

SUMMARY
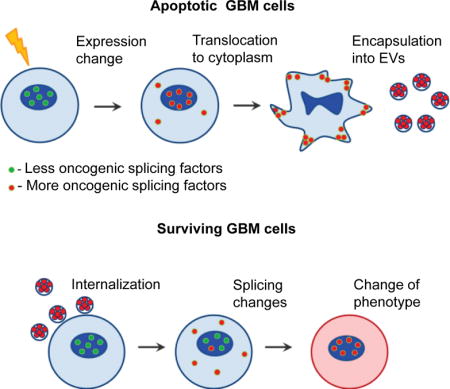
Aggressive cancers such as glioblastoma (GBM) contain intermingled apoptotic cells adjacent to proliferating tumor cells. Nonetheless, intercellular signaling between apoptotic and surviving cancer cells remain elusive. In this study, we demonstrate that apoptotic GBM cells paradoxically promote proliferation and therapy-resistance of surviving tumor cells by secreting extracellular vesicles (apoEVs) enriched with various components of spliceosomes. apoEVs alter RNA splicing in recipient cells thereby promoting their therapy-resistance and aggressive migratory phenotype. Mechanistically, we identified RBM11 as a representative splicing factor that is upregulated in tumors after therapy and shed in EVs upon induction of apoptosis. Once internalized in recipient cells, exogenous RBM11 switch splicing of MDM4 and Cyclin D1 towards the expression of more oncogenic isoforms.
Graphical abstract
INTRODUCTION
Glioblastoma (GBM) is characterized by highly invasive behavior, abrupt growth, and poor prognosis for patient survival. GBM can be divided into three transcriptomic subtypes: proneural (PN), classical, and mesenchymal (MES) (Phillips et al., 2006; Mao et al., 2013; Verhaak et al., 2010). Among them, PN cells are considered to be a GBM precursor that subsequently evolves into more heterogeneous tumors with one of the three subtypes (Ozawa et al., 2014). Recent pre-clinical and clinical data have consistently demonstrated that following therapy, GBM cells undergo phenotypic shift from less aggressive PN subtype to a more aggressive and therapy resistant MES phenotype (Bhat et al., 2013; Mao et al., 2013). Emerging evidence indicates that this phenotypic change occurs, at least in part, due to direct conversion of one cell type to another rather than selection of pre-existing therapy resistant tumor cells (Halliday et al., 2014).
Molecular mechanisms of MES transition in GBM remain unclear, however, multiple studies describe mechanisms of epithelial to mesenchymal transition (EMT) – a process that is frequently observed in non-brain epithelial cancers and might be analogous to PN-to-MES transition in GBM (Kiemer et al., 2001). EMT-like phenotypic changes allow cells to leave initial tumor lesions and invade into adjacent normal tissues. Several studies have demonstrated that EMT can be controlled by pre-mRNA spicing and changes in alternative splicing alone are sufficient to induce EMT program in certain experimental paradigms (Yang et al., 2016; Wang et al., 2014). In order for a cell to undergo EMT, it has to evolve endogenous gene expression changes, or receive exogenous signals coming from tumor microenvironment.
Because of the pressure of rapid expansion of tumor, intratumoral microenvironment becomes inevitably harsh, resulting in abundant apoptotic tumor cells intermingled with neighboring proliferating cells (Brat et al., 2004). Recent studies have suggested that signals from apoptotic cells may play an important role in the progression of various types of malignancies (Huang et al., 2011; Obenauf et al., 2015). Clinically, these data support the paradoxical observation that the high number of apoptotic cells within a tumor positively correlates with a more aggressive tumor phenotype and decreased patient survival (Qian et al., 2014). Various pathways may be involved in the transduction of signal between apoptotic and surviving tumor cells. One possible mechanism is an extracellular vesicles (EVs)-mediated inter-cellular transport, which allows cells to send and receive molecules that could not be transferred otherwise including proteins, DNA, mRNA, miRNA and even whole ribosomes (Crescitelli et al., 2013). Nonetheless, mechanisms involved in transduction of signal between apoptotic and surviving tumor cells remains elusive.
In this study, we investigated the hypothesis that extracellular vesicles secreted by apoptotic GBM cells (apoEVs) cause a phenotypic shift of the recipient surviving tumor cells to promote their aggressiveness due to the change of molecules transferred by EVs after induction of apoptosis.
RESULTS
EVs secreted by apoptotic GBM cells promote more aggressive phenotype of recipient cells
A defining pathological feature of GBM is central necrosis with adjacent apoptotic cells (Brat et al., 2004). Staining of GBM tissues with antibodies against activated caspase 3 and Ki67 demonstrates that apoptotic and proliferating GBM cells are located in a close proximity around necrotic areas of the tumors (Figure 1A and S1A). In order to investigate the functional association of apoptotic cells to the neighboring surviving counterparts, we first measured caspase-3/7 activity in five freshly dissociated patient GBM samples. Flow cytometry analysis showed that apoptotic cells represent 10-70% of total tumor cells (Figures 1B and S1B). To determine if these cells can affect “healthy” tumor cells, we used the mouse intracranial xenograft models established from patient-derived GBM (neuro)spheres (Mao et al., 2013). To model clinical tumors, we co-injected lethally-irradiated and untreated GBM157 or GBM1027 cells in 1:1 ratio into brains of immunocompromised mice and as control, we injected lethally-irradiated or untreated tumor cells alone. MRI and staining of mouse brain sections for human Nestin and human-specific nuclear antigen (HNA) demonstrated that apoptotic GBM cells, when co-injected with live tumor cells, promote tumor growth (Figures 1C and S1C) accompanied by the acquired MES-like cellular morphology and increased invasion into surrounding normal brain tissues (Figure 1D). Next, we incubated GBM157 cells with apoptotic cells (1:1 ratio) or with conditioned medium obtained from apoptotic cells. The results indicated that factors secreted by apoptotic GBM cells promoted growth of healthy GBM cells (Figures 1E and S1D), accompanied by an increased expression of representative of MES markers (N-cadherin, CD44, ALDH1A3 and С/EBP-β) (Mao et al., 2013) in vitro (Figure S1E), and in vivo (Figures S1F and S1G). Interestingly, this growth promoting signal was eliminated by filtration of conditioned medium (Figures S1C and S1D), indicating that the effect of apoptotic cells can be mediated by EVs.
Figure 1. apoEVs promote growth of GBM cells in vitro and in vivo.
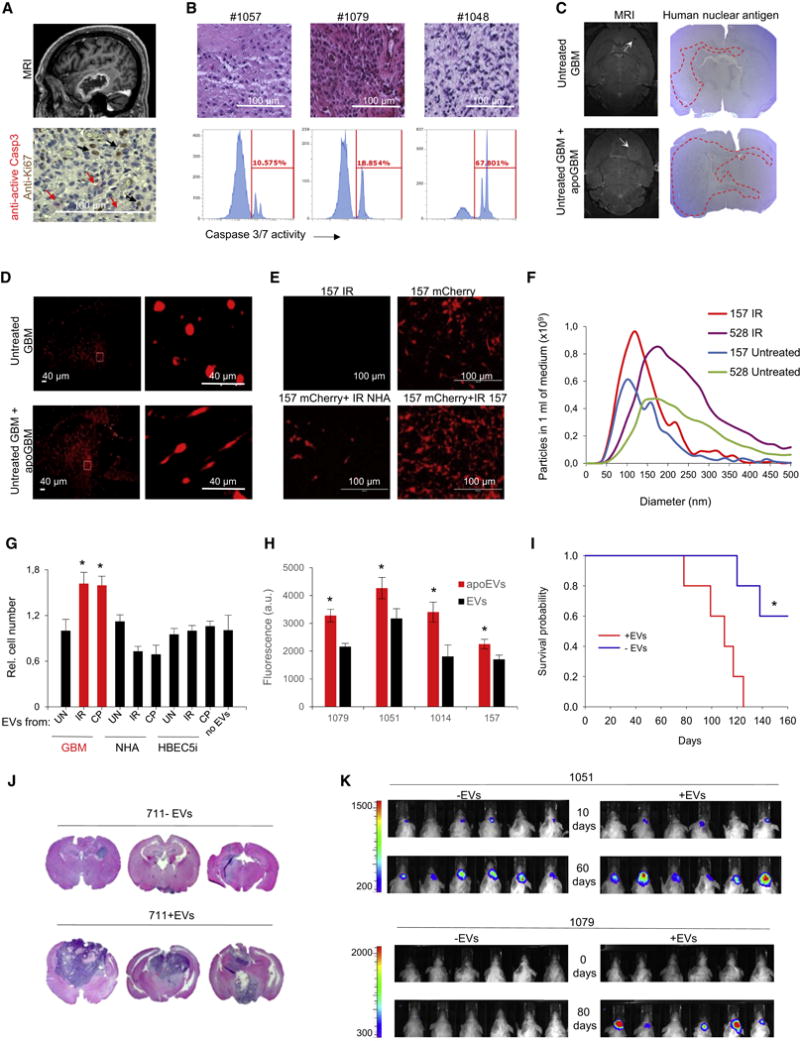
(A) MRI image of GBM tumor (top) and section of patient GBM stained for active Caspase 3 and Ki67 (bottom).
(B) H&E staining of sections from three representative GBM (top) and flow cytometry analysis of caspase 3/7 activity in freshly dissociated corresponding tumors (bottom).
(C) Representative MRI images and mouse brain sections stained for human nuclear antigen after intracranial transplantation of untreated glioma spheres (GBM1027 for MRI and GBM157 for brain sections) alone or together with lethally irradiated (12 Gy) spheres, ratio 1:1 (n = 5 mice per group).
(D) Representative images of mouse brain sections obtained as in “C” and stained for human Nestin.
(E) Co-culture of mCherry labeled GBM157 cells with unlabeled lethally irradiated normal human atrocities (NHA) or GBM157 cells (ratio 1:1).
(F) Nanoparticle tracking analysis of EVs produced by untreated or lethally irradiated GBM157 and GBM528 cells.
(G) In vitro cell growth assay of GBM157 spheres cultivated for 7 days with EVs from different sources: GBM157 spheres, normal human astrocytes (NHA) or immortalized brain endothelial cells (HBEC5i) that were left untreated (UN), lethally irradiated (IR) or treated with 50 μM of cisplatin (CP).
(H) In vitro cell growth assay of GBM157, GBM1079, GBM1051 and GBM1014 spheres cultivated for 7 days with EVs from lethally irradiated (apoEVs) or untreated (EVs) corresponding cells.
(I) Kaplan-Meier survival curves of mice injected with GBM711 spheres along or together with apoEVs (n = 5 mice per group, p<0.01, log-rank test).
(J) Representative images of H&E stained mouse brain sections after intracranial transplantation of GBM711 spheres alone or together with apoEVs from 711 cells (n= 5 mice per group).
(K) Representative bioluminescence images of mice intracranially injected with 2·105 luciferase labeled GBM1051 (upper panel) or GBM1079 (lower panel) neurospheres in the presence or absence of apoEVs produced by unlabeled lethally irradiated (12 Gy) cells from the same cells.
All quantitative data are average ± SD; *p < 0.01; See also Figures S1 and S2.
Nanoparticle tracking analysis (NTA) of conditioned medium demonstrated that upon induction of apoptosis, GBM cells shed a significantly higher number of EVs with larger average sizes (Figure 1F). We fluorescently labeled these EVs using Alexa Fluor 488 5-TFP ester and showed that surviving GBM cells can efficiently capture EVs both in vitro and in vivo (Figures S1H and S1I). Importantly in our experimental conditions we didn’t detect significant uptake of EVs by CD11b+ immune cells.
We then characterized EVs-mediated effects on the phenotype of surviving GBM cells. Apoptosis in EVs donor cells was induced ether by 6-12 Gy γ-irradiation (IR), 25-100 μM temozolomide (TMZ) or 20-50 μM cisplatin (CP) after that EVs were purified by ultracentrifugation and added to recipient cell in 50 μg/ml concentration. This concentration was considered clinically relevant as the concentration of exosomes in the blood of cancer patients was shown to be in the range of 20-100 μg/ml (Muller et al., 2014). We confirmed that our experimental conditions induce apoptosis, but not necrosis or autophagy, in the majority of EVs donor cells (Figures S2A and S2B). We found that EVs shed by apoptotic GBM cells (apoEVs), but not by normal human astrocytes (NHA) or immortalized brain endothelial cells (HBEC5i), enhanced growth of recipient GBM cells in vitro (Figures 1G and S2C). We confirmed this data using EVs produced by neurospheres from 5 different patients (Figure 1H and S2D). Interestingly, the growth promoting effect of apoEVs was diminished when EVs donor cells were pre-treated with a caspase inhibitor zVAD(OMe)fmk (Figure S2E). We also observed the pro-proliferative effect of apoEVs in vivo by comparing the tumor sizes and mouse survival periods after injection of GBM711 spheres with or without apoEVs into brains of immunocompromised mice (Figures 1I and 1J). To confirm these results, we intracranially co-injected luciferase-labeled GBM1079 or GBM1051 spheres in the presence or absence of EVs produces by unlabeled lethally irradiated or untreated corresponding cells. Data on Figures 1K and S2F show that the presence of apoEVs significantly promotes tumor growth.
We then used single cells derived from patient GBMs to measure cellular motility by time-lapse imaging on micropatterned polydimethylsiloxane surface that mimic structure of parenchymal stroma (Gallego-Perez et al., 2012). Comparison of recorded cellular tracks demonstrated that pre-incubation with apoEVs significantly increased cell motility in vitro (Figures 2A and S2G). Similar results were obtained with the wound healing assay (Figure S2H). To confirm this data in vivo, we analyzed xenograft tumors formed in the presence or absence of apoEVs. As shown in Figure 2B, the control samples demonstrated a clear tumor-normal tissue border, whereas, when co-injected with apoEVs, GBM711 cells invaded into adjacent mouse brain tissues. Next, we found that apoEVs significantly elevate the expression of the representative MES markers (Mao et al., 2013) in the recipient surviving GBM157 cells in vitro (Figures 2C, S2I, S2J and S2K). This shift towards MES phenotype was clearly observed in vivo, as GBM711 spheres co-injected with apoEVs formed brain tumors with prominent expression levels of CD44 and ALDH1A3 equivalent to those in GBM-like tumors formed by de novo MES tumor cells (Figures 2D and S3A). Previous reports demonstrated that an important hallmark of EMT is activation of aerobic glycolysis (the Warburg effect) (Prabhu et al., 2015). We therefore examined glucose metabolism in GBM157 cells cultivated with or without apoEVs and observed that apoEV significantly increase glycolysis in recipient cells (Figure 2E) but do not affect level of oxidative phosphorylation (Figures S3B). Next, we tested the effect of apoEVs on therapy sensitivity of GBM cells and found that pre-incubation with apoEVs significantly increases resistance of GBM spheres to temozolomide, cisplatin and γ-radiation (Figures 2F, S3C, S3D and S3E). Importantly, cytoprotective effect of apoEVs was dependent on its concentration (Figure S3F). Collectively, these data indicate that when GBM cells undergo apoptotic cell death, they paradoxically promote growth and gain of a MES phenotype of surviving GBM cells, thereby conferring therapy resistance.
