

Abstract
The germ tube burst method (GTBM) was employed to examine karyotypes of 33 Fusarium species representative of 11 species complexes that span the phylogenetic breadth of the genus. The karyotypes revealed that the nucleolar organizing region (NOR), which includes the ribosomal rDNA region, was telomeric in the species where it was discernible. Variable karyotypes were detected in eight species due to variation in numbers of putative core and/or supernumerary chromosomes. The putative core chromosome number (CN) was most variable in the F. solani (CN = 9‒12) and F. buharicum (CN = 9+1 and 18-20) species complexes. Quantitative real-time PCR and genome sequence analysis rejected the hypothesis that the latter variation in CN was due to diploidization. The core CN in six other species complexes where two or more karyotypes were obtained was less variable or fixed. Karyotypes of 10 species in the sambucinum species complex, which is the most derived lineage of Fusarium, revealed that members of this complex possess the lowest CN in the genus. When viewed in context of the species phylogeny, karyotype evolution in Fusarium appears to have been dominated by a reduction in core CN in five closely related complexes that share a most recent common ancestor (tricinctum and incarnatum-equiseti CN = 8-9, chlamydosporum CN = 8, heterosporum CN = 7, sambucinum CN = 4-5) but not in the sister to these complexes (nisikadoi CN = 11, oxysporum CN = 11 and fujikuroi CN = 10-12). CN stability is best illustrated by the F. sambucinum subclade, where the only changes observed since it diverged from other fusaria appear to have involved two independent putative telomere to telomere fusions that reduced the core CN from five to four, once each in the sambucinum and graminearum subclades. Results of the present study indicate a core CN of 4 may be fixed in the latter subclade, which is further distinguished by the absence of putative supernumerary chromosomes. Karyotyping of fusaria in the not too distant future will be done by whole-genome sequencing such that each scaffold represents a complete chromosome from telomere to telomere. The CN data presented here should be of value to assist such full genome assembling.
Keywords: accessory, chromosome, genome, NOR, pathogen, phylogeny, qPCR, RPB1, RPB2, supernumerary
INTRODUCTION
The genus Fusarium contains over 300 phylogenetically distinct species that occupy a broad array of ecological niches worldwide (Aoki et al. 2014). Many of these species are plant pathogens, causing serious diseases on agriculturally, horticulturally and silviculturally important plants, notably F. graminearum and F. oxysporum, which are ranked among the top five plant pathogenic fungi worldwide (Dean et al. 2012). Annually, fusarial diseases are responsible for multi-billion US dollar losses to the world’s agricultural economy. In addition, fusaria produce a plethora of mycotoxins, such as trichothecenes, fumonisins and zearalenone, which pose a significant threat to food safety and human health. Toxin contaminated food and feed is frequently unsuitable for consumption, resulting in additional losses to world agriculture (Munkvold 2017). Phylogenetically diverse fusaria are also capable of causing superficial or invasive, life-threatening opportunistic infections in humans and veterinary animals (O’Donnell et al. 2010, 2016). In contrast to most other mycotic agents, fusaria are broadly resistant to the spectrum of antifungals currently available (Guarro 2013, Al-Hatmi et al. 2016). Given their global impact on agriculture, and human and veterinary medicine, two web-accessible DNA sequence databases were constructed to facilitate strain typing via the internet: FUSARIUM-ID (http://isolate.fusariumdb.org/; Geiser et al. 2004) and Fusarium MLST (http://www.westerdijkinstitute.nl/fusarium/; O’Donnell et al. 2015).
Comparative phylogenetic and phylogenomic studies have begun to revolutionize our understanding of species limits, evolutionary relationships and mycotoxin potential in Fusarium. Such foundational information is essential for developing novel control strategies aimed at minimizing the threat that fusaria and their toxins pose to agricultural biosecurity. Phylogenetic species recognition based on genealogical concordance (GCPSR sensu Taylor et al. 2000), which is directed at identifying genealogically exclusive lineages by sequencing portions of several phylogenetically informative loci, has consistently exposed the severe limitations of morphological and biological species recognition in Fusarium and greatly accelerated species discovery in the genus. Currently, close to two-thirds of the 300 phylogenetically distinct fusaria were discovered via GCPSR studies conducted by scientists worldwide. Phylogenetic analyses of portions of the largest and second largest subunits of RNA polymerase (RPB1 and RPB2) have resolved a monophyletic Fusarium, which is strongly supported by the Fusarium scientific community (Geiser et al. 2013), with 22 clades referred to as species complexes (Laurence et al. 2011, O’Donnell et al. 2013, Zhou et al. 2016).
Following the pioneering Fusarium genomics studies by Cuomo et al. (2007), Ma et al. (2010), and others (reviewed in Ma et al. 2013), whole-genome sequences have been obtained from a broad range of fusaria (e.g., Kim et al. 2017). To extract full information from the genome, a new standard was proposed, where each contig represents a complete chromosome from telomere to telomere (Waalwijk et al. 2017). This approach was elegantly demonstrated for F. fujikuroi (Wiemann et al. 2013), where each of the 12 scaffolds corresponds to a chromosome. The advantages of a fully assembled genome are multifarious, including a complete inventory of effectors and intact gene clusters as well as structural rearrangements and genomic compartmentalization, as recently reviewed (Thomma et al. 2016). In this regard, Ma et al. (2010) demonstrated that the genomes of F. graminearum and F. oxysporum f. sp. lycopersici evolved, respectively, by chromosome fusion and acquisition of lineage-specific (LS) chromosomes.
To assist full genome assembling, prior knowledge of chromosome number (CN) of the organism is invaluable. Pulsed field gel electrophoresis (PFGE) has been used extensively since the 1980s to analyse CN in diverse fungi, including Fusarium (e.g. Boehm et al. 1994, Fekete et al. 1993). However, accurate determination of the CN with PFGE is restricted to species that contain small- to intermediate-sized chromosomes such as yeasts because ones larger than 6 Mbp typically cannot be resolved by this technique. Because chromosomes of Fusarium and other filamentous fungi are often too large to allow separation by PFGE, their CN has been underestimated in many cases using this technique (Taga et al. 1998). Although conventional light-microscopic techniques have been employed historically to determine CN of fungi, most of the published karyotypes of fusaria from this line of research appear to be underestimates (see Table 1). Moreover, these early species identifications without supporting molecular systematic data should be viewed with caution. Fortunately, this technical hurdle was overcome by development (Shirane 1988) and subsequent refinement of the germ tube burst method (GTBM; Taga et al. 1998, Tsuchiya & Taga 2001, Mahmoud & Taga 2012), whereby mitotic chromosomes are released from a disrupted germ tube and spread on a microscope slide, thus enabling accurate chromosome counts. After Taga et al. (1998) applied the technique to several species in the F. solani species complex, it was used to resolve four chromosomes in F. graminearum (Gale et al. 2005) and F. culmorum (Waalwijk et al. 2017) that are too large to be separated by PFGE.
Table 1.
