

Abstract
Triple-negative breast cancer (TNBC) is the most invasive form of breast cancer due to an absence of estrogen (ER), progesterone (PR), and human epidermal growth factor-2 (HER2) receptors on the cell surface. TNBC accounts for approximately 12 to 20 percent of all breast cancer cases. The absence of ER, PR, and HER2 receptors on TNBCs and its ability to develop drug resistance renders it difficult to eradicate or retrogress tumor growth with hormonal therapy and chemotherapy. Triple-negative breast cancer is associated with poorer prognosis, increased chance of relapse, and lower chance of survival. Patients with TNBC have poorer outcome to conventional treatments than patients with other types of breast cancer. Natural killer cell-mediated immunotherapy is a promising therapeutic option for patients with TNBC. Natural killer cells contribute to the immune system by recognizing tumor cells through interactions between ligands on tumor cells and natural killer cell receptors. NK cell function is regulated by a net balance of signals from activating and inhibitory receptors interacting with ligands on target cells. Lectin-like Transcript-1 (LLT1, CLEC2D, OCIL) is a ligand that interacts with NK cell receptor NKRP1A (CD161) and inhibits NK cell activation. In this study, we have identified expression of LLT1 on TNBC cell lines MDA-MB-231 and MDA-MB-436 through flow cytometry, western blot, and confocal microscopy. We have demonstrated that blocking LLT1 on TNBCs with antibodies disrupts interaction with NKRP1A and enhances lysis of TNBCs by primary natural killer cells. We have also shown that a gene knockdown of LLT1 decreases cell surface expression of LLT1 on TNBCs and increases NK cell-mediated lysis of these TNBCs. The results suggest that LLT1 on TNBCs function as a method of evasion from immunosurveillance by NK cells. Blocking LLT1-NKRP1A interaction activates lysis by NK cells and will potentially open a new immunotherapeutic strategy for treatment of TNBC.
Keywords: LLT1, triple-negative breast cancer, breast cancer, natural killer cells, NKRP1A (CD161), TNBC, LLT1-NKRP1A interaction, lectin-like transcript-1
Introduction
Breast cancer is the second most common type of cancer and the second leading cause of death of all cancers for women in the United States [1]. It is estimated that 252,710 new cases of breast cancer will be diagnosed in women and 40,610 women will die from breast cancer in 2017 [1]. Among the subtypes of breast cancers, triple-negative breast cancer (TNBC) is considered the most invasive subtype as patients with TNBC have poorer outcome and response to conventional chemotherapeutic treatments [2,3]. TNBC is a subtype of breast cancer that is characterized by the absence or low expression of estrogen (ER), progesterone (PR), and human epidermal growth factor-2 receptors (HER2) on the surface of these tumor cells [4]. Breast cancer cells with the triple-negative phenotype (ER- PR- HER2-) have limited response to endocrine therapy, trastuzumab, and chemotherapy due to the lack of these three receptors [2,4,5]. Although chemotherapy is still a standard treatment for TNBC with doxorubicin or taxane favored, patients in this group consistently have worse outcome after chemotherapy treatment in comparison with groups of patients diagnosed with other subtypes [5]. TNBC is estimated to account for approximately 12-20% of all cancer cases and has the poorest prognosis of all subtypes [5,6]. Considering the low success with standard chemotherapy and endocrine therapy, immunotherapy has emerged in further development as a novel treatment for this aggressive form of breast cancer such as immune checkpoint inhibitors and monoclonal antibodies [7].
Utilizing natural killer (NK) cells to target tumor cells has shown potential as an immunotherapeutic option in different types of cancers such as multiple myeloma and prostate cancer [8]. NK cells are part of the innate lymphoid cell family and are known to secrete interferon-γ (IFN-γ) when stimulated by specific interactions of ligands on target cells with NK cell receptors [9]. NK cell function is triggered and regulated by a balance of inhibitory and activating NK receptors interacting with ligands on target cells [10]. NK cells contribute to the immune system by recognizing tumor and infected cells through either deficiency of major histocompatibility complex (MHC) class I receptors or an increase of activating ligands [9]. NK cell inhibitory receptors such as killer cell immunoglobin-like receptors (KIRs), TIGIT, Tactile (CD96), and PD1 (CD279) recognize ligands on target cells and inhibit cytolytic lysis against the targeted cell [10,11]. In contrast, ligands on target cells binding to NK cell activating receptors such as FcγRIIIA (CD16a), NKp30, NKp44, NKp46, natural killer gene 2D (NKG2D), and CD2 subset 1 (CS1, CRACC) can overcome inhibitory signals and induce the NK cell to lyse the target cells [9-12].
There is an increasing interest in developing targeted therapies for patients with TNBC. Current chemotherapeutic treatments for this group of patients include anthracyclines such as doxorubicin, ixabepilone, taxanes, and platinum agents such as carboplatin and cisplatin [13-15]. Although it has been supported by various studies on neoadjuvant chemotherapy that TNBCs have increased sensitivity and susceptibility to chemotherapy compared with other subtypes, chemotherapy still brings adverse side effects from physical and immune complications to long-term cognitive impairment with some studies reporting executive function impairment [13,16-20]. Developing immune targeted therapies by stimulating natural killer cells to lyse TNBCs with monoclonal antibodies targeting specific ligands could open a novel strategy to treating TNBC with fewer and less adverse side effects [21].
One particular ligand of interest for NK cell targeting on TNBCs is Lectin-Like Transcript 1 (LLT1, CLEC2D, OCIL). LLT1 is part of the C-type lectin-like receptor superfamily (CTLR) which is encoded by CLEC2 genes within the human natural killer gene complex [22]. LLT1 is expressed on lymphocytes such as B cells, NK cells, and T cells as well as on activated dendritic cells [22,23]. Crystallography has revealed that LLT1 forms a homodimer at its cell surface [22,24]. This highly glycosylated homodimer enables LLT1 to serve as a ligand for the NKRP1A receptor [25,26]. At the gene expression, northern blot analysis conducted by Germain et al. have supported that LLT1 has five alternatively spliced variants (excluding isoform 3 which is a RNA decay product) of the CLEC2D gene [27]. Isoform 1 that codes for LLT1 was identified as a surface protein that interacts with NKRP1A receptor [27].
The receptor NKRP1A is encoded by a single gene KLRB1 and is expressed on NK cells, CD4+ and CD8+ T cells, invariant NKT cells, γδ-TCR+ T cells, and a subset of CD3+ thymocytes [26,28]. Studies have found that NKRP1A expression contribute to the role of differentiation of lymphocytes and can be acquired at the surface of T cells and NK cells by cytokines [29]. It was also shown that NKRP1A was expressed on both dendritic cells and during monocyte differentiation from both the bone marrow and precursors in the thymus [29]. From the same study by Poggi et al., functional analysis has shown that antigens binding to NKRP1A leads to an increase in intracellular calcium in human monocytes and dendritic cells and production of interleukins IL-1β and IL-12 by non-activated monocytes and dendritic cells [29]. The induced production of IL-12 further allows an upregulation of NKRP1A expression in human NK cells which can play a role in regulating activation of NK cells [30]. Interaction between LLT1 on target cells and natural killer cell receptor NKRP1A leads to inhibition of NK-cell mediated cytolytic targeting [26]. It was found that cross-linking of LLT1 with monoclonal antibodies induces production of interferon-gamma (IFN-γ) by natural killer cells through the ERK signaling pathway [31,32]. The role of interaction between LLT1 and NKRP1A in modulating immune responses was observed when upregulation of LLT1 was induced by pathogens and expression of NKRP1A was found on NK, Th1, and Th17 cells [33]. LLT1 expression on B cells inhibits NK cell function and cross-linking of NKRP1A with CD3 on T cells increases secretion of IL-17 [33]. Furthermore, overexpression of LLT1 was observed on prostate cancer cells and leads to inhibition of NK cell mediated cytolytic killing against these prostate cancer cells [8].
