

 Substituent exchange reactions of silylium ions can be steered in opposite directions. The judicious choice of the hydrosilane and the counteranion enables the selective formation of either triaryl- or trialkylsilylium ions.
Substituent exchange reactions of silylium ions can be steered in opposite directions. The judicious choice of the hydrosilane and the counteranion enables the selective formation of either triaryl- or trialkylsilylium ions.
Abstract
An in-depth experimental and theoretical study of the substituent exchange reaction of silylium ions is presented. Apart from the substitution pattern at the silicon atom, the selectivity of this process is predominantly influenced by the counteranion, which is introduced with the trityl salt in the silylium ion generation. In contrast to Müller's protocol for the synthesis of triarylsilylium ions under kinetic control, the use of Reed's carborane anions leads to contact ion pairs, allowing selective formation of trialkylsilylium ions under thermodynamic control. DFT calculations finally revealed an unexpected mechanism for the rate-determining alkyl exchange step, which is initiated by an unusual 1,2-silyl migration in the intermediate ipso-disilylated arenium ion. The resulting ortho-disilylated arenium ion can then undergo an alkyl transfer via a low-barrier five-centered transition state.
Introduction
Silylium ions (R3Si+) have recently emerged as useful and versatile catalysts for synthetically attractive transformations.1,2 The most commonly used approach to generate silylium ions is the Bartlett–Condon–Schneider reaction,3 that is the silicon-to-carbon hydride transfer from a hydrosilane to the trityl cation (Ph3C+) paired with a weakly coordinating counteranion.4,5 However, substituent redistribution of the hydrosilane starting material can occur under these highly Lewis acidic reaction conditions, leading to undesired mixtures of various silicon compounds.6–8 Hence, hydrosilanes containing three identical substituents, e.g. Et3SiH or iPr3SiH, are usually employed in this reaction.9 Conversely, Müller and co-workers have turned this unselective process into a useful synthetic route to triarylsilylium ions (Scheme 1, top).10 When sterically demanding methyl(diaryl)silanes MeAr2SiH are used in the hydride abstraction with Ph3C+[B(C6F5)4]–, the formation of otherwise difficult to prepare triarylsilylium ions Ar3Si+[B(C6F5)4]– is observed.11 Notably, the useof less bulky hydrosilanes such as MePh2SiH or Me(o-Tol)2SiH does not give triarylsilylium ions but mixtures of different silicon cations.12
Scheme 1. Divergence in the generation of silylium ions by substituent redistribution (x + y + z = 4).

Herein, we report that treatment of hydrosilanes of type Me2RSiH (R = aryl, benzyl) with Reed's carborane-based trityl salt Ph3C+[CHB11H5Br6]– (ref. 13) results in substituent exchange reactions selectively forming the elusive trimethylsilylium ion Me3Si+[CHB11H5Br6]– (Scheme 1, bottom). This method thus complements Müller's approach and offers a practical route to Me3Si+, avoiding the use of gaseous and highly flammable Me3SiH.14 A systematic experimental and computational investigation was performed to gain a full mechanistic picture of this phenomenon. DFT calculations revealed an unexpected mechanism and suggested an active role of the carborane counteranion in the outcome of these reactions.
Results and discussion
Generation of the trimethylsilylium ion by substituent redistribution
When a mixture of Me2PhSiH and Ph3C+[CHB11H5Br6]– in toluene was stirred overnight at room temperature, a white suspension was obtained. The solid was collected by filtration, washed with n-pentane, and dissolved in o-Cl2C6D4 for NMR spectroscopic analysis. Unexpectedly, only a singlet at 0.83 ppm was detected in the 1H NMR spectrum, while no aromatic resonances except for those of the deuterated solvent were observed. The low-field 29Si NMR chemical shift of 93 ppm in the corresponding 1H/29Si HMQC spectrum, which is characteristic of trialkylsilylium ions, indicated clean formation of Me3Si+[CHB11H5Br6]– (Fig. 1). The structural integrity of the carborane counteranion was confirmed by 11B NMR spectroscopy.
Fig. 1. 1H/29Si HMQC NMR spectrum (500/99 MHz, o-Cl2C6D4, 298 K, optimized for J = 7 Hz) of Me3Si+[CHB11H5Br6]– from the reaction of Me2PhSiH with Ph3C+[CHB11H5Br6]–.

Unambiguous evidence for the structure of Me3Si+[CHB11H5Br6]– was eventually provided by its crystallographic characterization (Fig. 2).15 Single crystals suitable for X-ray diffraction analysis were obtained by vapor diffusion with n-hexane from a solution of the silylium salt in o-F2C6H4 at room temperature. In accordance with reported molecular structures of silylium carboranes,16 one bromine atom at the pentagonal belt of the icosahedral anion is bound to the silicon cation. Both the Si–Br bond distance of 2.435(6) Å and the sum of all C–Si–C bond angles of 346.3(6)° are comparable to the larger Et3Si+[CHB11H5Br6]–.