Fusarium isolates used in this study with the chromosome (CN) numbers found.
| Species Complex1 | Figure | Fusarium Species2 | NRRL3 | Alternative accession nos4 | CN5 | Core Chromosomes | Non-core Chromosomes |
|---|---|---|---|---|---|---|---|
| dimerum | Fig. 1a | F. dimerum | 20691 | CBS 489.81 | 15 | 13 | 2 |
| dimerum | Fig. S1a | F. dimerum | 36130 | CBS 102613 | 15 | 13 | 2 |
| ventricosum | Fig. 1a | F. ventricosum-2 | 25729 | CBS 430.91 | 11 | 10 | 1 |
| ventricosum | Fig. S1a | F. ventricosum-1 | 13953 | CBS 830.85 | 11-15 | 10 or 11 | 1, 3 or 4 |
| solani | Fig. 1a | Fusarium sp. FSSC 11 | 66287 | ATCC 204495 | 15 | 12 | 3 |
| solani | Fig. S1a | Fusarium sp. FSSC 11 | 66287 | ATCC 204495 | 15 | 12 | 3 |
| solani | Fig. 1a | F. striatum | 22147 | BBA 64379 | 13 | 12 | 1 |
| solani | Fig. S1a | F. striatum | 22101 | BBA 64379 | 12 | 12 | 0 |
| solani | Fig. 1a | Fusarium sp. FSSC 10 | 22153 | ATCC 18099 | 9 | 9 | 0 |
| solani | Fig. S1a | Fusarium sp. FSSC 10 | 22165 | ATCC 18098 | 10 | 9 | 1 |
| buharicum | Fig. 1b | F. sublunatum | 13384 | CBS 189.34 = BBA 62431 | 18-20 | 18-20 | 0 |
| buharicum | Fig. S1b | F. sublunatum | 13384 | CBS 189.34 = BBA 62431 | 18-20 | 18-20 | 0 |
| buharicum | Fig. 1b | F. buharicum | 13371 | CBS 796.70 = DSM 62165 = FRC R-4955 | 10 | 9 | 1 |
| buharicum | Fig. S1b | F. buharicum | 13371 | CBS 796.70 = DSM 62165 = FRC R-4955 | 10 | 9 | 1 |
| lateritium | Fig. 1b | F. stilboides | 20429 | ATCC 15662 | 15 | 14 | 1 |
| lateritium | Fig. S1b | F. stilboides | 20429 | ATCC 15662 | 15 | 14 | 1 |
| nisikadoi | Fig. 1b | F. nisikadoi | 25203 | MAFF 237507 = BBA 69014 | 11 | 10 | 1 |
| nisikadoi | Fig. S1b | F. nisikadoi | 25308 | MAFF 237506 | 12 | 11 | 1 |
| fujikuroi | Fig. 1b | F. nygamai | 66293 | FRC M-7492 | 12 | 12 | 0 |
| fujikuroi | Fig. S1b | F. nygamai | 66291 | FRC M-5868 | 12 | 13 | 2 |
| fujikuroi | Fig. 1b | F. verticillioides | 66290 | MAFF 239106 | 11 | 10 | 1 |
| fujikuroi | Fig. S1b | F. verticillioides | 66290 | MAFF 239106 | 11 | 10 | 1 |
| fujikuroi | Fig. 1b | F. proliferatum | 66289 | ITEM 2287 | 12 | 10 | 2 |
| fujikuroi | Fig. S1b | F. proliferatum | 36220 | CBS 115.97 | 13 | 11 | 2 |
| fujikuroi | Fig. 1b | F. fujikuroi | 66288 | MAFF 238524 | 12 | 11 | 1 |
| fujikuroi | Fig. S1b | F. fujikuroi | 66292 | MAFF 238525 | 12 | 11 | 1 |
| heterosporum | Fig. 1c | F. heterosporum | 20693 | CBS 720.79 = PD 79/878 | 8 | 7 | 1 |
| heterosporum | Fig. S1c | F. heterosporum | 20693 | CBS 720.79 = PD 79/878 | 8 | 7 | 1 |
| heterosporum | Fig. 1c | F. graminum | 20692 | CBS 737.79 = BBA 62228 | 7 | 7 | 0 |
| heterosporum | Fig. S1c | F. graminum | 20692 | CBS 737.79 | 7 | 7 | 0 |
| tricinctum | Fig. 1c | F. tricinctum | 25481 | CBS 393.93 = BBA 64485 | 9 | 8 | 1 |
| tricinctum | Fig. S1c | Fusarium sp. | 36132 | CBS 102796 | 10 | 9 | 1 |
| tricinctum | Fig. 1c | F. acuminatum | 28652 | ITEM 865 | 10 | 9 | 1 |
| tricinctum | Fig. S1c | F. acuminatum | 28449 | CBS 214.77 | 9 | 8 | 1 |
| tricinctum | Fig. 1c | F. arthrosporioides | 26416 | CBS 303.95 | 11 | 8 | 3 |
| tricinctum | Fig. S1c | F. arthrosporioides | 26416 | CBS 303.95 | 11 | 8 | 3 |
| tricinctum | Fig. 1c | F. avenaceum | 36374 | CBS 239.94 = IPO 92-3 = PD 92/1185 | 9 | 8 | 1 |
| tricinctum | Fig. S1c | F. avenaceum | 26911 | CBS 408.86 = FRC R-8510 | 10 | 8 | 2 |
| incarnatum-equiseti | Fig. 1c | Fusarium sp. FIESC 24 | 36255 | CBS 145.44 = BBA 4095 | 9 | 8 | 1 |
| incarnatum-equiseti | Fig. S1c | Fusarium sp. FIESC 24 | 36255 | CBS 145.44 = BBA 4095 | 9 | 8 | 1 |
| incarnatum-equiseti | Fig. 1c | Fusarium sp. FIESC 16 | 20425 | CBS 131.73 = IMI 160602 | 9 | 9 | 0 |
| incarnatum-equiseti | Fig. S1c | Fusarium sp. FIESC 16 | 20425 | CBS 131.73 = IMI 160602 | 9 | 9 | 0 |
| sambucinum | Fig. 1d | F. longipes-4 | 13317 | FRC R-314 | 5 | 5 | 0 |
| sambucinum | Fig. S1d | F. longipes-4 | 13317 | FRC R-314 | 5 | 5 | 0 |
| sambucinum | Fig. 1d | F. cf. compactum | 13829 | FRC R-6784 | 5 | 5 | 0 |
| sambucinum | Fig. S1d | F. cf. compactum | 13829 | FRC R-6784 | 5 | 5 | 0 |
| sambucinum | Fig. 1d | F. poae | 66297 | TAPO 21 | 6 | 4 | 2 |
| sambucinum | Fig. S1d | F. poae | 66297 | TAPO 21 | 6 | 4 | 2 |
| sambucinum | Fig. 1d | F. kyushuense | 66296 | MAFF 240372 | 4 | 4 | 0 |
| sambucinum | Fig. S1d | F. kyushuense | 66296 | MAFF 240372 | 4 | 4 | 0 |
| sambucinum | Fig. 1d | F. sporotrichioides | 66295 | ITEM 3593 | 6 | 5 | 1 |
| sambucinum | Fig. S1d | F. sporotrichioides | 66295 | ITEM 3593 | 6 | 5 | 1 |
| sambucinum | Fig. 1d | F. pseudograminearum | 28065 | CBS 109954 = FRC R-6761 | 4 | 4 | 0 |
| sambucinum | Fig. S1d | F. pseudograminearum | 28065 | CBS 109954 = FRC R-6761 | 4 | 4 | 0 |
| sambucinum | Fig. 1d | F. lunulosporum | 13393 | BBA 62459 = FRC R-5822 | 4 | 4 | 0 |
| sambucinum | Fig. S1d | F. lunulosporum | 13393 | BBA 62459 = FRC R-5822 | 4 | 4 | 0 |
| sambucinum | Fig. 1d | F. cerealis | 13721 | CBS 110268 = KF-748 | 4 | 4 | 0 |
| sambucinum | Fig. S1d | F. cerealis | 25491 | CBS 589.93 | 4 | 4 | 0 |
| sambucinum | Fig. 1d | F. culmorum | 66294 | IPO 39 | 4 | 4 | 0 |
| sambucinum | Fig. S1d | F. culmorum | 66294 | IPO 39 | 4 | 4 | 0 |
| sambucinum | Fig. 1d | F. graminearum | 31084 | PH-1 | 4 | 4 | 0 |
| sambucinum | Fig. S1d | F. graminearum | 38154 | Fg820 | 4 | 4 | 0 |
1As defined in O’Donnell et al. (2013).
2Phylogenetic species within the Fusarium solani (FSSC) and F. incarnatum-equiseti (FIESC) species complexes are distinguished by a unique Arabic number. Fusarium sp. FSSC 11 was previously reported as F. solani f. sp. pisi (VanEtten et al. 1994), but F. solani corresponds to phylospecies FSSC 5 (Schroers et al. 2016). In addition, phylogenetically distinct species within the F. ventricosum and F. longipes clades are identified by unique numbers.
3NRRL, ARS Culture Collection, Peoria, IL.
