

Abstract
Hyperinsulinemia has been hypothesized to cause hypertension in obesity, type 2 diabetes, and metabolic syndrome through a renal mechanism. However, it has been challenging to isolate renal mechanisms in chronic experimental models due, in part, to technical difficulties. In this study, we tested the hypothesis that a renal mechanism underlies insulin hypertension. We developed a novel technique to permit continuous insulin infusion through the renal artery in conscious rats for 7 days. Mean arterial pressure increased by ~10 mmHg in rats that were infused intravenously (IV) with insulin and glucose. Renal artery doses were 20% of the intravenous doses and did not raise systemic insulin levels or cause differences in blood glucose. The increase in blood pressure was not different from the IV group. Mean arterial pressure did not change in vehicle-infused rats, and there were no differences in renal injury scoring due to the renal artery catheter. Glomerular filtration rate, plasma renin activity, and urinary sodium excretion did not differ between groups at baseline and did not change significantly with insulin infusion. Thus, by developing a novel approach for chronic, continuous renal artery insulin infusion, we provided new evidence that insulin causes hypertension in rats through actions initiated within the kidney.
Keywords: hyperinsulinemia, hypertension, intrarenal
INTRODUCTION
Hyperinsulinemia is a hallmark of obesity, type 2 diabetes, and metabolic syndrome. The association of hyperinsulinemia with hypertension in those conditions led to the hypothesis that insulin could play a role in causing the hypertension through renal, sympathetic nervous system, or other mechanisms (11, 14, 30, 35, 40). We tested that hypothesis in a series of studies in chronically instrumented rats, in which up to 7 days of hyperinsulinemia was induced by intravenous insulin infusion, accompanied by glucose to prevent hypoglycemia (5–8, 26, 27). Those studies revealed a modest but significant (~10 mmHg) hypertensive effect that was dependent on thromboxane synthesis (27) and an intact renin-angiotensin system (5, 26).
However, despite strong evidence that insulin stimulates renal tubular sodium transport (15, 28, 34, 42, 44), insulin-induced hypertension was characterized by systemic vasoconstriction rather than increased sodium balance (8). Moreover, the sympathetic nervous system has been increasingly implicated in the pathophysiology of hypertension in obesity and metabolic syndrome (16, 21, 37), but the intravenous infusion approach in the insulin hypertension studies could not isolate a renal mechanism independent of the sympathetic nervous system (16, 30, 37, 38). In addition, the same experimental approach in chronically instrumented dogs failed to report evidence of insulin-mediated hypertension or renal sodium retention (10, 17–20). Thus it has been difficult to gauge the translational significance of insulin hypertension and isolate a renal mechanism.
In the present study, we designed a novel approach that enabled sustained, 24 h/day insulin and glucose infusions into the kidney, using a customized catheter chronically implanted in the renal artery. This enabled us to test the hypothesis that the hypertensive response to chronic insulin plus glucose infusion is due to actions initiated within the kidney.
METHODS
Experiments were approved by the Institutional Animal Care and Use Committee of Augusta University. Male Sprague-Dawley rats (350–400 g) were purchased from Envigo (Madison, WI) and were instrumented with an abdominal aortic and left femoral vein catheter, as previously described (6, 8), under isoflurane anesthesia using aseptic technique. In addition, a custom-designed catheter (∼0.26-mm-OD tip) was inserted into the left renal artery via a puncture wound in the abdominal aorta (Fig. 1). The right kidney was removed to eliminate its confounding effect on measurement of GFR in the infused kidney. A Data Sciences International (DSI) PA-C10 telemeter was placed in the left femoral artery for continuous blood pressure monitoring. (The PA-C10 typically is used in mice, but its small size facilitated implantation in the rat femoral artery, which was necessary in this study because of the placement of the abdominal aortic and renal artery catheters.) The three catheters (20-gauge ID) were exteriorized at the scapulae through a stainless steel button, to which a 12-in. stainless steel spring was attached. The rats were housed in metabolic cages mounted with a DSI receiver. The spring was attached to a dual-channel Instech hydraulic swivel mounted above the cage that enabled chronic, 24 h/day infusion through the renal (IR) and femoral vein (IV) catheters, as well as undisturbed arterial blood sampling from the conscious rats as described previously (6, 8, 26). Rats were maintained in a temperature- and light-controlled environment with 12:12-h day/night cycle. Rats were allowed at least 5 days for recovery from surgery and acclimation to the metabolic cages before control measurements were begun, and had ad libitum access to food and water throughout the study. To control variation in sodium intake, rats were fed a sodium-deficient diet (Teklad CA.170950, Envigo) and were infused with sterile 0.9% saline 24 h/day IV to deliver ~2.8 mM Na+/day.
Fig. 1.

Representative image of the tip size of the custom-made renal catheter (top) and its placement in the renal artery (bottom).
Experimental protocol.
Rats were divided randomly into three groups: IR insulin, IV insulin, and vehicle. All rats received sterile water vehicle infusion in both the IR (sterile water) and IV (0.9% saline plus sterile water) catheters during the control period. During the 7-day insulin period, the IR and IV insulin-infused rats had the sterile water vehicle in the appropriate IR or IV infusion syringe replaced by insulin and 50% dextrose. The other syringe in those rats continued with sterile water vehicle infusion. The IV insulin (porcine, Wockhardt, Wrexham, UK) was infused at 1.5 mU·kg−1·min−1, and the IV 50% dextrose was coinfused at 20 mg·kg−1·min−1 as previously described (6, 8, 26). The IR infusion doses for insulin and glucose were set at 20% of the IV doses to approximate the amount of cardiac output that is delivered to the kidney. Blood samples were taken once during both the control (day 3) and insulin (day 3) periods for measurement of blood glucose levels, plasma insulin concentration, plasma renin activity (PRA) and glomerular filtration rate (GFR). Urine was collected daily for measurement of Na+ excretion.