Fig. 2. Molecular structure of Me3Si+[CHB11H5Br6]– (thermal ellipsoids at the 50% probability level; H atoms omitted for clarity).

In contrast to the clean formation of Me3Si+, the non-polar n-pentane filtrate contained several tri- and tetraorganosilanes, such as Ph4Si, MePh3Si, Ph3SiH, Me2Ph2Si, MePh2SiH, Me3PhSi, and Me2PhSiH, as verified by GC-MS analysis. Since silylium ions are known to promote substituent redistribution,8 this result did not come as a surprise but raised the question why Me3Si+ was selectively formed in this reaction mixture, whereas Müller's conditions cleanly afford sterically congested triarylsilylium ions.10
Influence of the substituent pattern at the silicon atom on the selectivity of the substituent redistribution reaction
To understand the differences between Müller's protocol10 and our findings, we systematically studied the hydride transfer reaction of various hydrosilanes of type MeAr2SiH and Me2ArSiH using trityl salts Ph3C+[B(C6F5)4]– and Ph3C+[CHB11H5Br6]– (Table 1). Depending on the counteranion, slightly modified procedures were applied for the generation of the silicon cations (see the ESI† for details). For all reactions, an excess of hydrosilane (4 equiv.) was used, thereby excluding any influence of stoichiometry on the product formation. In accordance with Müller's report, bulky methyl(diaryl)silane Me(C6Me5)2SiH was converted to the corresponding triarylsilylium ion, regardless of which counteranion was used (entries 1 and 2). In contrast, hydride abstraction from sterically less hindered MePh2SiH with Ph3C+[B(C6F5)4]– led to a complex reaction mixture as a result of anion decomposition (entry 3).12,17 The use of the carborane counteranion [CHB11H5Br6]– furnished the unscrambled silylium ion MePh2Si+[CHB11H5Br6]–, as confirmed by X-ray diffraction analysis (entry 4; see the ESI† for the molecular structure of MePh2Si+[CHB11H5Br6]–).15 However, the formation of the MePh2Si+ cation was accompanied by a substantial amount of a second silylium ion, which was found to be the Me2PhSi+ cation.18 Notably, longer reaction times (7 days) or elevated temperatures (50 °C for 72 h) did not significantly change the product ratio of ∼79 : 21 (not shown). In all cases, the generation of Me3Si+ was not observed. We then turned our attention to dimethyl(aryl)silanes (entries 5–8). The reaction of Me2PhSiH with Ph3C+[B(C6F5)4]– again resulted in decomposition of the borate counteranion (entry 5).17 Conversely, treatment of Me2PhSiH with trityl carborane Ph3C+[CHB11H5Br6]– exclusively afforded silylium salt Me3Si+[CHB11H5Br6]– without detectable formation of MePh2Si+ or Me2PhSi+ (entry 6). Strikingly, hydride abstraction from sterically more demanding Me2(C6Me5)SiH led to the corresponding triarylsilylium ion in the presence of the borate counteranion (entry 7), while substituent redistribution into the ‘opposite direction’ was induced by the carborane anion, now affording Me3Si+[CHB11H5Br6]– (entry 8).19 However, heating of the reaction at 50 °C for 72 h was necessary.
Table 1. Silylium ion generation by substituent redistribution: effect of the hydrosilane and counteranion (Si = triorganosilyl).
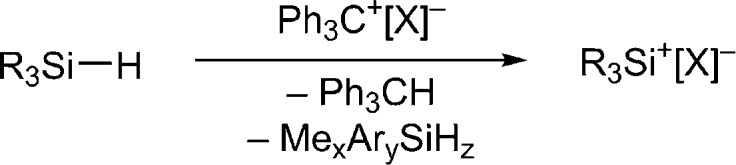
| ||||
| Entry a | Si–H (4 equiv.) | [X]– | Si+ | δ(29Si) b [ppm] |
| 1 | Me(C6Me5)2SiH | [B(C6F5)4]– | (C6Me5)3Si+ | 217 |
| 2 | Me(C6Me5)2SiH | [CHB11H5Br6]– | (C6Me5)3Si+ | 217 |
| 3 | MePh2SiH | [B(C6F5)4]– | — c | — |
| 4 | MePh2SiH | [CHB11H5Br6]– | MePh2Si+/Me2PhSi+ d | 57/76 |
| 5 | Me2PhSiH | [B(C6F5)4]– | — c | — |
| 6 | Me2PhSiH | [CHB11H5Br6]– | Me3Si+ | 93 |
| 7 | Me2(C6Me5)SiH | [B(C6F5)4]– | (C6Me5)3Si+ | 217 |
| 8 e | Me2(C6Me5)SiH | [CHB11H5Br6]– | Me3Si+ | 93 |
aAll reactions were performed according to General Procedure (GP) 1 for X– = [B(C6F5)4]– (C6D6, room temperature, 60 min) or GP 2 for X– = [CHB11H5Br6]– (toluene, room temperature, 18–24 h). See the ESI for details.
bMeasured in o-Cl2C6D4.
cA complex mixture was obtained as a result of counteranion decomposition.17
dRatio of 79 : 21 determined by 1H NMR spectroscopy.
eReaction performed at 50 °C for 72 h.