Figure 2. apoEVs promote more aggressive phenotype of GBM.
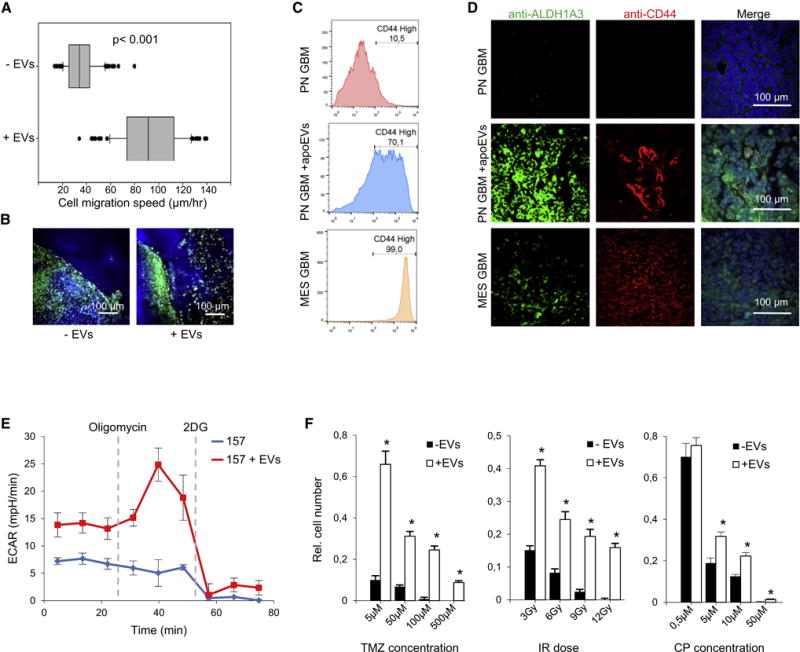
(A) Single cell motility assay of GBM157 cells that were either untreated or pretreated with apoEVs for 4 days. The line in the box is the median, the left and right of the box are the first and third quartiles, and the whiskers extend to 10th and 90th percentiles respectively.
(B) Representative images of mouse brain sections obtained as in “Figure 1I” and stained for human nuclear antigen (green) and DNA (blue) (n = 5 mice per group).
(C) FACS analysis for CD44 staining of control GBM157 spheres (PN) and spheres that were cultivated with apoEVs for 4 days. GBM267 spheres (MES) were used as a positive control.
(D) Representative images of mouse brain sections obtained as in “Figure 1I” and stained for DNA (blue), ALDH1A3 (green), and CD44 (red). Xenograft tumors formed by GBM267 spheres (MES) were used as a positive control (n = 5 mice per group).
(E) Extracellular acidification rate (ECAR) measured by a Seahorse Bioanalyzer in GBM157 cells that were either untreated or pretreated with apoEVs for 4 days
(F) In vitro cell viability assay of GBM157 spheres cultivated with or without apoEVs for 3 days, followed by treatment with indicated concentration of temozolomide, cisplatin or doses of γ-irradiation.
All quantitative data are average ± SD; *p < 0.01; See also Figures S2 and S3.
Lastly, we determined which type of vesicles is responsible for the observed effects. Fractionation experiments with subsequent Alamar Blue assay of cell proliferation demonstrated that the fractions with the strongest pro-proliferative and anti-apoptotic effect are apoEVs secreted 3-6 days after 6-12 Gy irradiation (Figures S3G and S3H), sedimentated by 120 000g centrifugation (Figure S3I and S3J), and positive for CD63 (Figure S3K). All of these are characteristic features of exosomes or exosome-like vesicles (Dieudé et al., 2015). It is important to note that the corresponding fraction of EVs secreted by untreated tumor cells had either no or much smaller effect on recipient cells. Therefore, we concluded that apoEVs has similar features to exosomes, however they are specifically produced by apoptotic GBM cells.
apoEVs are enriched with spliceosomal proteins and U snRNAs
To dissect the molecular mechanisms underlying the effect of apoEVs on recipient cells, we identified proteins that are present in EVs from both lethally-irradiated and untreated GBM spheres using liquid chromatography-tandem mass spectrometry (Table S1). Analysis of 1,089 identified proteins revealed that the clusters of proteasomal and spliceosomal proteins exhibit the most significant differences among these two samples (Figures 3A, 3B, S4A, S4B and S4C). To our knowledge, presence of spliceosomal proteins in EVs has not been described so far, while proteasomal proteins were previously identified only in EVs derived from apoptotic non-cancerous endothelial cells (Dieudé et al., 2015). We confirmed this result using western blotting (Figure 3C) and fluorescent NTA (Figure S4D). Importantly, neither western blotting nor mass spectrometry analysis display any enrichment of spliceosomal proteins in the microvesicle fraction produced by apoptotic cells (Figures 3C and S4E). Spliceosomal proteins are known to exist in a complex with small uridine-rich non-coding RNAs (U snRNA) (Kaida et al., 2010). Quantitative reverse transcription-PCR (qRT-PCR) showed that apoEVs are enriched with all types of spliceosomal U snRNAs (Figures 3D and S4F). In contrast, we did not observe corresponding enrichment in U7 and U8 snRNAs that are not involved in mRNA splicing. These proteomic and qRT-PCR data were consistent with our previous observation that ascitic fluid, formed in late-stages of ovarian cancer after courses of chemotherapy also enriched with several spliceosomal proteins and U snRNAs (Shender et al., 2014). Therefore, export of spliceosomal components may represent a common feature of multiple cancer types. Interestingly, we did not detect U1 snRNP related proteins in apoEVs using western blotting (Figure 3C) and qRT-PCR showed that U1 snRNA is the least enriched in apoEVs (Figure 3D), while it is typically the most abundant spliceosomal snRNA within the cell (Kaida et al., 2010). Consistently, we demonstrated that vesicles precipitated with anti-CD63 antibodies (a surface marker of exosomes) are enriched with U2, U4, U6 snRNAs but not with U1 snRNA (Figure 3E). Figure 3F summarizes proteins related to different parts of spliceosome that were identified in our study. Interestingly, much higher number of proteins were detected for U2 snRNP indicating that, there might be an export of specific spliceosomal complexes from the apoptotic cells.
Figure 3. apoEVs are enriched with spliceosomal proteins and U snRNAs.
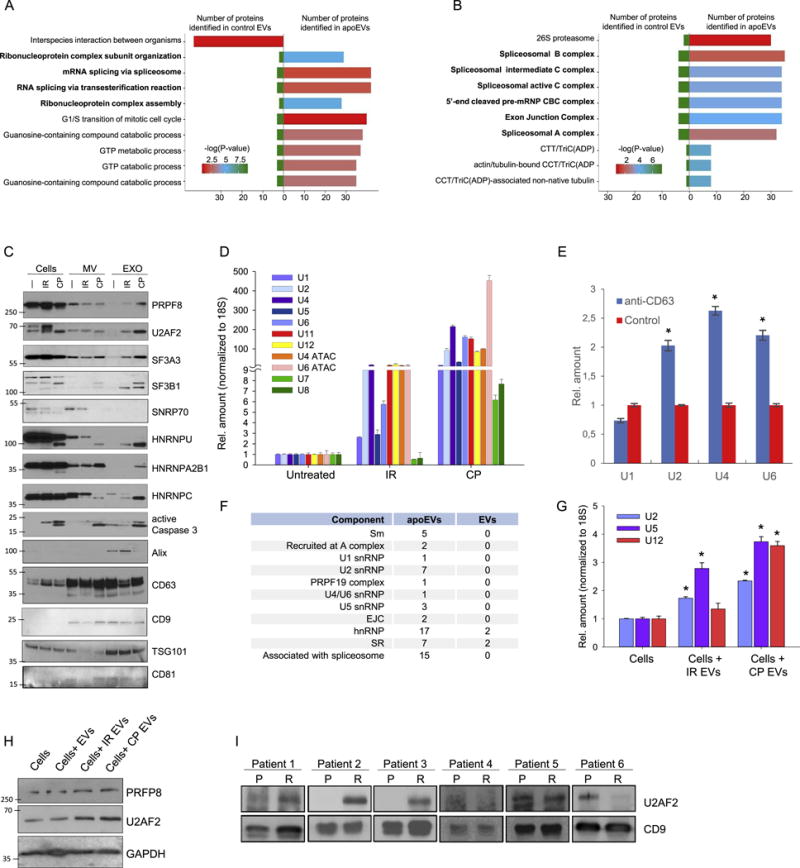
(A and B) Analysis of proteins identified by LC-MS/MS in apoEVs and control EVs with Gene Ontology Biological process database (A) or National Cancer Institute database (B).
(C) Western blotting analysis of exosome-like vesicles (EXO) and microvesicles (MV) purified from GBM157 spheres that were either untreated (—), lethally irradiated (IR), or treated with cisplatin (CP); PRPF8, U2AF2, SF3A3, SF3B1, SNRNP70, HNRNPU, HNRNPA2B1, HNRNPC – splicing factors; active Caspase 3 – apoptosis marker, Alix, CD63, CD9, TSG101, CD81 – exosome markers.
(D) qRT-PCR analysis of U snRNAs in vesicles purified as in “C”.
(E) qRT-PCR analysis of U snRNAs in vesicles purified by immunoprecipitation with anti-CD63 or control antibodies.
(F) Number of proteins related to different spliceosomal components that were identified in apoEVs and control EVs.
(G) qRT-PCR analysis of U snRNAs in GBM157 cells incubated for 10 hr with apoEVs from lethally irradiated or cisplatin treated cells, untreated cells were used as a control.
(H) Western blotting analysis of GBM157 neurospheres incubated with EVs from untreated cells and apoEVs purified from lethally irradiated or CP treated cells for 16 hr, untreated cells were used as a control.
(I) Western blotting analysis of exosomes isolated from blood serum samples obtained from GBM patients before (P) and after (R) post-surgical chemo- and radio-therapies. CD9 was used as a loading control.
All quantitative data are average ± SD; *p < 0.01; See also Figure S4 and Table S1.
To validate the transfer of spliceosomal proteins and U snRNAs through apoEVs to recipient cells, we incubated GBM cells with apoEVs for a short time, and showed that uptake of apoEVs significantly increases intracellular level of U2, U5 and U12 snRNAs (Figure 3G) as well as PRPF8 and U2AF2 spliceosomal proteins in recipient cells (Figure 3H). Finally, we purified EVs from the blood serum samples of six GBM patients obtained before and after post-surgical chemo- and radio-therapies. Consistent with our experimental data, we detected substantial increase of spliceosomal protein U2AF2 in samples after therapy in three cases (Figure 3I). Taken together, these results indicate that spliceosomal components might be specifically exported from apoptotic GBM cells via apoEVs.
Spliceosomal proteins are exported from apoptotic cells in a caspase dependent manner
Under physiologically normal conditions, spliceosomal proteins and U snRNAs are localized in the nucleus (Busà et al., 2010), which raised a question of how they translocate to EVs after apoptotic induction. We first co-transfected GBM cells with plasmids encoding RFP-tagged spliceosomal protein PRPF3 and GFP-tagged scaffold protein Coilin. At 24 hr after transfection, apoptosis was induced by cisplatin, staurosporine, or irradiation. Under all these conditions, we observed dissociation of PRPF3 from Coilin and its translocation to cytoplasm (Figure 4A). Similar data were obtained with endogenous SF3B1, HNRNPU and HNRNPA2B1 spliceosomal proteins (Figure S4G). Using fluorescent in situ hybridization (FISH), we also detected the translocation of U6 snRNA into cytoplasm (Figure 4A). Furthermore, we detected cytoplasmic localization of U2AF2, PRPF8 and NONO spliceosomal proteins in patient GBM cells positively stained for phospho-H2AX (Figure 4B) – a marker of DNA damage and early stages of apoptosis (Cook et al., 2009). Next, we asked if spliceosomal proteins are specifically exported to the cytoplasm or they translocated due to the breakdown of the nuclear envelop. We co-transfected GBM cells with plasmids encoding GFP with nuclear localization signal (GFP-NLS) and RFP-PRPF3. PRPF3 translocated to the cytoplasm at the early stages of apoptosis, while GFP-NLS retained predominantly in the nucleus even after nuclear fragmentation (Figure 4C). Importantly, we observed EVs with encapsulated RFP-PRPF3 using confocal microscopy (Figure 4D). We next asked if cytoplasmic export of spliceosomal proteins is dependent on caspase activity and found that EV-mediated export of spliceosomal proteins was blocked by the caspase inhibitor zVAD(OMe)fmk, indicating that this process is caspase-dependent (Figure 4E). Caspases are known to cleave multiple spliceosomal proteins during the early stages of apoptosis. This cleavage may lead to the changes in protein function and localization (Fischer et al., 2003). Interestingly, spliceosomal protein HNRNPU was exported only in a caspase cleaved form (Figures 3C and 4F). The cleaved HNRNPU did not contain N-terminal DNA binding domain, which is essential for chromatin interaction and nuclear localization (Göhring et al., 1997). To investigate how HNRNPU cleavage is associated with spliceosome export from apoptotic GBM cells, we designed a plasmid encoding HNRNPU with Yellow Fluorescence Protein (YFP) and Cyan Fluorescence Protein (CFP), fused to its N- and C-terminus, respectively (Figure 4G). We found that caspase cleavage allows the C-terminal portion of HNRNPU to be exported to the cytoplasm in a complex with other spliceosomal proteins (Figures 4H and S5A). Similar results were obtained, when we stained untransfected GBM cells with antibodies against the C-terminal region of endogenous HNRNPU protein (Figure S5B). All these data suggest that caspase activation may serve as one mechanism that facilitates nuclear export and subsequent apoEV encapsulation of spliceosomal components in apoptotic GBM cells.