To test whether glucose was required for the hypertensive response, a separate group of rats (IR insulin-only; n = 6) was run later, after the other experimental groups had been completed. They underwent the same preparation as the other groups, but during the insulin period had insulin added to the IR syringe without coinfusion of IR glucose. The IR insulin dose was set at 20% of the IV dose as with the previous IR insulin group, and the only difference was that sterile water vehicle was coinfused with the IR insulin.
Analytical methods.
Blood glucose concentrations were measured with an Accu-chek Active glucose meter (Roche Diabetes Care, Indianapolis, IN). Urine Na+ concentration was measured by atomic absorption (Perkin Elmer, Shelton, OH). Plasma insulin concentration was measured by ELISA (SPI Bio, Bertin Pharma, Montigny-le-Bretonneux, France). Plasma renin activity was measured by radioimmunoassay (Diasorin, Stillwater, MN). Glomerular filtration rate was measured by 125I-iothalamate clearance (Glofil, Iso-Tex Diagnostics, Friendswood, TX) after 24-h IV infusion to reach steady state (6, 8, 39). Telemetry signals for calculating mean arterial pressure (MAP) and average heart rate were sampled for 10 s every 1 min at 500 Hz, for 19 h/day from 1:00 PM to 8:00 AM.
Histology and injury scoring.
The left kidney was harvested at time of euthanasia and preserved in 10% neutral-buffered formalin, embedded in paraffin, sectioned, and then stained with either hematoxylin and eosin or Masson's trichrome. A renal pathologist was blinded to the identity of the kidney sections and scored them. Briefly, renal injury was assessed by scoring necrosis and sloughing off of renal tubular cells, flattened and regenerating tubular epithelium, tubular dilation, cast formation, glomerulosclerosis, or tubulointerstitial fibrosis on a scale 0 to 4 as follows: 0 = normal, 0.1 ≤1%; 0.5 = 1–5%; 1 = 5–25%; 2 = 25–50%; 3 = 50–75%; and 4 ≥75% areas in the kidney sections that displayed any of the aforementioned renal injury.
Statistics.
Results are presented as means ± SE. All data were graphed using Graphpad Prism software. Repeated-measures ANOVA was used for blood pressure, heart rate, and Na+ excretion. Two-way ANOVA was used for other comparisons among the three experimental groups. Student's t-test was used for comparisons within the IR insulin-only group. A probability value < 0.05 was considered to be significant.
RESULTS
Renal pathology.
Shown in Fig. 2A are representative cortical and medullary images of kidney sections from all three groups of the experimental rats that received a score of 1. Figure 2B shows that injury score averaged between ~0.5 and 1.0 for the three groups, with no significant differences. Kidney weight increased by ~25–30% based on comparison of the left kidney at time of euthanasia vs. the right kidney removed during surgery (Table 1), reflecting compensatory hypertrophy of the left kidney in response to removal of the right kidney. There were no differences in the compensatory renal hypertrophy between groups. The renal pathology data, combined with MAP, PRA, and GFR being in the normal range, support the validity and feasibility of this method for chronic renal-artery infusion in rats.
Fig. 2.

Representative images of the cortex and medulla that were scored as 1 for renal injury by a pathologist (A) and mean renal pathology scores (B) from vehicle-, IR insulin-, and IV insulin-infused rats. n = 6–10 rats per group.
Table 1.
Metabolic variables in rats infused with vehicle, IV insulin + glucose, and IR insulin + glucose
| Vehicle | IV Insulin | IR Insulin | |
|---|---|---|---|
| Body wt (BW), g) | |||
| Beginning | 396.9 ± 4.8 | 393.7 ± 5.9 | 384.3 ± 6.9† |
| Final | 406.6 ± 6.7 | 401.6 ± 5.5 | 400.3 ± 7.2 |
| Kidney wt (KW), g | |||
| UNX | 1.31 ± 0.04 | 1.31 ± 0.04 | 1.33 ± 0.05 |
| Remaining | 1.71 ± 0.12‡ | 1.68 ± 0.07‡ | 1.77 ± 0.09‡ |
| KW/BW | |||
| Beginning | 0.0033 ± 0.0001 | 0.0033 ± 0.0001 | 0.0034 ± 0.0001 |
| Final | 0.0042 ± 0.0003‡ | 0.0042 ± 0.0002‡ | 0.0043 ± 0.0002‡ |
| Blood glucose, mmol/l | |||
| Control | 7.2 ± 0.2 | 7.1 ± 0.2 | 6.9 ± 0.2 |
| Insulin | 6.8 ± 0.2 | 6.5 ± 0.2 | 7.3 ± 0.2 |
| Food intake, g/day | |||
| Control | 17.8 ± 2.0 | 18.3 ± 1.3 | 15.1 ± 1.1 |
| Insulin | 21.8 ± 1.2 | 12.4 ± 1.6* | 19.0 ± 1.1 |
| Water intake, ml/day | |||
| Control | 9 ± 4 | 9 ± 3 | 10 ± 3 |
| Insulin | 7 ± 4 | 11 ± 2 | 10 ± 3 |
| Plasma protein, g/dl | |||
| Control | 6.1 ± 0.1 | 6.0 ± 0.1 | 6.2 ± 0.1 |
| Insulin | 6.1 ± 0.1 | 6.0 ± 0.1 | 6.4 ± 0.1 |
| Hematocrit, % | |||
| Control | 38 ± 1 | 36 ± 1 | 37 ± 1 |
| Insulin | 36 ± 1 | 37 ± 1 | 38 ± 1 |
Data are means ± SE; n = 8–12 rats per group.