Overall, these results indicate that hydride abstraction from hydrosilanes of type Me2ArSiH with a carborane-based trityl salt tends to form the trimethylsilylium ion, whereas hydrosilanes of type MeAr2SiH with a bulky aryl substituent favor triarylsilylium ion generation.
Mechanism of the substituent redistribution reaction with Me2PhSiH
To gain insight into the reaction mechanism and to understand why the treatment of Me2PhSiH with Ph3C+[CHB11H5Br6]– exclusively gives Me3Si+[CHB11H5Br6]–, we constructed a complete reaction energy profile using DFT calculations at the M06/cc-pVTZ(-f)//6-31G** level of theory (Fig. 3; see the ESI† for details of the computational method).20 The hydride abstraction from Me2PhSiH with Ph3C+[CHB11H5Br6]– was found to have a barrier of 15.5 kcal mol–1 and is therefore expected to occur rapidly at room temperature (not shown). In the condensed phase, the resulting silylium ion Me2PhSi+ (6A), which is located at a relative free energy of 6.5 kcal mol–1, is stabilized through coordination by the solvent, another hydrosilane molecule, or by the counteranion (see the ESI† for a comparison of the association energies).8e,21 Coordination of one of the bromine atoms of the carborane counteranion to the silicon cation results in the highest binding energy, and the resulting ion pair 6A′ is predicted to be at a relative free energy of –24.1 kcal mol–1. Silylium ion 6A can also interact with another equivalent of Me2PhSiH to form hydride-bridged adduct 7A,21 located at –6.5 kcal mol–1. Note that these energies are not adjusted for the different concentrations of the components and assume normal conditions. Given that Me2PhSiH (1A) is present in excess, these normal energies suggest that adduct 7A will be encountered easily in significant quantities.
Fig. 3. Energy (kcal mol–1) profile of the substituent redistribution in the reaction of Me2PhSiH (1A) with Ph3C+[CHB11H5Br6]– (2A). The energies are relative to the starting materials 1A and 2A.

Hydride-bridged adduct ion 7A can undergo a phenyl group transfer to arrive at phenyl-bridged adduct 8A7c,8b,22 via the four-centered transition state 7A-TS, associated with a barrier of 13.4 kcal mol–1. Surprisingly, the subsequent methyl group transfer does not proceed via another typical four-membered transition state.23 Instead, our calculations suggest that 1,2-migration of the silicon group in 8A occurs via the low barrier transition state 8A-TS, leading to ortho-disilylated arenium ion 9A. This seemingly unfavorable intermediate is only 4.1 kcal mol–1 higher in energy than arenium ion 8A. Finally, 9A facilitates the exchange of one methyl group, passing through five-centered transition state 9A-TS with an overall barrier of 24.3 kcal mol–1 relative to 7A. This energetically most demanding reaction step forms methonium ion 10A, which is metastable and rapidly rearranges to hydride-bridged adduct 11Avia low barrier transition state 10A-TS. The hydrosilane-stabilized silylium ions 7A and 11A are almost isoenergetic (ΔG = 0.4 kcal mol–1), suggesting that both structures coexist in equilibrium. The formal dissociation of 11A gives either Me3Si+ or MePh2Si+, the former being calculated to be 2.8 kcal mol–1 higher in energy. However, coordination by the carborane anion changes the energy landscape decisively, as ion pair formation reverses the energy ordering. Me3Si+[CHB11H5Br6]– (12A′′), which is located at –28.5 kcal mol–1, is 2.9 kcal mol–1 lower in energy than MePh2Si+[CHB11H5Br6]– (13A′) and also 4.5 kcal mol–1 more stable than Me2PhSi+[CHB11H5Br6]– (6A′), thus predicting the silylium salt 12A′′ as the major product of the substituent redistribution reaction.