4ATCC, American Type Culture Collection, Manassas, VA; BBA, Biologische Bundesanstalt für Land-und Forstwirtschaft, Berlin, Germany; CBS, Westerdijk Fungal Biodiversity Institute, Utrecht, The Netherlands; DSMZ, Leibniz-Institut DSMA-Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany; FRC, Fusarium Research Center, The Pennsylvania State University, State College, PA; IMI, CABI Biosciences, Egham, Surrey, UK; IPO, IPO-Collection of Fungal Pathotypes, Wageningen, The Netherlands; ITEM, Agro-Food Microbial Culture Collection, Bari, Italy; KF, Fusarium collection at the Institute of Plant Genetics, Polish Academy of Sciences, Poznan, Poland, Institute of Food Technology Culture Collection, Agricultural University of Poznan, Poland; MAFF, Genebank Project, Ministry of Agriculture, Forestry and Fisheries, Tsukuba, Japan; PD, Dutch Plant Protection Service, Wageningen, The Netherlands; TAPO, Agricultural Biotechnology Center, Gödöllő, Hungary.
5CN, chromosome number.
Following the success of the Fusarium comparative genomics project (Cuomo et al. 2007, Ma et al. 2010), powerful platforms for whole-genome sequencing and subsequent assembly and annotation (e.g. CLC Bio Workbench, Aarhus, Denmark) have greatly accelerated progress in fungal genome research over the last decade. In support of these efforts, the present study was initiated to: (1) determine CN for a broad set of Fusarium species including representatives of 11 species complexes that span the phylogenetic breadth of the genus; (2) obtain an initial assessment of CN variability in key clades and species; (3) map kerryotypes on a robust phylogeny to develop hypotheses of CN evolution; and (4) assess the phylogenetic distribution of putative supernumerary chromosomes within the genus. The results should provide a valuable framework for future comparative phylogenomic research on the genus.
MATERIAL AND METHODS
Material studied
The strains used in this study and the collections in which they are preserved are detailed in Table 1. For convenience in this paper we refer to individual strains by the ARS Culture Collection (https://nrrl.ncaur.usda.gov/, NRRL) accession numbers.
Molecular phylogenetic analysis
Strains were grown in 20 mL of yeast-malt broth (YM: 20 g dextrose, 5 g peptone, 3 g yeast extract, and 3 g malt extract per L water; Difco, Detroit, MI) at 25 °C on a rotary shaker set at 200 rpm for 3-5 d. Mycelium was harvested over a Büchner funnel, freeze-dried overnight and then total genomic DNA was extracted from 50-100 mg of pulverized mycelium using a hexadecyltrimethyl-ammonium bromide (CTAB, Sigma-Aldrich, St Louis, MO) protocol (Gardes & Bruns 1993). Portions of the DNA-directed RNA polymerase II largest (RPB1) and second largest subunit (RPB2) were amplified by PCR and the resulting amplicons were sequenced following published protocols (O’Donnell et al. 2010). ABI 3730 sequence chromatograms were edited with Sequencher 5.2.4 (Gene Codes, Ann Arbor, MI) and the aligned consensus sequences were exported as NEXUS files (3383 bp alignment). Maximum likelihood (ML) analyses were conducted with GARLI 2.01 (Zwickl 2006) on the CIPRES Science Gateway TeraGrid (https://www.phylo.org/) using the GTR + I + Γ model of molecular evolution. Unweighted maximum parsimony (MP) analyses were conducted with PAUP* 4.0b10 (Swofford 2003), using the heuristic search option, tree bisection-reconnection branch swapping, and 1000 random addition sequences. Clade support was assessed by conducting 1000 ML and MP bootstrap pseudoreplicates of the data (ML-BS/MP-BS). DNA sequences were deposited in GenBank under accession numbers MG282363–MG282421.
Whole-genome sequencing
After total genomic DNA of F. buharicum 13371 and F. sublunatum 13384 was isolated using a ZR Fungal/bacterial DNA MiniPrepTM kit (Zymo Research, Irvine, CA), genomic DNA libraries were prepared using a NExtera XT DNA library Preparation Kit as specified by the manufacturer (Illumina, San Diego, CA), and then sequence reads were generated using an Illumina MiSeq platform at NCAUR. CLC Genomics Workbench (CLC bio, Qiagen, Aarhus, Denmark) was used to trim and assemble the reads and to analyse the assembled genome sequences.
Cytology
Strains were maintained as slant cultures using synthetic low nutrient agar (SNA, Nirenberg 1976), vegetable juice agar [10 % (v /v) mix vegetable juice (Kagome, Nagoya, Japan), 0.3 % (w /v) CaCO3, 1.5 % (w/v) agar] or potato dextrose agar (Difco, Detroit, Michigan). The germ tube burst method (GTBM) was used to prepare microscope slides containing mitotic metaphase chromosomes as previously described (Taga et al, 1998, Tsuchiya & Taga 2010, Mehrabi et al. 2012). To obtain macroconidia for the GTBM, fusaria were cultured on SNA containing small pieces of filter paper (Aoki & O’Donnell 1999), carnation leaf agar (CLA) (Nelson et al. 1983) or mung bean broth (MBB, Gale et al. 2005). After macroconidia were harvested, conidia were inoculated in potato dextrose broth (PDB, Difco) according to Taga et al. (1998) for strains cultured on SNA and CLA and according to Gale et al. (2005) for strains cultured in MBB. To obtain germ tubes, 100-200 μL of a conidial suspension (3-5 × 105 conidia/mL) was placed on a clean slide coated with poly-L-lysine (Sigma-Aldrich P7280, St. Louis, MO) and incubated under humid conditions at 25 °C for 5 -12 h until the germ tubes grew to double the length of the macroconidia or the germ tubes began to branch. Of the 44 strains karyotyped, Fusarium sp. 22153 FSSC 10 (Fig. 1a) was the only isolate that was treated with thiabendazole to arrest mitosis at metaphase as previously described (Mahmoud & Taga 2012). Once germ tubes had reached the desired length, a 17:3 mixture of methanol and acetic acid was used to burst the germlings and fix their chromosomes. After chromosomes were fixed, they were stained with 4’, 6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich D8417) dissolved in antifade mounting solution (Johnson & Araujo 1981) or Vectashield (Vector Laboratories, Burlingame, CA) at 1 μg/mL. Observations were made using an Olympus BH2 or Olympus BX60 (Olympus, Tokyo, Japan) epifluorescence microscope equipped with a 100X/N.A.1.3 or 1.35 oil immersion objective. Fluorescence images were captured on 400 ASA/ISO colour print film (Fujicolor Super HG400, Fuji Film, Tokyo, Japan) or recorded with a colour CCD camera (DP70, Olympus). Film images were digitized using a COOLSCAN IV ED film scanner (Nikon, Tokyo, Japan).
Fig. 1a–d.