P < 0.05 vs. vehicle;
P < 0.05 vs. vehicle beginning body weight;
P < 0.05 vs. uninephrectomized (UNX) kidney.
Plasma insulin and glucose.
Plasma insulin concentrations were not different between groups during the control period (Fig. 3, Table 2). IV insulin infusion caused a significant, approximately threefold, increase in plasma insulin concentration, which is consistent with the range we reported previously in this model (6, 8, 26). There were no statistically significant changes in plasma insulin concentration in either the IR insulin rats or vehicle-infused rats. Renal-vein insulin measurements could provide direct evidence that the IV and IR infusions yielded similar intrarenal insulin levels, but that approach was not possible in this chronic preparation. In this context, the blood pressure response served as a bioassay for efficacy of IR-infused insulin. The absence of an increase in plasma insulin in the IR group argued against spillover and the need to infuse the IR dose IV as a spillover-control. Plasma insulin in the IR insulin-only group did not change significantly between the control and insulin periods (Table 2).
Fig. 3.
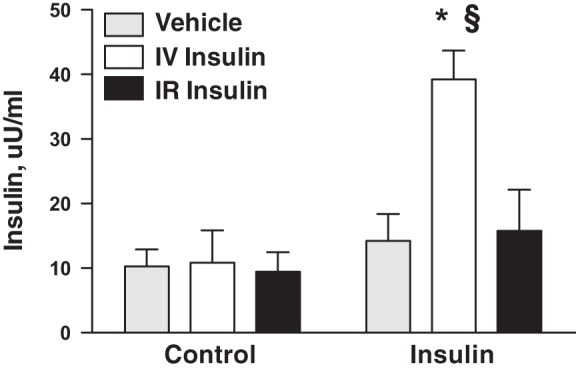
Plasma insulin levels during control and insulin-infusion periods in vehicle-, IR insulin-, and IV insulin-infused rats. n = 8–12 rats per group; *P < 0.05 vs. vehicle, §P < 0.05 vs. IR insulin.
Table 2.
Metabolic variables in rats infused with IR insulin without coinfusion of glucose
| IR Insulin Only | |
|---|---|
| Body wt (BW), g | |
| Beginning | 412.5 ± 6.0 |
| Final | 433.1 ± 7.6* |
| Kidney wt (KW), g | |
| UNX | 1.37 ± 0.02 |
| Remaining | 1.63 ± 0.06* |
| KW/BW | |
| Beginning | 0.0033 ± 0.0001 |
| Final | 0.0037 ± 0.0001* |
| Blood glucose, mmol/l | |
| Control | 7.3 ± 0.3 |
| Insulin | 7.4 ± 0.2 |
| Plasma insulin, µU/ml | |
| Control | 7.01 ± 2.95 |
| Insulin | 14.60 ± 9.98 |
| GFR, ml/min | |
| Control | 1.70 ± 0.10 |
| Insulin | 1.76 ± 0.13 |
| Food intake, g/day | |
| Control | 16.5 ± 2.7 |
| Insulin | 19.2 ± 2.0 |
| Water intake, ml/day | |
| Control | 5 ± 1 |
| Insulin | 7 ± 3 |
| Plasma protein, g/dl | |
| Control | 6.2 ± 0.1 |
| Insulin | 6.3 ± 0.1 |
| Hematocrit, % | |
| Control | 38 ± 1 |
| Insulin | 36 ± 1 |
Data are means ± SE; n = 6 rats per group.
P < 0.05 vs. beginning weight.
There were no significant differences in blood glucose concentration between groups during the control period, and no significant changes in any group during the insulin period (Table 1). Blood glucose levels in the IR insulin-only rats also did not differ between control and insulin infusion (Table 2). Food intake decreased in the IV insulin group during the insulin and glucose infusion period (Table 1), consistent with our previous reports (6, 8, 26) and due to the calories derived from the IV glucose (6). There were no changes in food intake in the IR insulin or vehicle groups, and this response is additional evidence against significant spillover of the IR infusion. Food consumption in the IR insulin-only group resembled that in the IR insulin and vehicle rats (Table 2).
Systemic hemodynamics and renal function.
Mean arterial pressure increased significantly by ~10 mmHg in the IV and IR groups over the 7-day insulin infusion period (Fig. 4A), similar in magnitude to our previous reports with IV insulin and glucose infusion (6, 8, 26). The MAP response was not different between the IR and IV groups, and there was no change in MAP in the vehicle group. Heart rate did not increase with insulin infusion or over the experimental period, but heart rate was significantly greater in the IV insulin rats than in vehicle (Fig. 4B). We have measured a modest increase in heart rate in some, but not all, previous studies with this IV insulin and glucose infusion (6, 8, 26). There also were no changes in heart rate in the IR or vehicle groups. There were no differences in urinary sodium excretion at baseline, and no significant changes or differences in sodium excretion or cumulative sodium balance during the insulin infusion period (Fig. 5). There also were no differences in GFR or PRA between groups during the control period, and no statistically significant changes in either variable during the insulin-infusion period (Fig. 6). MAP did not increase with insulin infusion in the IR insulin-only group (Fig. 7A), and there also were no changes in heart rate (Fig. 7B) or urinary sodium excretion (Fig. 7C) in those rats.
Fig. 4.
