

Abstract
Background:
Ischemia preconditioning (IPC) remains the most powerful intervention of protection against myocardial ischemia/reperfusion injury (IRI), but diabetes can weaken or eliminate its cardioprotective effect and detailed mechanisms remain unclear. In this study, we aimed to explore whether changes of autophagy in the diabetic condition are attributable to the decreased cardioprotective effect of IPC.
Methods:
Sixty diabetic male Sprague-Dawley rats were randomly divided into the control (C), IRI, rapamycin (R), wortmannin (W), rapamycin + IPC (R + IPC), and wortmannin + IPC (W + IPC) groups. The in vivo rat model of myocardial IRI was established by ligaturing and opening the left anterior descending coronary artery via the left thoracotomy. Durations of ischemia and reperfusion are 30 min and 120 min, respectively. Blood samples were taken at 120 min of reperfusion for measuring serum concentrations of troponin I (TnI) and creatine kinase isoenzyme MB (CK-MB) using the enzyme-linked immunosorbent assay. The infarct size was assessed by Evans blue and triphenyltetrazolium chloride staining. The expressions of LC3-II, beclin-1, phosphoinositide 3-kinase (PI3K), mammalian target of rapamycin (mTOR), and P-Akt/Akt ratio in the ischemic myocardium were assessed by Western blotting.
Results:
Compared to the IRI group, infarct size (56.1% ± 6.1% vs. 75.4 ± 7.1%, P < 0.05), serum cTnI (0.61 ± 0.21 vs. 0.95 ± 0.26 ng/ml, P < 0.05), and CK-MB levels (6.70 ± 1.25 vs. 11.51 ± 2.35 ng/ml, P < 0.05) obviously decreased in the W + IPC group. Compared with the C group, myocardial expressions of LC3-II (0.46 ± 0.04 and 0.56 ± 0.04 vs. 0.36 ± 0.04, P < 0.05) and beclin-1 (0.34 ± 0.08 and 0.38 ± 0.07 vs. 0.24 ± 0.03, P < 0.05) evidently increased, and myocardial expressions of mTOR (0.26 ± 0.08 and 0.25 ± 0.07 vs. 0.38 ± 0.06, P < 0.05), PI3K (0.29 ± 0.04 and 0.30 ± 0.03 vs. 0.38 ± 0.02, P < 0.05), and P-Akt/Akt ratio (0.49 ± 0.10 and 0.48 ± 0.06 vs. 0.72 ± 0.07, P < 0.05) markedly decreased in the IRI and R groups, indicating an increased autophagy. Compared with the IRI group, myocardial expression of beclin-1 (0.26 ± 0.03 vs. 0.34 ± 0.08, P < 0.05) significantly decreased, and myocardial expressions of mTOR (0.36 ± 0.04 vs. 0.26 ± 0.08, P < 0.05), PI3K (0.37 ± 0.03 vs. 0.29 ± 0.04, P < 0.05), and P-Akt/Akt ratio (0.68 ± 0.05 vs. 0.49 ± 0.10, P < 0.05) increased obviously in the W + IPC group, indicating a decreased autophagy.
Conclusions:
Increased autophagy in the diabetic myocardium is attributable to decreased cardioprotection of IPC, and autophagy inhibited by activating the PI3K-Akt-mTOR signaling pathway can result in an improved protection of IPC against diabetic myocardial IRI.
Keywords: Autophagy, Diabetes, Ischemia Preconditioning, Ischemia-Reperfusion Injury
摘要
背景:
缺血预处理(Ischemic preconditioning, IPC)仍然是心肌缺血/再灌注损伤(Ischemia/reperfusion injury,IRI)最强效的 保护干预措施,但糖尿病可减弱甚至消除其心肌保护作用,并且具体机制尚不明确。本研究旨在探讨糖尿病条件下自噬改变 是否参与了降低的IPC心肌保护作用。
方法:
将60只雄性糖尿病Sprague-Dawley大鼠随机分为对照组(C组)、IRI组、雷帕霉素组(R组)、渥曼青霉素组(W 组)、雷帕霉素+IPC组(R+IPC组)和渥曼青霉素+IPC组(W+IPC组)。通过左侧开胸结扎和开放左冠状动脉前降支建立 大鼠在体心肌IRI模型,心肌缺血和再灌注的时间分别是30分钟和120分钟。再灌注120 min时采集血样,采用酶联免疫吸 附法(enzyme-linked immunosorbent essay, ELISA)检测血清心肌肌钙蛋白I(cardiac troponin I,cTnI)和肌酸激酶同工酶 MB(creatine kinase isoenzyme MB,CK-MB);采用伊文氏蓝和2,3,5-三苯基氯化四氮唑双染色法测定心肌梗死面积;采用 Western-blotting检测LC3-II、Beclin-1、mTOR、PI3K心肌表达情况和P-Akt/Akt比率。
结果:
与IRI组相比,W+IPC组的心肌梗死面积(56.1%±6.1%比75.4±7.1%,P <0.05),血清cTnI浓度(0.61±0.21比 0.95±0.26 ng/ml,P <0.05)、血清CK-MB浓度(6.70±1.25比11.51±2.35 ng/ml,P <0.05)均明显降低;与C组相比,IRI和R组 LC3-II(0.46±0.04和0.56±0.04比0.36±0.04,P <0.05)和beclin-1(0.34±0.08和0.38±0.07比0.24±0.03,P <0.05)心肌表达明显 增强,mTOR(0.26±0.08和0.25 ± 0.07 比 0.38 ± 0.06, P < 0.05)和PI3K(0.29±0.04和0.30±0.03比0.38±0.02,P <0.05)心肌表达 以及P-Akt/Akt比率(0.49±0.10和0.48 ± 0.06比0.72 ± 0.07, P < 0.05)明显降低,表明自噬表达增强。与IRI组相比,W+IPC组 beclin-1(0.26±0.03比0.34±0.08,P <0.05)心肌表达明显降低,mTOR(0.36±0.04比0.26±0.08,P <0.05)和PI3K(0.37±0.03比 0.29±0.04,P <0.05)心肌表达以及P-Akt/Akt比率(0.68±0.05比0.49±0.10,P <0.05)明显增加,表明自噬表达降低。
结论:
糖尿病心肌自噬表达增强可减弱IPC的心肌保护作用,激活PI3K-Akt-mTOR信号转导通路抑制自噬可改善IPC对糖尿病心肌IRI的保护作用。
INTRODUCTION
Ischemic heart disease is a major cause of morbidity and mortality worldwide. Reperfusion is the only method to rescue salvageable myocardium, but reperfusion itself may induce further myocardial injury, a phenomenon has been termed as ischemia/reperfusion injury (IRI).[1] Myocardial IRI has been regarded as a leading cause of disability or even death in patients with ischemic heart disease.[2] Ischemia preconditioning (IPC) induced by one or more cycles of brief warm ischemia and reperfusion has been associated with local protection to subsequent ischemic injuries.[3,4] Furthermore, IPC remains the most powerful intervention of protection against myocardial IRI.
