

Abstract
Hepatorenal fibrocystic disease (HRFCD) is characterized by cysts in the kidney and liver with associated fibrosis and is the result of defects in proteins required for cilia function or assembly. Previous reports indicate that macrophages, mainly M2-like macrophages, contribute to HRFCD, although the origin of these cells (yolk sac-derived resident macrophages vs. bone marrow-derived infiltrating macrophages) and their contribution to the observed phenotypes are unknown. We utilize a congenital model of cilia dysfunction (IFT88Orpk) to study the importance of macrophages in HRFCD. Our data show a rapid expansion of the bile duct region and development of fibrosis between 2 and 4 wk of age. Immunofluorescence microscopy analysis reveals an accumulation of F4/80+ macrophages in regions exhibiting biliary hyperplasia in IFT88Orpk mice. Flow cytometry data show that cilia dysfunction leads to an accumulation of infiltrating macrophages (CD11bhi, F4/80lo) and a reduction of resident macrophage (CD11blo, F4/80hi) number. A majority of the infiltrating macrophages are Ly6chi profibrogenic macrophages. Along with the accumulation of immune cells, expression of proinflammatory and profibrotic transcripts, including TGF-β, TNF-α, IL-1β, and chemokine (C-C) motif ligand 2, is increased. Quantitative RT-PCR analysis of flow-sorted cells shows enhanced expression of CCL2 in cholangiocytes and enhanced expression of VEGF-A and IL-6 in Ly6chi macrophages. Genetic inhibition of Ly6chi macrophage accumulation in IFT88Orpk FVB CCR2−/− mice reduced biliary fibrosis but did not affect epithelial expansion. Collectively, these studies suggest that biliary epithelium with defects in primary cilia preferentially recruits Ly6chi infiltrating macrophages, which promote fibrotic progression in HRFCD pathogenesis.
NEW & NOTEWORTHY These studies are the first to address the contribution of the infiltrating and resident macrophage niche during progression of hepatorenal fibrocystic disease (HRFCD). We show that the number of infiltrating macrophages is significantly upregulated in HRFCD mouse models. Finally, we show that prevention of Ly6chi infiltrating macrophage accumulation significantly reduces biliary fibrosis, but not biliary hyperplasia, suggesting that this population may be responsible for the fibrotic progression of the disease in HRFCD patients.
Keywords: cholangiocyte, hepatorenal fibrocystic disease, liver, macrophage
INTRODUCTION
Hepatorenal fibrocystic disease (HRFCD), including polycystic kidney disease (PKD), is a commonly inherited group of genetic diseases that affect ~1 in 500 people (38). In patients with HRFCD, abnormal expansion of epithelial cells in the kidney and liver leads to cysts and fibrosis in both organs (39, 40). Mutations in proteins that are required for cilia formation [e.g., intraflagellar transport 88 (IFT88)] or localize to and function in the primary cilium [polycystic kidney and hepatic disease 1 (PKHD1) and polycystic kidney disease 1 (PC1) or polycystic kidney disease 2 (PC2)] are a major cause of HRFCD. Importantly, mutations in fibrocystin/polyductin (Pkhd1) are responsible for the autosomal-recessive form of PKD and show a phenotype similar to that observed in the IFT88Orpk mouse model of HRFCD, including periportal fibrosis and bile duct expansion (10, 16, 33). Unlike these mouse models, mice with mutations in Pkd1 or Pkd2 display severe bile duct expansion and cyst formation with minimal extracellular matrix production and fibrosis (31–33). The mechanisms by which dysfunctional primary cilia lead to the hepatic phenotypes in humans and rodent models remain unknown.
Recent data indicate that macrophages are associated with cyst formation and that liposomal clodronate-mediated depletion of phagocytic macrophages reduces cyst formation and fibrosis in the kidney and liver of HRFCD mouse models (5, 14, 16, 24, 35, 36, 42). In contrast, bindarit-mediated inhibition of chemokine (C-C) motif ligand 2 (CCL2), which is known to recruit infiltrating bone marrow-derived macrophages, did not reduce cyst formation in the kidney or liver of PCK rats, although this inhibitor did improve podocyte structure (44). Therefore, it was concluded that macrophages with high phagocytic capacity (resident-like macrophages) that are independent of CCL2-mediated recruitment from the bone marrow are the major drivers of renal cyst formation and fibrosis. In these studies, the contribution of infiltrating and resident macrophages in the liver and the impact of bindarit on these populations were not addressed.
Macrophages in the liver and other tissues can be distinguished on the basis of their origin and expression levels of specific cell surface markers. Tissue resident macrophages (F4/80hi, CD11blo cells) in the liver, termed Kupffer cells, are derived from the yolk sac before the onset of hematopoiesis (11, 28, 41). They are independent of the transcription factor Myb and maintain their population mainly through self-renewal (28). In contrast, infiltrating macrophages (CD11bhi, F4/80lo cells) originate from the hematopoietic lineage in the adult bone marrow, are dependent on the transcription factor Myb, and are recruited to tissues in response to injury or infection through cytokines, such as CCL2 binding to its cognate receptor on the immune cell (CCR2) (9, 18, 21, 28, 30).
Infiltrating macrophages in the liver can be further subtyped on the basis of cell surface expression of Ly6c, although the contribution of Ly6chi and Ly6clo macrophages in mouse models of liver injury is controversial as genetic or pharmacological inhibition of infiltrating macrophages can be both protective and damaging following liver injury (3, 6, 15, 20, 22). In contrast, the involvement of these macrophage subtypes in liver fibrosis is more clear. An elegant study by Ramachandran et al. demonstrated that Ly6chi macrophages were present during periods of maximal injury and fibrosis in the liver following CCl4 treatment (25). The increase in the number of Ly6chi macrophages correlated with a decrease in the number of resident macrophages and maximal production of the extracellular matrix genes collagen types 1 and 3 (Col1a2 and Col3a1) and collagen protein. These data are in agreement with data from other models of liver injury that show a reduction of the severity of biliary fibrosis as a result of genetic and pharmacological inhibition of Ly6chi macrophages (15, 20, 21, 30). This reduction in fibrosis occurs through a decrease in the number of Ly6chi macrophages, as well as decreased levels of secreted profibrotic cytokines, including TGF-β and PDGF-B. In contrast, Ly6clo macrophages are associated with periods of maximal fibrosis regression in the liver, and blockade of Ly6clo macrophage accumulation slowed the resolution of fibrosis (3, 20, 25, 26). The influence of Ly6clo macrophages on fibrosis resolution occurred through production of matrix-degrading enzymes (3, 25). The involvement of Ly6chi and Ly6clo macrophages in biliary hyperplasia and cyst formation remains to be determined.
