

Abstract
We reply to two recently published, multi-authored opinion papers by opponents of sequence-based nomenclature, namely Zamora et al. (IMA Fungus 9: 167–175,2018) and Thines et al. (IMA Fungus 9: 177–183, 2018). While we agree with some of the principal arguments brought forward by these authors, we address misconceptions and demonstrate that some of the presumed evidence presented in these papers has been wrongly interpreted. We disagree that allowing sequences as types would fundamentally alter the nature of types, since a similar nature of abstracted features as type is already allowed in the Code (Art. 40.5), namely an illustration. We also disagree that there is a high risk of introducing artifactual taxa, as this risk can be quantified at well below 5 %, considering the various types of high-throughput sequencing errors. Contrary to apparently widespread misconceptions, sequence-based nomenclature cannot be based on similarity-derived OTUs and their consensus sequences, but must be derived from rigorous, multiple alignment-based phylogenetic methods and quantitative, single-marker species recognition algorithms, using original sequence reads; it is therefore identical in its approach to single-marker studies based on physical types, an approach allowed by the Code. We recognize the limitations of the ITS as a single fungal barcoding marker, but point out that these result in a conservative approach, with “false negatives” surpassing “false positives”; a desirable feature of sequence-based nomenclature. Sequence-based nomenclature does not aim at accurately resolving species, but at naming sequences that represent unknown fungal lineages so that these can serve as a means of communication, so ending the untenable situation of an exponentially growing number of unlabeled fungal sequences that fill online repositories. The risks are outweighed by the gains obtained by a reference library of named sequences spanning the full array of fungal diversity. Finally, we elaborate provisions in addition to our original proposal to amend the Code that would take care of the issues brought forward by opponents to this approach. In particular, taking up the idea of the Candidatus status of invalid, provisional names in prokaryote nomenclature, we propose a compromise that would allow valid publication of voucherless, sequence-based names in a consistent manner, but with the obligate designation as “nom. seq.” (nomen sequentiae). Such names would not have priority over specimen- or culture-based names unless either epitypified with a physical type or adopted for protection on the recommendation of a committee of the International Commission on the Taxonomy of Fungi following evaluation based on strict quality control of the underlying studies based on established rules or recommendations.
Keywords: Aspergillus, Daldinia, Fusarium, IMC11, International Commission on the Taxonomy of Fungi, ITS barcoding marker, Morchella, nom. seq., Penicillium
BACKGROUND
Prior to the 19th International Botanical Congress in Shenzen (IBC 2017) in July 2017, a first proposal to formally adopt names based on sequence types for so-called “dark matter fungi” was put forward (Hawksworth et al. 2016). This proposal received moderate attention in the mycological community (Hibbett et al. 2016, Grube et al. 2017, Seifert 2017, Hibbett 2018, Ryberg & Nilsson 2018) and was rejected by the Nomenclatural Section at IMC 2017 but with the formation of a Special Committee to look into the matter across all groups covered by the Code (Hawksworth et al. 2017). That Committee is charged with reporting to the 20th Congress in Rio de Janeriro in 2023. With the decision that nomenclatural rules specific to Fungi are now under the responsibility of the International Mycological Congresses (IMCs) and form a separate chapter in the Code (Hawksworth et al. 2017, Turland et al. 2018), this matter in so far as it relates to fungi can be decided on at an International Mycological Congress. The original proposal was consequently published in revised form for consideration by the upcoming 11th International Mycological Congress (IMC11) in San Juan, Puerto Rico in July 2018 (Hawksworth et al. 2018), with a supporting positional paper on how to address potential challenges of this approach (Lücking & Hawksworth 2018). The mycological community has rather passionately responded to this second proposal, and since then, an intense discussion has developed including two opposingt papers in this issue, one by some members of the International Commission on the Taxonomy of Fungi (ICTF) and another with 412 authors (Thines et al. 2018, Zamora et al. 2018). While our co-author on the two proposals, David Hibbett, has responded separately in this issue to Seifert’s (2017) editorial on sequence-based nomenclature (Hibbett 2018), here we provide a reply to the two multi-authored papers, as we consider a clarification of certain misconceptions and incorrectly interpreted evidence regarding the original proposal and the nature of sequence-based nomenclature is required as a basis for informed debate in Puerto Rico.
RESPONSES
Part I. Reply to Zamora et al. “Considerations and consequences of allowing DNA sequence data as types of fungal taxa”
Species versus DNA sequences
We agree with what is outlined by Zamora et al. (2018: 169) on this matter in practically every detail. However, the arguments put forward highlight the limitations of a single marker for species delimitations, an issue that applies generally to DNA sequencing methods and to all markers (as those authors correctly state), and is not unique to the approach of sequence-based nomenclature. Therefore, it cannot stand as an argument against sequence-based nomenclature, while the same approach is regularly allowed for specimen-based nomenclature. The Code does not require new species to be based on multiple markers, or indeed any molecular data at all. A polyphasic approach to species delimitation, using multiple markers and an array of phenotypic characters, is certainly the desired “gold standard”. Yet, however complex evolutionary processes may be, in most groups of fungi, species are reasonably well-delimited using either a single barcoding marker (be it ITS or another locus) or a maximum of two to three combined markers (see below in Reply to Thines et al. Point 1).
For practical reasons, in a given group, sequence-based nomenclature has to be executed with a single marker, to enforce congruence and avoid parallel classifications (Lücking & Hawksworth 2018: 153), and in lieu of reasons for alternative loci (e.g. in Aspergillus, Fusarium, Penicillium; see below), the locus of choice is the ITS fungal barcoding marker, if not for the simple reason that almost all environmental fungal meta-barcoding sequences in the Sequence Read Archive (SRA) correspond to ITS – and there are now more than a billion (!) of them (Lücking & Hawksworth 2018: 144). Being fully aware of the limitations of using ITS as the single marker to perform sequence-based nomenclature, we reiterate here (and below) two important aspects of this approach:
• Sequence-based nomenclature does not aim at accurately resolving species, simply because in a substantial number of cases it cannot do that. Sequence-based nomenclature aims at naming sequences that represent unknown fungal lineages so that these sequences can serve as proper references and the untenable situation of an exponentially growing number of unlabeled fungal sequences that fill online repositories may be limited, and stop making taxonomic assessments of newly generated sequence data increasingly more difficult (Fig. 1).
• Being aware of the limitations of using ITS as a single marker, with the risks of insufficient resolution in some lineages (“false negatives”) or hypervariability in others (“false positives”), we consider these risks outweighed by the gains obtained by a reference library of named sequences facilitating communication across the full array of fungal diversity.
Fig. 1.

BLAST result of an unnamed sequence (GenBank accession HM069408) representing an unnamed species of Archaeorhizomyces (Rosling et al. 2011). The BLAST result gives no clue as to the phylogenetic relationships of this sequence.
Impact on nomenclatural types
Zamora et al. (2018: 170) state: “An acceptance of the proposal would fundamentally alter the meaning of nomenclatural types. This is because instead of using a physical object as the type of a name, we would just use information from a character of the organism as the type.” We disagree. Firstly, there is no fundamental change to the meaning of types, since precisely in fungi it is already possible to use an illustration of a character as type “... if there are technical difficulties of preservation or if it is impossible to preserve a specimen that would show the features attributed to the taxon ...” (Art. 40.5). This provision was used to establish the lichenicolous genus Lawreymyces, with seven species, using images of the ITS sequences (Hawksworth 2017, Lücking & Moncada 2017), the latter triggering the Nomenclatural Section of the Shenzen IBC to insert a new example in the Code making clear that such images of DNA sequences are “not depictions of features of the organism” (Turland et al. 2018: Art. 40.5 Ex. 6). While we disagree with this decision, as a DNA sequence is a feature of an organism, we find it hard to argue that a drawing of, for example spore characters, is allowable as a type but a DNA sequence is not. Both forms of types are conceptually similar in nature as they are impressions of features and are not photographic images of physical structures; to have a sequence type would not therefore be a fundamental change in our opinion. It is difficult to argue that an illustration is more diagnostic than a DNA sequence, as exemplified by the new chytrid Fimicolochytrium jonesii based on an illustration as type, but the authors themselves acknowledged that morphological characters in this case are “... not completely accurate in assigning chytrids to the correct genus or species, thus emphasizing the importance of molecular characters for identifying these taxa.” (Simmons & Longcore 2012: 1229).