For this study, we have observed an expression of LLT1 on TNBC cell lines MDA-MB-231 and MDA-MB-436 through flow cytometry, western blot, and confocal microscopy. Blocking LLT1 on the cell surface of TNBCs by anti-human LLT1 antibodies have increased cytolytic targeting by primary natural killer cells isolated from peripheral blood mononuclear cells (PBMCs). Knockdown of the gene LLT1 on MDA-MB-436 by transfection with small interference RNAs (siRNA) also has increased cytolytic killing by primary natural killer cells. Hence, we conclude that blocking LLT1-NKRP1A interaction and decreasing cell surface expression of LLT1 increases susceptibility of the TNBC cells to NK-cell mediated cytolytic killing and will potentially introduce a novel immunotherapeutic strategy for patients diagnosed with invasive and difficult-to-treat triple-negative breast cancer.
Materials and methods
Cell culturing
All cell lines used were acquired from American Type Culture Collection (ATCC). MDA-MB-231 and MDA-MB-436 were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Life Technologies, Carlsbad, CA) with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 10 ml of penicillin-streptomycin. MCF10A was cultured in Medium 171 (Life Technologies Corporation, Carlsbad, CA) supplemented with Mammary Epithelial Growth Supplement (Gibco Life Technologies Corporation, Carlsbad, CA). Cells were grown to 80 to 90% confluence on sterile culture flasks or well plates in a 37°C 5% CO2 incubator and then passaged by aspirating the cells with 1× PBS with ethylene-diamine-tetracetic acid (EDTA) or Trypsin-EDTA (for MCF10A) for further growth or use.
Isolation of human peripheral blood mononuclear cells (PBMCs) and primary natural killer cells
Peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-treated whole blood samples by Histopaque-1077 (Sigma-Aldrich, Saint Louis, MO) density centrifugation. Whole blood samples were collected from healthy individuals with consent and approval by the University of North Texas Health Science Center Institution Review Board. Primary natural killer cells were isolated from PBMCs by using a NK cell isolation kit (Miltenyi Biotec, San Diego, CA) which utilizes a column in the magnetic field of a MACS Miltenyi Biotec Separator to collect the flow-through fluid containing NK cells. Primary natural killer cells were cultured in 4+ Roswell Park Memorial Institute complete medium (RPMI medium 1640, Gibco Life Technologies, Carlsbad, CA) with 15% FBS.
Flow cytometry analysis
Surface expression of LLT1 was detected by flow cytometry analysis. Cells were grown to 80 to 90% confluence on sterile culture flasks (Thermo Scientific, Rockford, IL) or well plates (Corning, Inc., Corning, NY). Cells were aspirated with 1× PBS-EDTA for collection and count. Cells were checked for viability by staining with Trypan Blue and counted on a hemocytometer. All cell lines were incubated with Human Fc fragment (Rockland, Inc.) and then were stained with anti-human LLT1-PE antibodies (R&D Systems, Clone #402659, Minneapolis, MN) or isotype control mouse IgG1-PE antibodies (Biolegend, Clone #MOPC-21, San Diego, CA). Cells were then detected and analyzed by flow cytometry using the Beckman Coulter Cytomics FC500 Flow Cytometer (University of North Texas Health Science Center Flow Cytometry Core Facility, Fort Worth, TX). FlowJo v10 software (FlowJo, LLC, Ashland, OR) was used to analyze data collected from flow cytometry.
Transfection of cells with small interference RNAs (siRNAs) and confirmation of knockdown by flow analysis
MDA-MB-436 were grown to 80% to 90% confluence on sterile 96-well plates. SMARTpool: ON-TARGETplus CLEC2D (LLT1) 5 nmol small interference RNAs (siRNAs) and ON-TARGETplus non-targeting 5 nmol siRNA #2 (GE Healthcare Dharmacon, Inc., Lafayette, CO) were diluted from a 20 µM stock to a 5 µM working stock with 1× siRNA buffer (diluted from 5× siRNA buffer, GE Healthcare Dharmacon, Inc., Lafayette, CO). Transfection optimization conditions were determined by GE Healthcare Dhar macon. Manufacturer protocol on transfection was followed (Dharmacon). Cells were transfected with either CLEC2D siRNA or non-targeting siRNA for 63 hours in the 37°C 5% CO2 incubator. Final concentration of CLEC2D siRNA or non-targeting siRNA was 25 nM. After 63 hours of transfection, confirmation of knockdown of cell surface LLT1 on MDA-MB-436 was confirmed by flow cytometry using anti-human LLT1-PE antibodies or isotype control mouse IgG1-PE antibodies.
Immunofluorescence confocal microscopy
MDA-MB-231 and MDA-MB-436 were cultured on coverslips overnight and fixed with 2% paraformaldehyde. All samples were incubated with blocking solution containing human Fc fragment (R&D Systems, Minneapolis, MN) to prevent nonspecific binding. Cells were then stained with mouse anti-human LLT1-PE antibody (R&D Systems, Minneapolis, MN) overnight at 4°C. After washing in PBS, the coverslips were mounted on slides using Aqua-Mount (Thermo Scientific, Rockford, IL), and imaged on the Zeiss LSM 510 Confocal Laster Microscope using the 40×, 1.2 NA, 0.28 WD (water), C-Apochromat objective utilizing 488 nm wavelength.
51Cr release assay
MDA-MB-231, MDA-MB-436, MCF10A, and siRNA transfected MDA-MB-436 cells were incubated with 51Cr in a 37°C 5% CO2 incubator for 90 minutes. After incubation, 51Cr-labeled cells were treated with either 1 µg of unconjugated goat anti-human CLEC2D antibody (Thermo Fisher Scientific, Rockford, IL) or goat IgG isotype control antibody (Abcam, Cambridge, MA).
Primary NK cells were isolated from peripheral blood mononuclear cells from healthy individuals as previously stated. Primary NK cells were incubated with human Fc fragment and then were co-incubated with 51Cr-labeled target cells at effector-to-target cell ratios of 25:1, 5:1, and 1:1 for 3.5 hours in the 37°C 5% CO2 incubator. Supernatants were collected and quantified with a scintillation counter. Percent specific lysis were then calculated.