The goal of this study is to better define the contribution and importance of infiltrating and resident macrophages during the progression of HRFCD. Importantly, these are the first studies to investigate the contribution of these macrophage subsets in a mouse model of HRFCD. Our data indicate that a specific macrophage subpopulation, Ly6chi infiltrating macrophages, accumulates before the onset of severe biliary hyperplasia and fibrosis. Furthermore, inhibition of this population in IFT88Orpk FVB CCR2−/− mice led to reduced biliary fibrosis but did not impact biliary hyperplasia. These data suggest a role for the infiltrating macrophages in promoting fibrosis, but not biliary hyperplasia, and raise the possibility that targeting infiltrating macrophages may be an effective therapeutic strategy to reduce biliary fibrosis in human patients with HRFCD.
MATERIALS AND METHODS
Mice.
IFT88Orpk male and female mice on the FVB background were bred in-house at the University of Alabama at Birmingham (UAB). Animals were maintained in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International in accordance with Institutional Animal Care and Use Committee regulations at the UAB. An approximately equal number of male and female mice were analyzed at each time point.
FVB CCR2−/− mice were generated by targeting the Cas9 guide sequence to the 5′ end of exon 3 of the CCR2 gene, which comprises 1,122 bp. This led to a 1,074-bp deletion plus a 2-bp insertion in the following region: ATGGAAGACAATAATATGTTACCTCAGTTCATCCAtgGGTTGGGTTGTAA, where the lowercase letters represent the 2-bp insertion. Deletion of a majority of the CCR2 gene led to a functional knockout, as shown in Fig. 8.
Fig. 8.

Ly6chi macrophages and fibrosis are reduced in IFT88Orpk FVB CCR2−/− mice. A: PCR analysis of DNA extracted from wild-type (WT), heterozygous (het), and knockout (mut) mice for the CCR2 gene. B: representative fluorescence-activated cell sorting plots of infiltrating macrophage subtypes harvested from control (cont) and IFT88Orpk (mut) mice on control (cont) or CCR−/− (mut) background at 4 wk of age with quantification of Ly6chi and Ly6clo macrophages from their respective groups (n = 4–5). C: representative histological images of liver sections from 4-wk-old mice (n = 6–8). Cystic index was quantified for IFT88Orpk CCR2 control and IFT88Orpk CCR2−/− mice. Cholangiocyte number was quantified using flow cytometry [(cytokeratin 19 (Ck19)-positive cells] and quantitative RT-PCR for Ck19. D: representative picrosirius red-stained sections from 4-wk-old mice (n = 6–8) with quantification of picrosirius red-positive area and quantitative RT-PCR data for collagen types 1 and 3 (Col1a2 and Col3a1). Values are means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001.
Fixation and tissue processing.
Livers from control and IFT88Orpk cilia mutant mice were harvested at postnatal days 14 and 28 (2 and 4 wk of age). For harvesting, mice were anesthetized with tribromoethanol (Avertin) and a section of liver tissue was removed for histology. Mice were then perfused with 20 ml of PBS, and the remaining liver tissue was removed for further processing. The liver was fixed by immersion in 4% (wt/vol) paraformaldehyde overnight at 4°C.
Paraffin embedding and hematoxylin-eosin staining.
After overnight fixation, liver samples were dehydrated in 70% ethanol overnight and then embedded in paraffin according to standardized procedures at the UAB Comparative Pathology Laboratory. Paraffin-embedded tissue was sectioned at 5 µm and stained using hematoxylin and eosin. Picrosirius red staining of liver sections was performed by the UAB Comparative Pathology Laboratory.
Immunofluorescence microscopy.
After harvest and fixation, liver tissue was cryopreserved by immersion in 30% sucrose (wt/vol) in PBS, embedded in optimal cutting temperature compound (OCT), and frozen at −80°C. OCT-embedded liver tissue was sliced in a cryostat at −14°C into 8-µm-think sections. Immunofluorescence microscopy was performed on stained OCT-embedded sections. Briefly, cryosectioned liver tissue was washed with 1× PBS and fixed with 4% paraformaldehyde for 10 min. The sections were permeabilized with 1% Triton X-100 for 8 min and blocked in PBS with 1% BSA, 0.3% Triton X-100, 2% (vol/vol) donkey serum, and 0.02% sodium azide for 30 min at room temperature. The sections were incubated in primary antibody overnight at 4°C and then in the appropriate secondary antibody for 1 h at room temperature. Primary antibodies included F4/80 (1:200 dilution; catalog no.14-4801, clone BM8, eBioscience) cytokeratin 19 (1:150 dilution; catalog no. 52625, Abcam), and smooth muscle actin (SMA)-Cy3 (1:300 dilution; catalog no. C6198, Sigma). All primary and secondary antibody incubations were performed in 1× PBS with 1% BSA, 0.3% Triton X-100, 2% donkey serum, and 0.02% sodium azide. Secondary antibodies included Alexa Fluor 647-conjugated donkey anti-rat (1:250 dilution; catalog no. 712-606-153, Jackson ImmunoResearch) and fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit (1:250 dilution; catalog no. 711-096-152, Jackson ImmunoResearch). After addition of the secondary antibody, slides were washed and nuclei were stained by Hoechst nuclear stain (Sigma-Aldrich). Coverslips were mounted using Immu-Mount (Thermo Scientific). All fluorescence images were captured on a spinning-disk confocal microscope (model ERS 6FE, Perkin-Elmer), and images were processed and analyzed in Volocity software (version 6.1.1, Perkin-Elmer).
Quantification of cystic index.
For our data, we used ImageJ software to quantify epithelial expansion and cyst formation in the liver. Briefly, regions exhibiting bile duct hyperplasia were circled using the trace tool, and white spaces in those regions were quantified. For this quantification, portal vein regions and other nonepithelial regions were excluded based on pathology. For this analysis, we quantified all the bile duct regions in three ×10 images from each mouse. The average cystic indexes from the three ×10 images were averaged to generate the cystic index from each mouse. For these quantifications, reviewers were blinded to genotype.
Flow cytometry.