It should be noted that silylium ions are significantly more stabilized by coordination of the carborane counteranion than by formation of solvent adducts such as R3Si(toluene)+[CHB11H5Br6]–. Moreover, the energy differences between these arenium ions are small, predicting a mixture of different silylium ions in the absence of the carborane counteranion (see the ESI† for details).24 This result was supported by independent control experiments (Scheme 2). The hydride abstraction from Me2PhSiH with borate-based trityl salt Ph3C+[B(C6F5)4]– was repeated but stopped after stirring for 10 min in toluene (cf.Table 1, entry 5). NMR spectroscopic analysis of the polar phase in o-Cl2C6D4 revealed the formation of a mixture of Me3Si+[B(C6F5)4]– and Me2PhSi+[B(C6F5)4]– in a ratio of ∼51 : 49 along with small amounts of byproducts arising from counteranion decomposition. In contrast, stopping the reaction of Me2PhSiH with Ph3C+[CHB11H5Br6]– after stirring for 10 min in toluene furnished Me3Si+[CHB11H5Br6]– as the major product along with only small amounts of unscrambled Me2PhSi+[CHB11H5Br6]– (ratio ∼84 : 16). In both reactions, full conversion of the trityl salt was observed.
Scheme 2. Influence of the counteranion on the selectivity of the trimethylsilylium ion formation.

As shown in Fig. 4, the silylium ions can be bound either to the apical or one of the equatorial bromine atoms of the carborane counteranion, with a slight preference of 1.1 kcal mol–1 for the apical position in Me3Si+[CHB11H5Br6]– (12A′′). This result is in contrast to the molecular structure in the solid state, which shows the equatorial isomer (cf.Fig. 2). We speculate that either packing effects or a statistical preference for the equatorial isomer is the reason for this discrepancy. Notably, the equatorial isomer 12A′ is still 1.8 kcal mol–1 lower in energy than the equatorial isomer of MePh2Si+[CHB11H5Br6]– (13A′). The higher ion pairing energy in 12A′ can be ascribed to the low steric demand of Me3Si+, leading to a closer carborane coordination and to attractive van der Waals interactions between the methyl moieties and the carborane anion. Especially in the apical position, the methyl functionality can interact with the highly polarizable bromine atoms. In contrast, the molecular fit of the sterically more demanding silylium ions Me2PhSi+ (6A) and MePh2Si+ (13A) with the carborane counteranion is less tight, and the ion pairing is therefore slightly less favorable. This trend is reflected in the corresponding Si–Br bond lengths of these silylium carborane salts, which were computed to be shortest in both isomers of Me3Si+[CHB11H5Br6]– (12A′ and 12A′′). Hence, this ion pair is the most stable silylium salt despite the lack of stabilizing phenyl groups. Both isomers of Me2PhSi+[CHB11H5Br6]– (6A′ and 6A′′) are higher in energy than the corresponding MePh2Si+[CHB11H5Br6]– (13A′ and 13A′′), indicating that the stabilization of these silylium carborane salts is determined by a delicate balance of electronic and steric effects. It should also be noted here that the DFT optimized structures for Me3Si+[CHB11H5Br6]– (12A′) and MePh2Si+[CHB11H5Br6]– (13A′) are in good agreement with the corresponding molecular structures obtained by X-ray diffraction analysis (see the ESI† for details).
Fig. 4. Computed apical and equatorial isomers of Me2PhSi+[CHB11H5Br6]– (top), Me3Si+[CHB11H5Br6]– (middle) and MePh2Si+[CHB11H5Br6]– (bottom). Si–Br bond lengths are given in Å and relative free energy differences (kcal mol–1) are shown in parentheses.

Mechanism of the substituent redistribution reaction with MePh2SiH
To understand why the reaction of MePh2SiH with Ph3C+[CHB11H5Br6]– does not furnish Me3Si+[CHB11H5Br6]–, we constructed again a complete energy profile employing DFT simulations (Fig. 5). The initial hydride transfer of the hydrosilane to the trityl cation has a calculated barrier of 14.3 kcal mol–1 (not shown), which is 1.2 kcal mol–1 lower in energy compared to the case of Me2PhSiH due to the slightly higher hydride donor strength of MePh2SiH (see Table S1 in the ESI† for details). The resulting silylium ion MePh2Si+ (6B) with a relative free energy of 0.8 kcal mol–1 is almost isoenergetic to the reactant state. Adduct formation with another equivalent of MePh2SiH affords hydrosilane-stabilized silylium ion 7B, which undergoes a phenyl/methyl exchange reaction following a very similar reactivity pattern as described above, leading to scrambled hydride-bridged adduct 11B. The transformation of 7B to 11Bvia intermediates 8B, 9B, and 10B is again reversible, since 7B and 11B have similar free energies (ΔG = 0.7 kcal mol–1). As before, the methyl group transfer via five-membered transition state 9B-TS shows the highest barrier, which is 24.2 kcal mol–1 relative to 7B. In this equilibrium, unscrambled MePh2Si+[CHB11H5Br6]– (6B′) with a relative free energy of –25.9 kcal mol–1 is predicted to be the major species, followed by scrambled Me2PhSi+[CHB11H5Br6]– (12B′) and Ph3Si+[CHB11H5Br6]– (13B′′), which are basically isoenergetic at –24.6 kcal mol–1 and –24.7 kcal mol–1, respectively. This finding is in good agreement with the experimental observation of unscrambled MePh2Si+[CHB11H5Br6]– being the main product of the reaction (cf.Table 1, entry 4).25
Fig. 5. Energy (kcal mol–1) profile of the substituent redistribution in the reaction of MePh2SiH (1A) with Ph3C+[CHB11H5Br6]– (2B). The energies are relative to the starting materials 1B and 2B.