Figure 4. Spliceosomal proteins and U snRNAs are exported from apoptotic cells in a caspase-dependent manner.
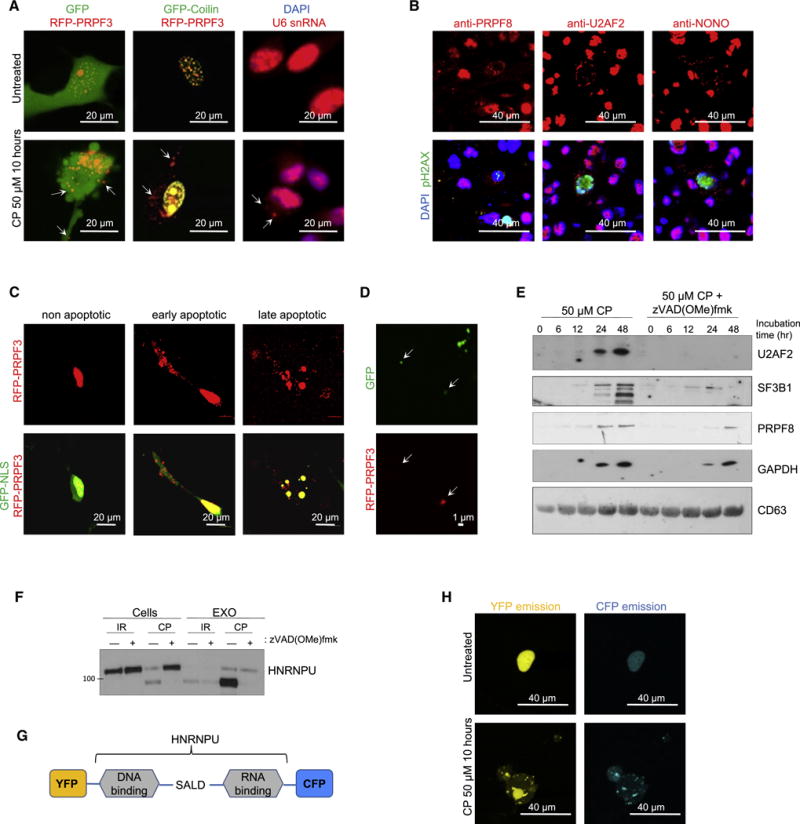
(A) Fluorescence images of GBM157 cells coexpressing RFP-PRPF3 and GFP (left) or GFP-Coilin (middle) after treatment with 50 μМ of cisplatin (CP) for 10 hr. Right: cisplatin-treated 157 cells were stained with DAPI and fluorescently-labeled probe for U6 snRNA.
(B) Immunofluorescent staining of human GBM tumor sections with DAPI (blue), antibodies against phosphorylated histone H2AX (green) and antibodies against splicing factors (red).
(C) Fluorescence images of GBM157 cells coexpressing RFP-PRPF3 and GFP-NLS after treatment with cisplatin.
(D) Fluorescence images of vesicles secreted into cultural medium by GBM157 cells coexpressing RFP-PRPF3 and GFP after treatment with cisplatin.
(E) Western blotting analysis of EVs purified from GBM157 cells treated for different periods of time with cisplatin in a presence or absence of pan-caspase inhibitor zVAD(OMe)fmk.
(F) Western blotting analysis of EVs and corresponding donor GBM157 cells treated with cisplatin (CP) or γ-irradiation in a presence or absence of zVAD(OMe)fmk.
(G) Schematic representation of fusion protein encoded by pEYFP-HNRNPU-CFP plasmid. SALD - Caspase3 cleavage site.
(H) Fluorescence images of CFP and YFP emission in GBM157 cells transfected with pEYFP-HNRNPU-CFP plasmid and treated with cisplatin.
See also Figures S4 and S5.
apoEVs induce mesenchymal-like splicing changes in recipient cells
Given that apoEVs transfer spliceosomal components, we asked whether the phenotypic shift seen in recipient GBM cells conferred by apoEVs is indeed mediated by changes in alternative splicing. We performed 100 bp paired-end RNA sequencing with untreated MES GBM sphere lines, untreated PN GBM sphere lines and PN GBM spheres pretreated for two days either with conditioned medium from lethally-irradiated spheres with or without filtration or with conditioned medium from control PN GBM spheres (n=3). Analysis of the RNA sequencing data (Table S2) demonstrated that apoEVs induce substantial changes in mRNA splicing. Upon comparison of MES and PN GBM spheres, more than 30% of alternative splicing events (ASE) affected by apoEVs overlap with those in the more therapy-resistant MES GBM spheres (Figure 5A). This result may indicate that apoEVs are in charge of promoting MES-like splicing changes in recipient PN cells for their phenotypic shift. In fact, gene ontology enrichment analysis revealed that apoEVs-induced ASE affects genes that regulate DNA damage repair and stress response (Figure 5B). This result correlates well with the data that apoEVs increase therapy resistance of recipient cells and raise an intriguing possibility that splicing factors secreted by apoptotic cells may prepare neighboring cells for subsequent insults. Comparison of our data with previously published datasets of ASE observed during EMT (Yang et al., 2016; Shapiro et al., 2011; Venables et al., 2013) showed a significant overlap indicating that the MES phenotype might be regulated by similar changes in the alternative splicing program in various types of cells beyond GBM (Figure 5C and Figure S5C). Specifically, we detected that apoEVs induce splicing changes in 9 out of 20 previously described key EMT related proteins such as CTNND1, SCRIB, ABI1, MPZL1, ITGA6, CD46, TCF7L2, PARD3 and LRRFIP2 (Pradella et al., 2017). To investigate the effect of apoEVs on gene expression in recipient cells, we incubated GBM157 spheres for four days with control EVs or apoEVs and then performed RNA sequencing. Data on Figure 5D demonstrate that treatment with apoEVs but not with control EVs decreased expression of PN markers and increased expression of MES markers (Mao et al., 2013; Phillips et al., 2006; Verhaak et al., 2010).
Figure 5. apoEVs promote mesenchymal-like splicing changes and rescue cells from the inhibition of endogenous splicing factors.
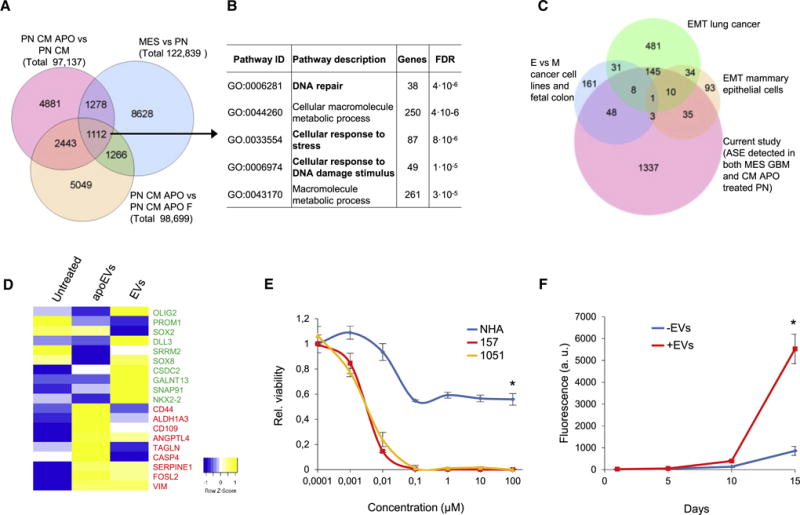
(A) Venn diagram representing alternative splicing events (ASE) detected in 3 different comparisons: MES cells versus PN cells; GBM157 (PN) cells cultivated for 4 days with conditional medium from lethally irradiated PN cells (CM APO) versus PN cells cultivated with conditional medium from untreated PN cells (CM); PN cells cultivated with CM APO versus PN cells cultivated with CM APO that were filtered (CM APO F) to remove vesicles.
(B) Gene ontology enrichment analysis of genes affected by ASE that were detected in all three comparisons.
(C) A Venn diagram representing genes affected by MES specific ASE induced by CM APO in GBM157 cells and genes affected by ASE during Epithelial-Mesenchymal Transition (EMT) described in three previous studies.
(D) Heatmap generated from RNAseq data showing expression of PN (green) and MES (red) markers in GBM157 neurospheres that were left untreated or treated for 4 days with apoEVs or control EVs produced by the cells from the same patient.
(E) In vitro viability assay of GBM spheres from two different patients and normal human astrocytes (NHA) treated with Pladienolide B.
(F) In vitro cell growth assay of GBM157 cells pretreated for two days with 20 nM of Pladienolide B and then cultivated in a fresh drug-free media in a presence or absence of apoEVs.
All quantitative data are average ± SD; See also Figure S5 and Table S2.
Given that apoEVs enriched with spliceosomal components can indeed affect mRNA splicing in recipient cells, we examined whether these EVs can rescue GBM cells from the inhibition of endogenous spliceosomal machinery. First, we tested the effect of two different splicing inhibitors: pladienolide B and isoginkgetin (Bonnal et al., 2012) on the viability of GBM cells and normal human astrocytes (NHA). Isoginkgetin did not noticeably affect cell proliferation up to the concentration of 100 μM (Figure S5D), whereas pladienolide B showed efficient cell killing in concentrations as low as 10 nM (Figure 5E). Of note, this inhibitor had significantly weaker effect on NHA cells as opposed to GBM cells. Next, we preincubated GBM cells with 20 nM of pladienolide B for 2 days and then plated them in a fresh media in the presence or absence of apoEVs. As shown on Figure 5F, apoEVs can efficiently restore cell proliferation after pladienolide B treatment. These data are consistent with our observation that apoEVs are enriched with SF3B1 – a direct target of pladienolide B (Figure 3C) and suggest that exogenous spliceosomal components from apoEVs can, at least partly, rescue GBM cells from inhibition of endogenous spliceosomal machinery. Interestingly, we observed slight decrease in molecular weight of SF3B1 upon induction of apoptosis (Figures 3C and 4E), however it was not inhibited by the caspase inhibitor (Figure S5E), raising a possibility that the exported in apoEVs protein might be extensively phosphorylated.
RBM11 is partly responsible for apoEV-mediated phenotypic changes of recipient cells
We next sought to identify an exogenous splicing factor that contributes to the apoEV-mediated phenotypic changes of recipient GBM cells. Supposedly, apoEVs can transfer only a very limited number of protein molecules due to their small sizes, therefore we speculated that splicing changes mediated by EVs occur because apoEVs deliver splicing factors that are otherwise not present (or present in very low amount) in recipient surviving GBM cells. To test this, we compared expression of genes that are known to regulate alternative splicing among PN, apoptotic PN and MES GBM spheres (Figures 6A, S6A and Table S3). We identified RBM11 as a MES specific splicing factor (Pedrotti et al., 2012) that is highly upregulated in PN cells after lethal irradiation. These data were validated by immunofluorescence, qRT-PCR, Western blot and bisulfite sequencing of the RBM11 promoter region (Figures 6B, S6B, S6C, S6D, S7A and S7B). Further analysis confirmed that the exogenously-expressed fusion protein GFP-RBM11 is encapsulated into apoEVs (Figure S7C), followed by transportation into recipient tumor cells (Figure 6C). We confirmed these data for endogenous RBM11 and demonstrated that it is present in apoEVs (Figure S7D) and that uptake of apoEVs significantly increases RBM11 levels in recipient cells (Figures 6D, S7E and S7F). Next, we examined the function of exogenous RBM11 in recipient cells. Overexpression of RBM11 resulted in the increased migration of GBM cells, accompanied by elevation of MES markers ALDH1A3 and Vimentin (Figures 6E, S7G and S7H). Analysis of glucose metabolism showed that exogenous RBM11 activates glycolysis in PN GBM cells to a similar level observed in apoEVs treated PN cells (Figure 6F and S3J). Unexpectedly, qRT-PCR and Western blotting detected the upregulation of endogenous RBM11 by exogenously expressed RBM11 at both mRNA and protein levels (Figures 6E and 6G). To differentiate whether this effect is induced by exogenous RBM11 protein or mRNA that encodes RBM11, we overexpressed frame-shift RBM11 mRNA that contains premature stop codon and therefore cannot produce protein however was nearly identical to the RBM11 mRNA coding region. As shown in Figure 6G, expression of this RNA mutant did not affect the levels of endogenous RBM11 mRNA indicating that exogenous RBM11 protein, but not mRNA, upregulates expression of endogenous RBM11. This result is consistent with previously published data that certain splicing factors can regulate their own expression both in cancer and normal cells (Saltzman et al., 2011; Takeo et al., 2009) and further suggests that exogenous RBM11 from apoEVs may provoke autocrine endogenous RBM11 activation (Figure 6H), thereby forming a positive feedback loop to amplify signals coming from apoEVs to recipient surviving tumor cells. Finally, we tested the effect of RBM11 knockdown on MES cells. Our data indicates that decrease in RBM11 expression leads to shift towards PN phenotype (Figures S7I and S7J) and dramatically increases sensitivity of MES cells to ɣ-irradiation (Figure 6I).
Figure 6. Splicing factor RBM11 is upregulated during apoptosis and subsequently transported by apoEVs to the recipient cells.