After perfusion with PBS, liver sections were removed and added to RPMI 1640 medium on ice. Harvested liver tissue was mechanically digested through a 70-µm mesh (Falcon, BD Biosciences), yielding single-cell suspensions. Cells were centrifuged at 1,300 rpm for 5 min, and red blood cells were lysed using ammonium-chloride-potassium (ACK) red blood cell lysis buffer (catalog no. 10128-802, Quality Biological) at 37°C for 5 min. Cells were spun at 1,300 rpm, resuspended in 1 ml of 1% BSA containing Fc blocking solution (1:200 dilution), and incubated for 30 min on ice. After cell counting with Trypan blue, ~2 × 106 cells were stained for 30 min at room temperature with conjugated primary antibodies, including phycoerythrin (PE)-Cy7-rat anti-mouse CD45 (1:300 dilution; catalog no. 552848, clone 30-F11, BD Bioscience), eFluor 450-rat anti-mouse F4/80 (1:200 dilution; catalog no. 48-4801, clone BM8, eBioscience), allophycocyanin (APC)-rat anti-mouse CD11b (1:200 dilution; catalog no. 17-0112, clone M1/70, eBioscience), APC/Cy7-rat anti-mouse Gr-1 (1:100 dilution; catalog no. 557661, clone RB6-8C5, BD PharMingen), and peridinin-chlorophyll protein complex (PerCP)-Cy5.5-rat anti-mouse Ly6c (1:200 dilution; catalog no. 560525, clone AL-21, BD PharMingen). After exposure to the primary antibody, cell suspensions were incubated with Foxp3 fixation/permeabilization solution (eBioscience) for 30 min at room temperature. Cells were washed twice with permeabilization buffer and incubated with rat anti-mouse Ki67-FITC (1:200 dilution; catalog no. 11-5698-82, clone SolA15, eBioscience) and rabbit anti-mouse cytokeratin 19 (1:250 dilution; catalog no. 52625, Abcam). Cells were washed with permeabilization buffer and incubated with anti-rabbit rhodamine (1:150 dilution; catalog no. 711-026-152, Jackson Laboratories). Cells were washed with permeabilization buffer and resuspended in 1× PBS. After immunostaining, the cells were analyzed on a flow cytometer (model LSRII, Becton Dickinson). Data were analyzed using FlowJo v10 software.
Fluorescein-activated cell sorting of individual populations for quantitative RT-PCR.
For flow sorting of individual cell populations from the whole liver, livers were harvested and digested in RPMI 1640 medium containing 0.5 mg/ml collagenase with 100 U/ml DNase I for 30 min at 37°C. A single-cell suspension was obtained, and cells were stained as described above. For isolation of cholangiocytes, we used the cell surface marker Dolichos biflorus agglutinin (catalog no. RL-1032, Vector Laboratories). Cells were sorted into individual tubes containing 1% BSA in PBS, spun at 1,200 rpm for 5 min, and resuspended in TRIzol.
RNA isolation and quantitative RT-PCR.
RNA was isolated and transcribed into cDNA, and quantitative RT-PCR (qRT-PCR) was performed using TaqMan real-time PCR. The following probes were used: collagen type 3 (Col3a1, Mm01254476_m1), collagen type 1 (Col1a2, Mm00483937_m1), PDGF-B (Mm00440677_m1), VEGF-A (Mm00437306_m1), cytokeratin 19 (Mm00492980_m1), IL-6 (Mm00446190_m1), SMA (Mm01546133_m1), TNF-α (Mm00443258_m1), IL-1β (Mm01336189_m1), CCL2 (Mm00441242_m1), TGF-β (Mm01178820_m1), hypoxanthine phosphoribosyltransferase (HPRT, Mm00446968_m1), and colony-stimulating factor 1 (CSF-1, Mm00432686_m1).
Statistics.
Values are means ± SE. ANOVA and Student’s t-tests were used for statistical analysis, and differences were considered significant at P ≤ 0.05.
RESULTS
IFT88Orpk cilia mutant mice have rapid expansion of biliary regions between 2 and 4 wk of age.
IFT88Orpk is a hypomorphic mutant with reduced levels of IFT88 expression, leading to short, malformed cilia (39, 40). These mice have hepatorenal dysfunction related to cholestasis and, on the FVB background, succumb to disease within 4–5 wk of age. In these studies we began by characterizing the biliary phenotype at 2 and 4 wk of age, which represent early and late periods of disease progression. At 2 wk, IFT88Orpk mice have mild expansion of the biliary region, presumably due to abnormal ductal plate closure (27). In contrast, at 4 wk, cilia mutant mice have a significant expansion of the biliary region (Fig. 1A). The increased cholangiocyte number in cilia mutant mice at 2 and 4 wk of age was confirmed by flow cytometry analysis of liver tissue using the cholangiocyte-specific marker cytokeratin 19 (Fig. 1B) (19). The percentage of cytokeratin 19-positive cholangiocytes relative to total cells decreased between 2 and 4 wk of age in IFT88Orpk mice as a result of significant liver growth in control and IFT88Orpk mice during this period; however, the number of cholangiocytes per gram of tissue increased nearly fivefold in 4-wk-old cilia mutant mice (Fig. 1, C and D) (2). The increase in cholangiocyte number in cilia mutant mice at 2 and 4 wk of age was due to increased cholangiocyte proliferation (Fig. 1E).
Fig. 1.

Rapid expansion of the biliary region in cilia mutant mice between 2 and 4 wk of age. A: hematoxylin-eosin-stained sections of liver tissue harvested from control and cilia mutant mice at 2 and 4 wk of age. Inset regions within black boxes in top mutant images are shown in bottom mutant images. B: quantification of cholangiocyte number in 2- and 4-wk-old mice (n = 5–10) as determined by flow cytometry for cytokeratin 19-positive cells. C: liver weight of control and cilia mutant livers at 2 and 4 wk (n = 5–7 mice). D: quantification of absolute numbers of cholangiocytes in control and cilia mutant livers at 4 wk (n = 4–5 mice). E: number of proliferating cholangiocytes as a percentage of total cells quantified using flow cytometry staining for Ki67, cytokeratin 19 double-positive cells (n = 5–7 mice). Values are means ± SE. *P < 0.05; **P < 0.01.
Cilia mutant mice develop periportal fibrosis at 4 wk of age.
Biliary fibrosis is a common manifestation in patients with hepatorenal dysfunction (12). Therefore, using picrosirius red staining, the hydroxyproline assay, and qRT-PCR data from whole liver tissue, we analyzed the involvement of periportal fibrosis in IFT88Orpk mutants. At 2 wk of age, cilia mutant mice had minimal accumulation of collagen proteins; however, 4-wk-old IFT88Orpk cilia mutant mice had extensive picrosirius red staining and collagen protein accumulation compared with controls (Fig. 2A). Increased collagen accumulation in 4-wk-old mice was confirmed using the hydroxyproline assay (Fig. 2B). Furthermore, analysis of mRNA from whole livers of 4-wk-old IFT88Orpk mice indicated an increase in expression of the profibrotic genes TGF-β and PDGF-B and increased levels of Col1a2, Col3a1, and α-SMA (Fig. 2C). Across all parameters tested, matrix production was increased in 4-wk-old IFT88Orpk mice compared with 4-wk-old control mice and 2-wk-old IFT88Orpk mice. Collectively, these results demonstrate a rapid onset of periportal fibrosis in the IFT88Orpk mutant mice between 2 and 4 wk of age.
