

Abstract
Decreased selenium-binding protein 1 (SBP1) is associated with increased invasion and poor prognosis of hepatocellular carcinoma (HCC). However, the underlying mechanism remains unknown. To unravel this mechanism, HCC cells expressing SBP1 were constructed and the impact on migration, invasion, and epithelial-mesenchymal transition (EMT) was evaluated. SBP1 expression reduced HCC cell migration and invasion by inhibiting EMT. Gene expression profiles of control and SBP1 expressing HCC cells revealed 186 differentially expressed genes, of which fibroblast growth factor 5, vascular endothelial growth factor receptor 1, and C-X-C motif chemokine receptor 4 (CXCR4) showed the greatest differences. CXCR4 expression was inhibited by SBP1 and restored the migration and invasion ability of HCC cells through activation of AKT signaling. Tumor samples from 200 HCC patients supported our in vitro findings and revealed an inverse correlation between SBP1 and CXCR4 expression. Patients with low SBP1 and high CXCR4 expression had the poorest prognosis and survival rate. Our results suggest that downregulation of SBP1 induces increased CXCR4 expression and results in EMT of HCC cells. Together, SBP1 and CXCR4 are promising potential biomarkers and therapeutic targets for HCC patients.
Keywords: Selenium-binding protein 1, CXCR4, epithelial-mesenchymal transition, hepatocellular carcinoma
Introduction
Selenium is an essential trace element that plays a vital role in cancer prevention. In the human body selenium has various functions in a diverse range of selenium-containing proteins. Of these proteins, selenium-binding protein 1 (SBP1, also known as hSP56 or SELENBP1) is a possible mediator of the anticancer functions of selenium [1-3]. Previous studies indicate that SBP1 downregulation is associated with poor prognosis in various cancers, including pancreatic cancer [4], breast cancer [5], colorectal cancer [6], esophageal adenocarcinoma [7], gastric cancer [8-10], hepatocellular carcinoma [11], and cervical carcinoma [12]. Although the tumor suppressive function of SBP1 has been validated by accumulating evidence, the mechanism still requires further investigation.
The expression of SBP1 is regulated by hypoxia inducible factor 1 alpha subunit (HIF1A), which is a fundamental mediator of cellular adaptation to micro-environmental stress [13]. Our previous work further demonstrated that SBP1 could counter-regulate the expression of HIF1A through the inhibition of glutathione peroxidase 1 (GPX1) activity [11]. We also demonstrated that decreased SBP1 in hepatocellular carcinoma (HCC) cells could lead to increased cancer cell migration and invasion, but the underlying mechanism was not fully understood.
HCC is the most commonly diagnosed liver cancer and the third leading cause of cancer related deaths worldwide [14]. It is characterized by its invasive and metastatic potential, which is closely associated with epithelial-mesenchymal transition (EMT) [15]. EMT is a complicated reprogramming process that leads to loss of cell-cell adhesions and enhanced invasiveness, both of which are related to tumor metastasis. EMT is often characterized by a loss of epithelial cadherin (E-cadherin) and an increase in mesenchymal markers, such as neural cadherin (N-cadherin) and vimentin (VIM, an intermediate filament) [16]. The improper induction of EMT can endow tumor cells with migration and invasion capabilities that then initiate the tumor metastasis cascade. The initiation of EMT has been studied intensively, and HIF1A was found to be capable of inhibiting the EMT phenotype of tumor cells through an unrevealed mechanism [13]. Based on our previous findings, the possible connection between HIF1A, SBP1, and EMT deserves further investigation. In this study, we aimed to uncover the underlying mechanism of SBP1 tumor suppression in HCC, and investigate the potential relationship between SBP1 expression and EMT.
Materials and methods
Clinical materials
The study participants were randomly selected from among consecutive patients with HCC who underwent curative resection from 2006 to 2007 at the Liver Cancer Institute of Fudan University (Shanghai, China). Clinicopathological parameters were described in Supplemental Table 1. Ethical approval was obtained from the research ethics committee of Zhongshan Hospital, and written informed consent was obtained from each patient. Follow-up data were summarized at the end of May 2012, with a median follow-up of 62 months and a range of 5 to 73 months.