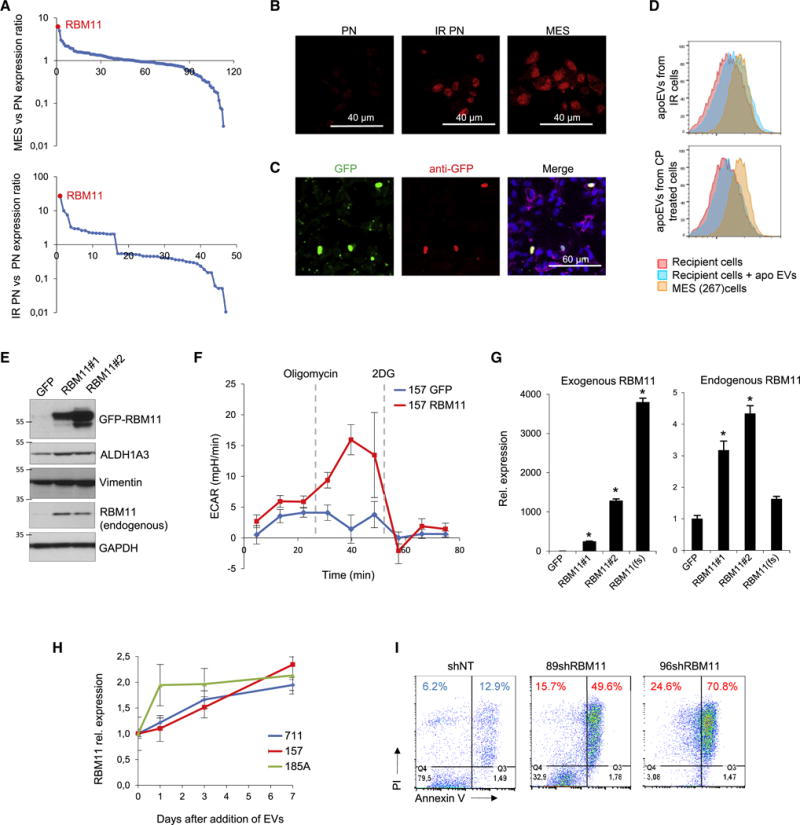
(A) Relative expression of genes involved in alternative splicing regulation in GBM157 (PN) versus MES glioma spheres (top) and in untreated versus lethally irradiated PN spheres (bottom), determined by microarray analysis.
(B) Immunofluorescent staining of untreated or lethally-irradiated GBM157 (PN) sphere with antibodies against RBM11. GBM267 spheres (MES) were used as a positive control.
(C) Immunofluorescent staining of GBM157 cells incubated for 24 hr with apoEVs from lethally-irradiated GBM157 cells overexpressing GFP-RBM11 protein with antibodies against GFP.
(D) FACS analysis for RBM11 staining of control GBM157 spheres or GBM157 spheres incubated for 10 hr with apoEVs from either lethally-irradiated or cisplatin treated GBM157 cells. GBM267 spheres (MES) were used as a positive control.
(E) Western blotting analysis of GBM157 cells stably expressing either GFP or GFP-RBM11 protein (two RBM11 isoforms were described so far: NM_144770 (#1) and NM_001320602 (#2), both were used in this study).
(F) Extracellular acidification rate (ECAR) measured by a Seahorse Bioanalyzer in GBM157 cells overexpressing GFP or GFP-RBM11.
(G) qRT-PCR analysis of expression of exogenous and endogeneous RBM11 (primers for 3′UTR) in GBM157 cells stably expressing GFP, GFP-RBM11 (#1 and #2 isoforms) or GFP-RBM11 frame-shift mutant RBM11(fs).
(H) qRT-PCR analysis of RBM11 expression in GBM157, GBM711 and GBM185 glioma spheres at different time points of cultivation with apoEVs from corresponding cell line.
(I) FACS analysis for Annexin V/PI staining of irradiated MES cells expressing non-target shRNA, or shRNA against RBM11.
All quantitative data are average ± SD; *p < 0.01; See also Figures S6 and S7 and Table S3.
Next, we investigated how exogenous RBM11 affects pre-mRNA splicing in recipient cells. We performed transcriptome microarray with PN cells overexpressing GFP-RBM11, or GFP as a control, and with MES cells infected with lentiviruses encoding shRNA against RBM11, or non-target sequence as a control, and found that RBM11 regulates a set of genes responsible for cell cycle progression and cell death (Figures 7A and Table S4). To gain insight into the functions of RBM11 in apoEVs recipient cells, we performed RNA immunoprecipitation with antibodies against RBM11 and found that RBM11 interacts with U1, U11 and U5 snRNPs (Figure S8A) confirming that RBM11 is directly involved in mRNA splicing regulation. In addition, this experiment revealed that RBM11 binds to mRNAs encoding Cyclin D1 - a key regulator of cell cycle progression, and MDM4 – an important regulator of apoptosis (Figures 7B, S8A and S8B). These data are in a good agreement with our observation that RBM11 affects cell cycle and cell death (Figure 7A). Finally, we identified that RBM11 can interact with its own mRNA confirming our hypothesis that exogenous RBM11 can affect expression of endogenous RBM11 (Figure 7B). We then sought to determine how RBM11 affects splicing of Cyclin D1 and MDM4 pre-mRNAs. The Cyclin D1 gene gives rise to two mRNA isoforms Cyclin D1a and Cyclin D1b and, and the former is known to promote replication checkpoint and DNA repair (Li et al., 2010). MDM4 mRNA encodes either a full length protein (MDM4f) or a short isoform (MDM4s), the latter has significantly higher anti-apoptotic activity than the former (Mancini et al., 2009). qRT-PCR showed that overexpression of RBM11 in PN cells promotes a shift toward MES-specific Cyclin D1a and MDM4s, whereas RBM11 knockdown in MES cells induces shifts toward a PN-specific Cyclin D1b and MDM4f (Figures 7C, 7D and S8C). Consistently, we found that apoEVs enhance Cyclin D1a and MDM4s expression in recipient PN cells (Figures 7C and 7D). Collectively, these data indicate that RBM11 binds to Cyclin D1 and MDM4 mRNAs, thereby regulating their splicing. Interestingly, we also found that overexpression of RBM11 increases PKM2/PKM1 isoform ratio (Figure S8D), which may explain the effect of apoEVs and RBM11 overexpression on the glucose metabolism of GBM cells (Pradella et al., 2017) (Figures 2E and 6F). However, we did not detect PKM mRNA in RBM11 immunoprecipitation samples, therefore PKM can be an indirect target of RBM11. We then analyzed The Cancer Genome Atlas (TCGA) database and found that GBM tumors with elevated expression of PKM2, MDM4s and Cyclin D1a were associated with poor prognoses for patients (Figures 8A, 8B and S8E). To further evaluate if exogenous RBM11 in apoEVs is responsible for the phenotypic shift in recipient cells, we compared the stimulatory effect of apoEVs secreted by RBM11-silenced or control cells on GBM spheres. apoEVs obtained after RBM11 silencing exhibited a substantially diminished growth-promoting effect both in vitro and in vivo (Figures 8C and 8D), indicating that RBM11 contributes, at least in part, to the effect of apoEVs on recipient GBM cells. To further confirm that exogenous RBM11 as well as other spliceosomal components encapsulated in apoEVs indeed plays an essential role in tumor aggressiveness, we co-injected luciferase labeled GBM1051 spheres into mouse brains together with irradiated unlabeled GBM1051 neurospheres with knockdown of RBM11 and/or core spliceosomal protein PRPF8. Our data indicated that apoptotic cells deficient in spliceosomal components have significantly diminished growth-promoting effect on surviving GBM cells in vivo (Figures 8E and 8F). Lastly, we assessed the clinical relevance of the experimental data of RBM11. First, we compared RBM11 immunoreactivity in 43 paired GBM specimens of primary and recurrent tumors from the matched patients. In most samples, RBM11 protein levels were substantially elevated in recurrent GBMs compared to the matched untreated tumors (Figure 8G). Next we used another patient cohort to analyze correlation of RBM11 expression levels with patient survival. We found that the higher RBM11 immunoreactivity correlates with the poor post-surgical survival of glioma patients (Figure 8H). Similar data were obtained using another set of GBM samples with another antibody against RBM11 (Figure S8F).
Figure 7. RBM11 regulates splicing of genes responsible for cell cycle progression and cell death.
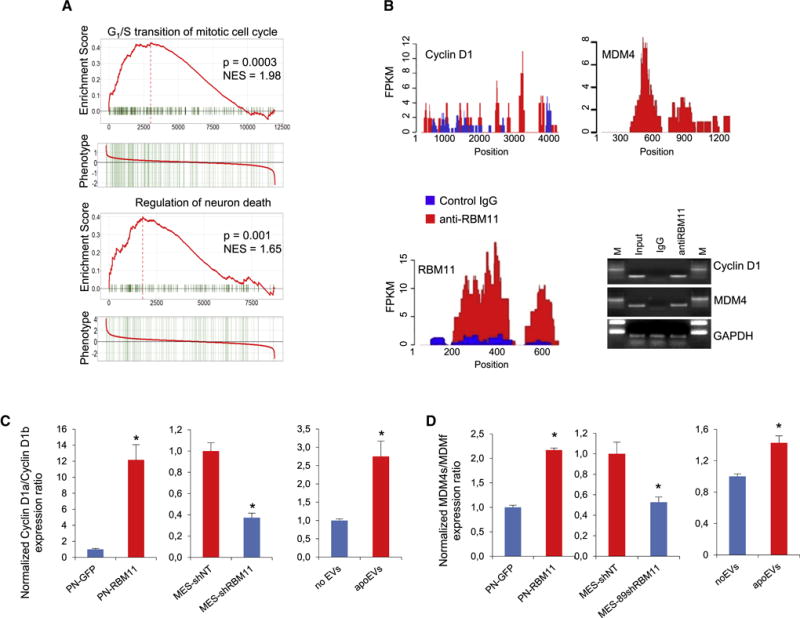
(A) GSEA of gene expression data of GBM157 cells (PN) expressing GFP versus GBM157 cells expressing GFP-RBM11 and of GBM267 cells (MES) expressing shRNA against RBM11 versus GBM267 cells expressing non target shRNA.
(B) RNA-IP enrichment profiles for antibodies against RBM11 (red) and a control IgG (blue); RT-PCR of corresponding samples with primers for Cyclin D1, MDM4 and GAPDH.
(C, D) qRT-PCR analysis of Cyclin D1 (C) or MDM4 (D) isoform expression ratios in GBM157 cells (PN) expressing GFP or GFP-RBM11, in GBM267 spheres (MES) expressing shRNA against RBM11 or non-target shRNA as a control, and in GBM157 cells cultivated for 7 days in a presence or absence of apoEVs. All quantitative data are average ± SD; *p < 0.01; See also Figure S8 and Table S4.
Figure 8. Exogenous RBM11 promotes malignancy of recipient tumor cells.
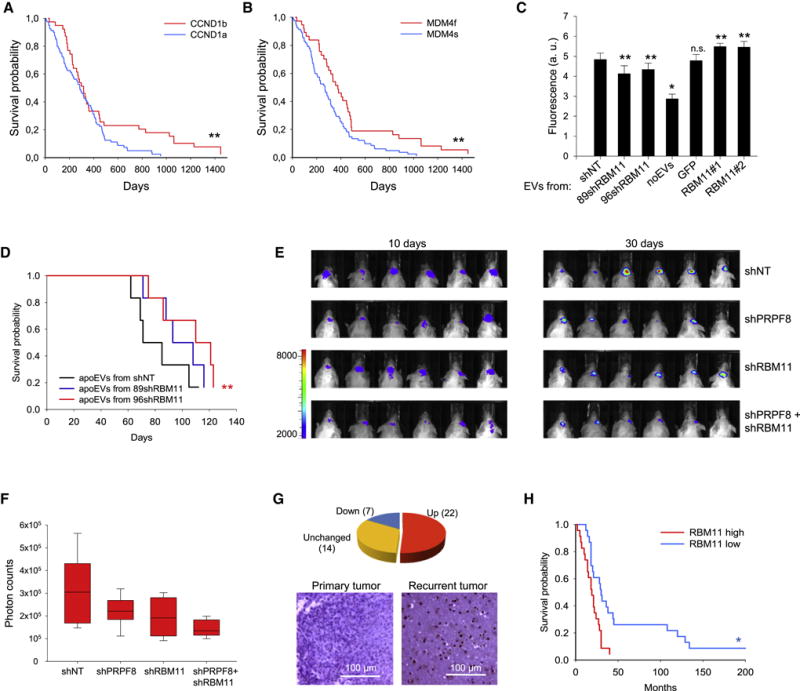
(A, B) Kaplan-Meier curve showing the overall survival of glioma patients subdivided based on the splicing of Cyclin D1 (A, p=0.0392, log-rank test) and MDM4 (B, p=0.0112, log-rank test), RNAseq data were obtained from TCGA database.
(C) In vitro cell growth assay of 157 spheres cultivated for 7 days with apoEVs from different sources: 157 cells stably expressing GFP, GFP-RBM11, two different shRNA against RBM11 or non-target shRNA (NT).
(D) Kaplan-Meier survival curves of mice injected with GBM711 spheres together with apoEVs from GBM711 cells stably expressing shRNA against RBM11 or NT shRNA as a control (n = 6 mice per group, p=0.13 and p=0.04, log-rank test).
(E) Representative bioluminescence images of mice intracranially coinjected with 2·105 luciferase labeled GBM1051 neurospheres and 2·105 lethally-irradiated (12 Gy) unlabeled GBM1051 neurospheres that were previously infected with lentiviruses encoding control shRNA (shNT) or shRNA against RBM11 (shRBM11) and/or shRNA against PRPF8 (shPRPF8).
(F) Quantification of luciferase signal in mice from (E). The horizontal line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 10th and 90th percentiles respectively.
(G) Analysis of RBM11 immunoreactivity in 43 paired GBM specimens of primary and recurrent tumors from the matched patients (upper) and representative images of immunohistochemical staining for RBM11 (lower).
(H) Kaplan-Meier curve showing the overall survival of glioma patients (n=45) subdivided in two groups based on RBM11 immunoreactivity (p=0.0018, log-rank test), data obtained from the tissue microarray.
All quantitative data are average ± SD; *p < 0.01, **p < 0.05; See also Figure S8.
DISCUSSION
According to our evaluation, apoptotic cells account for as high as 70% of the entire tumor cell population in GBM, suggesting a potential significance of understanding how signals produced by dying cells affect surviving tumor cell phenotypes. In agreement with earlier observations (Huang et al., 2011; Obenauf et al., 2015), this study demonstrated that apoptotic tumor cells promote growth and therapy resistance of the surviving counterparts and this phenomenon is, at least in part, mediated by intracellular transfer of apoEVs.