Fig. 2.

IFT88Orpk mice develop fibrosis in periportal regions at 4 wk of age. A: picrosirius red-stained sections of liver tissue from 2- and 4-wk-old control and cilia mutant mice. B: quantification of picrosirius red-stained area (n = 3–4 mice) and hydroxyproline assay of livers from 2- and 4-wk-old mice (n = 4–5 mice). C: quantitative RT-PCR determination of TGF-β, PDGF-B, collagen types 1 and 3 (Col1a2 and Col3a1), and α-smooth muscle actin (SMA) expression in mRNA isolated from whole liver of 2- and 4-wk-old control and IFT88Orpk mice (n = 5–8). For each gene analyzed, expression levels in control mice were set to 1. RQ, relative quantification; HPRT, hypoxanthine phosphoribosyltransferase. Values are means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Cilia mutant mice have accumulation of macrophages and SMA+ myofibroblasts in regions of biliary hyperplasia.
Macrophages have been identified as potential contributors to cyst formation and fibrosis in several mouse models of PKD (14, 16, 35). Therefore, using immunofluorescence microscopy and the pan-macrophage marker F4/80, we analyzed whether macrophages were present in liver from IFT88Orpk mice before and during disease progression. At 2 wk of age, IFT88Orpk cilia mutant mice show a mild accumulation of macrophages in regions of bile duct expansion and the absence of periportal SMA+ myofibroblasts (Fig. 3A). In contrast, at 4 wk of age, IFT88Orpk mice have extensive macrophage accumulation in regions of biliary dysmorphogenesis and an associated accumulation of SMA+ myofibroblasts (Fig. 3B). The large accumulation of macrophages and myofibroblasts in regions of cholangiocyte hyperplasia raises the possibility that loss of cholangiocyte cilia alters normal homeostatic communication between cholangiocytes, myofibroblasts, and macrophages, which promotes disease progression between 2 and 4 wk of age in IFT88Orpk mice.
Fig. 3.

F4/80+ macrophages accumulate in regions of α-smooth muscle actin (SMA)-positive myofibroblasts and cytokeratin 19 (Ck19)-positive cholangiocytes in 4-wk-old IFT88Orpk mice. Livers were harvested from 2- and 4-wk-old control and IFT88Orpk mice (n = 4–6) and stained with cytokeratin 19 (cholangiocytes), F4/80 (pan-macrophage marker), and SMA (myofibroblasts). A: immunofluorescence confocal microscopy shows mild accumulation of F4/80+ cells in regions of cholangiocyte expansion in 2-wk-old IFT88Orpk mutant mice. Inset region is depicted with a white box. B: immunofluorescence confocal microscopy shows severe accumulation of F4/80+ macrophages (white) in regions of cholangiocyte (green) expansion in IFT88Orpk mice. Accumulation of SMA+ myofibroblasts (red) in regions containing cholangiocyte expansion and macrophages suggests possible communication between these cells. A representative image is shown for each group of mice. Inset region is depicted with a white box. Arrows denote region containing numerous SMA+ myofibroblasts.
Livers from 2-wk-old IFT88Orpk mice demonstrate an increase in infiltrating Ly6chi macrophages that greatly expands in 4-wk-old IFT88Orpk mice.
To further classify the macrophage subtypes associated with disease progression in the IFT88Orpk mice, using F4/80 and CD11b to distinguish infiltrating bone marrow-derived macrophages (F4/80lo, CD11bhi) from tissue resident macrophages (F4/80hi, CD11blo), we performed flow cytometry analysis of liver tissue from control and IFT88Orpk cilia mutant mice (11, 28). A majority of macrophages present in steady-state conditions in wild-type mice at 2 and 4 wk of age are tissue resident Kupffer cells (10 and 24% of CD45+ cells, respectively) (Fig. 4, A–C). These data are in agreement with a previous report showing that the number of resident macrophages as a percentage of CD45+ cells increases during liver development (29). Remarkably, the macrophage profile of liver tissue from IFT88Orpk cilia mutant mice shifts dramatically at 4 wk of age, with infiltrating macrophages representing the predominant macrophage population both as a percentage of total cells and as a percentage of CD45+ immune cells (Fig. 4, A–C). The increased number of infiltrating macrophages as a percentage of total cells occurs before the onset of biliary fibrosis (Fig. 4B). Our data also show that the number of resident macrophages decreases in IFT88Orpk mice between 2 and 4 wk of age. Collectively, these data suggest that the transition in the macrophage populations between 2 and 4 wk of age in liver tissue from wild-type mice fails to occur normally when cilia are disrupted.
Fig. 4.

Flow cytometry reveals a shift in macrophage profile in IFT88Orpk mice during postnatal liver development and an accumulation of Ly6chi macrophages in IFT88Orpk mice before cholangiocyte expansion and periportal fibrosis. Livers were harvested from control and IFT88Orpk mice at 2 and 4 wk of age, stained, and live single cells were analyzed based on surface expression of CD11b and F4/80. A: representative fluorescence-activated cell sorting (FACS) plot is shown. B and C: quantification of infiltrating and resident macrophages gated as a percentage of total cells and as a percentage of immune cells (CD45+) (n = 5–10 mice). D: representative FACS plot of infiltrating macrophages that were further subgated based on expression of Ly6c. Ly6chi macrophages were quantified as a percentage of total infiltrating macrophages (n = 5–10). E: whole livers from 4-wk-old control and IFT88Orpk mice (n = 4–6) were sorted, and absolute numbers of Ly6chi infiltrating macrophages and tissue resident macrophages were determined by flow cytometry. Cell numbers were normalized to average liver weight, as shown in Fig. 1C. Values are means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001.
High levels of Ly6c expression on infiltrating macrophages are typically associated with the profibrotic gene expression, and previous data indicate that genetic or pharmacological inhibition of the Ly6chi population reduces liver disease and fibrosis (7, 21, 25). To evaluate whether there are differences in Ly6c subpopulations associated with the increased fibrosis in the IFT88Orpk mice, we conducted flow cytometry analysis on the infiltrating macrophages isolated from liver tissue of 2- and 4-wk-old IFT88Orpk and control mice. Our data show an increased proportion of Ly6chi macrophages in IFT88Orpk mutant mice compared with their controls by 2 wk of age (66 vs. 58%; Fig. 4D). This population further increased to 84% of the infiltrating macrophages in the livers of 4-wk-old IFT88Orpk mice compared with 62% in livers from control mice (Fig. 4D). The increase in Ly6chi infiltrating macrophages and the decrease in resident macrophage number in 4-wk-old IFT88Orpk mice compared with controls were validated by obtaining absolute macrophage cell numbers from the whole liver and normalizing to average liver weight (Fig. 4E).
mRNA expression of proinflammatory and macrophage chemoattractant cytokines is increased in IFT88Orpk mice.