Cell culture
Human hepatocellular carcinoma cells, Huh-7 and 7721, were obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (CAS). MHCC97-H (97H) was established at our Institute. The cell culture methodology is described in the previous study [11].
Total RNA isolation and gene expression profile
Total RNA from Huh-7 and 97H cells was isolated using Trizol Reagent (Invitrogen, USA) and subsequently reverse transcribed using PrimeScipt RT reagent Kit (Takara, Japan) as previously described [11]. The commercially available GeneChip clariom D solution for human (Invitrogen, USA) was used.
qPCR array
Primers were as follows: 3’-GACGACCGCTTCCTCTACTTCA-5’ and 3’-GGGCTCTGGCTGGGACTTTA-5’ for SBP1; 3’-CCGGACTGTGGCTGTGAAAA-5’ and 3’-TGGCCAATGTGGGTCAAGAT-5’ for vascular endothelial growth factor receptor 1 (VEGFR1); 3’-GCTGCCACTGATAGGAACCC-5’ and 3’-GAGGAGGAGGCAGAAGAGGA-5’ for fibroblast growth factor 5 (FGF5); 3’-GGATGGCAAGAGACCCACAC-5’ and 3’-ATATTGGGCGGGAGTGTCAG-5’ for C-X-C motif chemokine receptor 4 (CXCR4) 3’-GGTATGACAACGAATTTGGC-5’ and 3’-GAGCACAGGGTACTTTATTG-5’ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The results were analyzed using the 2-ΔΔCt method, and GADPH was used as a reference gene for normalizing. Statistical analyses were performed using the Student t-test, and a P value of less than 0.05 was considered to be statistically significant. Three independent experiments were performed for each gene.
Antibodies and western blot
Cells were lysed using RIPA buffer as previously described [11]. Equivalent amounts of protein were separated by SDS-PAGE, transferred onto PVDF membranes, and incubated with primary antibodies. The primary antibodies used in this study were anti-CXCR4 (Cell Signaling Technology [CST], USA, 1:1000), anti-AKT (CST, 1:1000), anti-phospho-AKT (CST, 1:1000), anti-FGF5 (Abcam, USA, 1:1000), anti-VEGFR1 (Abcam, 1:1000), anti-N-cadherin (CST, 1:1000), anti-E-cadherin (CST, 1:1000), and anti-vimentin (CST, 1:1000). The membranes were then incubated with horseradish peroxidase-conjugated antibodies and visualized using enhanced chemiluminescence assay.
Plasmid construction and transfection
The plasmids CMV-MCS-IRES-eGFP-SBP1 and CMV-MCS-IRES-eGFP-CXCR4 were purchased from Genomeditech Shanghai, China. Transient transfections of plasmids were performed according to the manufacturer’s instruction [11].
Migration and invasion assays
The migration and invasion assays were performed using the Transwell chamber (Corning, USA) following the protocol as described previously [11].
Immunohistochemistry
Tissue microarrays were conducted by Shanghai Outdo Co., Ltd. A two-step immunohistochemistry (IHC) protocol was used to detect protein expression as previously described [11]. The antibodies used were CXCR4 (1:100), SBP1 (1:200), E-cadherin (1:100), and vimentin (1:100).
Statistical analysis
The software package SPSS v20.0 (IBM, USA) was used for statistical analyses. Univariate and multivariate Cox proportional hazards models were used to identify relevant prognostic factors. Kaplan-Meier survival curves and the log-rank (Mantel-Cox) test were used to compare patient survival and recurrence probability between subgroups. All statistical tests were 2-sided, and a P value of less than 0.05 was considered statistically significant.