Zamora et al. (2018: 170) rightfully argue that even for fungi only known from sequences, it is not necessary to designate the sequence as type, since conceptually the underlying sample contains the type and there are also techniques that can make fungal structures containing the target sequences visible, and none of these require changes to the Code. We do not only agree, but all these possibilities were evaluated previously (Lücking & Hawksworth 2018: 146ff, Table 1). The proposal to allow the actual sequence as type is based on three arguments:
• Even if there is a specimen or culture as type, if sequence data are available, the relationships of the taxon will always be evaluated based on sequence data. Sequences from types (Schoch et al. 2014), not the physical types themselves, fix the application of a name in a phylogenetic framework, so type sequences essentially serve as proxies for physical types and from there, the step to have sequences as actual types is small.
• Fungi known from sequences only can be validly described under the current Code using a physical type, such as the underlying sample, as has been done with Piromyces cryptodigmaticus (Fliegerová et al. in Kirk 2012). However, this approach is not optimal as the type is ambiguous and just serves as a formality, whereas the species name will be evaluated based on its type sequence. Even a FISH type (e.g. as a permanent slide), while visualizing the actual fungus, for practical purposes is useless as a type in terms of assessment or reproducibility of characters.
• In voucherless fungi, a sequence type has several advantages over other forms of types, including broad accessibility and unlimited long-term storage of multiple copies without quality loss. This does not apply if the type is the underlying sample, DNA extract, or FISH type.
Impact on names of taxa and future taxonomic studies
The arguments in this section largely correspond to points (6) and (9) of Thines et al. (2018) and are addressed below, with reference to Lücking & Hawksworth (2018: 152f).
Reliability and extent of data
The arguments in this section largely correspond to points (2) to (6) of Thines et al. (2018) and are addressed below, with reference to Lücking & Hawksworth (2018: 148ff).
Candidate names
Zamora et al. (2018) propose to alternatively consider the approach of so-called “candidate names” as suggested in the International Code of Nomenclature of Prokaryotes (ICNP; Parker et al. 2015) for archaea and bacteria that cannot be cultured and so do not fulfil the requirements for valid description under that Code. We appreciate this suggestion as a constructive alternative. The ICNP has three provisions for candidate names (Box 1).
Box 1
Candidatus names (extracted from ICPN Appendix 11)
“(3) A name of an organism in the status of Candidatus consists of the word Candidatus, followed by a “vernacular epithet” that consists of either a genus name with a specific epithet, or only a genus name, or only a specific epithet. ... the word Candidatus, but not the vernacular epithet is printed in italics.”
“(4) A Candidatus name is by definition a preliminary name and therefore has no standing in prokaryote nomenclature.”
“(5) A list in the form of a codified record of organisms of the status Candidatus is kept by the Judicial Commisssion of the ICSP in cooperation with the Editorial Board of the IJSEM and is published in that journal in appropriate intervals.”
While this concept could be applied to the case of voucherless fungi known only from sequences, there is one aspect which we consider detrimental to the idea of avoiding “chaos”, as so vehemently advocated by the opponents of sequence-based nomenclature. The Candidatus names are invalid and have no standing under the prokaryote code. Further, for those unfamiliar with this system, this causes confusion as the italicized word “Candidatus” at the start of the species name gives the impression that these are taxa in a genus Candidatus, as in “Candidatus Liberibacter africanus” and “Candidatus Brocadia anammoxidans"; the nuances of type faces are not easily appreciated by editors and non-specialists and can be found all italicized in the literature.
With such a provision in fungal nomenclature, there would be no rules as to how to coin names beyond “Candidatus”, or any system to which authors should adhere in such nomenclatural acts. We fear this would generate more confusion than adoption of any formal sequence nomenclature, including the notion that parts of the mycological community could simply choose to ignore these names. Validity of names is an indispensable component of an ideal sequence-based nomenclature, since only then nomenclatural acts can be formally evaluated and judged against agreed rules.
The idea of using a unique prefix, such as “DNA”, for sequence-based fungal taxa and the provision that such names have no priority over specimen- or culture-based names are analogous to the Candidatus concept and essentially have the same effect (see below under point 9). Another potentially useful approach similar in concept to the Candidatus concept would be to develop rules for sequence-based names separately, perhaps in a Code of Practice agreed by the ICTF, rather than to change the Articles relating to types. That way, while formally binding, sequence-based nomenclature could be regulated outside the main body of the Code, with the possibility of including provision as to how sequence-based names are to be derived and evaluated.
There is an established nomenclatural practice, already familiar to many mycologists, to append a notation after a name to indicate nomenclatural status, such as “nom. inval.”, “nom. illegit.”, “nom. nud.”, “nom. nov.”, “nom. cons.”, “nom. sanct.”, or “nom. prov.”. The use of provisional names (“nom. prov.”) is analogous to the Candidatus concept in that it is used when an author wishes to have more material before formally validating a taxon name, but wants to have a label to discuss it. However, this has the disadvantage that this could encourage an explosion of invalid unregulated names. We suggest that a more acceptable alternative would be the use of an addition “nom. seq.” (“nomen sequentiae”) after any sequence-based name in a parallel manner, but ruling that such names would then be validly published but not have priority over names based on physical types (specimens or cultures), or illustrations.
Part II. Reply to Thines et al. “Ten reasons why a sequence-based nomenclature is not useful for fungi anytime soon”
1. The resolution of barcoding loci, especially ITS, varies among different groups
The authors correctly point out that the variation of the fungal barcoding marker ITS is not uniform at the same hierarchical level, in particular species, among different groups of fungi. Lücking & Hawksworth (2018: 150f, Box 4) never claimed that this would be the case, and discussed this problem in detail. Principally, there can be three outcomes: (a) ITS-based clades principally correspond to species-level clades; (b) ITS-based clades only resolve (generally monophyletic) species complexes (underestimation of actual species richness or “false negative”); or (c) ITS-based clades correspond to infraspecific lineages (overestimation of actual species richness or “false positive”).
In many fungi in which ITS phylogenies have been placed in the context of multi-locus approaches and/or phenotype variation, the ITS barcoding locus works reasonably well for species delimitation (e.g. Roy et al. 1998, Geml et al. 2006, Weir et al. 2012, Gomes et al. 2013, Walter et al. 2013, Moncada et al. 2014, Lücking et al. 2014a, 2017, Del Prado et al. 2016). Even in Oomycota, besides cox1 and cox2, ITS is routinely used for species delimitation (Thines & Kummer 2013). There are, however, also cases in which ITS only resolves species complexes (“false negatives”), including in genera such as Aspergillus, Fusarium, Morchella, Penicillium, and Pseudocercospora; in these instances, additional barcoding markers such as calmodulin (CAL), ß-tubulin (BenA) or translation elongation factor 1-alpha 1 (TEF1) have been proposed (Crous et al. 2013, Samson et al. 2014, Visagie et al. 2014, O’Donnell et al. 2015, Richard et al. 2015). In contrast, highly variable ITS within a species (potentially leading to “false positives”), such as in the lichen-forming Cetraria aculeata (Fernández-Mendoza et al. 2011), appears to be rare (and is also subject to interpretation of the species concept applied), which means that sequence-based nomenclature using the ITS barcoding locus would tend to be conservative, certainly a desirable attribute.
In spite of the many studies available, Thines et al. (2018: 178) cite a single paper (Stadler et al. 2014a, cited as “b”) for their argument of potentially conserved ITS and its presumed subpar performance compared to other markers: “The ITS regions are rather conserved in many species groups, in particular within the Sordariomycetes and other classes of Ascomycota (Stadler et al. 2014b).” We examined the cited study, a polyphasic revision of the genus Daldinia (Xylariaceae), and found the following statements:
• (p. 23) “The preliminary molecular phylogeny of Daldinia presented here is exclusively based on ITS rDNA gene sequence data.” The study relied on ITS as a single marker, even if presumably inappropriate.