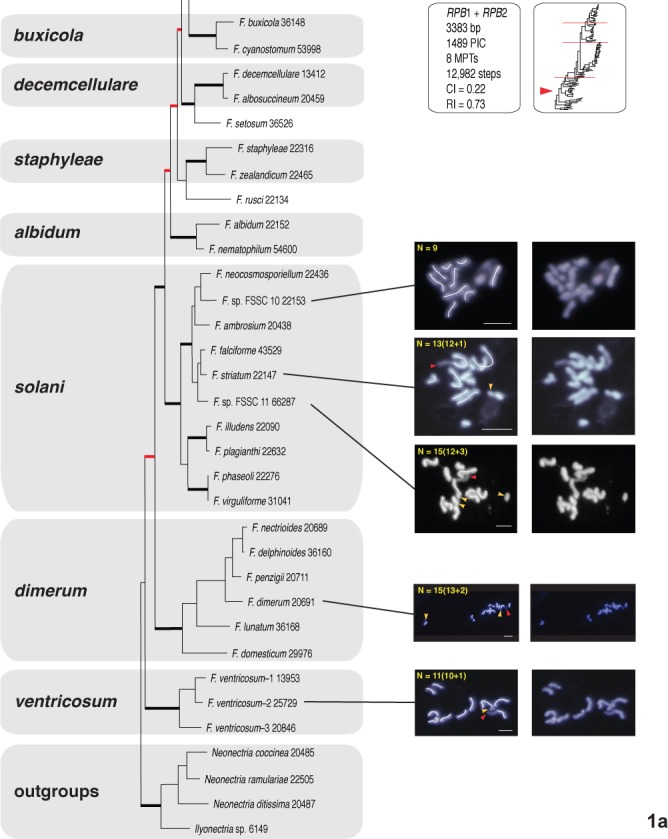
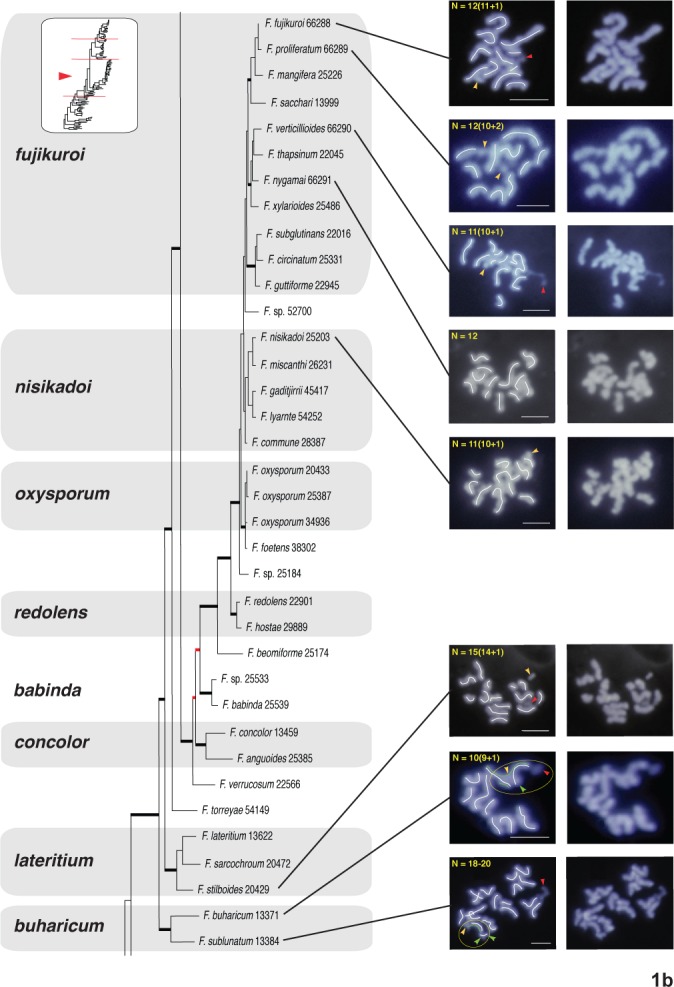
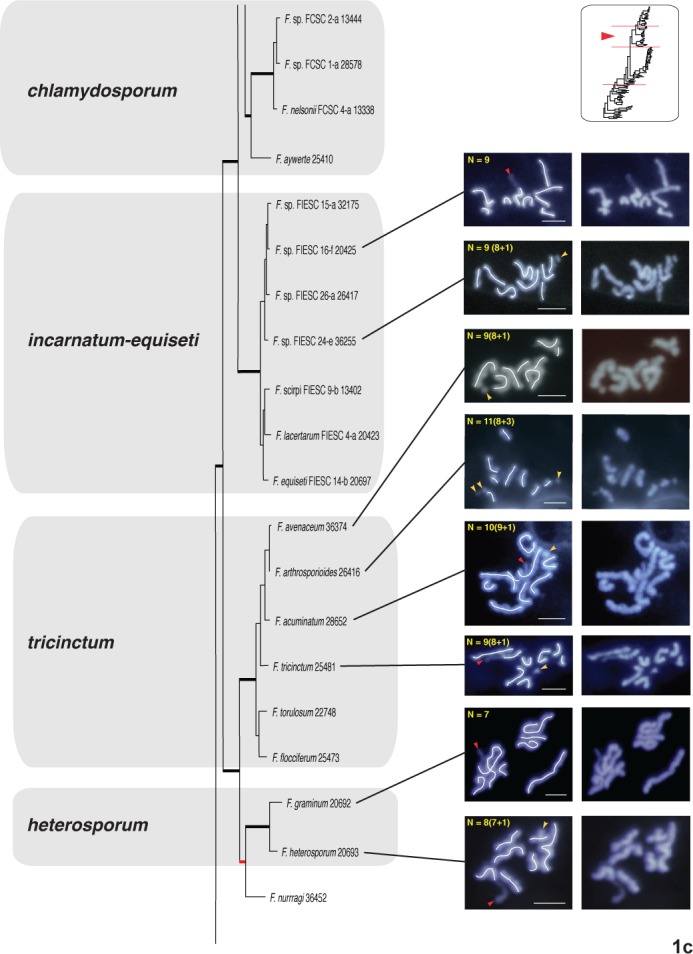
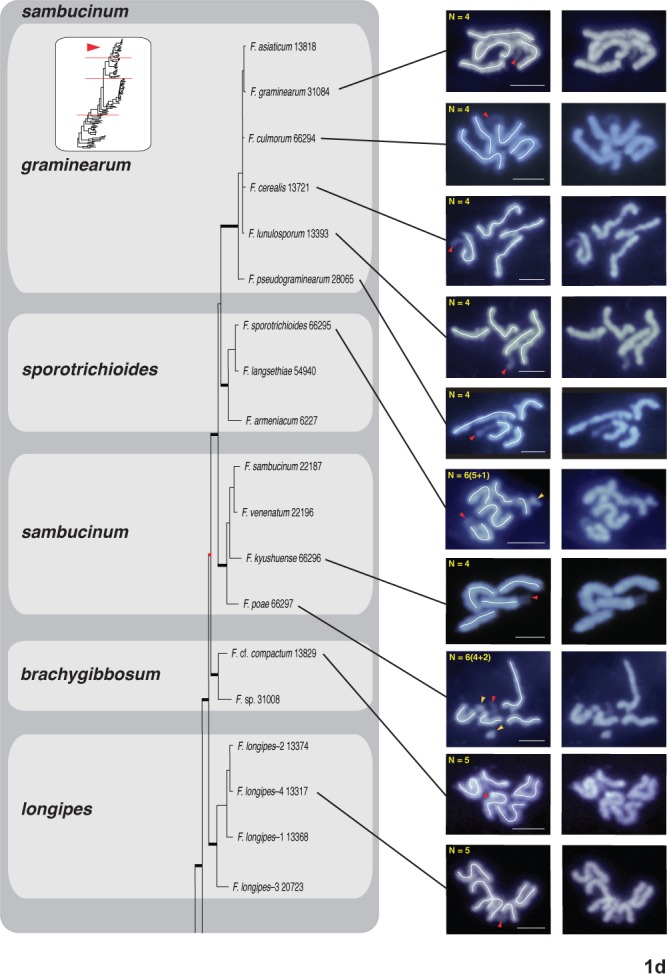
One of eight most-parsimonious phylograms, 12 982 steps in length, inferred from 3383 bp of aligned partial RPB1 and RPB2 sequences from 104 fusaria comprising 20 species complexes. The phylogram was rooted on outgroup sequences of Neonectria and Ilyonectria based on prior analyses (O’Donnell et al. 2013). ARS Culture Collection strains are identified by the 4-5 digit NRRL number. Thickened black nodes received ≥90 % ML-BS/MP-BS support, whereas the eight nodes in red received <70 % ML-BS/MP-BS. The chromosome number (CN) traced in the left panel for 31 species representing 11 species complexes was determined by the germ tube burst method and DAPI staining (Taga et al. 1998). Putative supernumerary chromosomes in 19 species spanning 11 species complexes are identified by a yellow arrowhead and the number following the + sign. A red arrowhead is used to specify NOR (rDNA), which is identifiable by the protrusion of chromatin from the apex of one of the chromosomes. A green trace line and green arrowheads are used to present an alternative interpretation of the karyotype of Fusarium buharicum and F. sublunatum. Bar = 2 μm
In filamentous fungi, including Fusarium, a thread- or rod-like chromatin protrusion from the apex of a metaphase chromosome has been proven to be nucleolar organizing region (NOR) representing the rDNA region (Taga & Murata 1994, Akamatsu et al. 1999, Taga et al. 2003). Accordingly we regarded the chromatin protrusion from a chromosome ends observed in this study as NOR (see red arrowheads in Figs 1 and S1).