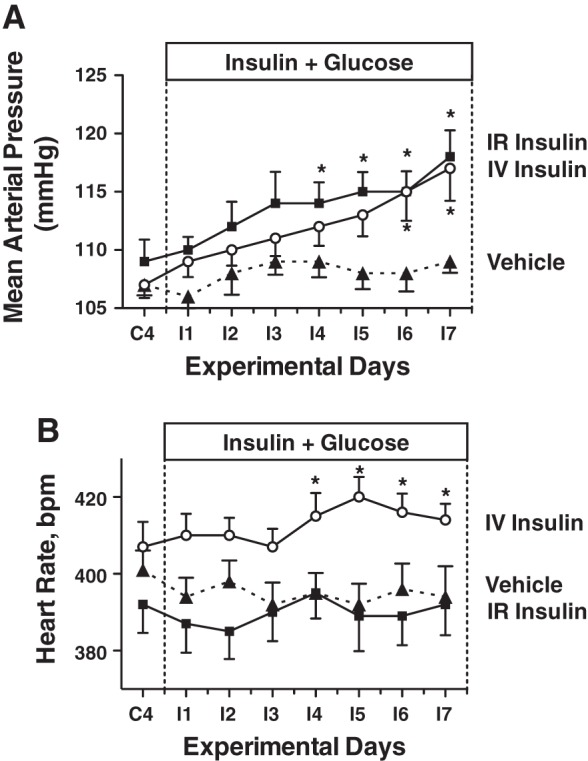
Mean arterial pressure (A) and heart rate (B) in IR insulin-, IV insulin-, and vehicle-infused rats during control and insulin infusion periods. n = 8–12 rats per group *P < 0.05 vs. vehicle.
Fig. 5.
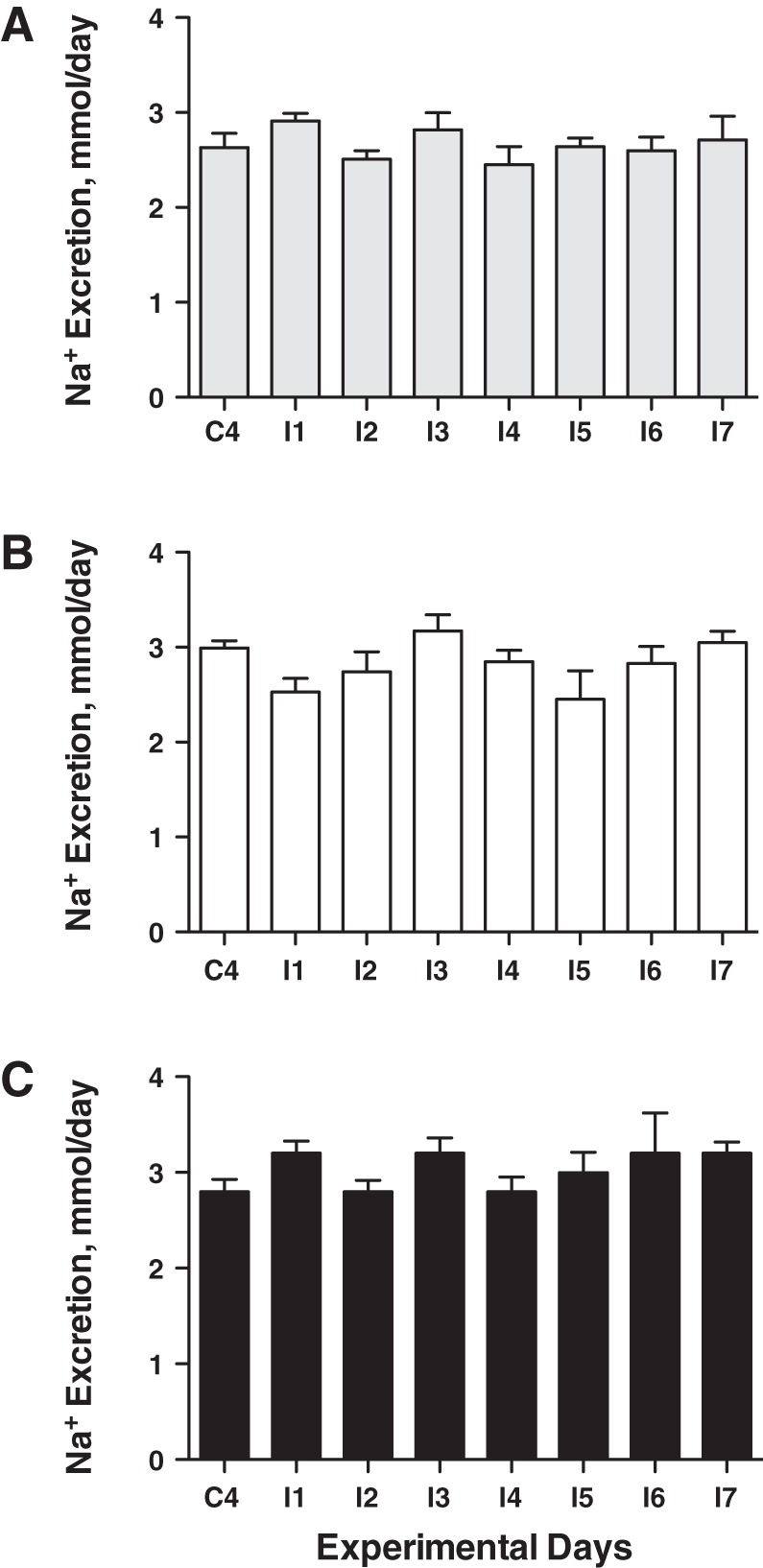
Na excretion in vehicle- (A), IV insulin- (B), and IR insulin-infused (C) rats during control and insulin infusion periods. n = 8–12 rats per group.
Fig. 6.