Results
SBP1 induction inhibits HCC cell migration and invasion by inhibiting EMT
SBP1 was barely detected in 97H (high metastasis) and Huh-7 (low metastasis) cell lines, consistent with our previous report [11]. To investigate the effect of SBP1 on HCC cells, a SBP1 expression vector was introduced into Huh-7 and 97H cells (Figure 1A). Huh-7 and 97H cells with an empty vector were used as controls and termed mock. In agreement with previous reports, we found a significant attenuation of migratory (Figure 1B) and invasive (Figure 1C) capacities in SBP1 expressing HCC cells [11]. To investigate the possibility that downregulation of SBP1 elicited EMT, we performed western blot analysis to examine the protein expression of epithelial marker E-cadherin and mesenchymal markers N-cadherin and vimentin (VIM). Although the expression of N-cadherin was not altered significantly, enhanced E-cadherin in parallel with decreased vimentin were observed in SBP1 expressing Huh-7 and 97H cells compared with mock Huh-7 and 97H cells (Figure 1D). As we previous reported [11], SBP1 expression in SMMC7721 cells was downregulated to a marginal level by siRNA treatment, and enhanced vimentin in parallel with decreased E-cadherin were observed (Supplemental Figure 1).
Figure 1.
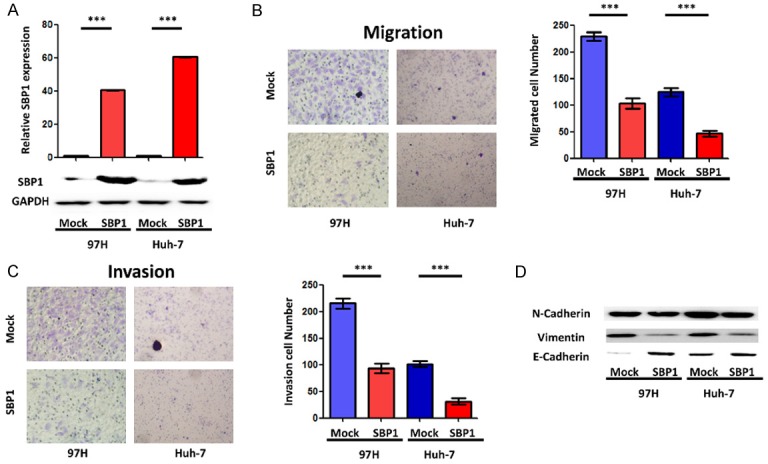
SBP1 induction inhibits HCC cell migration and invasion. A. The SBP1 mRNA and protein relative expression level in 97H-SBP1 and Huh-7-SBP1 cells by qPCR and western blot. B. Effects of SBP1 induction on migration of 97H and Huh-7 cells. C. Effects of SBP1 induction on invasion of 97H and Huh-7 cells. D. Effects of SBP1 induction on E-cadherin, vimentin, and N-cadherin protein expression. ***P<0.001.
Differentially expressed genes in SBP1 expressing HCC cells
To investigate the underlying mechanism of SBP1-mediated inhibition of tumor metastasis, we further analyzed the gene expression profiles of four different HCC cell lines: Huh-7-mock, Huh-7-SBP1, 97H-mock, and 97H-SBP1. The results showed 965 upregulated genes and 887 downregulated genes in 97H-SBP1 cells, and 1,718 upregulated genes and 910 downregulated genes in Huh-7-SBP1 cells. There were 186 differentially expressed genes in common, including 131 upregulated genes and 55 downregulated genes (Figure 2A). According to Gene Ontology analysis, the differentially expressed genes were classified into three groups: biological process, molecular function, and cellular component (Figure 2B). The main biological process categories that were enriched included cholesterol homeostasis, both low and high density lipoprotein particle remodeling, and ion transport. The primary enriched molecular functions were actin binding, phosphatase activity, lipid transporter activity, protein tyrosine phosphatase, and sodium-independent organic anion transmembrane transporter activity. Within the cellular component group, the differentially expressed genes were involved in extracellular space, apical plasma membrane, extracellular exosome, and extracellular region. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis reveald that differentially expressed genes were involved in the Rap1 signaling pathway, rheumatoid arthritis, and amoebiasis.
Figure 2.

Differentially expressed genes in SBP1 expressing 97H and Huh-7 cells. A. Differential expression of genes in SBP1 expressing 97H and Huh-7 cells. B. Gene Ontology and KEGG analysis of the common differentially expressed genes.