To address changes in the liver environment that may be responsible for the altered macrophage accumulation, biliary hyperplasia, and fibrosis in IFT88Orpk mice, whole liver tissue from 2- and 4-wk-old mice was analyzed using qRT-PCR. At 2 wk of age, IFT88Orpk mice have increased gene expression of the macrophage chemoattractant cytokine CCL2 (monocyte chemoattractant protein 1) compared with controls and elevated expression of the proinflammatory cytokine IL-1β (Fig. 5). These findings suggest active recruitment of infiltrating macrophages to the liver at 2 wk of age, although severe pathology is not yet present. In contrast, 4-wk-old IFT88Orpk mice have a robust and significant increase in gene expression of the macrophage recruitment chemoattractants CCL2 and CSF-1 and proinflammatory cytokines TNF-α and IL-1β (Fig. 5) (34, 43). The greater increase in proinflammatory gene expression in 4-wk-old compared with 2-wk-old IFT88Orpk mice is likely due to the persistently elevated levels of infiltrating macrophages.
Fig. 5.

Expression of proinflammatory and profibrotic cytokines is increased in IFT88Orpk mice. Livers from 2- and 4-wk-old mice were harvested and minced, and mRNA was extracted using TRIzol. Transcript levels of chemokine (C-C) motif ligand 2 (CCL2), IL-1β, colony-stimulating factor 1 (CSF-1), and TNF-α from whole liver tissue were determined by quantitative RT-PCR and normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase (HPRT). For each gene, expression levels in 2-wk-old control mice were set to 1. RQ, relative quantification. Values are means ± SE; n = 5–7. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Analysis of fluorescence-activated cell-sorted liver cells from IFT88Orpk mice shows increased expression of genes coding for immune recruitment cytokines in cilia mutant epithelial cells and increased expression of genes known to drive liver disease in macrophage populations.
To address the reciprocal communication between the biliary epithelium and the altered macrophage profiles in IFT88Orpk mice, we performed qRT-PCR on mRNA isolated from sorted cholangiocytes, Ly6chi macrophages, and resident macrophages (Kupffer cells). Importantly, the main cell type in the liver containing primary cilia is the bile duct epithelial cell (cholangiocyte) (17). Analysis of qRT-PCR data shows increased gene expression of CCL2 (Fig. 6), a chemoattractant involved in recruitment of infiltrating macrophages from the bone marrow, in cholangiocytes isolated from cilia mutant mice. In contrast, gene expression of CSF-1 was not different between control and cilia mutant epithelium (Fig. 6). While we did not detect differences in CSF-1 in cilia mutant epithelium, it is likely that the large increase in whole liver CSF-1 expression in IFT88Orpk mice at 4 wk of age (Fig. 5) is driven by an overall increase in the number of cholangiocytes or the production of this cytokine by other cell types such as liver sinusoidal endothelial cells (13).
Fig. 6.
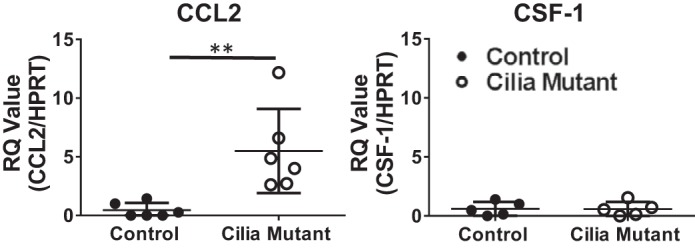
Expression of chemokine (C-C) motif ligand 2 (CCL2) is increased in epithelial cells from IFT88Orpk mice. Expression of CCL2 and colony-stimulating factor 1 (CSF-1) in fluorescence-activated cell-sorted control and cilia mutant epithelial cells was determined by quantitative RT-PCR. RQ, relative quantification; HPRT, hypoxanthine phosphoribosyltransferase. Values are means ± SE; n = 6. **P < 0.01.
To determine how changes in macrophage subpopulations may drive disease pathology in IFT88Orpk mice, we analyzed expression of proproliferative and profibrotic cytokines in macrophages isolated from 4-wk-old control and IFT88Orpk mutant mice. In Ly6chi macrophages from IFT88Orpk mutant mice, gene expression of IL-6 and VEGF-A, cytokines previously shown to drive cholangiocyte proliferation in cystic liver disease, is increased (Fig. 7) (1, 4, 31, 32, 37). We were surprised to find no difference in the level of TGF-β or PDGF-B mRNA expression between control and cilia mutant Ly6chi macrophages. While gene expression levels of TGF-β or PDGF-B are not increased in Ly6Chi macrophages, the number of Ly6chi macrophages is significantly higher in cilia mutant than control liver at this time point (Fig. 4) and, thus, likely contributes to the increased expression of these genes detected upon analysis of the whole liver. Analysis of mRNA expression in sorted resident macrophages shows no differences in TGF-β and PDGF-B expression but a small increase in VEGF-A expression, although not to a statistically significant level (P = 0.065; Fig. 7). Overall, these data suggest that the differences in gene expression intrinsic to the Ly6chi macrophage population, as well as changes in the number of Ly6chi macrophages, contribute to biliary hyperplasia and fibrosis.
Fig. 7.

Increased gene expression of proinflammatory and profibrotic cytokines in Ly6chi macrophages from IFT88Orpk mice. Gene expression of proproliferative cytokines (IL-6 and VEGF-A), profibrotic growth factors (TGF-β and PDGF-B), and Kdr, which encodes VEGF receptor 2 (VEGFR2), in cholangiocytes, infiltrating macrophages, and resident macrophages isolated from the liver of 4-wk-old control and cilia mutant mice was determined by quantitative RT-PCR. RQ, relative quantification; HPRT, hypoxanthine phosphoribosyltransferase. Values are means ± SE; n = 5–6. *P < 0.05; **P < 0.01.
IFT88Orpk FVB CCR2−/− mice have reduced Ly6chi macrophage accumulation and fibrotic disease.