Results
Triple-negative breast cancer cells express LLT1
Expression of LLT1 at the cell surface of TNBCs were identified through flow cytometry analysis. Non-tumorigenic breast cell line MCF10A and TNBC cell lines MDA-MB-231 and MDA-MB-436 were treated with human Fc fragment, then were subsequently stained with either anti-human LLT1-PE antibodies or isotype control IgG1-PE antibodies. Two TNBC cell lines’ cell surface LLT1 expression was compared to non-tumorigenic breast cell line MCF10A to determine if there is greater LLT1 expression on TNBCs than normal breast cells. In one representative, TNBC cell line MDA-MB-231 displayed the highest cell surface expression of LLT1 at median fluorescence intensity ratio (MFIR) of 1.84 with 13.3 percent of cells positive for cell surface LLT1 expression (Figure 1A). TNBC cell line MDA-MB-436 also displayed cell surface expression of LLT1 at MFIR of 1.36 with 4.4 percent of cells positive for LLT1 (Figure 1A). Both TNBC cell lines have significantly higher expression of LLT1 based on MFIR and percent of cells positive for LLT1 compared to normal breast cell line MCF10A.
Figure 1.
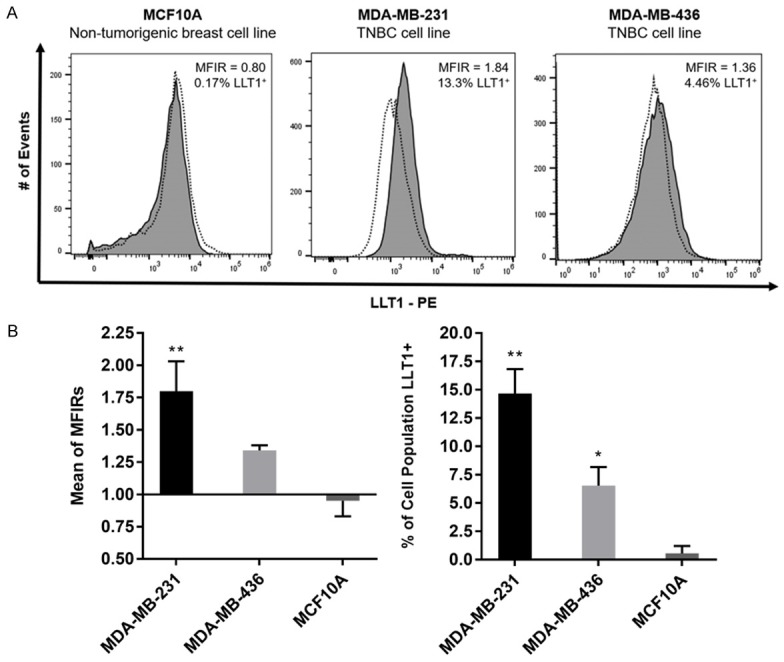
Triple-negative breast cancer cell lines display a higher expression of LLT1 at the cell surface than normal breast cells. A. Cell surface expression of LLT1 on TNBC cell lines MDA-MB-231 and MDA-MB-436 and non-tumorigenic breast cell line MCF10A was determined by flow cytometry analysis. Dotted lines (white shade) represents cells stained with isotype control IgG1-PE antibodies and solid line (dark gray shade) represents cells stained with anti-LLT1-PE antibodies. B. Median fluorescence intensity ratios (MFIRs) and percentage of cells displaying positive expression of LLT1 from 3 independent experiments were averaged. *P < 0.05 & **P < 0.01, One-way ANOVA with Dunnett’s multiple comparisons post-hoc.
To confirm consistency in detecting cell surface LLT1 expression, 3 independent experiments of flow cytometry analysis were performed for each cell line tested (Figure 1B). MFIRs and percentage of cells that were positive for LLT1 expression from these 3 independent experiments were averaged. We have observed that both TNBC cell lines MDA-MB-231 and MDA-MB-436 have significantly greater expression of cell surface LLT1 than normal breast cell line MCF10A (Figure 1B). MDA-MB-231 mean of MFIRs of 1.80 and its mean percent LLT1+ cells of 14.67% is statistically significantly higher (Mean of MFIRs and % LLT1+ **P < 0.01, Figure 1B) than non-tumorigenic breast cell line MCF10A’s respective values of 0.95 and 0.56% LLT1+ cells. MDA-MB-436 does show a higher expression of LLT1 based on mean of MFIRs of 1.34 and mean percent LLT1+ cells of 6.54% than MCF10A, but still much lower than MDA-MB-231 respective values. Hence, flow cytometry analysis under the same culture conditions and stained with the same anti-LLT1-PE antibodies have consistently shown that TNBCs show higher cell surface expression of LLT1 than non-tumorigenic breast cell line MCF10A. These results demonstrate that LLT1 may serve as a possible target of interest.
To further confirm expression of LLT1 on TNBCs, we performed immunofluorescent staining on TNBC cell lines MDA-MB-231 and MDA-MB-436. Both TNBCs were treated with human Fc fragment and then were stained with anti-human LLT1-PE antibodies. MDA-MB-231 also showed higher expression of LLT1 at the cell surface than MDA-MB-436 (Figure 2). These results suggest that expression of LLT1 as an inhibitory ligand on TNBCs could play a role in the TNBC cells evading NK cell-mediated lysis. Expression of LLT1 on TNBCs shown by both flow cytometry analysis and immunofluorescent staining leads to LLT1 as a target of interest in blocking LLT1-NKRP1A interaction between TNBCs and primary NK cells.
Figure 2.
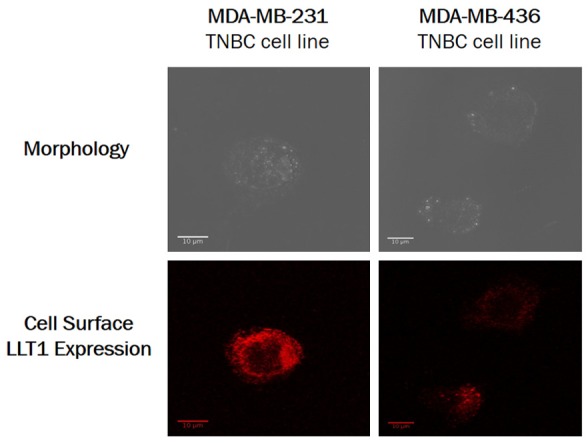
Triple-negative breast cancer cells expressed LLT1 at the cell surface. Triple-negative breast cancer cells MDA-MB-231 and MDA-MB-436 were fixed, blocked with human Fc fragment, and stained with anti-human LLT1-PE antibody. The cells were examined with a Zeiss LSM 510 Confocal Laster Microscope at 40× objective. Scale bar is 10 µm.
Blocking LLT1 with antibodies at the cell surface of TNBCs increased NK cell-mediated lysis against these tumor cells
To assess the function of LLT1, TNBC cell lines MDA-MB-231 and MDA-MB-436 and non-tumorigenic cell line MCF10A were radiolabeled with 51Cr and treated with anti-human LLT1 antibodies or isotype IgG antibodies. The labeled cells blocked with its antibodies were co-incubated with primary NK cells for 3.5 hours at effector-to-target ratios (E:T) of 25:1, 5:1, and 1:1. Cytolytic activity, percent of cells lysed by primary NK cells, were subsequently quantified. We used primary NK cells isolated from peripheral blood mononuclear cells derived from whole blood. Fc receptors on primary NK cells were blocked with human Fc fragment to prevent antibody-dependent cell-mediated cytotoxicity (ADCC) from occurring.