CXCR4 eliminates the EMT inhibition of SBP1 expression in HCC cells
Three promising genes, CXCR4, VEGFR1, and FGF5, were selected for further study. Quantitative RT-PCR analysis was performed to verify that CXCR4, VEGFR1, and FGF5 mRNA levels were significantly reduced in Huh-7-SBP1 and 97H-SBP1 HCC cells (Figure 3A). We performed western blot analysis to examine CXCR4, VEGFR1, and FGF5 protein expression in Huh-7-SBP1 and 97H-SBP1 HCC cells. Consistently, VEGFR1, FGF5, and CXCR4 proteins were markedly decreased in SBP1 expressing Huh-7 and 97H cells (Figure 3B). Interestingly, CXCR4 has been shown to be involved in malignancy of various cancers by triggering EMT [17,18]. Therefore, we pursued the possibility that SBP1 contributes to EMT by downregulating CXCR4.
Figure 3.
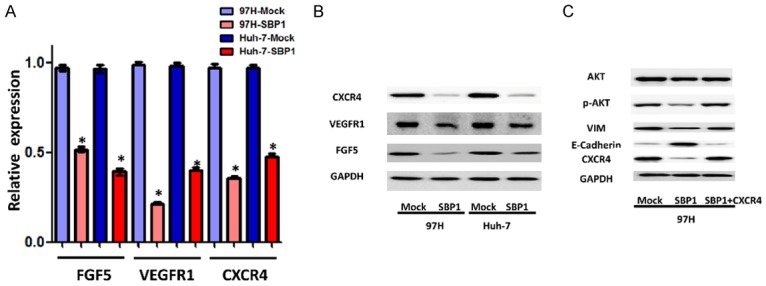
Downregulation of FGF5, VEGFR1, and CXCR4 in HCC cell lines expressing SBP1. A. Relative CXCR4, VEGFR1, and FGF5 expression level of SBP1 induction 97H and Huh-7 cells by qPCR. B. Effect of SBP1 induction on CXCR4, VEGFR1, and FGF5 protein expression in 97H and Huh-7 cells by western blot. C. Effect of CXCR4 overexpression on AKT, p-AKT, VIM, and E-cadherin protein expression in 97H-SBP1 cells by western blot. *P<0.05.
To address whether SBP1 inhibited the migration and invasion of HCC cells through suppression of CXCR4 expression, we introduced a CXCR4 expression vector into SBP1 expressing 97H cells. To further determine a possible causal connection between SBP1 and CXCR4 expression, western blot was performed to examine the influence of SBP1 and CXCR4 co-expression on CXCR4 downstream signaling targets AKT and p-AKT, as well as vimentin and E-cadherin. Although total AKT expression did not change significantly, decreased p-AKT expression in SBP1 expressing 97H cells was observed. Co-expression of SBP1 and CXCR4 upregulated the expression of VIM and p-AKT in comparison with SBP1 expressing 97H cells, and simultaneously downregulated the expression of E-cadherin (Figure 3C). Moreover, CXCR4 expression restored the reduced migration and invasion of 97H cells that was attenuated by SBP1 (Figure 4).
Figure 4.
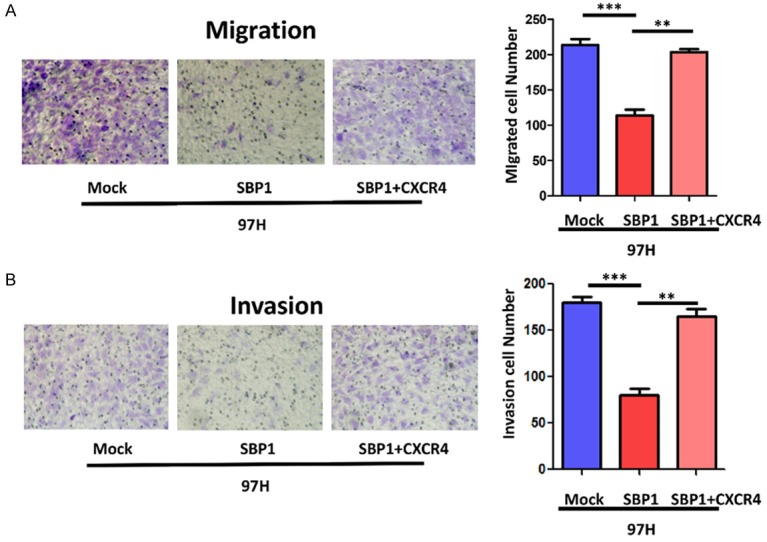
Effect of CXCR4 overexpression on migratory and invasive properties of SBP1 induction in 97H cells. A. Effect of SBP1 induction on migration in 97H cells. B. Effect of SBP1 induction on invasion in 97H cells. **P<0.01, ***P<0.001.