Our calculations suggest that a subsequent methyl exchange reaction leading to Me3Si+ is unlikely (11B → 18B, gray energy profile in Fig. 5). The transition state for this methyl group transfer, 16B-TS, is located 26.7 kcal mol–1 relative to 11B, which is 1.8 kcal mol–1 higher in energy than the barrier of the backward reaction via transition state 9B-TS. Consequently, the reaction of MePh2SiH with Ph3C+[CHB11H5Br6]– stops at the above-mentioned mixture of silicon cations rather than undergoing exhaustive substituent redistribution to furnish low energy Me3Si+[CHB11H5Br6]–.
This kinetic inhibition was further proven by another mechanistic control experiment (Scheme 3). When a mixture of Ph3C+[CHB11H5Br6]– and MePh2SiH in toluene was stirred overnight at room temperature, a pale yellow suspension was obtained, which is characteristic of silylium ions with aromatic substituents (cf.Table 1, entry 4). Addition of less bulky Me2PhSiH to this mixture resulted in a quick decolorization and formation of a white suspension. NMR spectroscopic analysis of the solid now confirmed exclusive formation of Me3Si+[CHB11H5Br6]–.
Scheme 3. Probing the kinetic inhibition in the substituent redistribution reaction with MePh2SiH.

Scope of the substituent redistribution reaction
The hydride abstraction from various dialkyl(phenyl)silanes with Ph3C+[CHB11H5Br6]– finally revealed that the redistribution reaction is not restricted to methyl groups (Table 2). Although Et2PhSiH reacted much slower compared to Me2PhSiH, exclusive formation of trialkylsilylium ion Et3Si+[CHB11H5Br6]– was observed (entries 1 and 2). Employing more bulky iPr2PhSiH led to clean generation of unscrambled dialkyl(aryl)silylium ion iPr2PhSi+[CHB11H5Br6]–, as verified by X-ray crystallography (entry 3; see the ESI† for the molecular structure of iPr2PhSi+[CHB11H5Br6]–).15 These results are in accordance with our calculations, predicting high energy barriers for the transfer of bulky alkyl groups. Sterically even more shielded tBu2PhSiH then completely thwarted the hydride abstraction, and only the trityl salt was recovered from the reaction mixture (entry 4).
Table 2. Silylium ion generation from hydrosilanes of type R2PhSiH.
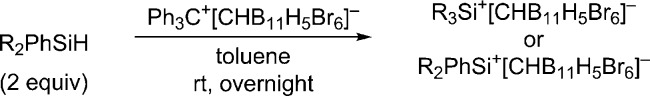
aAll reactions were performed according to GP 2. See the ESI for details.
bMeasured in o-Cl2C6D4.
cWith 4 equiv. of Et2PhSiH and 7 days reaction time.
dNo reaction; only Ph3C+[CHB11H5Br6]– was recovered.
To investigate whether the phenyl group in Me2PhSiH can be replaced by other ‘leaving groups’, we also tested a benzyl and an alkyl substituent in Me2RSiH (Table 3). As in the case of Me2PhSiH (entry 1), clean formation of Me3Si+[CHB11H5Br6]– was observed with Me2BnSiH (entry 2), showing that the phenyl group is not essential for the exchange process. In contrast, the bulky tert-butyl group in Me2tBuSiH completely prevented substituent redistribution, and silylium ion Me2tBuSi+[CHB11H5Br6]– was formed as the only product (entry 3). This result again demonstrates that the intermolecular substituent exchange reaction is sensitive towards sterically demanding alkyl groups (cf. entry 3 in Table 2).
Table 3. Silylium ion generation from hydrosilanes of type Me2RSiH.
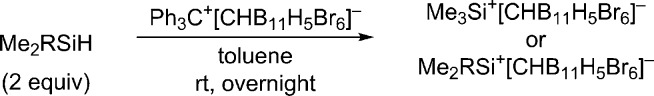
aAll reactions were performed according to GP 2. See the ESI for details.
bMeasured in o-Cl2C6D4.