To determine the importance of Ly6chi macrophages in biliary hyperplasia and fibrosis, we used CRISPR/Cas9 technology to generate a 1,074-bp deletion plus a 2-bp insertion in exon 3 of the CCR2 gene in FVB mice. CCR2 deletion was confirmed by PCR genotyping (Fig. 8A). Similar to previous reports, genetic deletion of CCR2 significantly reduced the number of Ly6chi macrophages in the liver, while Ly6clo macrophage number was unaffected (Fig. 8B) (21, 30). We were surprised to find that genetic inhibition of Ly6chi macrophages did not reduce epithelial pathology, cystic index, cholangiocyte number, or cytokeratin 19 mRNA levels (Fig. 8C), indicating that the Ly6chi macrophages do not have an obvious role in promoting cholangiocyte proliferation or bile duct hyperplasia. In contrast, IFT88Orpk CCR2−/− double mutants did not have a significant increase in collagen protein or levels of collagen 1 and 3 mRNA compared with control mice. IFT88Orpk CCR2−/− mice also had reduced collagen protein levels as determined by picrosirius red staining and reduced Col1a2 and Col3a1 gene expression compared with IFT88Orpk CCR2 control mice (Fig. 8D). Collectively, these data show that Ly6chi infiltrating macrophages in IFT88Orpk mice promote fibrotic disease but not epithelial expansion. Thus, targeting Ly6chi macrophages may be a potential target for intervention to reduce complications of fibrosis in patients with HRFCD.
DISCUSSION
The underlying mechanism through which dysfunctional cilia on epithelial cells lead to HRFCD is unknown. We have demonstrated rapid expansion of the bile duct region and development of periportal fibrosis in IFT88Orpk cilia mutant mice on the FVB background between 2 and 4 wk of age. This period of rapidly increasing liver pathology is preceded by increased levels of Ly6chi infiltrating macrophages and expression of the macrophage chemoattractant cytokine CCL2. qRT-PCR analysis of mRNA isolated from sorted cilia mutant cholangiocytes shows that this cell population is predominantly responsible for the increased expression of CCL2 in whole liver tissue. Furthermore, the data indicate that Ly6chi infiltrating macrophages produce growth factors and cytokines that can drive biliary fibrosis. Finally, we show that genetic inhibition of Ly6chi infiltrating macrophage accumulation in IFT88Orpk CCR2−/− mice significantly reduces liver fibrosis, but does not affect biliary hyperplasia, indicating that these two phenotypes likely occur through different mechanisms. Based on these data, we suggest a model whereby the presence of dysfunctional cilia on bile duct cholangiocytes leads to abnormal production of CCL2, possibly due to a perceived low level of injury. This results in increased recruitment of Ly6chi infiltrating macrophages and production of excess proinflammatory/profibrotic cytokines that stimulate cholangiocytes and myofibroblasts to produce extracellular matrix proteins, leading to biliary fibrosis (Fig. 9). Fibrosis is a major clinical complication of HRFCD; thus, targeting this specific macrophage compartment may be effective in reducing liver fibrosis in HRFCD patients.
Fig. 9.

Proposed mechanism responsible for enhanced bile duct expansion and fibrosis in IFT88Orpk liver. Schematic depicts reciprocal signaling between bile duct cholangiocytes, macrophages, and myofibroblasts during development of hepatic fibrocystic liver disease. In our model, the presence of dysfunctional primary cilia on cholangiocytes leads to persistent low levels of injury, increased production of macrophage chemoattractant cytokines [chemokine (C-C) motif ligand 2 (CCL2)], and increased numbers of Ly6chi infiltrating macrophages. Increased numbers Ly6chi infiltrating macrophages promote expansion of biliary fibrosis in 4-wk-old IFT88Orpk mice.
The presence of macrophages during disease pathogenesis in mouse models of PKD is known, although whether these cells are a cause or a consequence of the disease is controversial (14, 16, 35). Recently, Locatelli and colleagues identified phagocytic macrophages as important contributors to liver disease progression in a PKHD1 model of autosomal-recessive PKD (16). In agreement with this report, data from two previous studies demonstrated an improvement in renal cyst severity and fibrosis following depletion of phagocytic macrophages by liposomal clodronate (14, 35). Inhibition of macrophage recruitment (presumably infiltrating macrophages) by the pharmacological inhibitor bindarit did not rescue renal or hepatic cysts in the PCK cystic rat model, although the level of macrophage reduction only reached ~40% in this study (44).
Recent data indicate that unique macrophage populations in the liver can be identified through differential expression of CD11b and F4/80 (28). Bone marrow-derived macrophages express high levels of CD11b and low levels of F4/80 (CD11bhi, F4/80lo), whereas resident macrophages express high levels of F4/80 and low levels of CD11b (F4/80hi, CD11blo). Our data show that infiltrating macrophages are significantly increased in IFT88Orpk mutant mice. Importantly, in the IFT88Orpk mouse, accumulation of infiltrating macrophages occurs before any overt signs of biliary fibrosis and persists until our terminal time point of analysis at 4 wk of age. Further subtyping of this macrophage population shows a preferential recruitment of Ly6chi infiltrating macrophages to the liver in IFT88Orpk mutants at 2 wk of age that is further exacerbated at 4 wk of age.
We were surprised to find a decrease in the number of resident macrophages in cilia mutant mice at 2 and 4 wk of age, suggesting that functional primary cilia are needed for proper regulation of resident macrophage maintenance or accumulation in the liver. Also, resident macrophage number is reduced in other models of liver injury, supporting a model wherein the decrease in resident macrophages in the IFT88Orpk mouse is due to a persistent state of low-level liver injury (43). It is also possible that the reduction of resident macrophage number in cilia mutant mice is due to the inability of resident macrophages to seed the liver in the absence of a primary cilium or that primary cilia are needed to maintain resident macrophage survival. Conditional cilia mutant approaches are being utilized to address this question. These data indicate that primary cilia in the liver tightly regulate both the infiltrating and the resident macrophage niche in an attempt to regulate disease pathogenesis.
The IFT88Orpk mouse model of hepatorenal disease exhibits global cilia dysfunction. While a recent report suggested that mononuclear cells contain primary cilia, our laboratory and others have been unable to detect primary cilia on macrophages (8). In these studies and others, IFT88 gene expression in control resident macrophages was detected by qRT-PCR; however, immunofluorescence analysis of sorted macrophage populations indicates that this is likely a result of low-level contamination with CD45− epithelial or endothelial cells (unpublished observations). Furthermore, the IFT88Orpk mouse was generated through insertion of a 21-kb fragment into an intronic region near the 3′ end of the IFT88 gene (23). This insertion leads to alternative transcript production and hypomorphic levels of the IFT88 protein. The probes used to detect IFT88 in this assay were targeted to the region spanning exons 4–5; therefore, even if the mRNA is produced, as observed in cilia mutant macrophages, it is unlikely that a functional protein is produced. Overall, it is unlikely that the hepatic phenotype in these mice is due to intrinsic ciliary-mediated dysfunction on the immune cells or macrophages themselves. Our unpublished observations also indicate that specific deletion of cilia on cholangiocytes by backcrossing the IFT88 conditional line onto cytokeratin 19 promoter-driven cre recombinase (K19creERIFT88f/f) leads to abnormal immune cell accumulation following injury. Furthermore, qRT-PCR analysis of sorted cholangiocytes shows that cilia mutant epithelium produces elevated levels of macrophage chemoattractants, suggesting that cilia dysfunction on the cholangiocyte epithelium is the major driver of macrophage accumulation in IFT88Orpk mice.