qPCR experiment
A qPCR experiment was conducted to assess whether the two-fold difference in CN in F. buharicum 13371 (CN = 9+1) and F. sublunatum 13383 (CN = 18-20) was due to a difference in ploidy. Each strain was grown in YM broth, mycelium was lyophilized and then 30 mg of F. graminearum 29169 dry weight mycelium was added as an internal normalizing control to 50 mg of each strain as an internal normalizing control. Genomic DNA was extracted using the ZR Fungal/Bacterial DNA miniprep Kit followed by the Genomic DNA Clean and Concentrate Kit (Zymo Research, Irvine, CA) as prescribed by the manufacturer. The efficiency of qPCR primers targeting translation elongation factor 1-α (TEF1; qTEFf: CTCGGTAAGGGTTCCTTCAAGT × qTEFr: CCAATGACGGTGACATAGTAGC) and DNA-directed RNA polymerase II largest subunit (RPB1; qRPBf: GTGTTATTCCTCAGCCCGCTAT × qRPBr: TCCTTGCTGTCCGTACCATTGA) present in both F. buharicum 13371 and F. sublunatum 13383, and Tri6 (Tri6f: TAACCACATCGTCGGGACTG × Tri6r: GCCGACTTCTTGCAGGTCTT), which is only present in F. graminearum 29169, were determined by generating standard curves from a ten-fold dilution series (50 ng to 0.0005 ng) of mixed DNA for each primer pair. When qPCR was performed, the geometric mean of the two genes Cq values was determined and then normalized to Tri6 Cq, which allowed the fold-change in copy number of TEF1 and RPB1 in 13384 relative to 13371 to be calculated using the ΔΔCq method (Vandesompele et al. 2002, Schmittgen & Livak 2008, Brown et al. 2015).
RESULTS
The 44 strains karyotyped in this study were accessioned in the ARS Culture Collection (NRRL), but the acronym is not included with the 5-digit strain number to improve readability. Maximum likelihood and maximum parsimony bootstrapping (ML-BS/MP-BS) of the108 taxon partial RPB1 + RPB2 datasets (3383 bp) were conducted, respectively, with GARLI (Zwickl 2006) and PAUP* (Figs 1, S1). Phylogenies inferred for the 104 fusaria comprising the in-group were rooted on sequences of Neonectria and Ilyonectria based on more inclusive analyses (O’Donnell et al. 2013). Forty-six of the nodes received ≥90 % ML-BS/MP-BS support (identified by thickened black nodes), including representatives of 20 species complexes that were strongly supported as monophyletic. However, the eight nodes in red along the backbone of the phylogenies received <70 % ML-BS/MP-BS (Figs 1, S1).
The germ tube burst method with DAPI staining (Taga et al. 1998) was used to determine the chromosome number for 33 fusaria representing 11 species complexes.
When the nucleolar organizing region (NOR) representing the amplified rDNA region was visible, it was always telomeric on one chromosome as reported by Taga et al. (1998, see red arrowheads in Figs 1 and S1). Putative supernumerary chromosomes were detected in 19 species spanning 11 species complexes. These were indicated with yellow arrowheads and the number following the + sign on the images in the left panel (Figs 1, S1). The CNs are described as the number of putative core chromosomes + putative supernumerary chromosomes such as 10+1. The latter were defined based on estimated sizes < 2 Mb consistent with prior comparative genomic analyses of phylogenetically diverse fusaria (Coleman et al. 2009, Ma et al. 2010).
Fusarium ventricosum and F. dimerum species complexes
The Fusarium ventricosum and F. dimerum clades represent the two earliest diverging lineages of Fusarium. The CN of two phylospecies, F. ventricosum-2 25729 and F. ventricosum-1 13953, were 10+1 (Fig. 1a) and 10+1-to-11+4 (Fig. S1a), respectively. Fusarium ventricosum-1 13953 was the only strain karyotyped where variable numbers of core and supernumerary chromosomes were detected (i.e., 11(10+1), 14(11+3) and 15(11+4). Fusarium dimerum strains 20691 (Fig. 1a) and 36130 (Fig. S1a) both possessed 13 core and two putative supernumerary chromosomes (i.e., 13+2).
Fusarium solani species complex (FSSC)
Two unnamed phylospecies within this large species complex were analysed (i.e. FSSC 10 and 11, O’Donnell et al. 2008). Fusarium sp. FSSC 10 (formerly F. solani f. sp. cucurbitae and Nectria haematococca MPI) strains 22153 and 22165 contained 9 and 9+1 chromosomes, respectively (Figs 1a and S1a). By way of contrast, F. striatum FSSC 21 strain 22147 contained 12 and 22101 12+1 chromosomes. Fusarium sp. phylospecies FSSC 11 (formerly F. solani f. sp. pisi and N. haematococca MPVI) strain 66287 contained 12+3 chromosomes (Figs 1a and S1a).
Fusarium buharicum species complex
Two species in this complex, F. buharicum and F. sublunatum, were karyotyped. The CN of F. buharicum 13371 was 9+1. However, in marked contrast, the CN of the closely related species F. sublunatum 13384 was 18 to 20 (Figs 1b, S1b). To investigate the possible cause(s) of the differences in the karyotype of F. buharicum 13371 and F. sublunatum 13384, we generated whole-genome sequence data, which indicated the genome of F. sublunatum 13384 was 35.7 Mb (N50 = 102.3 kb) and F. buharicum 13371 36.1 Mb (N50 = 61.4 kb). The similar genome sizes of the two species indicate the larger number of chromosomes in F. sublunatum was not due to an increase in genome size. If F. sublunatum was diploid, single nucleotide polymorphisms (SNPs) could exist among homologous chromosomes even if homologous genomic regions had sufficient identity to be assembled into the same contig. Thus, SNPs should be evident in alignments of individual reads to contig sequences; that is, for a segment of the genome with a SNP, half the reads should have one base at the SNP position, while the other half of the reads should have a different base at the same position. However, SNP analysis indicated that the frequencies were similar: 0.028 % in F. sublunatum 13384 and 0.033 % in F. buharicum 13371. This finding is consistent with the two fungi having the same ploidy.
To further investigate whether the change in CN in F. sublunatum 13384 was due to diploidization, a qPCR experiment was conducted to assess ploidy of F. buharicum and F. sublunatum. After each strain was grown in yeast-malt (YM) broth, the filtered mycelium was lyophilized and then 30 mg of F. graminearum 29169 dry weight mycelium was added as an internal normalizing control to 50 mg of each strain (13384 and 13371) as an internal normalizing control. Following genomic DNA extraction, primer efficiency curves showed primer pairs targeting TEF1 (qTEFf x qTEFr) and RPB1 (qRPBf x qRPBr) were 97.1 % and 97.2 % efficient, respectively. Primers targeting the Tri6 gene (efficiency 99.3 %), which is only present in F. graminearum 29169, were used as a reference control for normalization to assess fold-changes in gene copy number between strains 13371 and 13384. The geometric mean of the two genes Cq value was then normalized to Tri6. The fold change in copy number of TEF1 and RPB1 in 13384 relative to 13371 was then calculated as 1.0017 ± 0.08. Thus, there is no indication of increased ploidy number given that each strain has approximately the same copy number of the two single copy nuclear genes tested.
Fusarium lateritium species complex
The CN of Fusarium stilboides 20429, the sole representative of the F. lateritium species complex, was 14+1 (Figs 1b, S1b).
Fusarium nisikadoi and F. fujikuroi species complexes
The chromosome complement of two strains of F. nisikadoi, 25203 and 25308, was determined as 10+1 and 11+1, respectively (Figs 1b, S1b). The closely related F. fujikuroi species complex (FFSC) consists of a large number of species with phytopathological and mycotoxilogical relevance. The FFSC is composed of species that cluster in three major clades that coincide with their putative geographic origin and/or the origin of their respective hosts in Africa, Asia and South America (O’Donnell et al. 1998). The CN of the Asian clade F. fujikuroi strains 66288 and 66292 was 11+1 (Figs 1b and S1b). The other Asian clade representative, F. proliferatum 66289 and 36220, possessed 10+2 and 11+2 chromosomes, respectively (Figs 1b and S1b). Two species in the African clade were karyotyped: F. verticillioides 66290 with 10+1 and F. nygamai 66291 and 66293 with 12 and 13+2 chromosomes, respectively (Figs 1b, S1b).