We have performed a treatment of TNBC cell lines MDA-MB-231 with anti-LLT1 antibodies at four concentrations to determine the concentration of antibody to use for targeting LLT1 on TNBCs (Figure 3). Based on the effects of lysis based on the different concentrations of anti-LLT1 antibody, we have then proceeded with targeting LLT1 on TNBC cell lines with treatment of 1 µg of anti-LLT1 antibody (Figure 4). Blocking LLT1 on both TNBCs MDA-MB-231 and MDA-MB-436 enhanced specific lysis by NK cells (Figure 4B, 4C). MDA-MB-231 and MDA-MB-436 had increased specific lysis by NK cells when LLT1 was blocked than cells treated with isotype IgG antibodies.
Figure 3.
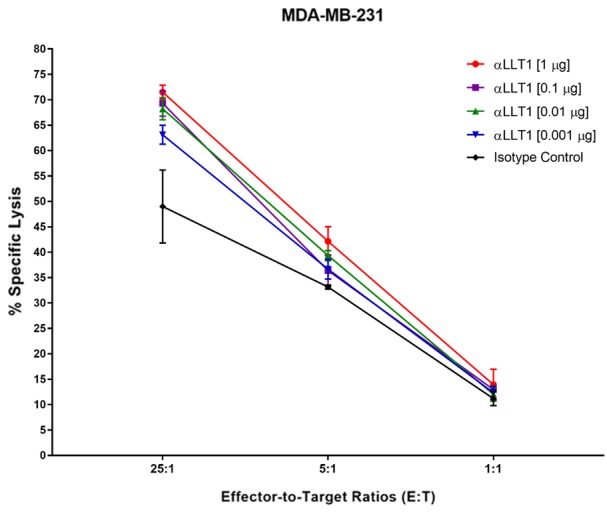
Anti-LLT1 antibody dose-dependent treatment on TNBC MDA-MB-231 demonstrates enhanced killing by primary NK cells. TNBC cell line MDA-MB-231 was treated with either anti-human LLT1 antibodies (αLLT1) or isotype control antibodies. MDA-MB-231 cells were treated with anti-LLT1 antibodies at four different concentrations. There is greatest lysis of TNBCs when treated with anti-LLT1 antibodies at 1 µg per well compared to other concentrations and isotype IgG control (P = 0.08, student paired t-test). Cells were labeled with 51Cr and then were co-incubated with primary NK cells isolated from PBMCs derived from whole blood of healthy volunteers at effector-to-target ratios (NK-to-5000 TNBCs) of 25:1, 5:1, and 1:1 for 3.5 hours. Specific lysis of labeled cells was subsequently quantified and calculated. This assay was performed in triplicates and error bars indicate standard deviations.
Figure 4.
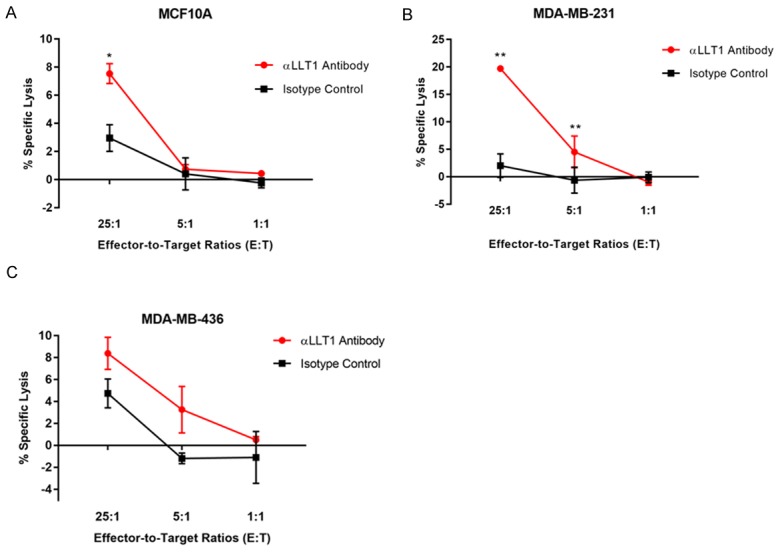
Blocking LLT1 on triple-negative breast cancer cells increased lysis of TNBCs by NK cells. A-C. Blocking LLT1 at the cell surface of TNBC cell lines MDA-MB-231 and MDA-MB-436 enhanced lysis of these cells by primary NK cells. MCF10A, MDA-MB-231, and MDA-MB-436 were blocked with anti-human LLT1 antibodies (αLLT1 in legend) or goat IgG isotype control antibodies (isotype control in legend). Cells were labeled with 51Cr and then were co-incubated with primary NK cells at effector-to-target (E:T) ratios of 25:1, 5:1, and 1:1 for 3.5 hours. Specific lysis of cells was subsequently quantified. These assays were performed in triplicates and error bars indicate standard deviations. *P < 0.05 & **P < 0.01, Student paired t-test compared to isotype control.
At 25:1 E:T ratio, there was a statistically significant difference in percent specific lysis of MDA-MB-231 cells between cells treated with anti-LLT1 antibodies compared to cells treated with IgG isotype antibody and cells not treated with antibodies (Figure 4B). At 25:1 ratio, 19.71% of MDA-MB-231 treated with anti-LLT1 antibodies were killed by primary NK cells compared to 2.02% of MDA-MB-231 cells treated with IgG isotype antibody. There was a distinct difference between the percent specific lysis of MDA-MB-231 cells treated with anti-LLT1 antibodies compared to non-tumorigenic breast MCF10A cells treated with the same anti-LLT1 antibody at the 25:1 E:T ratio. At 25:1 ratio, 7.54% of MCF10A cells treated with anti-LLT1 antibody were killed by primary NK cells compared to 2.96% of MCF10A cells treated with IgG isotype antibody. At 5:1 E:T ratio, there was a statistically significant difference in percent specific lysis of MDA-MB-231 cells treated with anti-LLT1 antibodies compared to treatment with isotype antibodies.
We also have tested the effects of blocking LLT1-NKRP1A interaction by treating TNBC cell line MDA-MB-436 with anti-LLT1 antibodies (Figure 4C). There was a lower percentage of MDA-MB-436 cells killed by NK cells than MDA-MB-231 cells at all the E:T ratios. The lower percentage of MDA-MB-436 cells killed can be attributed to the lower expression of cell surface LLT1 on this cell line in contrast to MDA-MB-231 LLT1 expression as shown in flow cytometry analysis. At 25:1 E:T ratio, 8.39% of MDA-MB-436 cells treated with anti-LLT1 antibodies were killed while 4.75% of cells treated with isotype antibodies were killed.