Negative correlation between SBP1 and CXCR4 expression in HCC tissues
SBP1 expression in HCC tissues was examined in previous studies and showed decreased expression in tumor tissues [11]. We further detected SBP1, CXCR4, vimentin, and E-cadherin expression levels in the cohort containing 200 HCC tumor tissues. Representative IHC images are shown in Figure 5.
Figure 5.
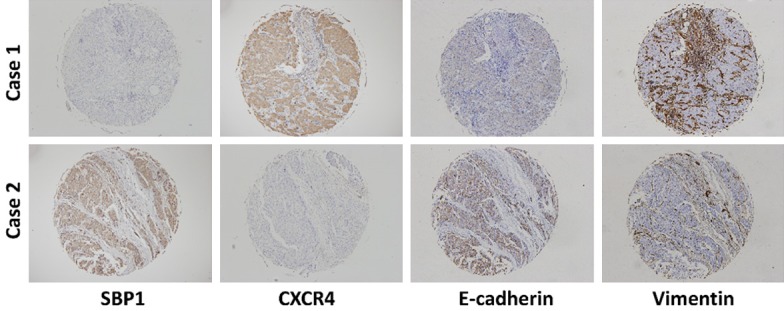
Representative IHC images of SBP1, CXCR4, E-cadherin, and vimentin expression in HCC tissues.
Generally, SBP1 expression showed an inverse relationship with CXCR4 expression in HCC tissues. Similarly, SBP1 expression positively correlated with E-cadherin expression and negatively correlated with vimentin expression in HCC (Figure 5). Kaplan-Meier analysis showed that downregulated SBP1 expression was associated with reduced overall survival time and cumulative recurrence rate (Figure 6A). Inversely, patients with high CXCR4 expression had poor prognoses (Figure 6B). Comprehensively, those patients with high SBP1 expression and low CXCR4 expression had the longest overall survival time. Meanwhile, a small portion of patients (n=35) with both low or both high SBP1 and CXCR4 expression had slightly poor prognoses. Those patients with low SBP1 expression and high CXCR4 expression had the poorest prognoses and survival rate (Figure 6C).
Figure 6.
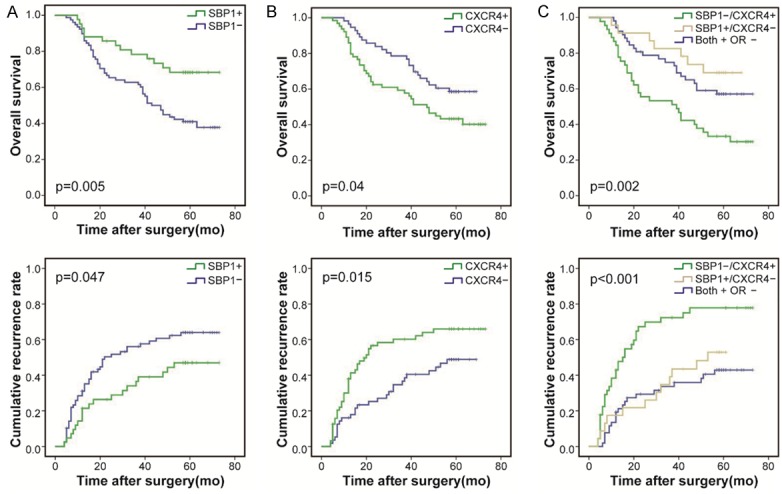
Impact of SBP1 and CXCR4 expression on HCC patient survival. A. Kaplan-Meier survival curves show that patients with negative expression of SBP1 in tumor cells experienced shorter overall survival (OS) periods (P=0.005) and higher recurrence rates (P=0.047) than patients with positive expression of SBP1. B. Kaplan-Meier survival curves show that patients with positive expression of CXCR4 in tumor cells experienced shorter OS periods (P=0.04) and higher recurrence rates (P=0.015) than patients with negative expression of CXCR4. C. HCC patients co-expressing negative expression of SBP1 and positive expression of CXCR4 had the poorest prognosis in terms of OS (P=0.002) and cumulative recurrence rate (P<0.001).