Conclusion
It has been known for decades that silylium ions can undergo redistribution reactions of their substituents.8 The present combined experimental and detailed computational study finally provides a full mechanistic picture of this phenomenon. The mechanism involves a series of phenyl and alkyl exchange reactions, the latter being calculated to be the energetically most demanding steps. While the transfer of phenyl groups proceeds via common four-centered transition states, the corresponding alkyl exchange was found to pass through unusual five-membered transition states. These are accessible after 1,2-silyl migration at the stage of the intermediate disilylated arenium ions.
Additionally, our DFT calculations revealed that the silicon cations are significantly more stabilized by ion pair formation with the carborane counteranion (R3Si+[CHB11H5Br6]–) than by formation of toluenium (R3Si(toluene)+[CHB11H5Br6]–) or hydrosilane-stabilized silylium ions ([R3Si–H–SiR3]+[CHB11H5Br6]–). More importantly, purely aliphatic silylium carboranes with small substituents, i.e., methyl or ethyl groups, were found to be distinctly lower in energy than the corresponding mixed aliphatic/aromatic or purely aromatic silylium ion pairs as a result of stronger attractive interactions (ΔG ≥ 2.9 kcal mol–1 for R = Me). These energy differences account for the highly selective formation of Me3Si+[CHB11H5Br6]– and Et3Si+[CHB11H5Br6]– from the reaction of the corresponding hydrosilanes R2PhSiH (R = Me, Et) with Ph3C+[CHB11H5Br6]– under thermodynamic control.
The phenyl group in Me2PhSiH turned out to be replaceable by other ‘leaving groups’, such as a benzyl or even a sterically demanding C6Me5 group. However, two alkyl groups must be preinstalled in the hydrosilane starting material to steer the reaction towards formation of Me3Si+[CHB11H5Br6]–. In contrast, hydride abstraction from MePh2SiH with only one alkyl substituent leads to a mixture of different silylium ions, as exhaustive scrambling to Me3Si+ is kinetically inhibited. Exchanging the phenyl groups in MePh2SiH by 2,6-disubstituted aryl groups (e.g. C6Me5) eventually provides access to sterically congested triarylsilylium ions, as previously demonstrated by Müller and co-workers.10
These general trends provide a solid foundation for the mechanistic understanding of the substituent redistribution of silylium ions, thereby enabling the prediction of the outcome of these exchange reactions. Thus, this process can be used as a reliable synthetic route not only to triaryl- but also to trialkylsilylium ions by deliberate choice of the hydrosilane and counteranion of the trityl salt.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
L. O. thanks the Fonds der Chemischen Industrie for a predoctoral fellowship (2015–2017), and M. O. is indebted to the Einstein Foundation (Berlin) for an endowed professorship. B. P. and M.-H. B. are grateful for the financial support (IBS-R10-D1) from the Institute for Basic Science. DFT calculations were performed using the high performance computing facility located on the KAIST campus.
Footnotes
†Electronic supplementary information (ESI) available: Experimental details, characterization, spectroscopic and crystallographic data, DFT calculation methods, energy data, and the coordinates of the calculated geometries. CCDC 1818576, 1818581, and 1818582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01833b
References
- For general reviews of silylium ion chemistry, see: ; (a) Lee V. Y. and Sekiguchi A., in Organosilicon Compounds, ed. V. Y. Lee, Academic Press, Oxford, 2017, vol. 1, pp. 197–230. [Google Scholar]; (b) Müller T., in Structure and Bonding, ed. D. Scheschkewitz, Springer, Berlin, 2014, vol. 155, pp. 107–162. [Google Scholar]; (c) Müller T., in Science of Synthesis Knowledge Updates 2013/3, ed. M. Oestreich, Thieme, Stuttgart, 2013, pp. 1–42. [Google Scholar]