Fusarium heterosporum species complex
The CNs of F. graminum 20693 and F. heterosporum 20692 were 7 and 7+1, respectively (Figs 1c, S1c).
Fusarium tricinctum species complex
The following five species in this complex were karyotyped: F. avenaceum 36374 and 26911 with 8+1 and 8+2 chromosomes, respectively; F. arthrosporioides 26416 with 8+3; F. acuminatum 28652 and 28449 with 9+1 and 8+1, respectively; F. tricinctum 25481 with 8+1 and Fusarium sp. 36132 with 9+1 (Figs 1c, S1c).
Fusarium incarnatum-equiseti species complex (FIESC)
The CNs of two unnamed phylospecies in this species-rich complex, FIESC 16-f (NRRL 20425) and FIESC 24-e (NRRL 36255) were 9 and 8+1, respectively (Figs 1c, S1c).
Fusarium sambucinum species complex
Ten species, including representatives of five subclades, were karyotyped within the F. sambucinum species complex (Figs 1d, S1d). The CN of F. longipes-4 13317 and F. cf. compactum 13829 in the longipes and brachygibbosum subclades, respectively, was five. The chromosome complement of F. poae 66297 and F. kyushuense 66296 in the sambucinum subclade was 4+2 and 4, respectively (Figs 1d, S1d). The CN of F. sporotrichioides 66295 in the subclade by the same name was 5+1. Lastly, the following five species in the graminearum subclade, also known as the B clade of trichothecene toxin-producing fusaria, possessed four chromosomes (Figs 1d, S1d): F. pseudograminearum 28065, F. lunulosporum 13393, F. cerealis 13721 and 25491, F. culmorum 66294, and F. graminearum 31084 and 38154. The nucleolar organizing region (NOR) representing the amplified rDNA region was visible as an extension of one of the four chromosomes in all 10 F. sambucinum clade species (indicated by red arrowhead, Figs 1d, S1d).
Karyotype reduction
Core CNs in species complexes where two or more karyotypes were obtained were comparable: ventricosum (CN = 10‒11), fujikuroi (CN = 10‒12), tricinctum and incarnatum-equiseti (CN = 8‒9), heterosporum (CN = 7), and sambucinum (CN = 4‒5). The latter was the most deeply sampled clade, resulting in the discovery that CNs of species in all five subclades appeared to be fixed: sporotrichioides, brachygibbosum and longipes with CN = 5 and sambucinum and graminearum with CN = 4. To test if this reduction in karyotype was due to chromosome fusions, genomes retrieved from public repositories were aligned, using MUMmer and Blast2. Large regions of synteny were identified between the genomes of F. graminearum, F. avenaceum, Fusarium sp. FSSC 11 (published as F. solani; Coleman et al. 2009) and F. verticillioides. (Fig. S2). The left half of chr I of F. graminearum showed synteny with the largest chromosome of F. verticillioides and the largest contig from the assemblies of F. avenaceum isolates FaLH27 and Fusarium sp. FSSC 11. The central part of F. graminearum chr I showed synteny with parts of chromosomes IV and VIII of F. verticillioides and with single contigs of F. avenaceum and Fusarium sp. FSSC 11. Finally, the distal part of F. graminearum chr I was syntenic with chr V of F. verticillioides, one contig in F. avenaceum and two contigs in Fusarium sp. FSSC 11 (Fig. S2). However, no remnants of telomere sequences were detected at the putative chromosomal junctions in F. graminearum chr I.
DISCUSSION
We used the GTBM to karyotype 44 strains comprising 33 Fusarium species, and these included representatives of 11 species complexes that spanned the phylogenetic breadth of the genus. When viewed within a robust evolutionary framework, karyotype evolution in five closely related clades (tricinctum and incarnatum-equiseti CN = 8-9, chlamydosporum CN = 8 [electrophoretic karyotype reported in Fekete et al. 1993], heterosporum CN = 7 and sambucinum CN = 4-5) appears to have been dominated by a reduction in core CN, consistent with previous reports in Fusarium (Ma et al. 2010), and the general trend of genome reduction in eukaryotes (Wolf & Koonin 2013). However, a core CN reduction in the sisters of the aforementioned clades was not detected (lateritium CN = 14, nisikadoi CN = 10-11, oxysporum CN = 11 (Ma et al. 2010) and fujikuroi CN = 10-13); their CNs are comparable to those identified in the three most basal clades in Fusarium we sampled (dimerum CN = 13, ventricosum CN = 10-11 and solani CN = 9-12). Given the divergence time estimate for Fusarium that places its origin at approximately 83 Mya in the Cretaceous (O’Donnell et al. 2013), our limited sampling suggests CN may be relatively stable within most clades with the notable exception of the F. solani (CN = 9‒12) and F. buharicum species complexes (F. buharicum CN = 9+1 and F. sublunatum CN = 18-20).
We postulated that the two-fold increase in CN observed in F. sublunatum might be due to diploidization, but were able to reject this hypothesis based on a comparative genomic analysis and a qPCR experiment that indicated these species share the same copy number of the two genes evaluated. Although the precise mechanism(s) that contributed to the two-fold difference in CN are unknown, they might be elucidated by whole-genome sequencing to sufficient depth that each scaffold corresponds to a whole chromosome as recently done for F. fujikuroi (Wiemann et al. 2013). To obtain a high quality assembly, SMRT sequencing might be necessary to assemble repetitive sequences (Vanheule et al. 2016). Preliminary analyses of in-house sequencing of F. buharicum 13371 and F. sublunatum 13384 suggests that the overall genome size in both species is ~36 Mb. This is in good agreement with the size of the core genome of most fusaria (Kim et al. 2017).
The present study also extends the initial discovery of variable numbers of supernumerary chromosomes in Fusarium sp. FSSC 11 (as F. solani f. sp. pisi) by Miao et al. (1991), and the eight fusaria karyotyped electrophoretically by Fekete et al. (1993), to four additional fusaria (F. ventricosum-1, F. striatum, Fusarium sp. FSSC 10 and F. nygamai). In this regard, putative supernumerary chromosomes were detected in one or more of the fusaria in all 11 clades sampled, but our data suggests they may be less prevalent in the F. sambucinum species complex where they were only detected in two of the 10 species karyotyped (i.e., F. poae and F. sporotrichioides), and none were detected within the graminearum subclade. Numerous studies have established that some fungal genomes are composed of core chromosomes, which are stable within a species and contain all functions that allow the organism to complete various aspects of its life-cycle, and additional chromosomes that are not involved in primary metabolism (summarized in Mehrabi et al. 2017: table 1). These additional chromosomes are usually small (< 2 Mb), frequently meiotically unstable, and they often have low gene density and a high number of repetitive sequences (Möller & Stukenbrock 2017). They have been referred to as conditionally dispensable (CD), mini- or B, accessory or supernumerary chromosomes (Covert et al. 1998)
In F. oxysporum these additional chromosomes were named lineage-specific (LS), because they possess effector genes that determine host specificity (Ma et al. 2010). We did not karyotype any representative of the F. oxysporum species complex, but whole-genome sequencing of the tomato vascular wilt pathogen, F. oxysporum f. sp. lycopersici, strain Fol 4287 identified 11 core and four supernumerary or LS chromosomes (Ma et al. 2010). The core chromosomes show high similarity among F. oxysporum isolates pathogenic to completely different hosts, as well as the putative non-pathogenic strain Fo47 with biocontrol potential (Ma et al. 2010). Furthermore, comparison of the tomato pathogen Fol4287 and cucurbit pathogen Forc016 showed that both strains share 11 highly syntenic core chromosomes with >98 % sequence similarity (Van Dam et al. 2017). In parallel with Fol4287, the cucurbit strain Forc016 contains three small chromosomes (2.4 Mb, 1.6 Mb and 1.2Mb, respectively) that can be lost without interference with in vitro growth. However, transfer of the 2.4 Mb chromosome to Fo47 revealed its role in pathogenicity towards cucumber, melon and watermelon (Van Dam et al. 2017). A core and accessory genome were also reported for F. poae, where four core chromosomes were highly syntenic with the four chromosomes of F. graminearum (Vanheule et al. 2016). In addition to these chromosomes, 8 Mb of extra DNA was detected via single-molecule real-time (SMRT) sequencing of F. poae. In contrast to members of the F. solani and F. oxysporum species complexes, the CG-content as well as the gene density of this accessory DNA was similar to that in the core genome. However, the accessory DNA of F. poae was very rich in transposable elements (25.6 % vs. only 2.1 % in the core genome) and it contained multiple gene duplications.