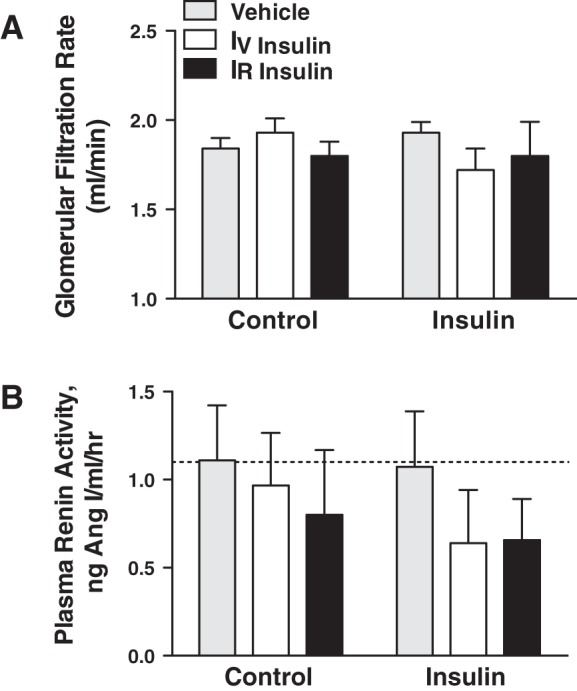
Glomerular filtration rate (A) and plasma renin activity (B) in IR insulin-, IV insulin-, and vehicle-infused rats during control and insulin infusion periods. n = 8–12 rats per group.
Fig. 7.
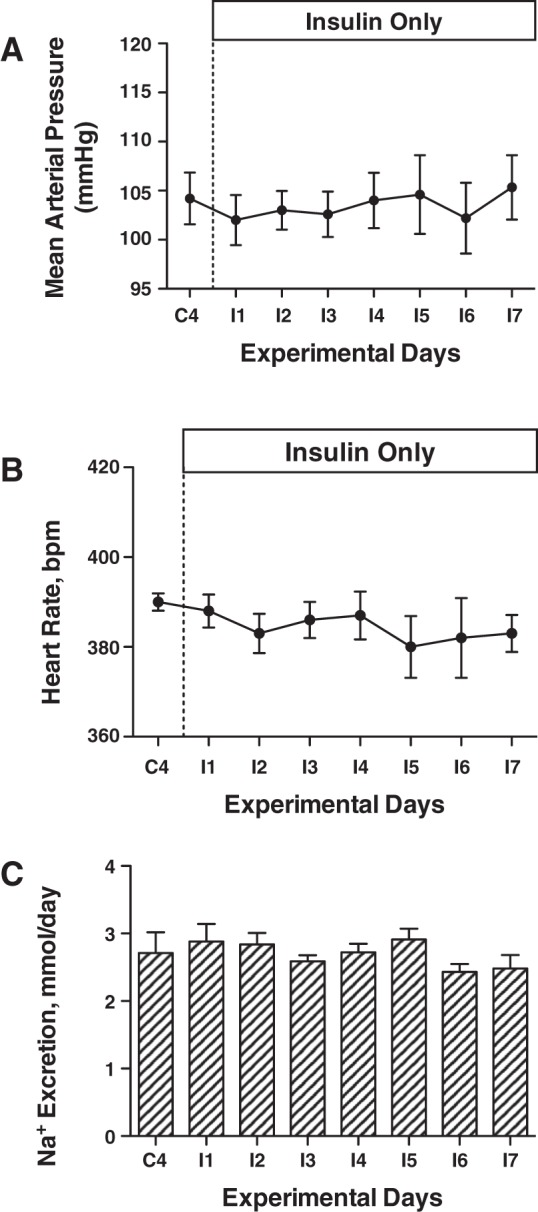
Mean arterial pressure (A), heart rate (B), and Na excretion (C) in IR insulin-only rats during control and insulin infusion periods. n = 6 rats.
DISCUSSION
This study developed a novel approach for chronic, continuous renal artery insulin infusion in uninephrectomized rats to test directly the hypothesis that the hypertensive response to chronic hyperinsulinemia in rats was due to a mechanism initiated within the kidneys. The IV insulin plus glucose infusion increased MAP and caused other systemic and renal responses; these results are consistent with previous relevant findings (5–8, 26, 27). Localizing that infusion to the kidney caused a virtually superimposable MAP response in the IR insulin group. The plasma insulin and glucose data provided no evidence of significant systemic spillover of the renal artery infusion in the IR rats. This strongly implicates the kidney as the site of the yet unidentified mechanism underlying the hypertensive response to systemic hyperinsulinemia in rats. In addition, the failure of IR insulin alone, i.e., without the coadministration of glucose, to affect blood pressure is consistent with our previous studies in dogs that suggest glucose is required for renal actions of insulin to be sustained (31, 32).
An effect of insulin on renal sodium handling has long been implicated based on the renal response of diabetic patients to insulin therapy (1), but the effects of insulin in diabetic patients cannot be separated from changes in blood glucose. DeFronzo demonstrated a direct effect of insulin to stimulate renal tubular sodium transport under conditions when changes in blood glucose were prevented (12), and hypothesized that if such an effect were sustained chronically it could contribute to the hypertension in obese patients with hyperinsulinemia (11). Many groups since have reported effects of insulin to stimulate renal sodium transport (15, 28, 34, 42, 44), but it nonetheless has been difficult to experimentally isolate a renal mechanism for chronic, insulin-induced hypertension.
Studies in rats from our laboratory (5–8, 26, 27) and others (33, 41) demonstrated a chronic hypertensive effect of hyperinsulinemia, but it was not associated with marked sodium retention or increased cumulative sodium balance. Plasma renin activity and GFR both tended to decrease, achieving statistical significance in some, but not all studies, which further complicated implicating the kidneys in the mechanism for insulin-induced hypertension. There was a parallel shift in the renal pressure-natriuresis relationship (6), which indicated that a sodium-retaining shift in kidney function occurred (3, 22, 23). However, there also was a significant increase in TPR and decrease in cardiac output as blood pressure increased (8), which indicated systemic vasoconstriction. This inability to identify a specific renal mechanism in rats with chronic, insulin-induced hypertension was the reason we designed the present study to isolate the chronic insulin plus glucose infusion to the kidneys. The IV group in the present study closely resembled the overall responses in our previous reports, i.e., the hypertensive response associated with tendencies for heart rate to increase and PRA and GFR to decrease, with no increase in sodium balance. By isolating the insulin plus glucose infusion to the renal artery in the IR group and getting the same hypertensive response, the present study provides strong evidence for a renal mechanism, albeit still unidentified, underlying the hypertensive effect of systemic hyperinsulinemia in rats.