We observed that apoEVs have substantial and sustainable effects on the phenotype of surviving GBM cells, which is in a good agreement with reported strong effect of vesicles produced after irradiation on the migration of recipient cells in different types of cancer (Jelonek et al., 2015; Arscott et al., 2013; Mutschelknaus et al., 2017). However, our study demonstrated that this effect may lead to global phenotypic changes of the recipient cells, not just increasing cell motility. Analysis of proteins in apoEVs revealed that apoEVs are enriched with various spliceosomal components. Importantly, our proteomic experiments did not detect abundant nuclear proteins such as histones and components of replication/transcription machinery in apoEVs. In addition, U7 and U8, the snRNAs unrelated with RNA splicing, were not significantly increased in apoEVs. These data suggest that splicing factors may be selectively exported from apoptotic tumor cells. It will be of interest to determine whether splicing proteins and snRNAs are exported separately or as a whole functional spliceosome complex. One intriguing possibility is that spliceosomes could be exported when they are assembled on partially spliced pre-mRNA molecules. If this is the case, export of spliceosomes will also allow delivery of alternatively spliced mRNAs directly to the recipient cells, which may further enhance the changes of the recipient cells’ phenotypes. Consistent with this hypothesis, recent studies demonstrated that normal glial cells can export polyribosomes assembled on mRNA molecules to promote survival of damaged axons (Court et al., 2008; Court et al., 2011). These studies indicate that the inter-cellular spliceosomal transport can possibly occur during normal organ development, and cancer cells may hijack this mechanism in order to promote their survival and therapy resistance.
A characteristic feature of GBM is a high degree of intratumoral heterogeneity. In this study, we investigated the effect of apoEVs on the PN subtype of GBM cells, because these cells were found in every examined GBM tumor regardless of a dominant tumor subtype (Patel et al., 2014) and are believed to serve as a precursor for all other subtypes of GBM (Ozawa et al., 2014). Notably, the higher proportion of non-PN cells within the tumor correlates with the worse patient survival (Patel et al., 2014). Therefore, transformation of PN cells induced by apoEVs may increase tumor heterogeneity, leading to a worse disease outcome. However, it will also be important to elucidate the effect of apoEVs on other subtypes of GBM cells. Of note, although we extensively evaluated our clinical samples, we could not distinguish how much causal contribution is coming from extracellular signals to induce the MES shift of the tumors. Multiple other intracellular mechanisms were proposed for MES transformation such as gene mutations and a direct effect of irradiation (Mao et al., 2013; Ozawa et al., 2014). These mechanisms may help to create a number of therapy resistant cells within the tumor, while apoEVs could disseminate malignant phenotype to the majority of less aggressive GBM cells. On the other hand, several recent studies revealed that EVs produced by cancer cells may dramatically change the properties of tumor associated endothelial cells (Huang et al., 2017) and immune cells (Muller et al., 2016), therefore it is highly possible that the effect of apoEVs in vivo might be associated not only with phenotypic changes of recipient tumor cells but also with modulation of microenvironmental function of normal cells within or adjacent to the tumor.
We demonstrated that apoEVs transfer spliceosomal components to recipient cells, which after internalization induces subsequent changes in mRNA splicing, and validated the role of one particular spliceosomal protein RBM11 in intercellular communication. Importantly, after internalization, RBM11 did not undergo lysosomal degradation and is transported to the nuclei of recipient cells. Increase in the intracellular level of RBM11 elevated the expression of MES markers and promoted glycolysis – a hallmark of the more malignant cancer phenotype. We further demonstrated the ability of the exogenous RBM11 protein to increase the expression of the endogenous RBM11 by binding to its mRNA. This positive feedback loop may explain a prolonged effect of apoEVs on recipient cells and may amplify otherwise weak signals coming from apoEVs alone. Consistently, analysis of paired GBM specimens revealed that after the initial treatments, GBM tumors elevate RBM11 expression. We further demonstrated that knockdown of RBM11 in apoEVs donor cells decreases the pro-tumorigenic effect of apoEVs on recipient tumor cells. However, the effect of RBM11 silenced apoEVs on tumor aggressiveness in mice was statistically-significant, yet limited. Therefore, the impact of apoEVs may not be fully caused by any single molecule. One possibility is that apoEVs contain multiple spliceosomal proteins with overlapping functional roles to affect the phenotype of recipient cells. Moreover, EVs are known to also transport various miRNA and proteins (Crescitelli et al., 2013), which may contribute to the inter-cellular signaling between apoptotic and surviving GBM cells.
Our investigation into the signaling outcome of RBM11-induced splicing changes in apoEVs recipient cells revealed that RBM11 changes the splicing of Cyclin D1 and MDM4 toward pro-oncogenic Cyclin D1a and MDM4s isoforms, both of which correlate with the worse survival of GBM patients. Consistent with these data, RBM11 expression itself serves as a worse prognostic marker for GBM. An open question remains regarding the contribution of RBM11-induced splicing changes in GBM therapy resistance.
In conclusion, this study revealed a mechanism of cancer cell communication based on apoEV-mediated transfer of spliceosomal proteins. It is likely that this mechanism is applicable to cancer types beyond GBM (Shender et al., 2014). Clinically, our data may provide the rationale to the molecular targeting of RNA splicing events or specific splicing factors to attenuate the gain of post-therapeutic malignant changes in GBM. Further studies will elucidate the critical alternative splicing events during cancer progression.
STAR METHODS
Detailed methods include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-active Caspase-3 | Abcam | Cat# ab32042 |
| anti-ALDH1A3 | Abcam | Cat# ab129815 |
| anti-Alix | Abcam | Cat# ab117600 |
| anti-CD9 | Abcam | Cat# ab92726 |
| anti-CD11b | Abcam | Cat# ab133357 |
| anti-CD44 | Cell Signaling | Cat# 3570 |
| anti-CD44-APC | Miltenyi Biotec | Cat# 130-098-110 |
| anti-CD63 | Abcam | Cat# ab134045 |
| anti-GAPDH | Cell Signaling | Cat# 2118 |
| anti-GFP | Abcam | Cat# ab290 |
| anti-HNRNPA2B1 | Abcam | Cat# ab6102 |
| anti-HNRNPC | Abcam | Cat# ab133607 |
| anti-HNRNPU | Abcam | Cat# ab20666 |
| anti-human Nestin | Abcam | Cat# ab22035 |
| anti-human nuclear antigen | EMD Millipore | Cat# MAB4383 |
| anti-Ki67 | Cell Signaling | Cat# 9449 |
| anti-LC3B | Cell Signaling | Cat# 3868 |
| anti-mouse IgG-Alexa Fluor 647 | Cell Signaling | Cat# 4410 |
| anti-mouse IgG-Horseradish peroxidase | GE Healthcare | Cat# NXA931 |
| anti-NONO | Abcam | Cat# ab70335 |
| anti-phospho-H2AX | EMD Millipore | Cat# 05-636 |
| anti-PRPF8 | Abcam | Cat# ab185547 |
| anti-rabbit IgG-Alexa Fluor 488 | Cell Signaling | Cat# 4412 |
| anti-rabbit IgG-Horseradish peroxidase | GE Healthcare | Cat# NA934V |
| anti-RBM11 | Abcam | Cat# ab173589 |
| anti-RBM11 | Abcam | Cat# ab69358 |
| anti-RBM11 | Sigma | Cat# HPA045885 |
| anti-SF3A3 | Abcam | Cat# ab156873 |
| anti-SF3B1 | Abcam | Cat# ab170854 |
| anti-SNRP70 | Abcam | Cat# ab51266 |
| anti-TAPA1 | Abcam | Cat# ab109201 |
| anti-TSG101 | Abcam | Cat# ab125011 |
| anti-U2AF1 | Abcam | Cat# ab172614 |
| anti-U2AF2 | Abcam | Cat# ab37530 |
| anti-Vimentin | Abcam | Cat# ab16700 |
| anti-αTubulin | Abcam | Cat# ab7291 |
| anti-β Actin | Cell Signaling | Cat# 3700 |
| Bacterial and Virus Strains | ||
| E. coli Stbl3 Competent Cells | ThermoFisher | Cat# C737303 |
| E. coli TOP10 Competent Cells | ThermoFisher | Cat# C404010 |
| Biological Samples | ||
| Human glioma tissue microarray | This study | N/A |
| Human glioblastoms tissues | This study | N/A |
| Human blood plasma samples | This study | N/A |
| Mouse tissues | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EGF | Peprotech | Cat# AF-100-15 |
| bFGF | Peprotech | Cat# AF-100-18B |
| B27 | ThermoFisher | Cat# 12587010 |
| Heparin | Sigma | Cat# H3149 |
| DMEM/F12 | ThermoFisher | Cat# 10565-018 |
| StemPro Accutase | ThermoFisher | Cat# A1110501 |
| Fetal bovine serum | ThermoFisher | Cat# 10438018 |
| Penicillin-Streptomycin | ThermoFisher | Cat# 15140122 |
| CO2 Independent Medium | ThermoFisher | Cat# 18045088 |
| Laminin | Sigma | Cat# 11243217001 |
| LysoTracker Deep Red | ThermoFisher | Cat# L12492 |
| N-(lissamine rhodamine B sulfonyl)-phosphatidylethanolamine | Avanti Polar Lipids | Cat# 810150P |
| Cisplatin | Sigma | Cat# 479306 |
| Temozolomide | Sigma | Cat# T2577 |
| Staurosporin | Sigma | Cat# S4400 |
| zVAD(OMe)fmk | Abcam | Cat# ab120487 |
| 5-Aza-2′-deoxycytidine | Sigma | Cat# A3656 |
| Pladienolide B | Santa Cruz Biotechnology | Cat# sc-391691 |
| Isoginkgetin | EMD Millipore | Cat# 416154 |
| Endothelial cell growth supplement | Sigma | Cat# E2759 |
| Lipofectamine 2000 | ThermoFisher | Cat# 11668019 |
| RIPA buffer | Sigma | Cat# R0278 |
| Phosphatase inhibitor cocktail | Sigma | Cat# P2850 |
| Protease inhibitor cocktail | Sigma | Cat# P8340 |
| Blotting Grade Blocker Non Fat Dry Milk | Bio-Rad | Cat# 1706404XTU |
| iScript reverse transcription supermix | Bio-Rad | Cat# 1708841 |
| SYBR Select Master Mix | ThermoFisher | Cat# 4472918 |
| Serum-free protein block solution | Dako | Cat# X090930-2 |
| Vectashield mounting medium containing DAPI | Vector Laboratories | Cat# H-1200 |
| DakoCytomation target retrieval solution pH 6 | Dako | Cat# S236984-2 |
| EnVision+ System–HRP labeled Polymer | Dako | Cat# K400211-2 |
| AlamarBlue reagent | ThermoFisher | Cat# DAL1100 |
| Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 membrane | EMD Millipore | Cat# UFC910096 |
| XF minimal basal medium | Seahorse Bioscience | Cat# 103335-100 |
| Alexa Fluor 488 5-TFP Ester | ThermoFisher | Cat# A30005 |
| XenoLight D-luciferin | PerkinElmer | Cat# 122799 |
| Polybrene | EMD Millipore | Cat# TR-1003-G |
| Puromycin | Sigma | Cat# P7255 |
| EcoRI | ThermoFisher | Cat# ER0271 |
| SalI | ThermoFisher | Cat# ER0642 |
| KpnI | ThermoFisher | Cat# ER0521 |
| BamHI | ThermoFisher | Cat# ER0051 |
| SacI | ThermoFisher | Cat# ER1132 |
| NheI | ThermoFisher | Cat# ER0975 |
| Critical Commercial Assays | ||
| HiSpeed Plasmid Midi Kit | Qiagen | Cat# 12643 |
| miRNeasy mini kit | Qiagen | Cat# 217004 |
| RNeasy mini kit | Qiagen | Cat# 74104 |
| DAB peroxidase substrate kit | Vector Laboratories | Cat# SK-4100 |
| Magna RIP RNA-Binding Protein Immunoprecipitation Kit | EMD Millipore | Cat# 17-700 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat# 69504 |
| EpiTect Bisulfite Kit | Qiagen | Cat# 59104 |
| CellEvent Caspase-3/7 Green Flow Cytometry Assay Kit | ThermoFisher | Cat# C10427 |
| Autophagy Detection Kit | Abcam | Cat# ab139484 |
| Quick Start Bradford protein assay | Bio-Rad | Cat# 5000201 |
| TruSeq Stranded mRNA-Seq Library Preparation Kit | Illumina | Cat# 20020594 |
| Deposited Data | ||
| RNA sequencing data | This study | GEO:PRJNA344648 |
| Microarray data | (Mao et al., 2013) | GSE67089 |
| Experimental Models: Cell Lines | ||
| Normal Human Astrocytes | Lonza | Cat# CC-2565 |
| HBEC5i | ATCC | Cat# CRL-3245 |
| GBM84 | (Mao et al., 2013) | See Table S5 |
| GBM157 | (Mao et al., 2013) | See Table S5 |
| MD30 | (Mao et al., 2013) | See Table S5 |
| GBM1016 | (Wang et al., 2017) | See Table S5 |
| GBM528 | (Mao et al., 2013) | See Table S5 |
| GBM185A | (Wang et al., 2017) | See Table S5 |
| GBM267 | (Wang et al., 2017) | See Table S5 |
| GBM28 | (Wang et al., 2017) | See Table S5 |
| GBM408 | (Wang et al., 2017) | See Table S5 |
| GBM1051 | This study | See Table S5 |
| GBM1027 | This study | See Table S5 |
| GBM711 | (Bhat, et al., 2013) | See Table S5 |
| GBM1079 | This study | See Table S5 |
| GBM1014 | This study | See Table S5 |
| GBM374 | (Wang et al., 2017) | See Table S5 |
| 293FT | ThermoFisher | Cat# R70007 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD scid | The Jackson Laboratory | Cat# 001303 |
| Oligonucleotides | ||
| Primers for qRP-PCR | This study | See Table S6 |
| Primers for splicing sensitive PCR | This study | See Table S6 |
| FISH probe | This study | See Table S6 |
| shRNA target sequence | This study | See Table S6 |
| Primers for cloning | This study | See Table S6 |
| Recombinant DNA | ||
| pTagRFP-C | Evrogen | Cat# FP141 |
| pTagRFP-PRPF3 | This study | N/A |
| pTagGFP2-C | Evrogen | Cat# FP191 |
| pTagGFP2-Coilin | This study | N/A |
| pEYFP-CFP | (Pavlyukov et al., 2012) | N/A |
| pEYFP-HNRNPU-CFP | This study | N/A |
| pTagGFP2-RBM11 | This study | N/A |
| pCDH‐EF1‐MCS‐IRES‐Puro | System Biosciences | Cat# CD532A-2 |
| pCDH-GFP-RBM1 | This study | N/A |
| pCDH-GFP | This study | N/A |
| pTagGFP-NLS | This study | N/A |
| pGEM-T | Promega | Cat# A3600 |
| pHAGE PGK-GFP-IRES-LUC-W | Addgene | Cat# 46793 |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| pLKO1-shNT | Sigma | Cat# SHC002 |
| pLKO1-89shRBM11 | Sigma | See Table S6 |
| pLKO1-96shRBM11 | Sigma | See Table S6 |
| pLKO1-shPRPF8 | Sigma | See Table S6 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | N/A |
| FlowJo | FLOWJO, LLC | N/A |
| samtools | http://www.htslib.org | N/A |
| AS Detector | http://www.novelbio.com/asd/ASD.html | N/A |
| STRING | https://string-db.org/ | N/A |
| ProteinPilot | SCIEX | N/A |
| X! Tandem | http://www.thegpm.org/tandem/ | N/A |
| Scaffold 4 | Proteome Software | N/A |
CONTACT FOR REAGENT AND RESOURCES SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ichiro Nakano (inakano@uabmc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo Intracranial Xenograft Tumor Models
6-week-old female NOD scid mice (Prkdcscid) were used for GSCs intracranial implantation. All animal experiments were carried out under an Institutional Animal Care and Use Committee (IACUC) approved protocol according to NIH guidelines. The GSCs suspension (2×105 cells in 3 μl of PBS) was injected into the brains of nude mice as previously described (Wang et al., 2017). When neuropathological symptoms developed, mice were sacrificed and perfused with ice-cold PBS and 4% paraformaldehyde (PFA). Mice brains were dissected, fixed in 4% PFA for 24 hr and then transferred to 10% formalin.