Previous reports identified VEGF receptor signaling as an important pathway promoting cyst formation in the liver. Inhibition of VEGF receptor signaling by SU-5416 significantly reduced liver cyst growth in both the PKD2WS25/− and the inducible PKD2 models of polycystic liver disease (1, 31, 32). In these reports, it was shown that VEGF and its receptors localized to the cystic biliary epithelium, similar to our observation (Fig. 7). However, our results indicate that Ly6chi infiltrating macrophages, but not cholangiocytes, are the predominant sources of VEGF-A in the IFT88Orpk model of HRFCD (Fig. 7). We were surprised to find that blockade of Ly6chi infiltrating macrophages did not reduce epithelial cyst formation, suggesting that VEGF-A is not the driving force behind epithelial cell accumulation in IFT88Orpk mice or that other cells produce different cytokines that drive epithelial expansion in the absence of Ly6chi infiltrating macrophages. The fact that prevention of Ly6chi macrophage accumulation, a major producer of VEGF-A, did not rescue epithelial expansion in the IFT88Orpk mouse likely indicates that the pathways responsible for epithelial expansion in models of cilia deficiency (Ift88) differ from mouse models of cilia dysfunction (Pkd2). Unravelling these differences will be a key factor in understanding the complex mechanisms driving biliary cyst formation and fibrosis. Overall, these data suggest that epithelial cell expansion and biliary fibrosis are likely driven through independent mechanisms, with fibrosis being controlled by the presence of hepatic Ly6chi infiltrating macrophages.
This work provides new insight into the involvement of macrophage subtypes during the initiation and progression of the biliary phenotype in a mouse model of HRFCD. We identify a specific subpopulation of macrophages that accumulate before the onset of severe liver pathology and show that blockade of this population reduces biliary fibrosis. These data lead us to propose that cilia dysfunction on cholangiocytes initiates a state of low-level injury that results in increased production of macrophage chemoattractant cytokines and enhanced accumulation of Ly6hi infiltrating macrophages. These macrophages participate in abnormal intercellular communication with myofibroblasts, leading to biliary fibrosis. Based on these data, we propose that targeting this population of macrophages may have therapeutic benefit in HRFCD patients with abnormal hepatic macrophage accumulation.
GRANTS
These studies were supported in part by Polycystic Kidney Disease Research Foundation Grant 214g16a (B. K. Yoder), a University of Alabama at Birmingham School of Medicine AMC21 Grant (B. K. Yoder), and National Institutes of Health Grant R01 DK-115752 (B. K. Yoder) and Training Grant in Basic Immunology and Immunologic Disease 2T32 AI-007051-38 (K. A. Zimmerman).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.A.Z., C.J.S., and B.K.Y. conceived and designed research; K.A.Z., C.J.S., N.G.-M., and Z.L. performed experiments; K.A.Z., C.J.S., N.G.-M., and Z.L. analyzed data; K.A.Z., C.J.S., N.G.-M., Z.L., and B.K.Y. interpreted results of experiments; K.A.Z. prepared figures; K.A.Z. and C.J.S. drafted manuscript; K.A.Z., C.J.S., N.G.-M., Z.L., and B.K.Y. edited and revised manuscript; K.A.Z., C.J.S., N.G.-M., Z.L., and B.K.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank members of the Yoder laboratory for suggestions and technical support, as well as Dr. Etty N. Benveniste and members of her laboratory.
REFERENCES
- 1.Amura CR, Brodsky KS, Groff R, Gattone VH, Voelkel NF, Doctor RB. VEGF receptor inhibition blocks liver cyst growth in pkd2WS25/− mice. Am J Physiol Cell Physiol 293: C419–C428, 2007. doi: 10.1152/ajpcell.00038.2007. [DOI] [PubMed] [Google Scholar]
- 2.Behrens A, Sibilia M, David JP, Möhle-Steinlein U, Tronche F, Schütz G, Wagner EF. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J 21: 1782–1790, 2002. doi: 10.1093/emboj/21.7.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brempelis KJ, Crispe IN. Infiltrating monocytes in liver injury and repair. Clin Transl Immunology 5: e113, 2016. doi: 10.1038/cti.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen LP, Qian YY, Li ZL, Bai HW, Cai M, Shi BY. [Role of IL-6/STAT3 in rat cholangiocyte proliferation induced by lipopolysaccharide]. Zhonghua Gan Zang Bing Za Zhi 17: 374–377, 2009. [PubMed] [Google Scholar]
- 5.Cowley BD Jr, Gudapaty S, Kraybill AL, Barash BD, Harding MA, Calvet JP, Gattone VH II. Autosomal-dominant polycystic kidney disease in the rat. Kidney Int 43: 522–534, 1993. doi: 10.1038/ki.1993.79. [DOI] [PubMed] [Google Scholar]
- 6.Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, Ransohoff RM, Charo IF, Jenne CN, Kubes P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med 212: 447–456, 2015. doi: 10.1084/jem.20141539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Möckel D, Baeck C, Hittatiya K, Eulberg D, Luedde T, Kiessling F, Trautwein C, Lammers T, Tacke F. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 63: 1960–1971, 2014. doi: 10.1136/gutjnl-2013-306294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol 11: 1332–1339, 2009. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, Ishiwata Y, Asano M, Wang H, Matsushima K, Takeya M, Kuziel WA, Mukaida N, Yokoyama H. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol 14: 2503–2515, 2003. doi: 10.1097/01.ASN.0000089563.63641.A8. [DOI] [PubMed] [Google Scholar]
- 10.Gallagher AR, Esquivel EL, Briere TS, Tian X, Mitobe M, Menezes LF, Markowitz GS, Jain D, Onuchic LF, Somlo S. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am J Pathol 172: 417–429, 2008. doi: 10.2353/ajpath.2008.070381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518: 547–551, 2015. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunay-Aygun M, Gahl WA, Heller T. Congenital hepatic fibrosis overview. In: GeneReviews, edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K. Seattle, WA: University of Washington, 1993. [PubMed] [Google Scholar]
