger MA, Tonn T, Grez M, Wels WS. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23:330–8. doi: 10.1038/mt.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, Wels W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100:1265–73. [PubMed] [Google Scholar]
- 12.Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, Hattingen E, Harter PN, Mittelbronn M, Tonn T, Steinbach JP, Wels WS. ErbB2/HER2-Specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv375. [DOI] [PubMed] [Google Scholar]
- 13.Gong J, Maki G, Kingemann H. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–8. [PubMed] [Google Scholar]
- 14.Laszlo G, Harrington K, Gudgeon C, Beddoe M, Fitzgibbon M, Ries R, Lamba J, McIntosh M, Meshinchi S, Walter R. Expression and functional characterization of CD33 transcript variants in human acute myeloid leukemia. Oncotarget. 2016;7:43281–94. doi: 10.18632/oncotarget.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Mao H, Zhang J, Chu J, Devine S, Caligiuri M, Yu J. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia. 2017;31:1830–4. doi: 10.1038/leu.2017.147. [DOI] [PMC free article] [PubMed] [Google Scholar]