Glioma Sphere Lines
Glioblastoma (GBM) specimens were collected during surgery under preoperative MRI-guided navigation and mechanically dissociated into pieces with 1-3 mm diameter. The samples were then treated with trypsin for 20 minutes at +37 °C to obtain single cells. Cell s uspensions were run through Lympholyte-H separation (Cedar Lane Labs) to remove Red Blood Cells and debris according to manufacturer’s specifications. Established cell lines were cultivated in serum-free Neurosphere media for no longer than 30 passages. For all cell lines, STR analysis was performed in order to confirm cell identity (Table S5). Some cell lines were subclassified as proneural or mesenchymal based on previously published microarray data (Bhat et al., 2013; Mao et al., 2013).
Cell Culture, Lentivirus Production and Transduction
Neurosphere cultures were established and cultivated in DMEM/F12 medium containing 2% B27 supplement, 1% Penicillin-Streptomycin solution, 2.5 μg/ml heparin, 20 ng/ml basic fibroblast growth factor (bFGF), and 20 ng/ml epidermal growth factor (EGF). bFGF and EGF were added twice a week and the cultural medium was changed every 7 days. Normal Human Astrocytes (Lonza) were cultivated in DMEM/F12 medium containing 10% FBS, 2% B27 supplement and 1% Penicillin-Streptomycin solution. Cerebral microvascular endothelium cells HBEC5i (ATCC) were cultivated in DMEM/F12 medium containing 10% FBS, 1% Penicillin-Streptomycin solution and 40 μg/ml Endothelial cell growth supplement. For all cell lines mycoplasma contamination was tested with Look Mycoplasma PCR Detection Kit (Sigma). For microscopy purposes cells were cultivated on laminin coated Lab-Tek II Chambered Coverglass.
Plasmid DNA was purified with HiSpeed Plasmid Midi Kit (Qiagen). Transfection was performed by using Lipofectamine 2000 reagent (Thermo scientific) according to the manufacturer’s protocol. Cells were examined 24-48 hr after transfection. Lentiviruses were produced as described previously (Pavlyukov et al., 2011). Briefly, HEK293FT packaging cells (Thermo scientific) were co-transfected with pLKO1 (Sigma; used shRNA sequences are listed in Table S6) or pCDHEF1-MCS-IRES-Puro (System Biosciences) lentiviral vector and two packaging plasmids psPAX2 and pMD2.G. Growth media was changed the following day and lentivirus-containing supernatants were harvested 72 hr later. One day before transduction GBM spheres were dissociated into single cells with accutase and seeded on laminin coated 6-well plates, 8×105 cells per well. Next day GBM cells were incubated with viral supernatants for 24 hr in the presence of 8 μg/ml polybrene (EMD Millipore). Two days after infection, transduced cells were selected with 1 μg/ml puromycin (Sigma) for 3 days.
Human Glioma Samples
This study was conducted under the approved Institutional Review Board (IRB) and Institutional Animal Care and Use Committee (IACUC) protocols in University of California Los Angeles (UCLA), MD Anderson Cancer Center (MDA), University of Alabama at Birmingham (UAB), and Ohio State University (OSU). The IRB Protocol number at UAB is #N151013001. Clinical glioma specimens and normal brain tissue samples were collected in the Department of Pathology and Laboratory Medicine at UCLA (samples 157, 408), MDA (sample, MD30, 267, 28, 711), UAB (samples 1027, 1051, 1079) and OSU (sample 83, 528, 84, 345, and 1016), and processed to the research laboratories after de-identification of the samples, as described previously (Wang et al., 2017). 43 paired GBM specimens were obtained from patients with the Institutional Review Board (IRB) of the Samsung medical center (2014-04-004 and 2013-10-072). The use of the de-identified tissues for the experiments involving isolation of glioma spheres, DNA and RNA isolation, and/or IHC on human tumors was exempt from requiring consent.
METHODS DETAILS
Extracellular Vesicles Isolation
Extracellular vesicles were isolated by ultracentrifugation as described previously (Crescitelli et al., 2013). Briefly, cells were removed by pelleting with centrifugation at 1000 g for 10 minutes. Supernatant was then filtered through 0.8 μm filter (EMD Millipore). In some experiments microvesicle fraction (MVs) were separately isolated by centrifugation at 16,500 g for 20 minutes, 4 °C. Extracellular vesicles were is olated by subsequent centrifugation at 120,000 g for 70 min, 4 °C. For in vitro experiment EVs pellet was resusp ended in fresh cultural medium for GBM spheres at 50 μg/ml concentration. This concentration was considered clinically relevant as the concentration of exosomes in the blood of canscr patients was shown to be in the range of 20-100 μg exosomal proteins in 1 ml of plasma samples (Muller et al., 2014).
For in vivo experiments EVs pellet was thoroughly resuspended in 30 ml of PBS, solution was centrifugated again at 120,000 g for 70 min, 4 °C. EVs pellet was resuspended in PBS and used for in vivo injection at 15 μg per mouse (Katakowski et al., 2013; Lee et al., 2013).
For mass spectrometry experiments EVs pellet was thoroughly resuspended in 30 ml of PBS, solution was centrifugated at 120,000 g for 70 min, 4 °C. EVs pe llet was resuspended in 30 ml of PBS and again centrifugated at 120,000 g for 70 min, 4 °C. PBS so lution was completely removed and pallet was used for subsequent mass spectrometry analysis.
To normalize amount of EVs purified from different donor cells EVs samples were assayed for total protein by the Bradford microassay method.
Western Blot
Cells were lysed for 30 min on ice in RIPA buffer (Sigma) containing 1% protease and 1% phosphatase inhibitor cocktails (Sigma). Lysates were pre cleaned by centrifugation at 16 000g, 15 min, 4 °C. Prot ein concentration was determined by Bradford method. Equal amounts of protein lysates (10 μg/lane) were fractionated by NuPAGE Novex 4-12% Bis-Tris Protein gel (Thermo scientific) and transferred to a PVDF membrane (Thermo scientific). Subsequently, the membrane was blocked with 5% Blotting Grade Blocker Non Fat Dry Milk (Bio-Rad) for 1 hr and then incubated with corresponding primary antibody overnight and next incubated with peroxidase conjugated secondary antibodies (GE Healthcare) for 1 hr. Staining was visualized with Amersham ECL Western Blot System (GE Healthcare).
RNA Isolation and Quantitative Real-Time PCR
mRNA was isolated by RNeasy mini kit (Qiagen), U snRNAs were isolated by miRNeasy mini kit (Qiagen). In both cases DNA digestion step was performed during RNA purification. RNA concentration was determined using a Nanodrop 2000 Spectrophotometer (Thermo scientific). cDNA was synthesized by using iScript reverse transcription supermix (Bio-Rad) according to the manufacturer’s protocol. qPCR was performed on StepOnePlus thermal cycler (Thermo scientific) with SYBR Select Master Mix (Thermo scientific). Cycling conditions were 95 °C for 5 min, and then 50 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C 30 s. Primer specificity was confirmed by visualizing DNA on an agarose gel following PCR. GAPDH or 18S RNA was used as an internal control. Primer sequences are shown in Table S6.
Immunocytochemistry
Immunocytochemistry (ICC) method was described previously (Wang et al., 2017). Briefly, cells were fixed with 4% PFA, permeabilized with 0.2% Triton-X, blocked with serum-free protein block solution (Dako) and incubated with corresponding primary antibodies for 1 hr at room temperature. Next cells were incubated with Alexa Flour-conjugated secondary antibody for 1 h at room temperature and mounted in Vectashield mounting medium containing DAPI (Vector Laboratories). Images were captured with Nikon A1 Confocal microscope (Nikon).
Immunohistochemistry
Immunohistochemistry (IHC) was performed as previously described Wang et al., 2017). Briefly, tumors embedded in paraffin blocks were deparaffinized, and hydrated through an ethanol series. After microwave antigen retrieval in DakoCytomation target retrieval solution pH 6 (Dako), slides were incubated in 0.3% hydrogen peroxide solution in methanol for 15 min at room temperature to inhibit internal peroxidase activity. Next samples were blocked with serum-free protein block solution (Dako) and incubated with corresponding primary antibodies overnight at 4°C. Next day slide s were stained with EnVision+ System–HRP labeled Polymer (Dako) and visualized with DAB peroxidase substrate kit (Vector Laboratories). IHC scoring was performed as described previously (Klein et al., 2001). Samples with scores more than 4 were considered as “high” expression group.
For frozen sections, fixation of the tissue sections was performed using 4% PFA. Samples were permeabilized with 0.2% Triton-X, blocked with serum-free protein block solution (Dako) and incubated with corresponding primary antibodies overnight at 4°C. Next slides were incubated with Alexa Flour-conjugated secondary antibody for 1 hr at room temperature and mounted in Vectashield mounting medium containing DAPI (Vector Laboratories). Images were captured with Nikon A1 Confocal microscope (Nikon).
Cell Proliferation Assay
Proliferation of GBM cells was determined using AlamarBlue reagent (Thermo scientific). Cells were seeded at 6 000 cells per well in a 96 well plate, after indicated period of time AlamarBlue reagent was added into each well and 4 hr later fluorescence was measured (Excitation 515-565 nm, Emission 570-610 nm) using Synergy HTX multi-mode reader (BioTek).
Nanoparticle Tracking Analysis
For nanoparticle tracking analysis (NTA) GBM cells were cultivated for 3 days in medium pre filtered through Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 membrane (EMD Millipore). Cells were pelleted by centrifugation at 500g, 20 min, 4 °C, supernatant w as diluted in PBS and used for NTA. Particle size distribution and concentration were measured on a NanoSight NS500 instrument (Nanosight) equipped with 532 nm laser. Videos of the particles Brownian motion were recorded at room temperature and processed with nanoparticle tracking analysis analytical software version 2.3. At least 10 individual measurements for 60 seconds, with a total of at least 5,000 tracks for individually tracked particles, were collected for each sample.
For fluorescent NTA supernatant obtained after 500g centrifugation was again centrifugated at 120,000g, 70 min, 4 °C. EVs pellet was thoroughly resuspended in 100 μl of PBS, EVs were fixed by addition of 900 μl of 4% PFA, permiabilized with 30 ml of 0.2% Triton-X and centrifugated at 120,000g, 70 min, 4 °C. EVs pel let was thoroughly resuspended in 100 μl of serum-free protein block solution (Dako) and 30 min later Alexa Fluor 555 labeled anti-U2AF2 antibodies were added. After 1 hr incubation 30 ml of PBS were added and solution was centrifugated at 120,000g, 70 min, 4 °C. EVs pellet was resuspended in 30 ml of PBS and again centrifugated at 120,000g, 70 min, 4 °C. EVs pellet was resuspended in 500 μl of PBS and used for NTA on a NanoSight NS500 instrument (Nanosight) equipped with 532 nm laser and 560 nm long-pass filter as described previously. All solutions used in this experiment were pre filtered through Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 membrane (EMD Millipore).
Single-Cell Motility Assay
Single-cell motility was performed as described previously (Gallego-Perez et al., 2012). Briefly, glioma spheres were pretreated with EVs for 4 days and then dissociated into single cells, plated, and allowed to adhere and on laminin coated spread micropatterned polydimethylsiloxane surfaces in serum-free, heparin-free, CO2-independent medium (Thermo scientific). Such surfaces were fabricated through a simple replica-molding process from a photolithographically patterned Si master. These substrates were sterilized in 70% ethanol prior to cell seeding. Cell motility was traced via time-lapse microscopy. Images were collected every 10 min during 24 hr, and analyzed using the manual tracker plugin in Fiji.
Wound Healing Assay
Wound healing (scratch) assay was performed as described previously (Wang et al., 2017). Briefly, glioma spheres were dissociated into single cells, plated on laminin coated 6 will plate, 1.5×106 cells per well. Next day cell cultures were scratched using a pipette tip. 48 hr later cells were visualized by light microscopy. These assays were performed three times.
RNA Immunoprecipitation
RBM11 protein-RNA complexes were purified from 157 glioma spheres stably expressing GFP-RBM11 with Magna RIP RNA-Binding Protein Immunoprecipitation Kit (EMD Millipore) according to the manufacturer’s protocol.