- 13.He H, Xu J, Warren CM, Duan D, Li X, Wu L, Iruela-Arispe ML. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood 120: 3152–3162, 2012. doi: 10.1182/blood-2012-04-422758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, Somlo S, Cantley LG. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 22: 1809–1814, 2011. doi: 10.1681/ASN.2011010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C, Tacke F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 50: 261–274, 2009. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 16.Locatelli L, Cadamuro M, Spirlì C, Fiorotto R, Lecchi S, Morell CM, Popov Y, Scirpo R, De Matteis M, Amenduni M, Pietrobattista A, Torre G, Schuppan D, Fabris L, Strazzabosco M. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 63: 965–982, 2016. doi: 10.1002/hep.28382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn 237: 2007–2012, 2008. doi: 10.1002/dvdy.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med 169: 1485–1490, 1989. doi: 10.1084/jem.169.4.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Means AL, Xu Y, Zhao A, Ray KC, Gu G. A CK19CreERT knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis 46: 318–323, 2008. doi: 10.1002/dvg.20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, Rénia L, Pol S, Mallet V, Gilgenkrantz H. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol 174: 1766–1775, 2009. doi: 10.2353/ajpath.2009.080632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 302: G1310–G1321, 2012. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, Heymann F, Kalthoff S, Lefebvre E, Eulberg D, Luedde T, Marx G, Strassburg CP, Roskams T, Trautwein C, Tacke F. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 64: 1667–1682, 2016. doi: 10.1002/hep.28682. [DOI] [PubMed] [Google Scholar]
- 23.Moyer JH, Lee-Tischler MJ, Kwon HY, Schrick JJ, Avner ED, Sweeney WE, Godfrey VL, Cacheiro NL, Wilkinson JE, Woychik RP. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 264: 1329–1333, 1994. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 24.Mrug M, Zhou J, Woo Y, Cui X, Szalai AJ, Novak J, Churchill GA, Guay-Woodford LM. Overexpression of innate immune response genes in a model of recessive polycystic kidney disease. Kidney Int 73: 63–76, 2008. doi: 10.1038/sj.ki.5002627. [DOI] [PubMed] [Google Scholar]
- 25.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, Williams MJ, Dunbar DR, Manning JR, van Rooijen N, Fallowfield JA, Forbes SJ, Iredale JP. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA 109: E3186–E3195, 2012. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rantakari P, Patten DA, Valtonen J, Karikoski M, Gerke H, Dawes H, Laurila J, Ohlmeier S, Elima K, Hübscher SG, Weston CJ, Jalkanen S, Adams DH, Salmi M, Shetty S. Stabilin-1 expression defines a subset of macrophages that mediate tissue homeostasis and prevent fibrosis in chronic liver injury. Proc Natl Acad Sci USA 113: 9298–9303, 2016. doi: 10.1073/pnas.1604780113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raynaud P, Tate J, Callens C, Cordi S, Vandersmissen P, Carpentier R, Sempoux C, Devuyst O, Pierreux CE, Courtoy P, Dahan K, Delbecque K, Lepreux S, Pontoglio M, Guay-Woodford LM, Lemaigre FP. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology 53: 1959–1966, 2011. doi: 10.1002/hep.24292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90, 2012. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 29.Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, Lippens S, Abels C, Schoonooghe S, Raes G, Devoogdt N, Lambrecht BN, Beschin A, Guilliams M. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun 7: 10321, 2016. doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF, Brenner DA. CCR2 promotes hepatic fibrosis in mice. Hepatology 50: 185–197, 2009. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M.. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology 138: 360–371, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology 51: 1778–1788, 2010. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strazzabosco M, Somlo S.. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology 140: 1855–1859 e1851, 2011. doi: 10.1053/j.gastro.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, Hughes MJ, Francis B, Wojtacha D, Man TY, Dear JW, Devey LR, Mowat AM, Pollard JW, Park BK, Jenkins SJ, Simpson KJ, Hume DA, Wigmore SJ, Forbes SJ. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology 149: 1896–1909 e1814, 2015. doi: 10.1053/j.gastro.2015.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, Wallace DP, Fields TA. Macrophages promote polycystic kidney disease progression. Kidney Int 83: 855–864, 2013. doi: 10.1038/ki.2012.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ta MH, Harris DC, Rangan GK. Role of interstitial inflammation in the pathogenesis of polycystic kidney disease. Nephrology (Carlton) 18: 317–330, 2013. doi: 10.1111/nep.12045. [DOI] [PubMed] [Google Scholar]
- 37.Urribarri AD, Munoz-Garrido P, Perugorria MJ, Erice O, Merino-Azpitarte M, Arbelaiz A, Lozano E, Hijona E, Jiménez-Agüero R, Fernandez-Barrena MG, Jimeno JP, Marzioni M, Marin JJ, Masyuk TV, LaRusso NF, Prieto J, Bujanda L, Banales JM. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut 63: 1658–1667, 2014. doi: 10.1136/gutjnl-2013-305281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson PD. Polycystic kidney disease. N Engl J Med 350: 151–164, 2004. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 39.Yoder BK, Richards WG, Sommardahl C, Sweeney WE, Michaud EJ, Wilkinson JE, Avner ED, Woychik RP. Differential rescue of the renal and hepatic disease in an autosomal recessive polycystic kidney disease mouse mutant. A new model to study the liver lesion. Am J Pathol 150: 2231–2241, 1997. [PMC free article] [PubMed] [Google Scholar]
- 40.Yoder BK, Richards WG, Sweeney WE, Wilkinson JE, Avener ED, Woychik RP. Insertional mutagenesis and molecular analysis of a new gene associated with polycystic kidney disease. Proc Assoc Am Physicians 107: 314–323, 1995. [PubMed] [Google Scholar]
- 41.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91, 2013. [Erratum in Immunity 38: 79–91, 2013. doi: 10.1016/j.immuni.2013.05.008] doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeier M, Fehrenbach P, Geberth S, Möhring K, Waldherr R, Ritz E. Renal histology in polycystic kidney disease with incipient and advanced renal failure. Kidney Int 42: 1259–1265, 1992. doi: 10.1038/ki.1992.413. [DOI] [PubMed] [Google Scholar]
- 43.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, Varol C. Infiltrating monocyte-derived macrophages and resident Kupffer cells display different ontogeny and functions in acute liver injury. J Immunol 193: 344–353, 2014. doi: 10.4049/jimmunol.1400574. [DOI] [PubMed] [Google Scholar]
- 44.Zoja C, Corna D, Locatelli M, Rottoli D, Pezzotta A, Morigi M, Zanchi C, Buelli S, Guglielmotti A, Perico N, Remuzzi A, Remuzzi G. Effects of MCP-1 inhibition by bindarit therapy in a rat model of polycystic kidney disease. Nephron 129: 52–61, 2015. doi: 10.1159/000369149. [DOI] [PubMed] [Google Scholar]