In summary, treating MDA-MB-231 and MDA-MB-436 cells with anti-LLT1 antibodies allowed an increase of killing by primary NK cells. Furthermore, MDA-MB-231 cells treated with anti-LLT1 antibodies had a greater percent of cells killed compared to non-tumorigenic breast cell line MCF10A treated with anti-LLT1 antibodies. The greater percentage of MDA-MB-231 cells killed when targeting LLT1 with antibodies than MCF10A cells was consistent with flow cytometry analysis demonstrating that MDA-MB-231 cells had a statistically significant higher expression of cell surface LLT1 in contrast to LLT1 cell surface expression on MCF10A. The lower percent of MCF10A being killed when targeting LLT1 with anti-LLT1 antibodies supports that LLT1 may serve as a possible target that would favor killing TNBCs while minimizing killing healthy breast cells. Blocking LLT1 with antibodies on TNBC cells increases cytolytic targeting by primary NK cells. Hence, blocking LLT1 interaction with NK cell receptor NKRP1A suppresses inhibitory signal transduction in NK cells. These results suggest that the function of LLT1 on TNBC cells inhibits NK cell activation of cytolytic function against TNBCs expressing LLT1 due to the LLT1-NKRP1A interaction. Expression of LLT1 on triple-negative breast cancer cells serves a role in evading immunosurveillance by NK cells.
Gene knockdown of LLT1 decreased cell surface expression of LLT1 on TNBCs and enhances NK cell-mediated lysis of TNBCs
To further assess the function of LLT1 as an inhibitory ligand on TNBC cells, we have performed a gene knockdown of LLT1 on the MDA-MB-436 cell line to decrease the expression of LLT1 at the cell surface. TNBC MDA-MB-436 was transfected with lipid-mediated small interference RNAs (siRNA) that targets the LLT1 gene for 63 hours. As a negative control, TNBC cell line MDA-MB-436 was transfected with non-targeting siRNAs (scramble siRNA) which does not target the LLT1 gene. Knockdown of cell surface LLT1 was confirmed by flow cytometry analysis after 63 hours of transfection before testing the transfected cells for killing by primary NK cells. MDA-MB-436 LLT1 siRNA-transfected or scramble siRNA-transfected cells were blocked with human Fc fragment and stained with anti-human LLT1-PE antibodies. MDA-MB-436 cells transfected with LLT1 siRNA showed negligible expression of LLT1 at the cell surface (Figure 5A, MFIR 0.31) versus MDA-MB-436 cells transfected with scramble siRNA (Figure 5A, MFIR 1.09). After confirming de creased expression of LLT1 at the cell surface of MDA-MB-436, we proceeded with testing that knockdown of LLT1 enhances natural killer cell-mediated lysis of these transfected triple-negative breast cancer cells.
Figure 5.
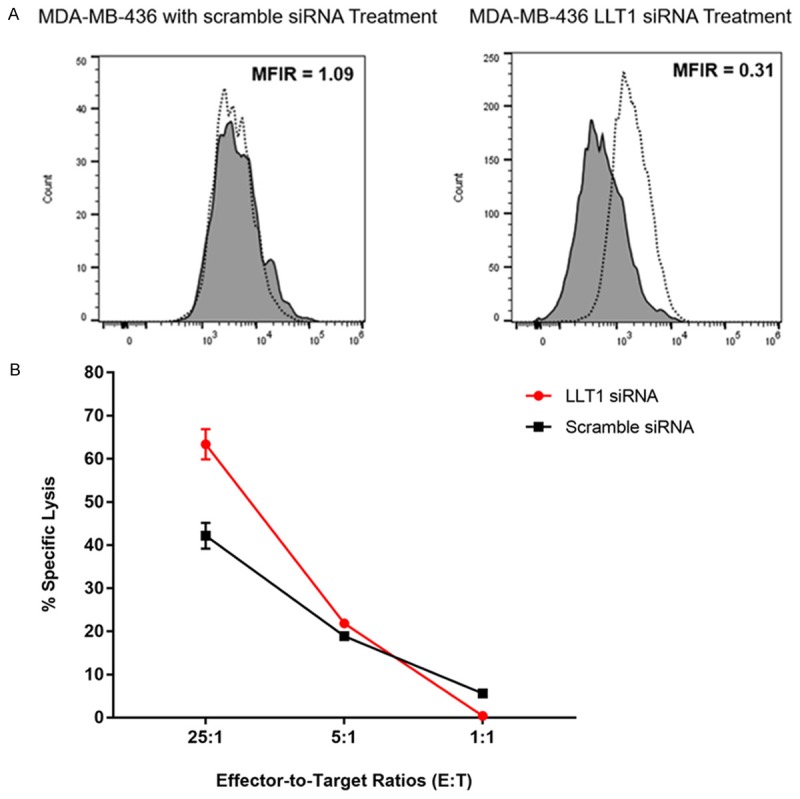
Knockdown of LLT1 at the cell surface of triple-negative breast cancer cells increased lysis of TNBCs by NK cells. A. TNBC cell line MDA-MB-436 was transfected for a period of 63 hours with scramble siRNA control or siRNA targeting LLT1 gene. Knockdown of LLT1 at the cell surface of MDA-MB-436 was confirmed by flow cytometry which displayed negligible expression of LLT1 at the cell surface (MFIR < 1.00). MFIR is median fluorescence intensity ratio. B. Transfected MDA-MB-436 cells with confirmed LLT1 knockdown at the cell surface were labeled with 51Cr and co-incubated with primary NK cells at E:T ratios of 25:1, 5:1, and 1:1 for 3.5 hours. Specific lysis of transfected MDA-MB-436 cells killed by NK cells was quantified. This assay was performed in triplicates and error bars represent standard deviations. P = 0.07 at 25:1 ratio, Student paired t-test compared to isotype control.
Upon confirmation of knockdown, to assess that knockdown of LLT1 increases killing of TNBC cells, MDA-MB-436 transfected with either LLT1 siRNA or scramble siRNA were labeled with 51Cr and then were co-incubated with primary NK cells for 3.5 hours MDA-MB-436 with confirmed knockdown of LLT1 had a higher percent specific lysis at both 25:1 and 5:1 E:T ratios in contrast to its scramble siRNA transfected cells (Figure 5B). At 25:1 E:T ratio, 63.38% of MDA-MB-436 LLT1 siRNA-transfected cells were killed by NK cells compared to 42.18% of MDA-MB-436 scramble siRNA-transfected cells killed (Figure 5B, P = 0.07 compared to scramble siRNA control). At 5:1 E:T ratio, 21.83% of MDA-MB-436 LLT1 siRNA-transfected were killed compared to 18.87% of scramble siRNA-transfected cells.
Hence, transfecting MDA-MB-436 cells with siRNA targeting the LLT1 gene decreases expression of LLT1 at the cell surface. Decreasing the expression of cell surface LLT1 prevents interaction of LLT1 with NKRP1A on primary NK cells. These results indicate that knockdown of LLT1 at the cell surface of TNBC cells enhances specific lysis by NK cells. Disrupting the LLT1-NKRP1A interaction prevents inhibitory signal transduction to NK cells thus favoring activation of natural killer cells to lyse these TNBC cells. These results further confirm that the function of LLT1 on the cell surface of TNBC cells allows evasion of immune system recognition and cytolytic targeting by NK cells.