Bisulfite Genomic Sequencing
Genomic DNA was purified with DNeasy Blood & Tissue Kit (Qiagen), DNA was bisulfite converted with EpiTect Bisulfite Kit (Qiagen) according to the manufacturer’s protocol with slight modifications. Bisulfite conversion cycling conditions were 95 °C for 5 min, 60 °C for 25 min, 95 °C for 5 min, 60 °C for 85 mi n, 95 °C for 5 min, 60 °C for 175 min, 95 °C for 5 min and t hen 60 °C for 120 min. A 719 bp segment of RBM11 promoter (nucleotide # -100 to # +619, translational start site = +1) was amplified from bisulfite-converted gDNA using Nested PCR. First round of PCR was performed with primers RBM11-biS_for and RBM11-biS_rev cycling conditions were 95 °C for 5 min, an d then 28 cycles of 95 °C for 30 s, 65 °C for 30 s, 72 °C 60 s and final extension at 72 °C for 7 min. Second round of PCR was performed with primers RBM11-biS_for2 and RBM11-biS_rev cycling conditions were 95 °C for 5 min, and then 22 cycles of 95 °C for 30 s, 67 °C for 30 s, 72 °C 60 s and final extension at 7 2 °C for 7 min. Amplicons were subcloned into the p GEM-T plasmid (Promega) and subsequently sequenced using vector-encoded T7 primer.
Flow Cytometry
For CD44 staining glioma spheres were dissociated into single cells and stained with anti-CD44-APC antibody (Miltenyi Biotec) according to manufacturer’s protocol. Cells without primary antibody were used for negative control.
For ALDEFLUOR staining glioma spheres were dissociated into single cells and stained at concentration 2×105 cells per ml with ALDEFLUOR Kit (StemCell Technologies) according to the manufacturer’s protocol. For RBM11 staining FACS analyses were performed as previously described (Pavlyukov et al., 2011). Briefly, cells were fixed by 4% PFA, permeabilized with 0.2% Triton-X, blocked with serum-free protein block solution (Dako) and incubated with primary antibodies for 1 h following incubation for an additional hr with Alexa Fluor 647 conjugated secondary antibodies.
For apoptosis assay cells were stained with CellEvent Caspase-3/7 Green Flow Cytometry Assay Kit (Thermo scientific) according to the manufacturer’s protocol.
For autophagy assay cells were stained with Autophagy Detection Kit (Abcam) according to the manufacturer’s protocol.
After staining samples were analyzed by Attune NxT Flow Cytometer (Thermo scientific) and the data were processed with FlowJo 10 software.
Cell Line STR Analysis
Services were provided by University of Arizona Genetics Core, Arizona Research Laboratories, Division of Biotechnology (http://uagc.arl.arizona.edu/). Genomic DNA was purified from individual cell lines and shipped on dry ice to Core Facility. PCR was performed and products were separated by capillary electrophoresis using an AB 3730 DNA Analyzer. Applied Biosystems Internal Size Standard (ISS) GeneScan500-LIZ was used to standardize allele size calls for each sample. The PCR product was diluted 1:50 with sterile water and prepared with POP7 Polymer per the manufacturer’s instructions (Applied Biosystems). Samples were run on a 36 cm capillary array (Applied Biosystems). For data analysis, electropherograms were analyzed from the .fsa files and allelic values assigned using Soft Genetics, Gene Marker Software Version 1.85. Alleles were matched to STR Profile recorded with DMSZ (when reference profile is available).
Organelle Labeling
Labeling of microvesicular bodies (MVBs) with the fluorescent phospholipid analog N-Rh-PE (N-(lissamine rhodamine B sulfonyl)-phosphatidylethanolamine) was performed as described previously (Fader et al., 2008). Briefly, N-Rh-PE was solubilized in absolute ethanol and injected into ice-cold medium with a Hamilton syringe while vigorously vortexing to a final concentration of 4 μM. Medium was then added to the cells and they were incubated for 60 min on ice. After incubation cells were extensively washed with cold PBS. Then cells were cultured in complete medium for 3 hr to allow internalized lipid to reach the MVBs.
Labeling of lysosomes was performed with LysoTracker Deep Red (Thermo scientific) according to the manufacturer’s protocol.
Measurement of Extracellular Acidification Rate (ECAR) and Oxygen Consumption Rate (OCR)
ECAR and OCR were measured using a Seahorse Bioscience XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) according to the manufacturer’s protocol. Cells were seeded in normal growth medium 24 hr before measurement at a density of 6×104 cells per well. The assay was performed in XF minimal basal medium (Seahorse Bioscience) supplemented with 11mM glucose and 2mM L-Glutamine (Life Technologies). Cells were washed two times and pre-incubated in this medium for 1 hr before measurement at 37°C in a humidified atmosphere with out CO2. Oligomycin A (0.5 μg/ml) and 2-DG (100mM) were used to evaluate glycolytic capacity. Data were analyzed using Seahorse software.
Fluorescent Hybridization in situ
Fluorescent hybridization in situ (FISH) was performed as described previously (Shishova et al., 2015) with FAM labeled probe for U6 snRNA (sequences are shown in Table S6).
Gene Expression Microarray Data Analysis
For microarray analysis two previously published datasets were used: GSE67089 datasets in the NCBI Gene Expression Omnibus for comparison of PN and MES cell lines and unpublished data for comparison of irradiated and non-irradiated PN cells. Gene expression microarray data were preprocessed and normalized with Affymetrix Expression Console software (V1.3.1) using RMA sketch method. Probes of microarray were excluded if they failed to achieve a detection p value 0.05. Quantile normalization of microarray value was performed using the preprocess Core R package. KEGG and GO term gene set enrichment analysis (GSEA) was done using clusterProfiler. The genes for GSEA were ordered by log fold change. Heatmap and clustering were performed with Cluster 3.0 software, using Pearson Correlation Coefficient as distance measure. The result was shown with Java Treeview software. Gene Enrichment Analysis was performed using STRING software (http://string-db.org/) as described previously (Szklarczyk et al., 2015).
Preparation of EVs Samples for Mass Spectrometry
EVs pellets were denaturated for 30 min in buffer containing 8M Urea, 2M Thiourea, 10 mM Tris (pH=8). Protein concentrations were determined using the Quick Start Bradford protein assay (Bio-Rad) according to the manufacturer’s protocol. Then protein disulfide bonds were reduced with 5 mm DTT at RT for 30 min and afterwards alkylated with 10 mm iodoacetamide at room temperature for 20 min in the dark. Next samples were diluted (1:4) with 50 mM ammonium bicarbonate buffer and digested with trypsin (0.01 μg of trypsin per 1 μg of protein) for 14 hr at 37 °C. After trypsin dig esting, reaction was stopped by addition of formic acid to the final concentration of 5%. Obtained tryptic fragments were desalted by Discovery DSC-18 50 mg microcolumns (Sigma), dried in vacuum and redissolved in 3% ACN with 0.1% FA solution prior to LC-MS/MS analysis.
LC-MS/MS Analysis
LC-MS/MS Analysis of EVs was performed in two biological and two technical replicates (Table S1) on a TripleTOF 5600+ mass-spectrometer with a NanoSpray III ion source (ABSciex) coupled with a NanoLC Ultra 2D+ nano-HPLC system (Eksigent). The HPLC system was configured in a trap-elute mode. For sample loading buffer and buffer A, the mixture of 98.9% water, 1% methanol, 0.1% formic acid (v/v) was used. Buffer B was 99.9% acetonitrile and 0.1% formic acid (v/v). Samples were loaded on a Chrom XP C18 trap column (3 μm 120 Å 350 μm*0.5 mm; Eksigent) at a flow rate of 3 μl/min for 10 min and eluted through a 3C18-CL-120 separation column (3 m, 120 Å, 75 m*150 mm; Eksigent) at a flow rate of 300 nl/min. The gradient was from 5 to 40% of buffer B in 90 min followed by 10 min at 95% of buffer B and 20 min reequilibration with 5% of buffer B. Between different samples, two blank 45 min runs consisting of 5*8 min waves (5%B-95%-95%-5%) were required to wash the system and to prevent carryover. Information-dependent mass-spectrometer experiment included 1 survey MS1 scan followed by 50 dependent MS2 scans. MS1 acquisition parameters were as follows: mass range for MS2 analysis was 300–1250 m/z, signal accumulation time was 250 ms. Ions for MS2 analysis were selected on the basis of intensity with the threshold of 200 cps and the charge state from 2 to 5. MS2 acquisition parameters were as follows: resolution of quadrupole was set to UNIT (0.7 Da), measurement mass range was 200–1800 m/z, signal accumulation time was 50 ms for each parent ion. Collision-activated dissociation was performed with nitrogen gas with collision energy ramping from 25 to 55 V within the signal accumulation time of 50 ms. Analyzed parent ions were sent to dynamic exclusion list for 15 sec in order to get an MS2 spectra at the chromatographic peak apex. β-Galactosidase tryptic solution (20 fmol) was run with 15 min gradient (5-25% of buffer B) every 2 samples and between sample sets to calibrate the mass-spectrometer and to control the overall system performance, stability and reproducibility.
LC-MS/MS Data Analysis
Raw LC-MS/MS data were converted to .mgf peaklists with the ProteinPilot (version 4.5). For this procedure we run ProteinPilot in identification mode with the following parameters: Cys alkylation by iodoacetamide, trypsin digestion, TripleTOF 5600 instrument, thorough ID search with detected protein threshold 95.0% against UniProt human Protein knowledgebase (version 2013_03, with 150600 entries). For thorough protein identification the generated peaklists were searched with the MASCOT (version 2.2.07) and the X! Tandem (CYCLONE, 2013.2.01) search engine against the UniProt human Protein knowledgebase (version 2013_03) with concatenated reverse decoy dataset (with 301200 entries altogether). Precursor and fragment mass tolerance were set at 20 ppm and 0.04 Da, respectively. Database searching parameters included the following: tryptic digestion with 1 possible miss cleavage, static modifications for carbamidomethyl (C) and dynamic/flexible modifications for oxidation (M). For X! Tandem we also selected parameters that allowed a quick check for protein N-terminal residue acetylation, peptide N-terminal glutamine ammonia loss or peptide N-terminal glutamic acid water loss. Result files were submitted to the Scaffold 4 software (version 4.0.7) for validation and meta analysis. We used LFDR scoring algorithm with standard experiment wide protein grouping. For the evaluation of peptide and protein hits, a false discovery rate of 5% was selected for both. False positive identifications were based on reverse database analysis. Lists of identified proteins from the apoEVs and control samples were analyzed using the STRING online service with KEGG, GO Cell Component, GO Biological Process, and NCI databases.
Labeling of apoEVs with Alexa Fluor 488 5-TFP Ester
EVs pellet was thoroughly resuspended in 30 ml of PBS, solution was centrifugated at 120 000 g for 70 min, 4 °C. EVs pellet was resuspended in 1 mL of 0.1 M s odium bicarbonate buffer (pH 9.0) and 100 μl of 10 mg/ml Alexa Fluor 488 5-TFP Ester solution in DMSO was added. Reaction was incubated for 1 hr at room temperature on shaker. Next 30 ml of PBS was added and solution was centrifugated at 120,000 g for 70 min, 4 °C. EVs pellet was thoroughly resuspended in 30 ml cultural media, solution was centrifugated at 120,000 g for 70 min, 4 °C. EVs pellet was resuspen ded in 15 μl of PBS and used for in vivo injection or in vitro experiments.
In vivo Bioluminescent Imaging
To monitor tumor growth in living animals, GBM spheres were transduced with lentiviral particles (pHAGE PGK-GFP-IRES-LUC-W) for co-expressesion of GFP and luciferase, and then GFP-expressing cells were sorted by FACS. GBM spheres expressing luciferase were intracranially transplanted into immuno-compromised mice. To examine the tumor growth, animals were administrated intraperitoneally with 2.5 mg/100ul solution of XenoLight D-luciferin (PerkinElmer) and anesthetized with isoflurane for the imaging analysis. The tumor luciferase images were captured by using an IVIS 100 imaging system (PerkinElmer).
RNA Sequencing and Analysis of Alternative Splicing
Analysis of alternative splicing events (ASE) was performed as described previously (Zhou et al., 2014). Briefly, complementary DNA (cDNA) libraries for paired end sequencing were prepared using TruSeq Stranded mRNA-Seq Library Preparation Kit (Illumina) according to the manufacturer’s protocol. Samples were sequenced with an llumina HiSeq 2000 system (Illumina) and 100 bp paired-end reads were generated. The raw sequence data and processed data have been submitted to Gene Expression Omnibus with accession number PRJNA344648. Sequence reads in fastq format were imported into a local instance of galaxy (galaxy.uabgrid.uab.edu). Quality control was performed by trimming each end of reads using read quality score, vendor sequencing adaptors were removed, and then reads were aligned against human genome (hg19) using STAR aligner algorithm. Resulting bam files were used for analysis of alternative splicing events after removing PCR duplicates using samtools (http://www.htslib.org). Analysis was performed by using AS Detector software (http://www.novelbio.com/asd/ASD.html). ASE were sorted according to adjusted p value and top 10% of ASE were used for comparison between datasets. Events related to alternative mRNA splicing (alternative 5′ splice sites, alternative 3′ splice sites, mutually exclusive exons, cassette exons, multy cassette exons and retained introns) that were detected in all 3 datasets were analyzed using the STRING online service with GO Biological Process database.