Discussion
Accumulating evidence has revealed the profound effect of EMT in tumor migration and invasion [19]. Our previous work validated the role of SBP1 as a tumor suppressor in HCC by suppressing tumor cell migration and invasion [11]. However, the potential relationship between SBP1 and EMT was not investigated. In this study, decreased vimentin and enhanced E-cadherin expression were observed in SBP1 expressing HCC cells, thus suggesting that SBP1 induction could inhibit HCC cell migration and invasion through the inhibition of EMT. This notion was further supported by the positive correlation of SBP1 and E-cadherin expression and the negative correlation of SBP1 and vimentin expression in a cohort of 200 HCC patients in this study.
To understand the mechanism of SBP1 inhibition of EMT, we further analyzed the gene expression profiles of SBP1 expressing and control cells. Three genes were identified as possible mediators, including VEGFR1, FGF5, and CXCR4. VEGFR1 is a receptor tyrosine kinase that binds to VEGFR-A and VEGFR-B. The tumor promoting role of VEGFR1 has been highlighted in various studies, and is strongly linked to its influence on angiogenesis [20]. FGF5 is a member of the fibroblast growth factor family and is closely related to cell proliferation. Accumulating evidence points to an oncogenic role for FGF5 in various cancers, including pancreatic cancer and hepatocellular carcinoma [21,22]. VEGFR1 and FGF5 could partially explain the tumor suppressive function of SBP1 in our findings, yet the connection between CXCR4 and EMT was most convincing. CXCR4 was found to be an important tumor promoter and is overexpressed in many different human cancers, while low or undetectable in normal tissue [23]. It has been implicated in various key steps of cancer development, especially the induction of EMT. Previous studies have already confirmed that the CXCR4/AKT pathway regulates NF-kB, and the enhanced transcriptional activity of NF-kB could directly induce cancer cell EMT [24]. In this study, we found that SBP1 induction in HCC cells resulted in decreased expression of CXCR4. CXCR4 exerts its function through activation of intracellular signaling pathways, including FAK, AKT, and ERK. We found that phospho-AKT expression was reduced in SBP1 expressing cells, indicating that the activation of CXCR4/AKT signaling may contribute to EMT in HCC in the absence of SBP1. To further validate this conclusion, we introduced a CXCR4 expression vector into SBP1 expressing HCC cells and found that CXCR4 was able to restore the migration and invasion capacity that had been attenuated by SBP1 overexpression. The CXCR4/AKT pathway may be the crucial mechanism of the anti-tumor activity of SBP1. These results were further validated in clinical samples, and SBP1 expression was consistently inversely correlated with CXCR4 expression in a cohort of 200 HCC patients. We also estimated the prognostic value of SBP1 and CXCR4. Both decreased SBP1 and increased CXCR4 expression were remarkable indicators of poor prognosis for HCC patients as shown by our previous work and other researchers [11,23]. However, our evaluation of the combination of SBP1 and CXCR4 showed increased significance. Patients with low SBP1 expression and high CXCR4 expression had extremely poor prognosis. Thus, in these patients, more aggressive therapeutic and follow-up strategies should be adopted.
On the other hand, SBP1 is regulated by HIF1A through a hypoxia response element in its promoter region, and can also counter-regulate the expression of HIF1A through inhibition of GPX1 activity. HIF1A is a fundamental mediator of cellular adaptation to reactive oxygen species (ROS). The paradoxical role of HIF1A in cancer biology had been recognized and attributed to its diverse downstream signaling pathways. It has been reported that HIF1A can suppress EMT and inhibit malignant tumor conversion [13]. CXCR4 is one target gene of HIF1A [25]. SBP1 may regulate the expression of CXCR4 through HIF1A. Combined with our findings, a ROS/HIF1A/SBP1/CXCR4/AKT/EMT pathway could be established.