Discussion
Utilizing NK cells to target tumor cells has shown great potential in immunotherapy in treating several types of cancers [8-10]. NK cells employ its ability to recognize and lyse tumor cells through its receptor interacting with ligands on the surface of tumor cells [45]. Interaction between NK cell receptors and ligands on tumor cells sends transducing signals that either inhibits or activates NK cell cytolytic function [9,10,45]. As a result of ligand-receptor interactions, NK cell functions in antitumor immune response include cytokine secretion, inducing cytotoxicity to lyse tumor cells, and mediating anti-dependent cell-mediated cytotoxicity (ADCC) with its CD16 (FcγRIIIa) receptors [9,46]. Our study characterized the role of expression of LLT1 on TNBCs interacting with NK cell receptor NKRP1A. We have demonstrated that LLT1-NKRP1A interaction sends an inhibitory signal to NK cells which suppresses cytolytic activity against TNBCs. Blocking the interaction of LLT1 with NKRP1A with anti-LLT1 antibodies and gene knockdown of LLT1 enhances activation of NK cells to lyse TNBCs. Interestingly, we have determined that the function of LLT1 on TNBCs is similar to the role of LLT1 expressed on activated B cells and dendritic cells in inhibiting NK cell function [22,23,33].
The role of LLT1-NKRP1A interaction centers around modulating immune functions and responses [26]. There have been numerous studies that show the expression and function of LLT1 on immune cells such as germinal center B cells, plasmacytoid dendritic cells, NK cells, and T cells [33,47,51,52]. Immune cells express and upregulate LLT1 to enhance its own function of targeting pathogens, presenting antigens to other cells, secrete cytokines, or enhance its receptor interactions with ligands on immune cells with intent for co-stimulation [31,33,47,52]. In addition, certain immune cells have anti-tumor effects such as CD8+ T cells and NK cells by recognizing tumor-associated antigens presented on the major histocompatibility complex I of tumor cells [53]. It may sound convenient for immune systems to recognize every tumor cell by its tumor-associated antigens being presented, but this is not always the case. The cancer immunoediting process describes how tumor cells can develop resistance to the immune system by acquiring mutations, ligands, receptors, and producing cytokines that will prevent immune systems from recognizing them [54]. For the case of the interaction between NK cells and tumor cells, the expression of inhibitory ligands such as LLT1 on cancer cells allows cancer cells to escape immune surveillance.
A study conducted by Llibre et al. showed that LLT1 was highly expressed on germinal center B cells in tonsillar tissue, spleen, lymph nodes, and Peyer’s patches [47]. The same study observed that NKRP1A was expressed on activated follicular dendritic cells and determined that NKRP1A on follicular dendritic cells interacts with LLT1 on germinal center B cells which allows activation and maturation of B cells within the germinal center [47]. Mathew et al. demonstrated that the expression of LLT1 on NK cells induces interferon-γ (IFN-γ) production by NK cells without inducing activation of cytolytic function of NK cells which suggests that induction of IFN-γ production by LLT1 upregulation plays a role in early innate immune response to pathogens [31-33]. LLT1 expression on immune cells and its overall function in modulating immune response may provide an advantage for tumor cells expressing LLT1 in evading from immunosurveillance. There have been studies showing expression of LLT1 on tumor cells including germinal center-derived B-cell non-Hodgkin’s lymphomas, glioblastoma, and prostate cancer [8,48,49]. Germain et al. showed that LLT1 expressed on germinal center-derived B-cell lymphomas functions as an inhibitory ligand interacting with NKRP1A and blocking this LLT1-NKRP1A interaction increased NK cell degranulation, increased IFN-γ production, and enhanced NK cell-mediated lysis [48]. Roth et al. reported that LLT1 expressed on malignant glioma cells counteracted NK cell targeting and that transforming growth factor-β upregulated LLT1 expression [49]. Mathew et al. showed that overexpression of LLT1 on prostate cancer cells inhibits NK cell-mediated lysis against prostate cancer cells [8]. These findings support that LLT1 on tumor cells function as an inhibitory ligand against NK cell activation and serves as a suitable candidate to target with NK cell-mediated immunotherapy such as antibodies.
We have observed expression of LLT1 at the cell surface of TNBCs by flow cytometry analysis and immunofluorescent confocal studies. We have tested for LLT1 expression on two TNBC cell lines and compared its expression to non-tumorigenic breast cell line MCF10A. In order to determine if LLT1 could serve a possible candidate for targeting with antibodies, comparing the expression of LLT1 on TNBCs to MCF10A was needed to minimize off-target effects against normal breast cells. Based on flow cytometry analysis of three independent experiments under the same culturing conditions and using the same anti-LLT1 antibody, the non-tumorigenic breast cells MCF10A expressed significantly low levels of LLT1 than all the breast cancer cell lines tested.
Based on our findings, we are interested in determining the function of LLT1 expressed on TNBCs. Therefore, we have used an anti-LLT1 antibody specific for binding to LLT1 on the TNBC cell lines in a chromium-release cytotoxicity with primary NK cells to determine the effects of blocking LLT1-NKRP1A interaction. Based on the previous studies detailing the function of the LLT1-NKRP1A interaction, we have hypothesized that LLT1 functions as an inhibitory ligand on TNBCs and that blocking LLT1-NKRP1A interaction will enhance killing of TNBCs. We have observed that anti-LLT1 antibodies binding to LLT1 on TNBC cell lines have prevented LLT1 from interacting with NKRP1A on primary NK cells. Utilizing antibodies to block interaction between LLT1 and NKRP1A has increased lysis of TNBC cell lines MDA-MB-231 and MDA-MB-436. Furthermore, targeting LLT1 on TNBCs has shown greater killing than normal breast cells MCF10A being treated with anti-LLT1 antibodies. Our results have demonstrated that the lower killing of normal breast cells and greater killing of TNBCs indicate that using LLT1 as a target can be distinguished between normal breast cells versus TNBCs due to this difference in expression.
We have also utilized another method of blocking LLT1-NKRP1A interaction by using small interference RNA targeting the LLT1 gene. We have determined that decrease of LLT1 cell surface expression have prevented LLT1 from interacting with NKRP1A on primary NK cells. By preventing this interaction, we have observed an increase in killing of TNBCs. Using siRNA-mediated downregulation of LLT1 has been shown to be successful in NK-mediated killing of glioma cells [49]. Likewise, when we used siRNA-mediated downregulation (CLEC2D siRNA purchased from Dharmacon), there was an increase in killing of TNBC MDA-MB-436 LLT1-siRNA-transfected cells by 21% compared to the same cell line treated with scramble siRNA with LLT1 expressed (Figure 5B).