Plasmid Construction
The DNA fragment encoding PRPF3 was amplified from 157GBM cDNA by PCR technique using primer pair EcoRI-PRPF3/PRPF3-SalI and cloned into EcoRI/SalI sites of pTagRFP-C plasmid (Evrogen) to generate pTagRFP-PRPF3 plasmid. The DNA fragment encoding Coilin was amplified from 157GBM cDNA by PCR technique using primer pair KpnI-Coilin/Coilin-BamHI and cloned into KpnI/BamHI sites of pTagGFP2-C plasmid (Evrogen) to generate pTagGFP2-Coilin plasmid. The DNA fragment encoding HNRNPU was amplified from 157GBM cDNA by PCR technique using primer pair EcoRI-HNRNPU/HNRNPU-SalI and cloned into EcoRI/SalI sites of pEYFP-CFP plasmid (Pavlyukov et al., 2012) to generate pEYFP-HNRNPU-CFP plasmid. The DNA fragment encoding RBM11 was amplified from 157GBM cDNA by PCR technique using primer pair SacI-RBM11/RBM11-BamHI and cloned into SacI/BamHI sites of pTagGFP2-C plasmid (Evrogen) to generate pTagGFP2-RBM11 plasmid. RBM11(fs) mutant was generated by not-in frame insertion of the RBM11 encoding sequence into pTagGFP2-C plasmid, the DNA encoding RBM11 was amplified from pTagGFP2-RBM11 plasmid by PCR technique using primer pair SacI-RBM11fs/RBM11-BamHI and cloned into SacI/BamHI sites of pTagGFP2-C plasmid. To generate lentiviral plasmids encoding GFP-RBM11 or GFP as a control, pTagGFP2-RBM11 or pTagGFP2-C plasmids were digested with NheI/BamHI and resulting DNA fragments was cloned into NheI/BamHI sites of pCDH‐EF1‐MCS‐IRES‐Puro plasmid (System Biosciences). Obtained plasmids were designated as pCDH-GFP-RBM11 and pCDH-GFP respectively. The DNA fragment encoding GFP-NLS was amplified from pTagGFP2-C plasmid (Evrogen) by PCR technique using primer pair GFP_for/GFP-NLS_rev and cloned into NheI/EcoRI sites of pTagGFP2-C plasmid (Evrogen) to generate pTagGFP-NLS plasmid. In all cases, the absence of unwanted mutations in the inserts and vector-insert boundaries was verified by sequencing.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as mean ± SD. The number of replicates for each experiment was stated in the figure legend. For data with replicates below 10, we assumed normal distribution on the basis of the appearance of the data and analyzed with Student’s tailed t-test. The statistical significance of Kaplan–Meier survival plot was determined by log-rank analysis. Statistical analysis was performed by Prism 6 (Graphpad Software), unless mentioned otherwise in figure legend. p < 0.05 was considered as statistically significant. No statistical method was used to predetermine sample size, and sample size (number of mice) was determined on the basis of our previous studies (Wang et al., 2017; Mao et al., 2013). No blinding or randomization was used. No samples, mice or data points were excluded from the reported analyses. Unless otherwise noted, all western blots, FACS data and microscopy images are representative of three biologically independent experiments.
DATA AND SOFTWARE AVAILABILITY
RNA sequencing data have been deposited in the Gene Expression Omnibus (GEO) under the accession code PRJNA344648. Results of RNAseq data analysis are present in Table S2. Proteomic data are in Table S1. Microarray data could be found in the NCBI Gene Expression Omnibus under the accession code GSE67089 and in Tables S3 and S4.
Supplementary Material
Table S1 (Related to Figure 3). Results of LC-MS/MS protein identification in EVs.
Table S2 (Related to Figure 5). List of alternative splicing events identified by RNA sequencing.
Table S3 (Related to Figure 6). Expression of splicing factors determined by microarray.
Table S4 (Related to Figure 7). Effect of RBM11 overexpression and knockdown on gene expression.
Table S5 (Related to STAR Methods). Characterization of glioma cell lines used in this study.
Table S6 (Related to STAR Methods). List of oligonucleotides and their sequences used in this study.
SIGNIFICANCE.
Alterations in mRNA splicing have been associated with cancer progression. Here we unexpectedly identified that splicing changes in rapidly proliferating GBM cells are induced in part by exogenous spliceosomal components transferred via EVs from neighboring apoptotic cancer cells. Uptake of apoEVs results in a switch of mRNA splicing toward the expression of multiple protein isoforms in recipient cells. Clinically, the elevated expression of these isoforms in GBM is associated with a worse patient prognosis. Taken together, these findings reveal a role of spliceosomal proteins in intercellular communication between apoptotic and surviving tumor cells and suggest that targeting of apoEV-mediated splicing changes may be exploited to decrease tumor aggressiveness that would eventually improve cancer patient outcome.
HIGHLIGHTS.
apoEVs promote more aggressive and therapy resistant phenotype of GBM
apoEVs are specifically enriched with spliceosomal proteins and snRNAs
apoEVs affect mRNA splicing in recipient GBM cells
RBM11 is partly responsible for apoEVs mediated phenotypic changes of recipient cells
Pavlyukov et al. show that apoptotic GBM cells secrete vesicles enriched with components of spliceosomes to alter RNA splicing in surviving tumor cells and promote their aggressiveness. They identify RBM11 as one such factor that switches MDM4 and cyclinD1 towards the more oncogenic isoforms in recipient cells.
Acknowledgments
We thank all our respective lab mates for their helpful discussion. This work was supported by NIH grants R01NS083767, R01NS087913, R01CA183991, R01CA201402 (I.N.), the Russian Foundation for Basic Research grants 16-04-01414 (V.O.S.) and 16-04-01209, 17-29-06056 (M.S.P.), the Russian Science Foundation grants 17-75-20205 (V.O.S.; for the LС-MS/MS analysis) and 16-14-10335 (M.I.S.; for the in vitro experiments), the Scholarships of the President of the Russian Federation SP-4811.2018.4 (V.O.S.) and by grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute HI14C3418 (D.N.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
M.S.P., D-H.N. and I.N. designed the experiments; M.S.P., S.B., H.Y., S.Z., S.Y., H.A.A., M.M., J.S., J.W., S.K., V.O.S., E.G.E., N.B.P., M.I.S., Y.L., Y.J.S. and N.V.A. performed experiments; M.S.P., D.C., K.A., S-H.K., L.J.L., G.P.A., G.V. and I.N. analyzed data; M.S.P., A.M. and I.N. wrote manuscript.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
References
- Arscott WT, Tandle AT, Zhao S, Shabason JE, Gordon IK, Schlaff CD, Zhang G, Tofilon PJ, Camphausen KA. Ionizing radiation and glioblastoma exosomes: implications in tumor biology and cell migration. Transl Oncol. 2013;6:638–648. doi: 10.1593/tlo.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–927. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- Busà R, Geremia R, Sette C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010;38:3005–3018. doi: 10.1093/nar/gkq004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Hendriks WT, MacGillavry HD, Alvarez J, van Minnen J. Schwann cell to axon transfer of ribosomes: toward a novel understanding of the role of glia in the nervous system. J Neurosci. 2008;28:1024–11029. doi: 10.1523/JNEUROSCI.2429-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Midha R, Cisterna BA, Grochmal J, Shakhbazau A, Hendriks WT, Van Minnen J. Morphological evidence for a transport of ribosomes from Schwann cells to regenerating axons. Glia. 2011;59:1529–1539. doi: 10.1002/glia.21196. [DOI] [PubMed] [Google Scholar]
- Crescitelli R, Lässer C, Szabó TG, Kittel A, Eldh M, Dianzani I, Buzás EI, Lötvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles. 2013;2 doi: 10.3402/jev.v2i0.20677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieudé M, Bell C, Turgeon J, Beillevaire D, Pomerleau L, Yang B, Hamelin K, Qi S, Pallet N, Béland C, et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med. 2015;7:318ra200. doi: 10.1126/scitranslmed.aac9816. [DOI] [PubMed] [Google Scholar]
- Fader CM, Sánchez D, Furlán M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Perez D, Higuita-Castro N, Denning L, DeJesus J, Dahl K, Sarkar A, Hansford DJ. Microfabricated mimics of in vivo structural cues for the study of guided tumor cell migration. Lab Chip. 2012;12:4424–4432. doi: 10.1039/c2lc40726d. [DOI] [PubMed] [Google Scholar]
- Göhring F, Schwab BL, Nicotera P, Leist M, Fackelmayer FO. The novel SAR-binding domain of scaffold attachment factor A (SAF-A) is a target in apoptotic nuclear breakdown. EMBO J. 1997;16:7361–7371. doi: 10.1093/emboj/16.24.7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, Holland EC. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci U S A. 2014;111:5248–5253. doi: 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–866. doi: 10.1038/nm.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Feng Y. Exosomes Derived From Hypoxic Colorectal Cancer Cells Promote Angiogenesis Through Wnt4-Induced β-Catenin Signaling in Endothelial Cells. Oncol Res. 2017;25:651–661. doi: 10.3727/096504016X14752792816791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelonek K, Wojakowska A, Marczak L, Muer A, Tinhofer-Keilholz I, Lysek-Gladysinska M, Widlak P, Pietrowska M. Ionizing radiation affects protein composition of exosomes secreted in vitro from head and neck squamous cell carcinoma. Acta Biochim Pol. 2015;62:265–72. doi: 10.18388/abp.2015_970. [DOI] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakowski M, Buller B, Zheng X, Lu Y, Rogers T, Osobamiro O, Shu W, Jiang F, Chopp M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013;335:201–204. doi: 10.1016/j.canlet.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer AK, Takeuchi K, Quinlan MP. Identification of genes involved in epithelial-mesenchymal transition and tumor progression. Oncogene. 2001;20:6679–6688. doi: 10.1038/sj.onc.1204872. [DOI] [PubMed] [Google Scholar]
- Klein M, Vignaud JM, Hennequin V, Toussaint B, Bresler L, Plénat F, Leclère J, Duprez A, Weryha G. Increased expression of the vascular endothelial growth factor is a pejorative prognosis marker in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2001;86:656–658. doi: 10.1210/jcem.86.2.7226. [DOI] [PubMed] [Google Scholar]
- Lee JK, Park SR, Jung BK, Jeon YK, Lee YS, Kim MK, Kim YG, Jang JY, Kim CW. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One. 2013;8:e84256. doi: 10.1371/journal.pone.0084256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, Wang M, Soutoglou E, Knudsen ES, Pestell RG. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res. 2010;70:8802–8811. doi: 10.1158/0008-5472.CAN-10-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini F, Di Conza G, Moretti F. MDM4 (MDMX) and its Transcript Variants. Curr Genomics. 2009;10:42–50. doi: 10.2174/138920209787581280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller L, Hong CS, Stolz DB, Watkins SC, Whiteside TL. Isolation of biologically-active exosomes from human plasma. J Immunol Methods. 2014;411:55–65. doi: 10.1016/j.jim.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller L, Mitsuhashi M, Simms P, Gooding W, Whiteside TL. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci Rep. 2016;6:20254. doi: 10.1038/srep20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutschelknaus L, Azimzadeh O, Heider T, Winkler K, Vetter M, Kell R, Tapio S, Merl-Pham J, Huber SM, Edalat L, et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci Rep. 2017;7:12423. doi: 10.1038/s41598-017-12403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlyukov MS, Antipova NV, Balashova MV, Vinogradova TV, Kopantzev EP, Shakhparonov MI. Survivin monomer plays an essential role in apoptosis regulation. J Biol Chem. 2011;286:23296–23307. doi: 10.1074/jbc.M111.237586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlyukov MS, Antipova NV, Balashova MV, Shakhparonov MI. Detection of Transglutaminase 2 conformational changes in living cell. Biochem Biophys Res Commun. 2012;421:773–779. doi: 10.1016/j.bbrc.2012.04.082. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrotti S, Busà R, Compagnucci C, Sette C. The RNA recognition motif protein RBM11 is a novel tissue-specific splicing regulator. Nucleic Acids Res. 2012;40:1021–1032. doi: 10.1093/nar/gkr819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Prabhu A, Sarcar B, Miller CR, Kim SH, Nakano I, Forsyth P, Chinnaiyan P. Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro Oncol. 2015;17:1220–1230. doi: 10.1093/neuonc/nou369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradella D, Naro C, Sette C, Ghigna C. EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression. Mol Cancer. 2017;16:8. doi: 10.1186/s12943-016-0579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 2011;25:373–384. doi: 10.1101/gad.2004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shender VO, Pavlyukov MS, Ziganshin RH, Arapidi GP, Kovalchuk SI, Anikanov NA, Altukhov IA, Alexeev DG, Butenko IO, Shavarda AL, et al. Proteome-metabolome profiling of ovarian cancer ascites reveals novel components involved in intercellular communication. Mol Cell Proteomics. 2014;13:3558–3571. doi: 10.1074/mcp.M114.041194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishova KV, Khodarovich YM, Lavrentyeva EA, Zatsepina OV. High-resolution microscopy of active ribosomal genes and key members of the rRNA processing machinery inside nucleolus-like bodies of fully-grown mouse oocytes. Exp Cell Res. 2015;337:208–218. doi: 10.1016/j.yexcr.2015.07.024. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeo K, Kawai T, Nishida K, Masuda K, Teshima-Kondo S, Tanahashi T, Rokutan K. Oxidative stress-induced alternative splicing of transformer 2beta (SFRS10) and CD44 pre-mRNAs in gastric epithelial cells. Am J Physiol Cell Physiol. 2009;297:330–338. doi: 10.1152/ajpcell.00009.2009. [DOI] [PubMed] [Google Scholar]
- Venables JP, Brosseau JP, Gadea G, Klinck R, Prinos P, Beaulieu JF, Lapointe E, Durand M, Thibault P, Tremblay K, et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol. 2013;33:396–405. doi: 10.1128/MCB.01174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cheng P, Pavlyukov MS, Yu H, Zhang Z, Kim SH, Minata M, Mohyeldin A, Xie W, Chen D, et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J Clin Invest. 2017;127:3075–3089. doi: 10.1172/JCI89092. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang Y, Chen D, Qian H, Tsai YS, Shao S, Liu Q, Dominguez D, Wang Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell. 2014;26:374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Park JW, Bebee TW, Warzecha CC, Guo Y, Shang X, Xing Y, Carstens RP. Determination of a Comprehensive Alternative Splicing Regulatory Network and Combinatorial Regulation by Key Factors during the Epithelial-to-Mesenchymal Transition. Mol Cell Biol. 2016;36:1704–1719. doi: 10.1128/MCB.00019-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wu W, Li H, Cheng Y, Wei N, Zong J, Feng X, Xie Z, Chen D, Manley JL, et al. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 2014;42:4019–4030. doi: 10.1093/nar/gkt1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Related to Figure 3). Results of LC-MS/MS protein identification in EVs.
Table S2 (Related to Figure 5). List of alternative splicing events identified by RNA sequencing.
Table S3 (Related to Figure 6). Expression of splicing factors determined by microarray.
Table S4 (Related to Figure 7). Effect of RBM11 overexpression and knockdown on gene expression.
Table S5 (Related to STAR Methods). Characterization of glioma cell lines used in this study.
Table S6 (Related to STAR Methods). List of oligonucleotides and their sequences used in this study.