We reviewed papers reporting karyotypes of fusaria employing classical aceto-carmine, aceto-orcein and HCl-Giemsa staining techniques but they are not discussed in detail here because, with the notable exception of CN = 4 in F. graminearum (Howson et al. 1963), most of the numbers reported appear to be underestimates. For example, Howson et al. (1963) reported CN = 4 in F. fujikuroi and F. stilboides whereas we discovered CN = 11+1 and 14+1, respectively, in these two species. In contrast, Punithalingam (1972) observed of eight chromosomes in F. culmorum, which is an overestimate based on our data, which unambiguously show this species possesses four chromosomes. Following its introduction in the late 1980s, pulsed field gel electrophoresis has been used extensively to karyotype diverse fusaria. Results of the present study match the findings of Fekete et al. (1993) who reported karyotypes that are identical to four of the fusaria we typed (i.e. F. avenaceum CN = 8, F. poae CN = 6, F. sporotrichioides CN = 6 and F. tricinctum CN = 9). In contrast to the electrophoretic karyotype study by Xu et al. (1995), who reported 35 strains representing six species in the F. fujikuroi species complex all possessed 12 chromosomes, our data indicate the CN of different strains of two of these species (F. verticillioides 66290 and F. proliferatum 36220) is 11 and 13, respectively. The CN of 11 in F. verticillioides 66290 might be due to loss of chromosome 12, which has been reported to be dispensable and meiotically unstable (Xu & Leslie 1996, Migheli et al. 1993). This chromosome was also absent from the whole genome sequence of F. verticillioides 20956 (Ma et al. 2010). Lastly, highly reproducible electrophoretic karyotypes were obtained for 15 VCGs of F. oxysporum f. sp. cubense, where CN ranged from nine to 14 (Boehm et al.1994). The wide variation in CN in F. oxysporum f. sp. cubense, however, is likely due to the polyphyletic origins of this forma specialis (O’Donnell et al. 2000).
The first Fusarium comparative genomics study included whole-genome analyses of F. graminearum, F. verticillioides and F. oxysporum (Ma et al. 2010). These authors discovered the core chromosomes of F. verticillioides and F oxysporum were highly syntenic, and they reported the four chromosomes of F. graminearum were composed of regions syntenic to multiple chromosomes of F. verticillioides and F. oxysporum. Additional lines of evidence support their hypothesis that the low chromosome number of four in F. graminearum and related species in the sambucinum subclade resulted from chromosomal fusions in their ancestors. Cuomo et al. (2007) reported that SNP frequencies were elevated and a higher density of genes coding for secreted proteins and genes expressed in planta were present at the putative junctions of the ancestral fused chromosomes. These junctions were also shown to contain genes that exhibited lower conservation and expression (Zhao et al. 2014). Moreover, gene clusters involved in specialized metabolism were overrepresented in these non-conserved regions. This finding supports the hypothesis that multiple chromosome fusion events may have occurred during the evolution of the genus, leading to karyotypes as small as CN = 4 in F. graminearum, which is one of the lowest observed in filamentous fungi. Integration of whole-genome sequence data from different sequencing platforms offers the promise of complete assemblies where contigs correspond to individual chromosomes with telomeric repeats on both ends as was shown for F. poae (Vanheule et al. 2016), F. fujikuroi (Wiemann et al. 2013) and F. subglutinans and F. temperatum (Waalwijk et al. 2017). The CN data presented here should provide a valuable reference for future comparative genomics studies of this agriculturally and medically important genus.
Acknowledgments
We are pleased to acknowledge the skilled technical assistance of Gail Doehring, Nathane Orwig, Amy McGovern, Jennifer Teresi, and Ahmed M. Nhmoud in various aspects of this study. Gert H.J. Kema (Wageningen Plant Research) is acknowledged for initiating the research reported here. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. CW received financial support from H2020 project MycoKey (project no. 678781).
Supplementary Material
REFERENCES
- Akamatsu H, Taga M, Kodama M, Johnson R, Otani H, Kohmoto K. (1999) Molecular karyotypes for Alternaria plant pathogens known to produce host-specific toxins. Current Genetics 35: 647–656. [DOI] [PubMed] [Google Scholar]
- Al-Hatmi AMS, Meis JF, de Hoog GS. (2016) Fusarium: molecular diversity and intrinsic drug resistance. PLoS Pathogens 12: e1005464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki T, O’Donnell K. (1999) Morphological and molecular characterization of Fusarium pseudograminearum sp. nov., formerly recognized as the group 1 population of F. graminearum. Mycologia 91: 597–609. [Google Scholar]
- Aoki T, O’Donnell K, Geiser DM. (2014) Systematics of key phytopathogenic Fusarium species: current status and future challenges. Journal of General Plant Pathology 80: 189–201. [Google Scholar]
- Boehm EWA, Ploetz RC, Kistler HC. (1994) Statistical analysis of electrophoretic karyotype variation among vegetative compatibility groups of Fusarium oxysporum f. sp. cubense. Molecular Plant-Microbe Interactions 7: 196–207. [Google Scholar]
- Brown DW, Lee SH, Kim LH, Ryu JG, Lee S, et al (2015) Identification of a 12-gene fusaric acid biosynthetic gene cluster in Fusarium species through comparative and functional genomics. Molecular Plant-Microbe Interactions 28: 319–332. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, et al (2009) The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genetics 5: e1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert SF. (1998) Supernumerary chromosomes in filamentous fungi. Current Genetics 33: 311–319. [DOI] [PubMed] [Google Scholar]
- Cuomo CA, Güldener U, Xu JR, Trail F, Turgeon BG, et al. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317: 1400–1402. [DOI] [PubMed] [Google Scholar]
- Dean R, Van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A. (2012) The top 10 fungal pathogens in molecular plant pathology. Molecular Plant Pathology 13: 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete C, Nagy R, Debets AJM, Hornok L. (1993) Electrophoretic karyotypes and gene mapping in eight species of the Fusarium sections Arthrosporiella and Sporotrichiella. Current Genetics 24: 500–504. [DOI] [PubMed] [Google Scholar]
- Gale LR, Bryant JD, Calvo S, Giese H, Katan T, et al (2005) Chromosome complement of the fungal plant pathogen Fusarium graminearum based on genetic and physical mapping and cytological observations. Genetics 171: 985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardes M, Bruns TD. (1993) ITS primers with enhanced specificity for basidiomycetes–application to the identification of mycorrhizae and rusts. Molecular Ecology 2: 112–118. [DOI] [PubMed] [Google Scholar]
- Geiser DM, del Mar Jiménez-Gasco M, Kang S, Makalowska I, Veeraraghavan N, et al, (2004) FUSARIUM-ID v. 1.0: A DNA sequence database for identifying Fusarium. European Journal of Plant Pathology 110: 473–479. [Google Scholar]
- Geiser DM, Aoki T, Bacon CW, Baker SE, Bhattacharyya MK, et al (2013) One Fungus, One Name: defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathology 103: 400–408. [DOI] [PubMed] [Google Scholar]
- Guarro J. (2013) Fusariosis, a complex infection caused by a high diversity of fungal species refractory to treatment. European Journal of Clinical Microbiology and Infectious Diseases 32: 1491–1500. [DOI] [PubMed] [Google Scholar]
- Howson WT, McGinnis RC, Gordon WL. (1963) Cytological studies on the perfect stages of some species of Fusarium. Canadian Journal of Genetics and Cytology 5: 60–64. [Google Scholar]
- Johnson GD, Araujo GM. (1981) A simple method of reducing the fading of immunofluorescence during microscopy. Journal of Immunological Methods 43: 349–350. [DOI] [PubMed] [Google Scholar]
- Kim H-S, Proctor RH, Brown DW. (2017) Comparative genomic analysis of secondary metabolite gene clusters in 207 isolates of Fusarium. 29th Fungal Genetics Conference Abstracts: 170.