• (p. 23) “Finally, most taxa of xylarioid Xylariaceae ... were omitted, since their ITS regions sequences [sic] were found to contain too many DNA portions that could not be aligned with certainty.” This points to variable, rather than conserved, ITS; in most Fungi, ITS is not well alignable between genera but more or less readily between species, and according to these authors, Xylariaceae do not seem to be an exception.
• (p. 27) “With few exceptions, the molecular phylogeny based on ITS nrDNA data largely supports this concept, and in one case (D. andina) the molecular data even gave hints where to place the respective fungus.”. This phrase refers to infrageneric divisions of Daldinia predicted by phenotype features including natural products, i.e. there is a high level of congruence between ITS data and phenotype features.
• (p. 132) “The species groups outlined in this monograph were mostly recognised as reasonably well supported groupings by the ITS rRNA gene phylogeny.” The authors recognized species complexes based on ITS and proceeded in establishing new taxa, such as Daldinia starbaeckii, within these complexes, based for instance on variation of ascospore size and chemical products, although the ITS did not provide any such separation, for instance from D. eschscholtzii.
Thines et al. (2018: 178) use these results as argument for conserved ITS in Daldinia, although in the absence of evidence from other markers we find this is a circular conclusion; also, in other groups of Fungi, analogous variation has been interpreted as intraspecific, e.g. chemical variation in species of Usnea (Mark et al. 2016). Therefore, until more markers are analyzed, such as in the genera cited above, we cannot agree with this specific example and consider other, better studied genera such as Fusarium more appropriate examples of conserved ITS. However, to consider insufficient resolution a failure of the concept of a barcoding marker is in our view ill-defined. Species are not fixed entities in time but emerge, evolve, and speciate, or become extinct. This process does not happen overnight but may take geological time spans; therefore, we cannot expect that the ITS, or any other marker, will resolve clades in the same way across all taxa, a point elaborated by Zamora et al. (2018: 169; see above). Species complexes that radiated recently are likely to exhibit low resolution, and ancient species already in the process of subsequent speciation are likely to have highly variable ITS. That does not mean that ITS undergoes different evolutionary mechanisms in different clades, but simply that some species-level clades are older or younger than others. Recently radiating species complexes could also be interpreted as infraspecific lineages, leading to the question of whether ITS properly resolves species depends on the ranking of such lineages (e.g. James et al. 2001, Onut̨-Brännström et al. 2017).
Thines et al. (2018: 177) state that “The idea of using sequence similarity as a measure of defining taxa is tempting ...”. There appears to be a widespread misconception that sequence-based nomenclature, in particular when based on ITS, should rely on sequence similarity, a concept derived from clustering techniques and employed, for instance, in the UNITE species hypotheses (Kõljalg et al. 2013). Lücking & Hawksworth (2018: 154ff) explicitly state that clustering techniques and pre-defined similarity thresholds are inappropriate to delimit lineages, and that instead multiple alignment-based phylogenetic approaches and quantitative species delimination methods should be used. These methods are independent of any a priori similarity threshold level, and instead the similarity between (sister) lineages is determined a posteriori and usually variable between clades and dependent on the time of speciation and other evolutionary parameters (such as population size and structure). In our proposed approach, sequence similarity is not at a measure to be considered (see also below under point 7).
2. There is a high risk of introducing artefacts as new species
We do agree that there is a risk, and this was addressed in Lücking & Hawksworth (2018: 148ff). However, based on published evidence, this risk is low and manageable (e.g. Lindner et al. 2013). Thines et al. (2018) cite erroneous base calls stemming from careless editing of Sanger contigs (particularly in terminal regions) or from sequencing errors, including TAQ polymerase errors, as one of the high risk factors. According to these authors, “... most widely used polymerases ... have a high rate of incorporating wrong nucleotides ...” (Thines et al. 2018: 178). In reality, reported TAG polymerase errors are less than 0.1% of replicated bases (Chen et al. 1991, Keohavong & Thilly 1998), on average less than one base in a full ITS sequence of 500 to 600 bases, a proportion that falls well within intraspecific variation and in a phylogenetic context provides no risk whatsoever of resulting in artifactual species recognition. The authors are correct in stating that terminal portions of Sanger sequences are often of subpar quality (actually much more so in protein-coding markers than in ribosomal DNA), but such portions can be easily recognized and trimmed, reducing the probability of any artifactual effect to practically zero.
Thines et al. (2018) fail to recognize the most important single nucleotide sequencing errors in high throughput sequencing, namely CAFIE errors, addressed in Lücking & Hawksworth (2018: 149ff). CAFIE errors on average are at levels an order of magnitude higher than TAG polymerase errors, at around 1 % (Lücking et al. 2014b), corresponding to about 5–6 bases in a full length ITS sequence. However, due to the stochastic distribution of these errors within the ITS (including the highly conserved 5.8S region), multiple alignment-based phylogenetic methods are robust against such errors and recover species-level clades accurately (Lücking & Hawksworth 2018: 155). In contrast, clustering methods are highly susceptible to sequencing errors, which largely account for a substantial overestimation of OTUs in environmental studies relying on clustering approaches (Lücking & Hawksworth 2018: 154ff).
We agree with Thines et al. (2018) that chimeras are the most critical source of artifactual ITS sequences, both in Sanger and high-throughput sequencing. However, there are methods of automatically filtering chimeric sequences that reduce the potential risk to about 1 % (Edgar et al. 2011, Quince et al. 2011, Schloss et al. 2011, Porazinska et al. 2012, Kim et al. 2013, Mysara et al. 2015, Edgar 2016), a proportion that certainly cannot be considered high. In addition, Sanger-generated chimeras can rather easily be detected as they can form long but unsupported branches, since they combine unique sequence patterns from unrelated ITS1 and ITS2 portions (hence a long stem branch) with affinities to two distinct, separate clades (hence with low support since bootstrapping will pull individual sequences to one of these other clades based on subsampling). High-throughput sequencing (HTS) chimeras are more difficult to detect but are also more easily filtered, since chimeric PCR products mostly result in sequences with subpar signal quality and so rarely pass through quality filters. If they do, they are extremely difficult to recognize as chimeras, but defining taxa through sequences originating from independent samples reduces this risk considerably, since the probability of congruent chimeras originating from two or more independent samples is close to zero.
Thines et al. (2018) mention intragenomic divergence of ITS sequences as a risk factor, a problem already addressed in their first point, with a study on Fusarium (O’Donnell & Cigelnik 1997). Another case cited is Xylaria hypoxylon (Peršoh et al. 2009, Stadler et al. 2014b), for which Stadler et al. (2014b: 65) state: “Remarkably, three cultures obtained independently from cultures derived from the same perithecium of the epitype material gave three slightly different ITS sequences. This indicates that DNA sequencing will not always lead to 100 % reproducible results, and special care should be taken not to overestimate the value of molecular techniques for estimation of species numbers and diversity.” We examined the three cited epitype sequences (AM993141, AM993142, AM993144). All three align for a total length of 512 bases, and we found the only difference to be a single base call in position 86 of sequence AM993144, exhibiting an A instead of a G. This variation amounts to 0.06 % of all base calls in the three sequences, i.e. less than the average of reported error levels of TAG polymerase. It could therefore be a simple sequencing error or else variation to be expected in the presence of concerted evolution; such variation is not rare in Sanger sequences (evidenced by double peaks in individual positions), but has no real impact on species recognition. It cannot be taken as a “high risk of introducing artefacts as new species.”