- For silylium ions in catalysis, see: ; (a) Klare H. F. T. ACS Catal. 2017;7:6999–7002. [Google Scholar]; (b) Schulz A., Villinger A. Angew. Chem., Int. Ed. 2012;51:4526–4528. doi: 10.1002/anie.201108320. [DOI] [PubMed] [Google Scholar]; (c) Klare H. F. T., Oestreich M. Dalton Trans. 2010;39:9176–9184. doi: 10.1039/c003097j. [DOI] [PubMed] [Google Scholar]
- (a) Bartlett P. D., Condon F. E., Schneider A. J. Am. Chem. Soc. 1944;66:1531–1539. [Google Scholar]; (b) Corey J. Y., West R. J. Am. Chem. Soc. 1963;85:2430–2433. [Google Scholar]; (c) Corey J. Y. J. Am. Chem. Soc. 1975;97:3237–3238. [Google Scholar]
- For recent reviews of weakly coordinating anions, see: ; (a) Riddlestone I. M., Kraft A., Schaefer J., Krossing I. Angew. Chem., Int. Ed. 2018;57 doi: 10.1002/anie.201710782. [DOI] [PubMed] [Google Scholar]; (b) Engesser T. A., Lichtenthaler M. R., Schleep M., Krossing I. Chem. Soc. Rev. 2016;45:789–899. doi: 10.1039/c5cs00672d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For further strategies to generate silylium ions, see: (allyl-leaving-group approach); (ring-opening protonolysis); (silylene protonation); (cyclohexadienyl-leaving-group approach); ; (a) Lambert J. B., Zhao Y., Wu H., Tse W. C., Kuhlmann B. J. Am. Chem. Soc. 1999;121:5001–5008. [Google Scholar]; (b) MacLachlan M. J., Bourke S. C., Lough A. J., Manners I. J. Am. Chem. Soc. 2000;122:2126–2127. [Google Scholar]; (c) Schäfer A., Reißmann M., Schäfer A., Schmidtmann M., Müller T. Chem.–Eur. J. 2014;20:9381–9386. doi: 10.1002/chem.201402874. [DOI] [PubMed] [Google Scholar]; (d) Simonneau A., Biberger T., Oestreich M. Organometallics. 2015;34:3927–3929. [Google Scholar]; (e) Chen Q.-A., Klare H. F. T., Oestreich M., J. Am. Chem. Soc., 2016, 138 , 7868 –7871 , (hydrosilane protonation) . [DOI] [PubMed] [Google Scholar]
- For a review of substituent redistribution reactions at silicon, see: Weyenberg D. R., Mahone L. G., Atwell W. H., Ann. N. Y. Acad. Sci., 1969, 159 , 38 –55 . [Google Scholar]
- For Lewis acid-catalyzed substituent redistribution reactions of hydrosilanes, see: ; (a) Speier J. L., Zimmerman R. E. J. Am. Chem. Soc. 1955;77:6395–6396. [Google Scholar]; (b) Khandelwal M., Wehmschulte R. J. Angew. Chem., Int. Ed. 2012;51:7323–7326. doi: 10.1002/anie.201201282. [DOI] [PubMed] [Google Scholar]; (c) Feigl A., Chiorescu I., Deller K., Heidsieck S. U. H., Buchner M. R., Karttunen V., Bockholt A., Genest A., Rösch N., Rieger B. Chem.–Eur. J. 2013;19:12526–12536. doi: 10.1002/chem.201203139. [DOI] [PubMed] [Google Scholar]; (d) Wehmschulte R. J., Saleh M., Powell D. R. Organometallics. 2013;32:6812–6819. [Google Scholar]; (e) Labbow R., Reiß F., Schulz A., Villinger A. Organometallics. 2014;33:3223–3226. [Google Scholar]; (f) Chen J., Chen E. Y.-X. Angew. Chem., Int. Ed. 2015;54:6842–6846. doi: 10.1002/anie.201502400. [DOI] [PubMed] [Google Scholar]; (g) Ma Y., Zhang L., Luo Y., Nishiura M., Hou Z. J. Am. Chem. Soc. 2017;139:12434–12437. doi: 10.1021/jacs.7b08053. [DOI] [PubMed] [Google Scholar]
- For substituent redistribution reactions of silicon cations, see: ; (a) Eaborn C., Lickiss P. D., Najim S. T., Stańczyk W. A. J. Chem. Soc., Chem. Commun. 1987:1461–1462. [Google Scholar]; (b) Choi N., Lickiss P. D., McPartlin M., Masangane P. C., Veneziani G. L. Chem. Commun. 2005:6023–6025. doi: 10.1039/b513203g. [DOI] [PubMed] [Google Scholar]; (c) Lühmann N., Hirao H., Shaik S., Müller T. Organometallics. 2011;30:4087–4096. [Google Scholar]; (d) Müther K., Hrobárik P., Hrobáriková V., Kaupp M., Oestreich M. Chem.–Eur. J. 2013;19:16579–16594. doi: 10.1002/chem.201302885. [DOI] [PubMed] [Google Scholar]; (e) Connelly S. J., Kaminsky W., Heinekey D. M. Organometallics. 2013;32:7478–7481. [Google Scholar]; (f) Ref. 7e; ; (g) Albers L., Rathjen S., Baumgartner J., Marschner C., Müller T. J. Am. Chem. Soc. 2016;138:6886–6892. doi: 10.1021/jacs.6b03560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For seminal reports, see: ; (a) Lambert J. B., Zhang S., Stern C. L., Huffman J. C. Science. 1993;260:1917–1918. doi: 10.1126/science.260.5116.1917. [DOI] [PubMed] [Google Scholar]; (b) Reed C. A., Xie Z., Bau R., Benesi A. Science. 1993;262:402–404. doi: 10.1126/science.262.5132.402. [DOI] [PubMed] [Google Scholar]