- Laurence MH, Summerell BA, Burgess LW, Liew ECY. (2011) Fusarium burgessii sp. nov. representing a novel lineage in the genus Fusarium. Fungal Diversity 49: 101–112. [Google Scholar]
- Ma LJ, Van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, et al. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-J, Geiser DM, Proctor RH, Rooney AP, O’Donnell K, et al. (2013) Fusarium pathogenomics. Annual Review of Microbiology 67: 399–416. [DOI] [PubMed] [Google Scholar]
- Mahmoud AM, Taga M. (2012) Cytological karyotyping and characterization of a 410 kb mini-chromosome in Nectria haematococca MPI. Mycologia 104: 845–856. [DOI] [PubMed] [Google Scholar]
- Mehrabi R, Taga M, Aghaee M, de Wit PJGM, Kema GHJ. (2012) Karyotyping methods for fungi. In: Plant Fungal Pathogens. [Methods in Molecular Biology vol; 835.] (Bolton MD, Thomma BPHJ, eds): 591–602. Clifton, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Mehrabi R, Gohari AM, Kema GHJ. (2017) Karyotype variability in plant-pathogenic fungi. Annual Review of Phytopathology 55: 483–503. [DOI] [PubMed] [Google Scholar]
- Miao VP, Covert SF, VanEtten DK. (1991) A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science 254: 1773–1776. [DOI] [PubMed] [Google Scholar]
- Migheli Q, Berio T, Gullino ML. (1993) Electrophoretic karyotypes of Fusarium spp. Experimental Mycology 17: 329–337. [Google Scholar]
- Möller M, Stukenbrock EH. (2017) Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology: 10.1038/nrmicro.2017.76 [DOI] [PubMed] [Google Scholar]
- Munkvold GP. (2017) Fusarium species and their associated mycotoxins. In: Mycotoxigenic Fungi: methods and protocols (Moretti A, Susca A, eds): 51–106. New York: Humana Press. [DOI] [PubMed] [Google Scholar]
- Nelson PE, Toussoun TA, Marasas WFO. (1983) Fusarium Species: an illustrated manual for identification. University Park, PA: Pennsylvania State University Press. [Google Scholar]
- Nirenberg H. (1976) Untersuchungen uber die morphologische und biologische Differenzierung in der Fusarium-Sektion Liseola. Biologischen Bundesanstalt für Land- und Forstwirtschaft, Berlin-Dahlem 169: 1–117. [Google Scholar]
- O’Donnell K, Kistler HC, Cigelnik E, Ploetz RC. (1998) Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proceedings of the National Academy of Sciences, USA 95: 2044–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Rooney AP, Proctor RH, Brown DW, McCormick SP, et al. (2013) Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genetics and Biology 52: 20–31. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Fothergill A, McCarthy D, Rinaldi MG, et al. (2008) Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. Journal of Clinical Microbiology 46: 2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Rinaldi MG, Gueidan C, Sarver BAJ, et al. (2010) Internet-accessible DNA sequence database for identifying fusaria from human and animal infections. Journal of Clinical Microbiology 48: 3708–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Wiederhold N, Robert VARG, Crous PW, Geiser DM. (2016) Veterinary fusarioses within the United States. Journal of Clinical Microbiology 54: 2813–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Ward TJ, Robert VARG, Crous PW, Geiser DM, Kang S. (2015) DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43: 583–595. [Google Scholar]
- Punithalingam E. (1972) Cytology of Fusarium culmorum. Transactions of the British Mycological Society 58: 225–230. [Google Scholar]
- Schmittgen TD, Livak KJ. (2008) Analyzing real-time PCR data by the comparative CT method. Nature Protocols 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schroers H-J, Samuels GJ, Zhang N, Short DPG, Juba J, Geiser DM. (2016) Epitypification of Fusisporium (Fusarium) solani and its assignment to a common phylogenetic species in the Fusarium solani species complex. Mycologia 108: 806–819. [DOI] [PubMed] [Google Scholar]
- Shirane N, Masuko M, Hayashi Y. (1988) Nuclear behavior and division in germinating conidia of Botrytis cinerea. Phytopathology 78: 1627–1630. [Google Scholar]
- Swofford DL. (2003) PAUP*: phylogenetic analysis using parsimony (and other methods). Version 40b10. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Taga M, Murata M. (1994) Visualization of mitotic chromosomes in filamentous fungi by fluorescence staining and fluorescence in situ hybridization. Chromosoma 103: 408–413. [DOI] [PubMed] [Google Scholar]
- Taga M, Murata M, Saito H. (1998) Comparison of different karyotyping methods in filamentous ascomycetes – a case study of Nectria haematococca. Mycological Research 102: 1355–1364. [Google Scholar]
- Tatusova TA, Madden TL. (1999) BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiology Letters 174: 247–250. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, et al (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genetics and Biology 31: 21–32. [DOI] [PubMed] [Google Scholar]
- Thomma BPHJ, Seidl MF, Shi-Kunne X, Cook DE, Bolton MD, et al. (2016) Mind the gap: seven reasons to close fragmented genome assemblies. Fungal Genetics and Biology 90: 24–30. [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Taga M. (2001) Cytological karyotyping of three Cochliobolus spp. by the germ tube burst method. Phytopathology 91: 354–360. [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Taga M. (2010) Fluorescence in situ hybridization for molecular cytogenetic analysis in filamentous fungi. In: Molecular and Cell Biology Methods for Fungi (Sharon A, ed.): 235–257. Clifton, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Van Dam P, Fokkens L, Ayukawa Y, van der Gragt M, ter Horst A, et al. (2017) A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Science Reports 7: 9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanEtten H, Funnell-Baerg D, Wasmann C, McCluskey K. (1994) Location of pathogenicity genes on dispensable chromosomes in Nectria haematococca MP VI. Antonie van Leeuwenhoek 65: 263–267. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 3: 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanheule A, Audenaert K, Warris S, Van de Geest H, Schijlen E, et al. (2016) Living apart together: crosstalk between the core and supernumerary genomes in a fungal plant pathogen. BMC Genomics 17: 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waalwijk C, Vanheule A, Audenaert K, Zhang H, Warris S, et al (2017) Fusarium in the age of genomics. Tropical Plant Pathology 42: 184–189. [Google Scholar]
- Wiemann P, Sieber CMK, von Bargen KW, Studt L, Niehaus E-M, et al. (2013) Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathogens 9: e1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Koonin EV. (2013) Genome reduction as the dominant mode of evolution. Bioessays 35: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JR, Leslie JF. (1996) A genetic map of Gibberella fujikuroi mating population A (Fusarium moniliforme). Genetics 143: 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JR, Yan K, Dickman MB, Leslie JF. (1995) Electrophoretic karyotypes distinguish the biological species of Gibberella fujikuroi (Fusarium section Liseola). Molecular Plant-Microbe Interactions 8: 74–84. [Google Scholar]
- Zhao C, Waalwijk C, de Wit PJGM, Tang D, van der Lee T. (2014) Relocation of genes generates non-conserved chromosomal segments in Fusarium graminearum that show distinct and co-regulated gene expression patterns. BMC Genomics 15: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, O’Donnell K, Aoki T, Smith JA, Kasson MT, Cao Z-M. (2016) Two novel Fusarium species that cause canker disease of prickly ash (Zanthoxylum bungeanum) in northern China form a novel clade with Fusarium torreyae. Mycologia 108: 668–681. [DOI] [PubMed] [Google Scholar]
- Zwickl DJ. (2006) Genetic algorithm approaches for the phylogenetic analysis of large biological sequence data sets under the maximum likelihood criterion. PhD dissertation, University of Texas, Austin. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.