Thines et al. (2018) list further examples to underline their point of aberrant intragenomic behaviour of ITS repeats. However, the studies of Won & Renner (2005) and Harpke & Peterson (2008), as evidence for ITS degeneration, refer to vascular plants in Gnetum and Cactaceae This can hardly be used to assess evolutionary processes concerning the ribosomal DNA cistron, including the ITS, in Fungi, particularly since speciation based on hybridization and allopolyploidy, cited in those studies as a potential cause of ITS degeneration, is widespread in vascular plants, but is of uncertain frequency in Fungi. A more appropriate reference for Fungi would have been Li et al. (2013), which according to Thines et al. (2018) underlines a potential lack of concerted evolution, but mostly deals with presumed ITS pseudogenes in Ophiocordiceps sinensis. Pseudogenes evolve within the genome of one lineage and explore new phylospace independent from other lineages; they therefore should not cluster with ITS variants of other lineages. An example is the study of Lindner & Banik (2011) on Laetiporus, in which a single species, L. cincinnatus, was found to contain multiple ITS variants. All these cluster with support with other species, in particular L. sulphureus (see also Lücking & Hawksworth 2018: 151ff), and the only explanation for such a pattern is hybridization and introgression, not ITS degeneration and pseudogene formation. The distinction between pseudogenes and hybridization is critical, since the latter does not “... produce artefact shadow taxa ...” as Thines et al. (2018: 178) claim, but corresponds to real species, independent whether of their ITS is detected in the hybrid genome of another species. Implementating ITS meta-barcoding in Candida s.lat., Colabella et al. (2018) found, depending on mapping procedures, identity values for reads from strains of two different species to the expected ITS sequence (from Sanger sequencing) of between 97.9% and 99.8%. These values were interpreted as intragenomic variation, but could at least in part be due to sequencing errors, as the proportions compare with those found by Lücking et al. (2014b); nevertheless, the suggested mapping procedures resulted in rather high accuracy to detect the correct species. The authors also found that some reads were “... highly homologous ... to the rDNA of other species ...” (Colabella et al. 2018: 99), further supporting the hypothesis that intragenomic ITS variation is chiefly due to hybridization rather than pseudogene formation.
3. There is no consensus regarding the data type or amount needed for species delimitation
We wholeheartedly agree with this point! However, such a consensus is not necessary. A scientific approach that relies on a pre-defined consensus, such as clustering based on similarity thresholds, is flawed. Rather, scientific analysis should be independent and not be based on a priori assumptions and instead enable us to test assumptions a posteriori. There are numerous analytical methods to properly analyze sequence data, elaborated in Lücking & Hawksworth (2018), and statistical approaches can be employed to determine how many independent sequences of a certain length are needed to render clades statistically reliable (see below under point 7).
4. Voucherless data are not reproducible
Thines et al. (2018) argue that DNA sequence types without physical voucher specimens are not reproducible. In addition, DNA sequence types do not allow the assessment of other characters, including phenotype and other molecular markers. While the latter statement is correct (and discussed further below and in Lücking & Hawksworth 2018: 146ff), this has little to do with reproducibility. The latter refers to the original data, i.e. the sequence(s) used to define a species and in particular the sequence type, and not the assessment of additional features. This issue has rarely been questioned in Sanger sequences, although the problem of reproducibility applies equally to both Sanger and HTS data and both have analogous underlying sources. Sanger and HTS sequences come from specimens or environmental samples, respectively, and these can always be restudied (if stored properly) or other specimens or samples can be gathered under the same conditions. Both generate DNA extracts and PCR products that can be reanalyzed, and both result in sequences (reads) based on trace files that can be reassessed. Unfortunately, in most cases neither specimens nor samples, or DNA extracts and PCR products, or trace files, are readily accessible to investigators other than those who produced the data, and proper storage is often not guaranteed. However, reproducibility of the original sequence data is possible in quite an analogous way in both Sanger and HTS sequences, so potential problems of reproducibility equally apply to both. Therefore, this cannot be used as an argument against sequence-based nomenclature.
In addition, sequences are evaluated in an alignment-based phylogenetic context and potential problems can be detected that way. An example is GenBank accession AF356664, which caused an entire class of Fungi, Eurotiomycetes, to erroneously appear nested within another class, Lecanoromycetes, due to a “multilocus chimera” (Lücking & Nelsen 2018). Highly congruent sequences originating from independent sources have an astronomically low possibility to be artifactual, which provides a means of testing the data without the need to go back to the source and reproduce the actual sequence (see below). The same approach can (and must) be used in sequence-based nomenclature (Hawksworth et al. 2016, 2018, Lücking & Hawksworth 2018: 149ff).
5. Sequence-based types cannot be verified
We are not sure why the authors list this as a separate point, as it is fully congruent with their previous argument. As mentioned above, a simple but effective means of testing a sequence is whether the same or a highly congruent sequence occurs in independent samples. A reliable statistical test can be employed to compute the probability that N sequences of length L from X independent samples are so similar that they cluster with strong support in a monophyletic clade, but instead of being of common descent represent sequencing artifacts. It can be shown that for N and X ≥ 5 and L ≥ 200 (e.g. separate ITS1 or ITS2 regions), this probability becomes astronomically small. Even if the sequences are from the same sample or run, the same principle applies, since HTS sequences are generated from independent PCR products in separate wells and the repetition of the same stochastic sequencing error, including chimeras, in separate PCR products and wells is highly unlikely and substantially decreases with increasing number of congruent sequences. Ergo, HTS-derived sequence types can be effectively tested, and so can the resulting clades.
6. Sequence-based types are not relatable
Thines et al. (2018) again argue that sequence types cannot be attached to specimens (which is indeed the essence of sequence-based nomenclature) and that therefore other characters cannot be assessed, a valid argument but essentially repeating points (4) and (5). Sequence-based nomenclature needs to be based on the molecular data of a single marker in order to work, and this is indeed a shortcoming of this approach, acknowledged by Lücking & Hawksworth (2018: 146ff). However, this issue relates in part to the potential limitations of ITS as a single barcoding marker (see also reply to Zamora et al. above), many of which are perceived rather than real, as shown in Lücking & Hawksworth (2018: 150ff), and do not relate to the use of a sequence type per se. While some limitations exist, the gain in producing named reference sequences across a broad range of unclassified fungal lineages surely far surpasses its problems.
We do agree with Thines et al. (2018: 179) that: “Presently about 120 000 species are acknowledged, but there are more than 400 000 names ... Only a mere fraction of the 120 000 accepted species have DNA sequences deposited. If species were named based on environmental sequences, and they were given the same status as species with specimens, the risk would arise that all work done before the first DNA sequences were deposited in GenBank, in 1991, would be deliberately ignored.” Since priority only applies within the same rank, we have to be concerned with approximately 240 000 species level names in Fungi as of this date (not 400 000), but this number is still extremely high considering that only a fraction, approximately 35 000 names or 15 %, have sequence data attached to them (Lücking & Hawksworth 2018: 152). This is in our view the core problem of sequence-based nomenclature, and Lücking & Hawksworth (2018: 152ff) considered this at some length, not only quantifying the problem but also offering solutions, something not acknowledged by Thines et al. (2018). Lücking & Hawksworth (2018: 153 Table 2) computed the average statistical synonym error rate, i.e. the proportion of new sequence-based species actually conspecific with previously established names lacking sequence data, as function of predicted overall species richness of Fungi. The proportion of inadvertent synonyms among new names based on sequences only ranges from about 20 % (assuming 700 000 fungal species), to about 10 % (assuming 1.5 million), to about 5 % (assuming 3 million). If currently there are 240 000 species level names in fungi, with 120 000 species accepted, and the number of true synonyms among the remaining 120 000 names ranges between zero and 60 000, the historical error rate of inadvertent synonymy lies between 33 % and 50 %. While a projected synonym error rate in sequence-based nomenclature between 5 % and 20 % compares quite favorably to the historical error rate based on physical types, this rate is still too high; Lücking & Hawksworth (2018: 153, Box 5) therefore proposed: “For a new species based on a sequence type, without a physical voucher specimen, to be validly established ... available names in the containing genus [must] have been linked to a phylogenetically defined and named clade different from that with the new species ... or must have been established as valid species or synonyms in other genera. [In addition] Names based on sequence types are not given priority over names based on physical types ..., unless later epitypified with a matching specimen or culture.”
Finally, Thines et al. (2018: 179) argue that “... sequence data do not relate to any real-world object.” This is the same as saying that spoors, even if distorted, are not caused by actual animals but are artifacts of tracking. The entire community of biodiversity researchers, evolutionists, geneticists, etc., may hopefully join us in disagreeing with this statement.