Perhaps the strongest argument against renal actions of insulin having a chronic hypertensive effect has been the negative results from dog studies (10, 17–20); in particular, the failure of chronic renal artery infusion of insulin to cause significant sodium retention in normal dogs (18), despite stimulating sodium reabsorption acutely in anesthetized dogs (13). Patients with insulinoma also are not hypertensive (36, 43). However, we reported recently that the same renal artery insulin infusion dose previously found to be ineffective in normal dogs (18) had an extremely powerful antinatriuretic effect in diabetic dogs (32): It completely reversed the natriuresis and diuresis caused by 6 days of diabetes (~22.2 mmol/l). This was consistent with our earlier report providing evidence that the sustained natriuresis and diuresis in dogs with type 1 diabetes was due to the loss of insulin rather than the hyperglycemia (31). This suggests that the sustained sodium-retaining effect of insulin may be linked mechanistically to glucose. In fact, Koopmans et al. (29) reported that chronic IV insulin infusion in rats did not cause hypertension when a significantly lower dose of glucose than we used was co-infused IV. In the present study, we tested the potential role of glucose by repeating the renal artery insulin infusion protocol, but without the coinfusion of glucose. There was no effect of the IR insulin-only infusion on blood pressure or sodium excretion. Whether an interaction between insulin and glucose is due to glucose modulation of insulin-activation of ENaC (2, 24, 42), insulin modulation of glucose-activation of SGLT, or some other mechanism, is not known.
The renal mechanism of action of insulin during chronic infusion, and why it leads to hypertension in rats but not dogs, is not known. The shift in the renal-pressure natriuresis relationship in insulin-hypertensive rats (6) indicates a sodium-retaining shift in kidney function (3, 22, 23), and the response to renal-artery infusion in the present study further supports a renal mechanism; however, the absence of significant changes in GFR or sodium balance continues to keep the identity of that mechanism a mystery. Even though PRA does not increase, we reported that insulin hypertension in rats was dependent on angiotensin II (5, 26), possibly mediated through stimulation of thromboxane synthesis (27). We subsequently showed that chronic glucose infusion in dogs caused hyperinsulinemia and mild hypertension when COX-2 was inhibited chronically, and arachidonic acid testing confirmed that COX-2 inhibition had blocked vasodilatory prostanoid production (4). Those results raise the possibility of differing prostaglandin/thromboxane profiles during hyperinsulinemia in rats vs. dogs that remains to be tested. More accurate evaluation of renal vascular resistance than can be achieved with our current single-point GFR measurement would be beneficial in that regard. Continuous measurement from renal artery flow probes could determine whether the increase in TPR we previously reported (8) includes significant renal vasoconstriction and also might provide indirect evidence regarding changes in tubular sodium reabsorption.
The present results show that the hypertensive response to chronic IV insulin infusion is due to a mechanism initiated within the kidneys. In addition, this effect seems dependent on the presence of glucose, although the threshold plasma glucose level and mechanism are not known. This putative role for glucose is consistent with our recent discovery in dogs that insulin has a sustained antinatriuretic effect only under diabetic conditions. This suggests there may be similar renal tubular sodium transport effects of insulin in rats and dogs in the presence of glucose. However, the chronic blood pressure response to hyperinsulinemia may depend on other factors that modulate systemic hemodynamics and renal function, such as the prostaglandin/thromboxane profile or the renal injury and insulin resistance that accompany progression through obesity and type 2 diabetes. The uninephrectomy in the normal rats in the present study is noteworthy in that regard.
Perspectives.
We previously hypothesized that the increase in plasma insulin that occurs during the evolution of obesity, metabolic syndrome, and overt type 2 diabetes may be a physiological mechanism to help minimize hyperglycemia-induced renal salt and volume loss (9). Thus the sodium-retaining effect of insulin would not be limited to the all-or-none effect that is evident in type 1 diabetes. In support of this hypothesis, that the sodium-retaining effect of insulin may be part of our normal control system for sodium and volume homeostasis, we recently reported that the normal increase in insulin caused by a meal may be required to prevent excessive postprandial renal salt and volume losses (25). Whether or not hyperinsulinemia can cause hypertension may depend on the presence of other factors that would exacerbate the physiological effect of insulin and cause a pathophysiological response. The effects of thromboxane (27) and COX-2 inhibition (4) on the blood pressure response to insulin or hyperglycemia, for example, suggest that the hormonal environment could play such a role, and the progressive loss of renal function that accompanies the progression to and through type 2 diabetes could have a similar influence. By developing a novel approach in the present study, we report that insulin causes hypertension in rats through actions initiated within the kidney. Future studies are needed to identify and quantify the relative contributions of renal tubular and renal and systemic hemodynamic mechanisms.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-56259 and HL-56259-S1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.L.I. performed experiments; D.L.I. analyzed data; D.L.I., J.-K.C., and M.W.B. interpreted results of experiments; D.L.I. prepared figures; D.L.I. and M.W.B. drafted manuscript; D.L.I. and M.W.B. edited and revised manuscript; D.L.I., J.-K.C., and M.W.B. approved final version of manuscript; M.W.B. conceived and designed research.
ACKNOWLEDGMENTS
We thank A. D. Duggan, R. Alaisami, and A. Washington for technical assistance with this project.