In conclusion, our findings provide novel insight into the inhibitory mechanism of SBP1 in HCC. Downregulation of SBP1 may cause increased CXCR4 expression and subsequent induction of an EMT phonotype in HCC cells. SBP1, in combination with CXCR4, could be a potential biomarker and possible therapeutic target for HCC patients.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [No. 81602513, 81502006 and 81572298].
Disclosure of conflict of interest
None.
Abbreviations
- SBP1
selenium-binding protein 1
- HCC
hepatocellular carcinoma
- CXCR4
C-X-C motif chemokine receptor 4
- EMT
epithelial-mesenchymal transition
- HIF1A
hypoxia inducible factor 1 alpha subunit
- GPX1
glutathione peroxidase 1
- ROS
reactive oxygen species
Supporting Information
References
- 1.Bansal MP, Oborn CJ, Danielson KG, Medina D. Evidence for two selenium-binding proteins distinct from glutathione peroxidase in mouse liver. Carcinogenesis. 1989;10:541–546. doi: 10.1093/carcin/10.3.541. [DOI] [PubMed] [Google Scholar]
- 2.Bansal MP, Mukhopadhyay T, Scott J, Cook RG, Mukhopadhyay R, Medina D. DNA sequencing of a mouse liver protein that binds selenium: implications for selenium’s mechanism of action in cancer prevention. Carcinogenesis. 1990;11:2071–2073. doi: 10.1093/carcin/11.11.2071. [DOI] [PubMed] [Google Scholar]
- 3.Pol A, Renkema GH, Tangerman A, Winkel EG, Engelke UF, de Brouwer APM, Lloyd KC, Araiza RS, van den Heuvel L, Omran H, Olbrich H, Oude Elberink M, Gilissen C, Rodenburg RJ, Sass JO, Schwab KO, Schafer H, Venselaar H, Sequeira JS, Op den Camp HJM, Wevers RA. Mutations in SELENBP1, encoding a novel human methanethiol oxidase, cause extraoral halitosis. Nature genetics. 2018;50:120–129. doi: 10.1038/s41588-017-0006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caba O, Irigoyen A, Jimenez-Luna C, Benavides M, Ortuño FM, Gallego J, Rojas I, Guillen-Ponce C, Torres C, Aranda E, Prados J. Identification of gene expression profiling associated with erlotinib-related skin toxicity in pancreatic adenocarcinoma patients. Toxicol Appl Pharmacol. 2016;311:113–116. doi: 10.1016/j.taap.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Zhang S, Li F, Younes M, Liu H, Chen C, Yao Q. Reduced selenium-binding protein 1 in breast cancer correlates with poor survival and resistance to the anti-proliferative effects of selenium. PLoS One. 2013;8:e63702. doi: 10.1371/journal.pone.0063702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang N, Chen Y, Yang X, Jiang Y. Seleniumbinding protein 1 is associated with the degree of colorectal cancer differentiation and is regulated by histone modification. Oncol Rep. 2014;31:2506–2514. doi: 10.3892/or.2014.3141. [DOI] [PubMed] [Google Scholar]
- 7.Silvers AL, Lin L, Bass AJ, Chen G, Wang Z, Thomas DG, Lin J, Giordano TJ, Orringer MB, Beer DG, Chang AC. Decreased selenium-binding protein 1 in esophageal adenocarcinoma results from posttranscriptional and epigenetic regulation and affects chemosensitivity. Clin Cancer Res. 2010;16:2009–21. doi: 10.1158/1078-0432.CCR-09-2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Zhan N, Dong WG. Altered expression of selenium-binding protein 1 in gastric carcinoma and precursor lesions. Med Oncol. 2011;28:951–7. doi: 10.1007/s12032-010-9564-6. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Dong WG, Lin J. Reduced seleniumbinding protein 1 is associated with poor survival rate in gastric carcinoma. Med Oncol. 2011;28:481–7. doi: 10.1007/s12032-010-9482-7. [DOI] [PubMed] [Google Scholar]