- (a) Schäfer A., Reißmann M., Schäfer A., Saak W., Haase D., Müller T. Angew. Chem., Int. Ed. 2011;50:12636–12638. doi: 10.1002/anie.201106582. [DOI] [PubMed] [Google Scholar]; (b) Schäfer A., Reißmann M., Jung S., Schäfer A., Saak W., Brendler E., Müller T. Organometallics. 2013;32:4713–4722. [Google Scholar]
- (a) Lambert J. B., Zhao Y. Angew. Chem., Int. Ed. Engl. 1997;36:400–401. [Google Scholar]; (b) Kim K.-C., Reed C. A., Elliot D. W., Mueller L. J., Tham F., Lin L., Lambert J. B. Science. 2002;297:825–827. doi: 10.1126/science.1073540. [DOI] [PubMed] [Google Scholar]; (c) Lambert J. B., Lin L. J. Org. Chem. 2001;66:8537–8539. doi: 10.1021/jo010772t. [DOI] [PubMed] [Google Scholar]
- Hydride abstraction from MePh2SiH with Ph3C+[B(C6F5)4]– was reported as a clean reaction: Lambert J. B., Zhang S., Ciro S. M., Organometallics, 1994, 13 , 2430 –2443 , . However, this result could not be reproduced by Müller (cf.ref. 10b) and us. . [Google Scholar]
- (a) Reed C. A. Acc. Chem. Res. 1998;31:133–139. [Google Scholar]; (b) Reed C. A. Acc. Chem. Res. 2010;43:121–128. doi: 10.1021/ar900159e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the synthesis and crystallographic characterization of related trimethylsilylium salts, see: ; (a) Me3Si+[CRB11F11]– (R = H, Et): Küppers T., Bernhardt E., Eujen R., Willner H., Lehmann C. W., Angew. Chem., Int. Ed., 2007, 46 , 6346 –6349 . [DOI] [PubMed] [Google Scholar]; (b) Me3Si(arene)+[B(C6F5)4]–: Ibad M. F., Langer P., Schulz A., Villinger A., J. Am. Chem. Soc., 2011, 133 , 21016 –21027 . [DOI] [PubMed] [Google Scholar]
- CCDC 1818576 for Me3Si+[CHB11H5Br6]–, CCDC ; 1818582 for MePh2Si+[CHB11H5Br6]–, and CCDC ; 1818581 for iPr2PhSi+[CHB11H5Br6]– contain the supplementary crystallographic data for this paper.
- Xie Z., Bau R., Benesi A., Reed C. A. Organometallics. 1995;14:3933–3941. [Google Scholar]
- The decomposition of the [B(C6F5)4]– counteranion is likely to proceed via an SEAr reaction of the formed silylium ions with the borate. The formation of B(C6F5)3 was verified by 19F NMR spectroscopic analysis, and GC-MS analysis revealed formation of several silanes containing a C6F5 unit
- The generated silylium ions were converted to the corresponding fluorosilanes using (C6F5)3PF2 (1.0 equiv.), thereby facilitating product characterization by both NMR spectroscopic and GC-MS analysis. For the preparation of (C6F5)3PF2, see: Caputo C. B., Hounjet L. J., Dobrovetsky R., Stephan D. W., Science, 2013, 341 , 1374 –1377 .24052304 [Google Scholar]
- Small amounts of the triarylsilylium ion (C6Me5)3Si+[CHB11H5Br6]– were also detected (cf.ref. 10)
- The mechanism of intermolecular substituent exchange reactions at related ferrocene-stabilized silylium ions had already been studied by quantum-chemical analyses (cf.ref. 8d). However, the calculated barriers for the transition states were relatively high. For the calculated mechanism of an intramolecular substituent exchange reaction at a silicon cation with a rigid naphthalene-1,8-diyl backbone, see: ref. 8c
- (a) Hoffmann S. P., Kato T., Tham F. S., Reed C. A. Chem. Commun. 2006:767–769. doi: 10.1039/b511344j. [DOI] [PubMed] [Google Scholar]; (b) Nava M., Reed C. A. Organometallics. 2011;30:4798–4800. doi: 10.1021/om200636u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R., Werner K., Müller T. Chem.–Eur. J. 2002;8:1163–1171. doi: 10.1002/1521-3765(20020301)8:5<1163::aid-chem1163>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- We were not able to locate a four-centered transition state from 8A to directly arrive at 10A. See Fig. S67 in the ESI for geometric scan calculations
- Me3Si(toluene)+[CHB11H5Br6]– was calculated to be only 0.7 kcal mol–1 lower in energy than MePh2Si(toluene)+[CHB11H5Br6]– (see the ESI for details)
- Although our calculations predict formation of small amounts of Ph3Si+[CHB11H5Br6]– in the reaction of MePh2SiH with Ph3C+[CHB11H5Br6]–, we were not able to detect this silylium ion by 1H/29Si HMQC NMR spectroscopy.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.