7. Sequences of reported OTUs are derived, not actual sequences
This is another point based on a misconception. While studies based on environmental sequence data often operate with cluster-derived OTUs, only in some cases are these reported as consensus sequences; in reality, each OTU is the cluster of original sequence reads contained therein. Data from environmental sequencing studies deposited in the SRA are exclusively original reads, not OTU consensus sequences. These reads are as real as Sanger sequences; in fact, HTS reads are raw sequences whereas Sanger sequences are typically consensus contigs of two or more raw sequences, so the argument of “derived” consensus sequences applies more to Sanger sequences than to HTS reads. While we agree with the statement that “OTU sequences do not need to correspond to an actual sequence found in an organism, as they are derived sequences” (Thines et al. 2018: 180), this argument is not pertinent since a DNA sequence type cannot be a cluster- or clade-based consensus but must always be the actual sequence best representing the clade.
Since Lücking & Hawksworth (2018) unmistakably advocated that clustering methods are inappropriate for delimiting sequence-based taxa (see above), we are astonished by the statement that “... it would be unclear where to draw boundaries between the different OTUs as there will always be the potential for overlap between OTUs if they are derived from rather similar sequences.” (Thines et al. 2018: 180). The authors apparently ignore the fundamental principles of multiple alignment-based phylogenetic analyses and quantitative species recognition methods now routinely applied to delimit clades, whether based on single or multiple markers, methods used by themselves on multiple occasions (e.g. Peršoh et al. 2009, Thines & Kummer 2013, Choi et al. 2015, Liu et al. 2016, Kijpornyongpan & Aime 2017, Hongsanan et al. 2017, Raja et al. 2017, Réblová et al. 2018).
8. Sequence-based types favour well-funded large mycology labs and leave researchers in developing countries behind
This statement appears to assume that sequence-based nomenclature is introduced by the same researchers that produce the environmental sequence data. This contradicts the statement made on the same page under their point (9): “If it is possible to publish new species from the computer just on the basis of a DNA sequence ...”
Sequence-based nomenclature is essentially a computational exercise; it is by no means trivial and requires considerable skill and understanding, but in terms of logistics, all that is needed is a computer and access to the internet, with freely available software and multi-core servers such as CIPRES (Miller et al. 2010), as well as the corresponding data repositories (SRA, GenBank). The SRA currently holds more than a billion fungal ITS reads (Lücking & Hawksworth 2018: 144) and just analysing these in a solid phylogenetic context would keep numerous researchers occupied for quite some time without any new environmental sequence data being generated. Such analyses can be done in virtually any part of the world which, quite to the contrary, gives researchers in less developed countries access to an entirely new dimension of fungal biodiversity research, research that almost nobody has been doing to this point, so why keep others from doing it?
9. Allowing sequence-based types would be detrimental for mycology as a discipline
We respectfully disagree. Citing Nilsson et al. (2016), Thines et al. (2018: 180) state: “If the act of publishing a sequence could be seen as the formal act of introducing a new species, there is a high risk that interest in the actual discovery of the organism would diminish, as the discovery of the actual organism would become the equivalent of an epitypification, which would probably be done for only a few highly prevalent or interesting organisms.” We find it difficult to follow this argument. If bigfoot would be formally named based on its footprints (some authors relate it to the extinct ape genus Gigantopithecus, so it might already have a name), people would not stop looking for the creature (even if DNA data suggest that its Himalayan counterpart, the yeti, may just be a bear; see Sykes et al. 2014, Gutiérrez et al. 2015). Also, mycology cannot be reduced to naming things. Fungal nomenclature is a part of mycology, but the essence of mycology is elucidating the role of fungi in ecosystems, their impact as pathogens of crops and humans, and their innumerable potential applications in food, pharmaceutics, acid and enzyme production, and biological control. None of this can be done with sequences; the physical fungus is always required, and hence naming sequences is not a threat to any other field of mycology.
Notably, the authors apparently inadvertently make this argument themselves: “Another problematic issue is that if sequence data were accepted as type, specimens might be seen as obsolete ... This could herald the end of fungaria and the decline of culture collections, even though these might hold the key for substances of unpredictable value for human welfare, such as antibiotics, therapeutically relevant metabolites, as well as platform chemicals and enzymes for biotechnology.” Since (ITS) sequence data are only useful to define clades and hold no information on features or properties, let alone possible importance and applications, there would be no reason or pressure to reduce or eliminate fungaria or culture collections. Naming sequences does not change the need for having the actual fungus at hand; to the contrary, providing a formal nomenclatural framework for fungi known only from sequences makes obtaining funding for the study of these and assessments of their potential properties more likely, especially based on the context in which the sequences were detected. With sequence-based nomenclature, other fields of mycology would not stop or slow down, but we would not have to wait for centuries until a substantial portion of the fungi on Earth has been named.
We agree that: “There is also the risk that in systems where quantity in research is valued higher than quality, massive amounts of names without detailed quality checks would be published, flooding fungal nomenclature with tens of thousands of meaningless names that would need to be sorted out in future decades or centuries.” (Thines et al. 2018: 180). Together with the risk of naming species that already have a name not attached to a sequence, this is certainly the most critical issue of this approach, and therefore we advocate strict quality control (e.g. Lücking & Hawksworth 2018: 156, Box 6). Such quality control can be enforced, and while nomenclaturists argue that rules how to perform science should not be part of the Code, complementary guidance can be agreed and provided by international bodies such as the ICTF. We see a viable solution in giving sequence-based names a unique identifier, such as a “nom. seq.” suffix (see above), which are accepted as valid but do not have priority over specimen-, culture-, or illustration-based names. Such names could then be fully incorporated into the fungal system (i.e. removing the suffix and the priority limitation) through two mechanisms: (a) epitypification with a physical specimen or culture; or (b) periodical evaluation of a list of names by a committee operating under the ICTF and applying rigorous quality control.
Interestingly, Thines et al. (2018: 180) also appear to argue for some sort of sequence-based nomenclature: “... nonmycologists ... often tend to assign the species or genus name according to the most similar DNA sequence found in a BLAST search. This has led to manifold inaccuracies, which has prompted ... to encourage a more accurate treatment of the taxonomy of the species. A DNA based typification would send the wrong signal also to the scientists of other communities who, for a correct interpretation of their results, rely on mycologists providing sound species concepts using polyphasic methodology.” The issue is not that identifications are naively based on BLAST searches, but that BLAST searches increasingly return results such as “uncultured fungus” and similar unspecified designations (Fig. 1). If these entities do not start to be formally named in some way, the problem will intensify exponentially.
Thines et al. (2018) postulate that mycologists will eventually provide “... sound species concepts using polyphasic methodology”, which is certainly a desirable goal, but is not bound to happen within a reasonable time frame, due to the enormous number of fungal species that need to be named. Assuming a reasonable time frame of 50 to 100 years (Lücking & Hawksworth 2018: 145, Box 1) and a predicted number of between 1.5 and 3 million fungal species, the current rate of little over 2000 new species per year would have to be increased to between 15 000 and 60 000 new species per year (Fig. 2). Since 2008, the number of new species described each year has increased on average by 100; projecting this increase would result in 7500 new species per year by 2068 (50 years) and 12 500 new species per year by the year 2118 (100 years). By then, we would have described an impressive 757 000 new species and surpassed plants. However, not only would this linear increase not approach the minimum rate to describe 1.5 million new species within 100 years, but it would be unrealistic. The current increase of about 100 additional new species per year is largely based on more effective approaches to detect and describe new fungal species, including an increased number of mycology students particularly in tropical regions of the world, such as at the Federal University of Pernambuco in Brazil (https://www3.ufpe.br/ppgbf/index.php?option=com_content&view=article&id=445&Itemid=246), and the Mushroom Research Centre in Thailand (http://www.mushroomresearchcentre.com), and the Key State Laboratory of Mycology in the Chinese Academy of Sciences. However, this tendency may reach a plateau, because of a lack of posts in mycology for the emerging students. Also, mycology and other fields are increasingly shifting towards applied high-tech areas, and the support for alpha-taxonomy, biodiversity inventories, and naming organisms can sadly be expected to further decrease. We therefore predict that the annual rate of new fungal species being described from physical specimens and cultures will rise but level off at below 5000 species per year, possibly even below 3000. Of the “50 most wanted fungi” based on environmental sequencing data (Nilsson et al. 2016), one has been formally described based on cultures (James & Seifert 2017, Torres-Cruz et al. 2017), so “... the hard work of finding and describing these unknowns ...” (James & Seifert 2017: 362) indeed proceeds at a slow pace. However strong the desire may be to name fungi based only on physical specimens or cultures, for hundreds of thousands to millions of species this is not possible, unless we are content to wait several more centuries, when most of the habitats potentially yielding new species will have vanished.