Hence, our results have tested for the expression and function of LLT1 on TNBCs. We have determined that there was greater LLT1 expression on TNBC cell lines than normal breast cells. The significant difference in cell surface expression of LLT1 between TNBCs and normal breast cells indicates that LLT1 can be used to target TNBCs while sparing majority of normal breast cells based on our findings. This difference in cell surface LLT1 expression between TNBCs and normal breast cells has been tested when TNBCs and normal breast cells were treated with anti-LLT1 antibodies. Our findings have demonstrated that cell surface LLT1 expression on TNBCs serves as an inhibitory ligand that suppresses NK cell activation when it interacts with NKRP1A. We have also shown that blocking LLT1-NKRP1A interaction by two methods, with anti-LLT1 antibodies and siRNA, have enhanced killing of TNBCs by primary NK cells. Furthermore, much like what other studies have shown when LLT1 was expressed on other types of cancers, TNBCs utilize LLT1 as one of its mechanisms in evading immunosurveillance from NK cells. Therefore, targeting LLT1 on TNBCs with monoclonal antibodies may introduce another strategy for patients diagnosed with TNBC.
There is still a major challenge that conventional treatments options, such as hormonal therapy, surgery, radiation therapy, and chemotherapy, has not been as successful in treating TNBC due to the absence of the ER, PR, and HER2 receptors [13,37]. Chemotherapy has been successful in treating first-time patients with TNBC; however, over time TNBCs develop resistance to these conventional treatments increasing occurrences of relapse and metastasis [38-42]. Mechanisms of how TNBCs develop resistance to chemotherapy is largely debated, but it is well known that TNBCs have chromosomal instability enhances its ability to adapt to future chemotherapy treatments [42-44]. Another limitation of chemotherapy is that chemotherapy presents adverse side effects from physical and immune complications to long-term cognitive impairment [5,13,16-20]. Considering the low success and side effects of hormonal therapy and chemotherapy, NK cell-mediated immunotherapy has become a promising novel strategy in tumor immunology [9,10]. We have demonstrated that LLT1 expression on TNBC cells function as an inhibitory ligand that interacts with natural killer cell receptor NKRP1A. By blocking LLT1-NKRP1A interaction with anti-LLT1 antibodies and siRNA-mediated decrease of LLT1 expression, cytolytic activity by NK cells increased in lysing TNBCs. Hence, targeting LLT1 on TNBC cells with antibodies will activate lysis by NK cells and could potentially lead to a new immunotherapeutic treatment for patients diagnosed with TNBC.
Acknowledgements
We thank Dr. Xiangle Sun of UNT Health Science Center Flow Cytometry Core Facility and Mrs. I-Fen Chang of Microscopy Core Facility for access, training, and assistance to all equipment used for our experiments. We strongly appreciate assistance from our lab assistant Rashmi Deshmukh in setting up parts of our experiments. This work was supported by NIH grant NS101481 to PM and UNTHSC Seed Grant to PC.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malorni L, Shetty PB, De Angelis C, Hilsenbeck S, Rimawi MF, Elledge R, Osborne CK, De Placido S, Arpino G. Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term followup. Breast Cancer Res Treat. 2012;136:795–804. doi: 10.1007/s10549-012-2315-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irvin WJ Jr, Carey LA. What is triple-negative breast cancer? Eur J Cancer. 2008;44:2799–2805. doi: 10.1016/j.ejca.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 5.Foulkes WD, Smith IE, Reis-Filho JS. Triplenegative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 6.Yeh J, Chun J, Schwartz S, Wang A, Kern E, Guth AA, Axelrod D, Shapiro R, Schnabel F. Clinical characteristics in patients with triple negative breast cancer. Int J Breast Cancer. 2017;2017:1796145. doi: 10.1155/2017/1796145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khosravi-Shahi P, Cabezon-Gutierrez L, Custodio-Cabello S. Metastatic triple negative breast cancer: optimizing treatment options, new and emerging targeted therapies. Asia Pac J Clin Oncol. 2018;14:32–39. doi: 10.1111/ajco.12748. [DOI] [PubMed] [Google Scholar]
- 8.Mathew SO, Chaudhary P, Powers SB, Vishwanatha JK, Mathew PA. Overexpression of LLT1 (OCIL, CLEC2D) on prostate cancer cells inhibits NK cell-mediated killing through LLT1-NKRP1A (CD161) interaction. Oncotarget. 2016;7:68650–68661. doi: 10.18632/oncotarget.11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- 10.Pahl J, Cerwenka A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology. 2017;222:11–20. doi: 10.1016/j.imbio.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Thielens A, Vivier E, Romagne F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol. 2012;24:239–245. doi: 10.1016/j.coi.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Mathew SO, Rao KK, Kim JR, Bambard ND, Mathew PA. Functional role of human NK cell receptor 2B4 (CD244) isoforms. Eur J Immunol. 2009;39:1632–1641. doi: 10.1002/eji.200838733. [DOI] [PubMed] [Google Scholar]
- 13.Gregorio AC, Lacerda M, Figueiredo P, Simoes S, Dias S, Moreira JN. Therapeutic implications of the molecular and immune landscape of triple-negative breast cancer. Pathol Oncol Res. 2017 doi: 10.1007/s12253-017-0307-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14.Cardoso F, Costa A, Norton L, Senkus E, Aapro M, Andre F, Barrios CH, Bergh J, Biganzoli L, Blackwell KL, Cardoso MJ, Cufer T, El Saghir N, Fallowfield L, Fenech D, Francis P, Gelmon K, Giordano SH, Gligorov J, Goldhirsch A, Harbeck N, Houssami N, Hudis C, Kaufman B, Krop I, Kyriakides S, Lin UN, Mayer M, Merjaver SD, Nordstrom EB, Pagani O, Partridge A, Penault-Llorca F, Piccart MJ, Rugo H, Sledge G, Thomssen C, Van’t Veer L, Vorobiof D, Vrieling C, West N, Xu B, Winer E. ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2) Breast. 2014;23:489–502. doi: 10.1016/j.breast.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Twelves C, Jove M, Gombos A, Awada A. Cytotoxic chemotherapy: still the mainstay of clinical practice for all subtypes metastatic breast cancer. Crit Rev Oncol Hematol. 2016;100:74–87. doi: 10.1016/j.critrevonc.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Yao C, Bernstein LJ, Rich JB. Executive functioning impairment in women treated with chemotherapy for breast cancer: a systematic review. Breast Cancer Res Treat. 2017;166:15–28. doi: 10.1007/s10549-017-4376-4. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen CM, Yamada TH, Beglinger LJ, Cavanaugh JE, Denburg NL, Schultz SK. Cognitive features 10 or more years after successful breast cancer survival: comparisons across types of cancer interventions. Psychooncology. 2013;22:862–868. doi: 10.1002/pon.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hermelink K, Untch M, Lux MP, Kreienberg R, Beck T, Bauerfeind I, Munzel K. Cognitive function during neoadjuvant chemotherapy for breast cancer: results of a prospective, multicenter, longitudinal study. Cancer. 2007;109:1905–1913. doi: 10.1002/cncr.22610. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Zhu C, Li J, Qiu L, Zhang L, Yu F, Ye R, Zhang J, Wang K. Dissociation of decision making under ambiguity and decision making under risk in breast cancer patients receiving adjuvant chemotherapy: a neuropsychological study. Brain Res. 2013;1533:63–72. doi: 10.1016/j.brainres.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 20.Jim HS, Donovan KA, Small BJ, Andrykowski MA, Munster PN, Jacobsen PB. Cognitive functioning in breast cancer survivors: a controlled comparison. Cancer. 2009;115:1776–1783. doi: 10.1002/cncr.24192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. 2017;31:37–54. doi: 10.1016/j.smim.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Llibre A, Klenerman P, Willberg CB. Multifunctional lectin-like transcript-1: a new player in human immune regulation. Immunol Lett. 2016;177:62–69. doi: 10.1016/j.imlet.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Rosen DB, Cao W, Avery DT, Tangye SG, Liu YJ, Houchins JP, Lanier LL. Functional consequences of interactions between human NKRP1A and its ligand LLT1 expressed on activated dendritic cells and B cells. J Immunol. 2008;180:6508–6517. doi: 10.4049/jimmunol.180.10.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kita S, Matsubara H, Kasai Y, Tamaoki T, Okabe Y, Fukuhara H, Kamishikiryo J, Krayukhina E, Uchiyama S, Ose T, Kuroki K, Maenaka K. Crystal structure of extracellular domain of human lectin-like transcript 1 (LLT1), the ligand for natural killer receptor-P1A. Eur J Immunol. 2015;45:1605–1613. doi: 10.1002/eji.201545509. [DOI] [PubMed] [Google Scholar]
- 25.Aldemir H, Prod’homme V, Dumaurier MJ, Retiere C, Poupon G, Cazareth J, Bihl F, Braud VM. Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol. 2005;175:7791–7795. doi: 10.4049/jimmunol.175.12.7791. [DOI] [PubMed] [Google Scholar]
- 26.Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, Lanier LL. Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol. 2005;175:7796–7799. doi: 10.4049/jimmunol.175.12.7796. [DOI] [PubMed] [Google Scholar]
- 27.Germain C, Bihl F, Zahn S, Poupon G, Dumaurier MJ, Rampanarivo HH, Padkjaer SB, Spee P, Braud VM. Characterization of alternatively spliced transcript variants of CLEC2D gene. J Biol Chem. 2010;285:36207–36215. doi: 10.1074/jbc.M110.179622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanier LL, Chang C, Phillips JH. Human NKR-P1A. A disulfide-linked homodimer of the C-type lectin superfamily expressed by a subset of NK and T lymphocytes. J Immunol. 1994;153:2417–2428. [PubMed] [Google Scholar]
- 29.Poggi A, Rubartelli A, Moretta L, Zocchi MR. Expression and function of NKRP1A molecule on human monocytes and dendritic cells. Eur J Immunol. 1997;27:2965–2970. doi: 10.1002/eji.1830271132. [DOI] [PubMed] [Google Scholar]
- 30.Poggi A, Costa P, Tomasello E, Moretta L. IL-12-induced up-regulation of NKRP1A expression in human NK cells and consequent NKRP1A-mediated down-regulation of NK cell activation. Eur J Immunol. 1998;28:1611–1616. doi: 10.1002/(SICI)1521-4141(199805)28:05<1611::AID-IMMU1611>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Mathew PA, Chuang SS, Vaidya SV, Kumaresan PR, Boles KS, Pham HT. The LLT1 receptor induces IFN-gamma production by human natural killer cells. Mol Immunol. 2004;40:1157–1163. doi: 10.1016/j.molimm.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 32.Bambard ND, Mathew SO, Mathew PA. LLT1-mediated activation of IFN-gamma production in human natural killer cells involves ERK signalling pathway. Scand J Immunol. 2010;71:210–219. doi: 10.1111/j.1365-3083.2009.02367.x. [DOI] [PubMed] [Google Scholar]
- 33.Germain C, Meier A, Jensen T, Knapnougel P, Poupon G, Lazzari A, Neisig A, Hakansson K, Dong T, Wagtmann N, Galsgaard ED, Spee P, Braud VM. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-gamma contributes to modulate immune responses. J Biol Chem. 2011;286:37964–37975. doi: 10.1074/jbc.M111.285312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Criscitiello C, Azim HA Jr, Schouten PC, Linn SC, Sotiriou C. Understanding the biology of triple-negative breast cancer. Ann Oncol. 2012;23(Suppl 6):vi13–18. doi: 10.1093/annonc/mds188. [DOI] [PubMed] [Google Scholar]
- 35.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109:1721–1728. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 36.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lumachi F, Luisetto G, Basso SM, Basso U, Brunello A, Camozzi V. Endocrine therapy of breast cancer. Curr Med Chem. 2011;18:513–522. doi: 10.2174/092986711794480177. [DOI] [PubMed] [Google Scholar]
- 38.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 39.Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M, Anderson K, Hess KR, Stec J, Ayers M, Wagner P, Morandi P, Fan C, Rabiul I, Ross JS, Hortobagyi GN, Pusztai L. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11:5678–5685. doi: 10.1158/1078-0432.CCR-04-2421. [DOI] [PubMed] [Google Scholar]
- 40.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 41.Tang H, Qiao J, Fu YX. Immunotherapy and tumor microenvironment. Cancer Lett. 2016;370:85–90. doi: 10.1016/j.canlet.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 43.Wein L, Loi S. Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC) Breast. 2017;34(Suppl 1):S27–S30. doi: 10.1016/j.breast.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 44.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 45.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 47.Llibre A, López-Macías C, Marafioti T, Mehta H, Partridge A, Kanzig C, Rivellese F, Galson JD, Walker LJ, Milne P, Phillips RE, Kelly DF, Freeman GJ, El Shikh ME, Klenerman P, Willberg CB. LLT1 and CD161 expression in human germinal centers promotes B cell activation and CXCR4 downregulation. J Immunol. 2016;196:2085–2094. doi: 10.4049/jimmunol.1502462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Germain C, Guillaudeux T, Galsgaard ED, Hervouet C, Tekaya N, Gallouet AS, Fassy J, Bihl F, Poupon G, Lazzari A, Spee P, Anjuere F, Pangault C, Tarte K, Tas P, Xerri L, Braud VM. Lectin-like transcript 1 is a marker of germinal center-derived B-cell non-Hodgkin’s lymphomas dampening natural killer cell functions. Oncoimmunology. 2015;4:e1026503. doi: 10.1080/2162402X.2015.1026503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roth P, Mittelbronn M, Wick W, Meyermann R, Tatagiba M, Weller M. Malignant glioma cells counteract antitumor immune responses through expression of lectin-like transcript-1. Cancer Res. 2007;67:3540–3544. doi: 10.1158/0008-5472.CAN-06-4783. [DOI] [PubMed] [Google Scholar]
- 50.Llibre A, Garner L, Partridge A, Freeman GJ, Klenerman P, Willberg CB. Expression of lectin-like transcript-1 in human tissues. F1000Res. 2016;5:2929. doi: 10.12688/f1000research.10009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boles KS, Barten R, Kumaresan PR, Trowsdale J, Mathew PA. Cloning of a new lectin-like receptor expressed on human NK cells. Immunogenetics. 1999;50:1–7. doi: 10.1007/s002510050679. [DOI] [PubMed] [Google Scholar]
- 52.Asselin-Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–465. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, Wang L, Shifrin N, Raulet DH. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 2014;122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]