REFERENCES
- 1.Atchley DW, Loeb RF, Richards DW, Benedict EM, Driscoll ME. On diabetic acidosis: a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest 12: 297–326, 1933. doi: 10.1172/JCI100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blazer-Yost BL, Liu X, Helman SI. Hormonal regulation of ENaCs: insulin and aldosterone. Am J Physiol Cell Physiol 274: C1373–C1379, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Brands MW. Chronic blood pressure control. Compr Physiol 2: 2481–2494, 2012. doi: 10.1002/cphy.c100056. [DOI] [PubMed] [Google Scholar]
- 4.Brands MW, Hailman AE, Fitzgerald SM. Long-term glucose infusion increases arterial pressure in dogs with cyclooxygenase-2 inhibition. Hypertension 37: 733–738, 2001. doi: 10.1161/01.HYP.37.2.733. [DOI] [PubMed] [Google Scholar]
- 5.Brands MW, Harrison DL, Keen HL, Gardner A, Shek EW, Hall JE. Insulin-induced hypertension in rats depends on an intact renin-angiotensin system. Hypertension 29: 1014–1019, 1997. doi: 10.1161/01.HYP.29.4.1014. [DOI] [PubMed] [Google Scholar]
- 6.Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Hypertension during chronic hyperinsulinemia in rats is not salt-sensitive. Hypertension 19, Suppl: I83–I89, 1992. doi: 10.1161/01.HYP.19.1_Suppl.I83. [DOI] [PubMed] [Google Scholar]
- 7.Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Sustained hyperinsulinemia increases arterial pressure in conscious rats. Am J Physiol Regul Integr Comp Physiol 260: R764–R768, 1991. [DOI] [PubMed] [Google Scholar]
- 8.Brands MW, Lee WF, Keen HL, Alonso-Galicia M, Zappe DH, Hall JE. Cardiac output and renal function during insulin hypertension in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol 271: R276–R281, 1996. [DOI] [PubMed] [Google Scholar]
- 9.Brands MW, Manhiani MM. Sodium-retaining effect of insulin in diabetes. Am J Physiol Regul Integr Comp Physiol 303: R1101–R1109, 2012. doi: 10.1152/ajpregu.00390.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brands MW, Mizelle HL, Gaillard CA, Hildebrandt DA, Hall JE. The hemodynamic response to chronic hyperinsulinemia in conscious dogs. Am J Hypertens 4: 164–168, 1991. doi: 10.1093/ajh/4.2.164. [DOI] [PubMed] [Google Scholar]
- 11.DeFronzo RA. The effect of insulin on renal sodium metabolism. A review with clinical implications. Diabetologia 21: 165–171, 1981. doi: 10.1007/BF00252649. [DOI] [PubMed] [Google Scholar]
- 12.DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest 55: 845–855, 1975. doi: 10.1172/JCI107996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeFronzo RA, Goldberg M, Agus ZS. The effects of glucose and insulin on renal electrolyte transport. J Clin Invest 58: 83–90, 1976. doi: 10.1172/JCI108463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrannini E, Buzzigoli G, Bonadonna R, Giorico MA, Oleggini M, Graziadei L, Pedrinelli R, Brandi L, Bevilacqua S. Insulin resistance in essential hypertension. N Engl J Med 317: 350–357, 1987. doi: 10.1056/NEJM198708063170605. [DOI] [PubMed] [Google Scholar]
- 15.Frindt G, Palmer LG. Effects of insulin on Na and K transporters in the rat CCD. Am J Physiol Renal Physiol 302: F1227–F1233, 2012. doi: 10.1152/ajprenal.00675.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grassi G, Mark A, Esler M. The sympathetic nervous system alterations in human hypertension. Circ Res 116: 976–990, 2015. doi: 10.1161/CIRCRESAHA.116.303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall JE, Brands MW, Kivlighn SD, Mizelle HL, Hildebrandt DA, Gaillard CA. Chronic hyperinsulinemia and blood pressure. Interaction with catecholamines? Hypertension 15: 519–527, 1990. doi: 10.1161/01.HYP.15.5.519. [DOI] [PubMed] [Google Scholar]
- 18.Hall JE, Brands MW, Mizelle HL, Gaillard CA, Hildebrandt DA. Chronic intrarenal hyperinsulinemia does not cause hypertension. Am J Physiol Renal Fluid Electrolyte Physiol 260: F663–F669, 1991. [DOI] [PubMed] [Google Scholar]
- 19.Hall JE, Brands MW, Zappe DH, Dixon WN, Mizelle HL, Reinhart GA, Hildebrandt DA. Hemodynamic and renal responses to chronic hyperinsulinemia in obese, insulin-resistant dogs. Hypertension 25: 994–1002, 1995. doi: 10.1161/01.HYP.25.5.994. [DOI] [PubMed] [Google Scholar]
- 20.Hall JE, Coleman TG, Mizelle HL, Smith MJ Jr. Chronic hyperinsulinemia and blood pressure regulation. Am J Physiol Renal Fluid Electrolyte Physiol 258: F722–F731, 1990. [DOI] [PubMed] [Google Scholar]
- 21.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res 116: 991–1006, 2015. doi: 10.1161/CIRCRESAHA.116.305697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall JE, Guyton AC, Brands MW. Pressure-volume regulation in hypertension. Kidney Int Suppl 55: S35–S41, 1996. [PubMed] [Google Scholar]
- 23.Hall JE, Mizelle HL, Hildebrandt DA, Brands MW. Abnormal pressure natriuresis. A cause or a consequence of hypertension? Hypertension 15: 547–559, 1990. doi: 10.1161/01.HYP.15.6.547. [DOI] [PubMed] [Google Scholar]