Fig. 2.
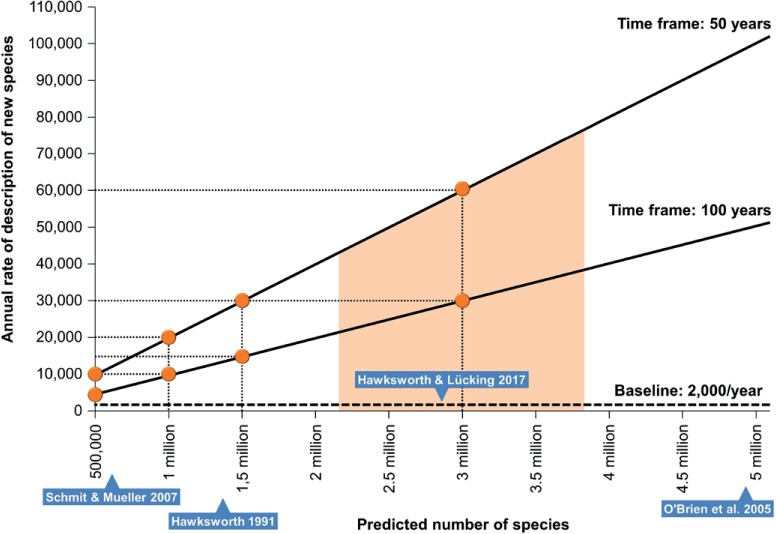
Necessary increase in the rate of newly described fungal species per year to reach a certain predicted number within reasonable time frames of 50 and 100 years, respectively.
10. An introduction of sequence-based nomenclature is impossible at present due to the fast pace at which sequencing technologies develop
The development of new technology has shaped fungal classifications (and all other fields of science) virtually since the beginning of time, factually since the start of fungal nomenclature in 1753 (Crous et al. 2015). In no single case when new technologies developed, such as the light microscope, tools to analyze chemical products, the electron microscope, and finally DNA sequencing, the mycological community first evaluated the new methodologies before they started to be used as tools in the formal classification of fungi. We do not see the reason why this should not be the case now.
At the start of formal fungal nomenclature, fungi were essentially classified based on their morphology and substrate ecology (including hosts).Today we know that a classification based on a polyphasic approach including molecular data, anatomy, chemistry, morphology, etc. is much more robust. Yet, even if we sometimes may wish away the additional work required to assess old types and protologues, we never seriously considered a provision in the Code that fungal nomenclature should have started officially in 1990. Even if we did: molecular sequencing is constantly developing, having started with a few selected markers and techniques that now seem ancient and obsolete; yet there was no movement proposing to wait in formalizing results from molecular studies until techniques became more advanced. Why now? We cannot help to see this reluctance as based on irrational fear, rather than scientific arguments. Even if we waited for entire genome sequences to be derived from environmental DNA, these likely would not result in significant advances over studies using selected markers, rendering the considerable amount of additional resources to obtain and analyse genomes from numerous species almost futile. Wherever technology will be taking us, the bulk of species delimitation studies and environmental meta-barcoding approaches will continue to use few, selected barcoding markers (e.g. Quaedvlieg et al. 2012). Also, if clades are clearly defined and supported based on single or few markers, no new technology in the future will essentially chance this.
We agree with Thines et al. (2018: 181) in saying: “At present, any such approaches are probably as useful as it had been to define communication standards for current mobile phones when the first portable telephones appeared in the late 80’s.” Indeed, relating to this analogy: we do not propose to set future communication standards, we propose to start using portable phones! Why? Because only their use, however primitive, fosters their advancement. The computational community is already putting substantial resources into phylogeny-based analyses of environmental sequences that compete with clustering methods in speed but far surpass them in accuracy (Berger et al. 2011, Zhang et al. 2013, Carbone et al. 2017, Barbera et al. 2018). Allowing formal sequence nomenclature would stimulate this field in unprecedented ways and all mycologists would profit from this.
PROPOSALS
In order to progress this issue at IMC11, rather than let it drift and be a potential source of confusion and frustration for at least another four years, we suggest two alternatives be considered as amendments "from the floor" of the Nomenclature Session:
(1) In the event of the proposals being accepted, to add the following Note to Chapter F of the Code under Art. F.4.2:
Note: Sequence-based names are to be registered in one of the approved repositories and allocated an identifier, but indicated by the addition of the suffix “nom. seq.” (nomen sequentium) after the name to indicate the special status of those names: Such names have priority over other sequence-based names, but do not have priority over names based on physical types (including cultures) or illustrations, until epitypified by a matching specimen or culture or included in a list of protected names.
(2) In the event of the formal proposals F-005 and F-006 (Hawksworth et al. 2018) not being accepted, to add the following new Note and an Example to Chapter F of the Code under Art. F. 5.5 (which deals with registration matters):
“Note X: In the case of designations based on molecular sequence data where there is no specimen or illustration available to serve as a nomenclatural type, the designations are to be registered in one of the approved repositories and allocated an identifier, but when released after effective publication such designations are to have “nom. seq.” (nomen sequentium) appended to indicate that the names are effectively published but remain not validly published until typified as required by this Code.
Example X: The designation Hawksworthiomyces sequentia de Beer & al. (in Fungal Biology 120: 1332. 2016) was assigned the identifier MB815690, but as it lacks a Code-compliant type it is to be referred to as H. sequentia de Beer & al. nom. seq. or H. sequentianom. seq., but not H. sequentia. The designation would remain available for use but not be validly published until typified by a specimen or illustration, and priority would date from the final act of validation, the later typification.
The International Code of Nomenclature for algae, fungi, and plants has always been careful not to make rules relating to taxonomic practice in order not to constrain scientific approaches, but we see advantage in having some additional guidance available which is provided by the international scientific community, and in the case of fungi this could be the ICTF. Such guidance could be on the lines of what is included here in Box 2, which would be analogous to Appendix 11 in the prokaryote Code.
Box 2
Possible topics and guidance that might be included in a Code of Practice on the introduction of sequence-based names.
In fungal nomenclature, when novel lineages are detected based on environmental (or analogous) sequence data alone, without a physical specimen or illustration, designations of new taxa can be formally introduced under the following conditions:
Sequence-based names must always be used with the agreed designation indicating their special status, for example Neoarchaeorhizomyces nom. seq. and; Neoarchaeorhizomyces paradoxus nom. seq.
Limitations of priority in relation to specimen- and illustration-based names.
Sequence-based names should not be introduced in genera that contain names not linked to phylogenetically defined clades.
Registration in the mandated repositories is required.
-
Sequence-based names should be introduced in accordance with the following protocol:
(a) Full ITS as the barcoding marker.
(b) Multiple alignment-based phylogenetic analysis in combination with quantitative, single-marker species recognition methods; clustering methods with preset similarity thresholds are not allowed.
(c) A clade formally recognized as species must contain at least five sequences from five independent samples (to be identified by their SRA sample accession numbers or GB accession numbers).
(d) The type sequence is not the clade consensus but the individual sequence best matching the clade consensus (to be determined quantitatively using an identify matrix).
(e) The underlying phylogeny used to establish new species in a given genus should contain all other species previously established in the genus.
Acknowledgments
We appreciate the frank and open contributions to this debate provided by many of our mycological colleagues around the world, which have assisted us in the clarification of our ideas and the formulation of the modified proposals and provisions now made.