- 10.Gong J, Li L. Sodium selenite inhibits proliferation of gastric cancer cells by inducing SBP1 expression. Tohoku J Exp Med. 2016;239:279–85. doi: 10.1620/tjem.239.279. [DOI] [PubMed] [Google Scholar]
- 11.Huang C, Ding G, Gu C, Zhou J, Kuang M, Ji Y, He Y, Kondo T, Fan J. Decreased selenium-binding protein 1 enhances glutathione peroxidase 1 activity and downregulates HIF-1alpha to promote hepatocellular carcinoma invasiveness. Clin Cancer Res. 2012;18:3042–53. doi: 10.1158/1078-0432.CCR-12-0183. [DOI] [PubMed] [Google Scholar]
- 12.Zhao C, Zeng H, Wu RT, Cheng WH. Loss of selenium-binding protein 1 decreases sensitivity to clastogens and intracellular selenium content in HeLa cells. PLoS One. 2016;11:e0158650. doi: 10.1371/journal.pone.0158650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scortegagna M, Martin RJ, Kladney RD, Neumann RG, Arbeit JM. Hypoxia-inducible factor-1alpha suppresses squamous carcinogenic progression and epithelial-mesenchymal transition. Cancer Res. 2009;69:2638–46. doi: 10.1158/0008-5472.CAN-08-3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 15.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 16.Kalluri R, Weinberg RA. The basics of epithelialmesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, Bhat K, Wang F, Wu E, Wang Z. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012;322:169–76. doi: 10.1016/j.canlet.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aversa I, Zolea F, Ieranò C, Bulotta S, Trotta AM, Faniello MC, De Marco C, Malanga D, Biamonte F, Viglietto G, Cuda G, Scala S, Costanzo F. Epithelial-to-mesenchymal transition in FHCsilenced cells: the role of CXCR4/CXCL12 axis. J Exp Clin Cancer Res. 2017;36:104. doi: 10.1186/s13046-017-0571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suh Y, Yoon CH, Kim RK, Lim EJ, Oh YS, Hwang SG, An S, Yoon G, Gye MC, Yi JM, Kim MJ, Lee SJ. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene. 2013;32:4873–82. doi: 10.1038/onc.2012.505. [DOI] [PubMed] [Google Scholar]
- 20.Meng J, Liu Y, Han J, Tan Q, Chen S, Qiao K, Zhou H, Sun T, Yang C. Hsp90beta promoted endothelial cell-dependent tumor angiogenesis in hepatocellular carcinoma. Mol Cancer. 2017;16:72. doi: 10.1186/s12943-017-0640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang F, Chang RM, Yu L, Lei X, Xiao S, Yang H, Yang LY. MicroRNA-188-5p suppresses tumor cell proliferation and metastasis by directly targeting FGF5 in hepatocellular carcinoma. J Hepatol. 2015;63:874–85. doi: 10.1016/j.jhep.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Kornmann M, Ishiwata T, Beger HG, Korc M. Fibroblast growth factor-5 stimulates mitogenic signaling and is overexpressed in human pancreatic cancer: evidence for autocrine and paracrine actions. Oncogene. 1997;15:1417–24. doi: 10.1038/sj.onc.1201307. [DOI] [PubMed] [Google Scholar]
- 23.Zhao H, Guo L, Zhao H, Zhao J, Weng H, Zhao B. CXCR4 over-expression and survival in cancer: a system review and meta-analysis. Oncotarget. 2015;6:5022–40. doi: 10.18632/oncotarget.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103:1671–9. doi: 10.1038/sj.bjc.6605968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valsecchi R, Coltella N, Belloni D, Ponente M, Ten Hacken E, Scielzo C, Scarfò L, Bertilaccio MT, Brambilla P, Lenti E, Martinelli Boneschi F, Brendolan A, Ferrero E, Ferrarini M, Ghia P, Tonon G, Ponzoni M, Caligaris-Cappio F, Bernardi R. HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood. 2016;127:1987–97. doi: 10.1182/blood-2015-07-657056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.