- 24.Ilatovskaya DV, Levchenko V, Brands MW, Pavlov TS, Staruschenko A. Cross-talk between insulin and IGF-1 receptors in the cortical collecting duct principal cells: implication for ENaC-mediated Na+ reabsorption. Am J Physiol Renal Physiol 308: F713–F719, 2015. doi: 10.1152/ajprenal.00081.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irsik DL, Blazer-Yost BL, Staruschenko A, Brands MW. The normal increase in insulin after a meal may be required to prevent postprandial renal sodium and volume losses. Am J Physiol Regul Integr Comp Physiol 312: R965–R972, 2017. doi: 10.1152/ajpregu.00354.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keen HL, Brands MW, Smith MJ Jr, Hall JE. Maintenance of baseline angiotensin II potentiates insulin hypertension in rats. Hypertension 31: 637–642, 1998. doi: 10.1161/01.HYP.31.2.637. [DOI] [PubMed] [Google Scholar]
- 27.Keen HL, Brands MW, Smith MJ Jr, Shek EW, Hall JE. Inhibition of thromboxane synthesis attenuates insulin hypertension in rats. Am J Hypertens 10: 1125–1131, 1997. doi: 10.1016/S0895-7061(97)00217-3. [DOI] [PubMed] [Google Scholar]
- 28.Kirchner KA. Insulin increases loop segment chloride reabsorption in the euglycemic rat. Am J Physiol Renal Fluid Electrolyte Physiol 255: F1206–F1213, 1988. [DOI] [PubMed] [Google Scholar]
- 29.Koopmans SJ, Ohman L, Haywood JR, Mandarino LJ, DeFronzo RA. Seven days of euglycemic hyperinsulinemia induces insulin resistance for glucose metabolism but not hypertension, elevated catecholamine levels, or increased sodium retention in conscious normal rats. Diabetes 46: 1572–1578, 1997. doi: 10.2337/diacare.46.10.1572. [DOI] [PubMed] [Google Scholar]
- 30.Landsberg L. Insulin-mediated sympathetic stimulation: role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J Hypertens 19, Suppl: 523–528, 2001. doi: 10.1097/00004872-200103001-00001. [DOI] [PubMed] [Google Scholar]
- 31.Manhiani MM, Cormican MT, Brands MW. Chronic sodium-retaining action of insulin in diabetic dogs. Am J Physiol Renal Physiol 300: F957–F965, 2011. doi: 10.1152/ajprenal.00395.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manhiani MM, Duggan AD, Wilson H, Brands MW. Chronic intrarenal insulin replacement reverses diabetes mellitus-induced natriuresis and diuresis. Hypertension 59: 421–430, 2012. doi: 10.1161/HYPERTENSIONAHA.111.185215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meehan WP, Buchanan TA, Hsueh W. Chronic insulin administration elevates blood pressure in rats. Hypertension 23: 1012–1017, 1994. doi: 10.1161/01.HYP.23.6.1012. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura M, Satoh N, Suzuki M, Kume H, Homma Y, Seki G, Horita S. Stimulatory effect of insulin on renal proximal tubule sodium transport is preserved in type 2 diabetes with nephropathy. Biochem Biophys Res Commun 461: 154–158, 2015. doi: 10.1016/j.bbrc.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Niskanen LK, Uusitupa MI, Pyörälä K. The relationship of hyperinsulinaemia to the development of hypertension in type 2 diabetic patients and in non-diabetic subjects. J Hum Hypertens 5: 155–159, 1991. [PubMed] [Google Scholar]
- 36.Pontiroli AE, Alberetto M, Pozza G. Patients with insulinoma show insulin resistance in the absence of arterial hypertension. Diabetologia 35: 294–295, 1992. doi: 10.1007/BF00400934. [DOI] [PubMed] [Google Scholar]
- 37.Rahmouni K. Cardiovascular regulation by the arcuate nucleus of the hypothalamus: neurocircuitry and signaling systems. Hypertension 67: 1064–1071, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark AL, Haynes WG. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest 114: 652–658, 2004. doi: 10.1172/JCI21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawicki PT, Heinemann L, Starke A, Berger M. Hyperinsulinaemia is not linked with blood pressure elevation in patients with insulinoma. Diabetologia 35: 649–652, 1992. doi: 10.1007/BF00400257. [DOI] [PubMed] [Google Scholar]
- 40.Skarfors ET, Lithell HO, Selinus I. Risk factors for the development of hypertension: a 10-year longitudinal study in middle-aged men. J Hypertens 9: 217–223, 1991. doi: 10.1097/00004872-199103000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Song J, Hu X, Riazi S, Tiwari S, Wade JB, Ecelbarger CA. Regulation of blood pressure, the epithelial sodium channel (ENaC), and other key renal sodium transporters by chronic insulin infusion in rats. Am J Physiol Renal Physiol 290: F1055–F1064, 2006. doi: 10.1152/ajprenal.00108.2005. [DOI] [PubMed] [Google Scholar]
- 42.Tiwari S, Nordquist L, Halagappa VK, Ecelbarger CA. Trafficking of ENaC subunits in response to acute insulin in mouse kidney. Am J Physiol Renal Physiol 293: F178–F185, 2007. doi: 10.1152/ajprenal.00447.2006. [DOI] [PubMed] [Google Scholar]
- 43.Tsutsu N, Nunoi K, Kodama T, Nomiyama R, Iwase M, Fujishima M. Lack of association between blood pressure and insulin in patients with insulinoma. J Hypertens 8: 479–482, 1990. doi: 10.1097/00004872-199005000-00014. [DOI] [PubMed] [Google Scholar]
- 44.Zaika O, Palygin O, Tomilin V, Mamenko M, Staruschenko A, Pochynyuk O. Insulin and IGF-1 activate Kir4.1/5.1 channels in cortical collecting duct principal cells to control basolateral membrane voltage. Am J Physiol Renal Physiol 310: F311–F321, 2016. doi: 10.1152/ajprenal.00436.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]