REFERENCES
- Barbera P, Kozlov AM, Czech L, Morel B, Darriba D, et al. (2018) EPA-ng: massively parallel evolutionary placement of genetic sequences. BioRxiv: 291658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge SA, Krompass D, Stamatakis A. (2011) Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Systematic Biology 60: 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone I, White JB, Miadlikowska J, Arnold AE, Miller MA. et al. (2017) T-BAS: Tree-Based Alignment Selector toolkit for phylogenetic-based placement, alignment downloads and metadata visualization: an example with the Pezizomycotina tree of life. Bioinformatics 33: 1160–1168. [DOI] [PubMed] [Google Scholar]
- Chen J, Sahota A, Stambrook PJ, Tischfield JA. (1991) Polymerase chain reaction amplification and sequence analysis of human mutant adenine phosphoribosyltransferase genes: the nature and frequency of errors caused by Taq DNA polymerase. Mutation Research 31: 169–176. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Klosterman SJ, Kummer V, Voglmayr H, Shin HD, Thines M. (2015) Multi-locus tree and species tree approaches toward resolving a complex clade of downy mildews (Straminipila, Oomycota), including pathogens of beet and spinach. Molecular Phylogenetics and Evolution 86: 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colabella C, Corte L, Roscini L, Bassetti M, Tascini C, et al. (2018) NGS barcode sequencing in taxonomy and diagnostics, an application in “Candida” pathogenic yeasts with a metagenomic perspectiv. IMA Fungus 9: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crous PW, Braun U, Hunter GC, Wingfield MJ, Verkley GJM, et al. (2013) Phylogenetic lineages in Pseudocercospora. Studies in Mycology 75: 37–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crous PW, Hawksworth DL, Wingfield MJ. (2015) Identifying and naming plant pathogenic fungi: past, present, and future. Annual Review of Phytopathology 53: 247–267. [DOI] [PubMed] [Google Scholar]
- Del-Prado R, Divakar PK, Lumbsch HT, Crespo AM. (2016) Hidden genetic diversity in an asexually reproducing lichen forming fungal group. PloS One 11 (8): e0161031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. (2016) UCHIME2: improved chimera prediction for amplicon sequencing. bioRxiv: 074252. [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Mendoza F, Domaschke S, García MA, Jordan P, Martín MP, Printzen C. (2011) Population structure of mycobionts and photobionts of the widespread lichen Cetraria aculeata. Molecular Ecology 20: 1208–1232. [DOI] [PubMed] [Google Scholar]
- Geml J, Laursen GA, O’Neill K, Nusbaum HC, Taylor DL. (2006) Beringian origins and cryptic speciation events in the fly agaric (Amanita muscaria). Molecular Ecology 15: 225–239. [DOI] [PubMed] [Google Scholar]
- Gomes RR, Glienke C, Videira SIR, Lombard L, Groenewald JZ, Crous PW. (2013) Diaporthe: a genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 31: 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube M, Gaya E, Kauserud H, Smith AM, Avery SV, et al. (2017) The next generation fungal diversity researcher. Fungal Biology Reviews 31: 124–130. [Google Scholar]
- Gutiérrez EE, Pine RH. (2015) No need to replace an “anomalous” primate (Primates) with an “anomalous” bear (Carnivora, Ursidae). ZooKeys 487: 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpke D, Peterson A. (2008) Extensive 5.8 S nrDNA polymorphism in Mammillaria (Cactaceae) with special reference to the identification of pseudogenic internal transcribed spacer regions. Journal of Plant Research 121: 261–270. [DOI] [PubMed] [Google Scholar]
- Hawksworth DL. (2017) DNA sequences as types: a potential loophole in the rules discovered. IMA Fungus 8: (4). [Google Scholar]
- Hawksworth DL, Hibbett DS, Kirk PM, Lücking R. (2016) (308–310) Proposals to permit DNA sequence data to serve as types of names of Fungi. Taxon 65: 899–900. [Google Scholar]
- Hawksworth DL, May TW, Redhead SA. (2017) Fungal nomenclature evolving: changes adopted by the 19th International Botanical Congress in Shenzhen 2017, and procedures for the Fungal Nomenclature Session at the 11th International Mycological Congress in Puerto Rico 2018. IMA Fungus 8: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawksworth DL, Hibbett DS, Kirk PM, Lücking R. (2018) (F-005-006) Proposals to permit DNA sequence data to be used as types of names of fungi. IMA Fungus 9: v–vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbett D. (2018) More than a handful of dirt: sequence-based species description and the role of the ICN (a response to Seifert). IMA Fungus 9: (20)–(22). [Google Scholar]
- Hibbett D, Abarenkov K, Koljalg U, Öpik M, Chai B, et al. (2016) Sequence-based classification and identification of Fungi. Mycologia 108: 1049–1068. [DOI] [PubMed] [Google Scholar]
- Hongsanan S, Maharachchikumbura SS, Hyde KD, Samarakoon MC, Jeewon R, et al. (2017) An updated phylogeny of Sordariomycetes based on phylogenetic and molecular clock evidence. Fungal Diversity 84: 25–41. [Google Scholar]
- James TY, Seifert KA. (2017) Description of Bifiguratus adelaidae: The hunt ends for one of the” Top 50 Most Wanted Fungi”. Mycologia 109: 361–362. [DOI] [PubMed] [Google Scholar]
- James TY, Moncalvo JL, Li S, Vilgalys R. (2001) Polymorphism at the ribosomal DNA spacers and its relation to breeding structure of the widespread mushroom Schizophyllum commune. Genetics 157: 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keohavong P, Thilly WG. (1989) Fidelity of DNA polymerases in DNA amplification. Proceedings of the National Academy of Sciences, USA 86: 9253–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijpornyongpan T, Aime MC. (2017) Taxonomic revisions in the Microstromatales: two new yeast species, two new genera, and validation of Jaminaea and two Sympodiomycopsis species. Mycological Progress 16: 495–505. [Google Scholar]
- Kim M, Lee KH, Yoon SW, Kim BS, Chun J, Yi H. (2013) Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics & Informatics 11: 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk PM. (2012) Nomenclatural novelties. Index of Fungi 1: 1. [Google Scholar]
- Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AF, et al. (2013) Towards a unified paradigm for sequence-based identification of Fungi. Molecular Ecology 22: 5271–5277. [DOI] [PubMed] [Google Scholar]
- Li Y, Jiao L, Yao YJ. (2013) Non-concerted ITS evolution in Fungi, as revealed from the important medicinal fungus Ophiocordyceps sinensis. Molecular Phylogenetics and Evolution 68: 373–379. [DOI] [PubMed] [Google Scholar]
- Lindner DL, Banik MT. (2011) Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in the genus Laetiporus. Mycologia 103: 731–740. [DOI] [PubMed] [Google Scholar]
- Lindner DL, Carlsen T, Nilsson HR, Davey M, Schumacher T, Kauserud H. (2013) Employing 454 amplicon pyrosequencing to reveal intragenomic divergence in the internal transcribed spacer rDNA region in Fungi. Ecology and Evolution 3: 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Wang M, Damm U, Crous PW, Cai L. (2016) Species boundaries in plant pathogenic fungi: a Colletotrichum case study. BMC Evolutionary Biology 16: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking R, Hawksworth DL. (2018) Formal description of sequence-based voucherless Fungi: promises and pitfalls, and how to resolve them. IMA Fungus 9: 143–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking R, Moncada B. (2017) Dismantling Marchandiomphalina into Agonimia (Verrucariaceae) and Lawreymyces gen. nov. (Corticiaceae): setting a precedent to the formal recognition of thousands of voucherless Fungi based on type sequences. Fungal Diversity 84: 119–138. [Google Scholar]
- Lücking R, Nelsen MP. (2018) Ediacarans, protolichens, and lichen-derived Penicillium: a critical reassessment of the evolution of lichenization in fungi. In Transformative Paleobotany (Krings M. et al. (eds.): in press. Amsterdam: Elsevier. [Google Scholar]
- Lücking R, Dal Forno M, Sikaroodi M, Gillevet PM, Bungartz F, et al. (2014a) A single macrolichen constitutes hundreds of unrecognized species. Proceedings of the National Academy of Sciences, USA 111: 11091–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking R, Lawrey JD, Gillevet PM, Sikaroodi M, Dal Forno M, Berger SA. (2014b) Multiple ITS haplotypes in the genome of the lichenized basidiomycete Cora inversa (Hygrophoraceae): fact or artifact? Journal of Molecular Evolution 78: 148–162. [DOI] [PubMed] [Google Scholar]
- Lücking R, Dal Forno M, Moncada B, Coca LF, Vargas-Mendoza LY, et al. (2017) Turbo-taxonomy to assemble a megadiverse lichen genus: seventy new species of Cora (Basidiomycota: Agaricales: Hygrophoraceae), honouring David Leslie Hawksworth’s seventieth birthday. Fungal Diversity 84: 139–207. [Google Scholar]
- Mark K, Saag L, Leavitt SD, Will-Wolf S, Nelsen MP, et al. (2016) Evaluation of traditionally circumscribed species in the lichen-forming genus Usnea, section Usnea (Parmeliaceae, Ascomycota) using a six-locus dataset. Organisms, Diversity & Evolution 16: 497–524. [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE): 1–8. New Orleans. [Google Scholar]
- Moncada B, Lücking R, Suárez A. (2014) Molecular phylogeny of the genus Sticta (lichenized Ascomycota: Lobariaceae) in Colombia. Fungal Diversity 64: 205–231. [DOI] [PubMed] [Google Scholar]
- Mysara M, Saeys Y, Leys N, Raes J, Monsieurs P. (2015) CATCh, an ensemble classifier for chimera detection in 16S rRNA sequencing studies. Applied and Environmental Microbiology 81: 1573–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson RH, Wurzbacher C, Bahram M, Coimbra VR, Larsson E, et al. (2016) Top 50 most wanted fungi. MycoKeys 12: 29–40. [Google Scholar]
- O’Donnell K, Cigelnik E. (1997) Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Molecular Phylogenetics and Evolution 7: 103–116. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Ward TJ, Robert VARG, Crous PW, Geiser DM, Kang S. (2015) DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43: 583–595. [Google Scholar]
- Onut̨-Brännström I, Tibell L, Johannesson H. (2017) A worldwide phylogeography of the whiteworm lichens Thamnolia reveals three lineages with distinct habitats and evolutionary histories. Ecology and Rvolution 7: 3602–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CT, Tindall BJ, Garrity GM. (2015) International Code of Nomenclature of Prokaryotes. International Journal of Systematic and Evolutionary Microbiology: DOI 10.1099/ijsem.0.000778. [Google Scholar]
- Peršoh D, Melcher M, Graf K, Fournier J, Stadler M, Rambold G. (2009) Molecular and morphological evidence for the delimitation of Xylaria hypoxylon. Mycologia 101: 256–268. [DOI] [PubMed] [Google Scholar]
- Porazinska DL, Giblin-Davis RM, Sung W, Thomas WK. (2012) The nature and frequency of chimeras in eukaryotic metagenetic samples. Journal of Nematology 44: 18–25. [PMC free article] [PubMed] [Google Scholar]
- Quaedvlieg W, Groenewald JZ, de Jesús Yáñez-Morales M, Crous PW. (2012) DNA barcoding of Mycosphaerella species of quarantine importance to Europe. Persoonia 29: 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. (2011) Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja HA, Miller AN, Pearce CJ, Oberlies NH. (2017) Fungal identification using molecular tools: a primer for the natural products research community. Journal of Natural Products 80: 756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réblová M, Miller AN, Réblová K, Štěpánek V. (2018) Phylogenetic classification and generic delineation of Calyptosphaeria gen. nov., Lentomitella, Spadicoides, Torrentispora and other morphologically similar fungi based on a six-locus dataset (Sordariomycetes). Studies in Mycology 89: 1–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard F, Bellanger JM, Clowez P, Hansen K, O’Donnell K, et al. (2015) True morels (Morchella, Pezizales) of Europe and North America: evolutionary relationships inferred from multilocus data and a unified taxonomy. Mycologia 107: 359–382. [DOI] [PubMed] [Google Scholar]
- Rosling A, Cox F, Cruz-Martinez K, Ihrmark K, Grelet GA, et al. (2011) Archaeorhizomycetes: unearthing an ancient class of ubiquitous soil Fungi. Science 333: 876–879. [DOI] [PubMed] [Google Scholar]
- Roy BA, Vogler DR, Bruns TD, Szaro TM. (1998) Cryptic species in the Puccinia monoica complex. Mycologia 90: 846–853. [Google Scholar]
- Ryberg M, Nilsson RH. (2018) New light on names and naming of dark taxa. MycoKeys 30: 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson RA, Visagie CM, Houbraken J, Hong SB, Hubka V, et al. (2014) Phylogeny, identification and nomenclature of the genus Aspergillus. Studies in Mycology 78: 141–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Gevers D, Westcott SL. (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PloS One 6 (12): e27310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch CL, Robbertse B, Robert V, Vu D, Cardinali G, et al. (2014). Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database 2014: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert KA. (2017) When should we describe species? IMA Fungus 8: (37)–(39). [Google Scholar]
- Simmons DR, Longcore JE. (2012) Thoreauomyces gen. nov., Fimicolochytrium gen. nov. and additional species in Geranomyces. Mycologia 104: 1229–1243. [DOI] [PubMed] [Google Scholar]
- Stadler M, Læssøe T, Fournier J, Decock C, Schmieschek B, et al. (2014a) A polyphasic taxonomy of Daldinia (Xylariaceae). Studies in Mycology 77: 1–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Hawksworth DL, Fournier J. (2014b) The application of the name Xylaria hypoxylon, based on Clavaria hypoxylon of Linnaeus. IMA Fungus 5: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes BC, Mullis RA, Hagenmuller C, Melton TW, Sartori M. (2014) Genetic analysis of hair samples attributed to yeti, bigfoot and other anomalous primates. Proceedings of the Royal Society of London B 281: 20140161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thines M, Crous PW, Aime MC, Aoki T, Cai L, et al. (2018) Ten reasons why a sequence-based nomenclature is not useful for fungi anytime soon. IMA Fungus 8: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thines M, Kummer V. (2013) Diversity and species boundaries in floricolous downy mildews. Mycological Progress 12: 321–329. [Google Scholar]
- Torres-Cruz TJ, Billingsley Tobias TL, Almatruk M, Hesse C, Kuske CR, et al. (2017) Bifiguratus adelaidae gen. et sp. nov., a new member of Mucoromycotina with endophytic and soil-dwelling habitats. Mycologia 109: 363–378. [DOI] [PubMed] [Google Scholar]
- Turland NJ, Wiersema JH, Barrie FR, Greuter W, Hawksworth DL, et al. (2018) International Code of Nomenclature for algae, fungi, and plants (Shenzhen Code) adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017. [Regnum Vegetabile no. 159.] Oberreifenberg: Koeltz Botanical Books. [Google Scholar]
- Visagie CM, Houbraken J, Frisvad JC, Hong SB, Klaassen CHW, et al. (2014). Identification and nomenclature of the genus Penicillium. Studies in Mycology 78: 343–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther G, Pawłowska J, Alastruey-Izquierdo A, Wrzosek M, Rodriguez-Tudela JL, et al. (2013) DNA barcoding in Mucorales: an inventory of biodiversity. Persoonia 30: 11–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Johnston PR, Damm U. (2012) The Colletotrichum gloeosporioides species complex. Studies in Mycology 73: 115–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Renner SS. (2005) The internal transcribed spacer of nuclear ribosomal DNA in the gymnosperm Gnetum. Molecular Phylogenetics and Evolution 36: 581–597. [DOI] [PubMed] [Google Scholar]
- Zamora JC, Svensson M, Kirschner R, Olariaga I, Ryman S, et al. (2018) Considerations and consequences of allowing DNA sequence data as types of fungal taxa. IMA Fungus 9: 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kapli P, Pavlidis P, Stamatakis A. (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29: 2869–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]