The diabetes is a common comorbidity of patients with ischemic heart disease. It has been shown that diabetes can not only aggravate myocardial IRI but also weaken or even eliminate the cardioprotective effects of many interventions including IPC.[5,6,7,8,9] Much efforts have been made, but the detailed mechanisms that diabetes affects the severity of myocardial IRI and efficacy of cardioprotective interventions are not well elucidated. Autophagy refers that intracellular impaired, denatured, senescent proteins and organelles are transported to the lysosomes, in which they are digested. It is characterized by the appearance of cytoplasmic autophagosomes and can be judged by the bilayer membrane structure of the autophagosome, encapsulating part of the cytoplasm and organelles.[10] The available evidence shows that autophagy is involved in the occurrence of myocardial IRI.[11,12,13,14] Furthermore, autophagy expression changes significantly in the diabetic myocardium and plays different roles in the processes of myocardial IRI between the normal and diabetic conditions.[12,13,14,15,16] However, few studies have assessed whether changes of autophagy in the diabetic condition are attributable to the decreased cardioprotective effect of IPC.
The phosphoinositide 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) signaling pathway is the only known inhibitory pathway of autophagy regulation.[17] Rapamycin, an allosteric inhibitor of mTORC1, can form a gain-of-function complex together with the intracellular protein FKBP12, which directly interacts with the FKBP12-rapamycin-binding domain of mTOR.[18] By inhibiting mTOR signaling pathway, rapamycin can induce and promote the occurrence of autophagy. In contrast, wortmannin, a PI3K inhibitor, can specifically block the formation of autophagosome and has been widely used as an inhibitor of autophagy.[19] Thus, this experiment was designed to determine the roles of autophagy changes in the protective effect of IPC against diabetic myocardial IRI using rapamycin and wortmannin interventions.
METHODS
Experiment protocols were approved by the Animals Ethics Committee of our institution. Twelve-week-old male Sprague-Dawley rats (290–320 g) were obtained from Beijing Vital River Laboratory Animal Technology Company (Beijing, China). The rats were housed in a temperature- and humidity-controlled room (24°C ± 1°C and 65% ± 10%, respectively) under a 12-h light-dark cycle. They were fasted for 12 h before the experiment, but kept drinking water.
Diabetic rat and in vivo myocardial ischemia/reperfusion injury models
Diabetic rat model was established according to the previous study.[20] Briefly, 12-week-old rats were bred for 1 week with the high-fat and high-sugar diet, which contains sucrose, milk powder, peanut, lard, and egg yolk on the basis of the standard full price diet. Heat content of the diet is 26.8 KJ/kg. Afterward, the rats were injected intraperitoneally with streptozotocin (50 mg/kg in 0.1 mol/L citric acid buffer, pH 4.2–4.5) for 2 days. The control rats were treated with the same volume of citric acid buffer. Diabetic condition was verified 48 h later by evaluating blood glucose levels. Rats with blood glucose level of 16.65 mmol/L or greater were considered as diabetic condition. If the blood glucose level was <16.65 mmol/L, an intraperitoneal injection of streptozotocin 30 mg/kg was given again. If the rat blood glucose was still lower than 16.65 mmol/L, establishment of diabetic condition was considered a failure. The rats with blood glucose level of 16.65 mmol/L or greater were bred with the high-fat and high-sugar diet for an additional 8 weeks.
The in vivo rat model of myocardial IRI was performed as previously described.[21] The rat was anesthetized with an intraperitoneal injection of 3% pentobarbital sodium (70 mg/kg). After tracheal intubation, the rat was ventilated with room air using an animal respirator. The ventilation rate was adjusted to 60–80 breaths/min, with the tidal volume of 2–3 ml/100 g body weight and the inspiratory/expiratory ratio of 1:1. The internal jugular vein was cannulated for blood sampling to assay serum concentrations of troponin I (TnI) and creatine kinase isoenzyme MB (CK-MB). The carotid artery was cannulated for monitoring heart rate (HR), systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean artery pressure (MAP) with a MP150 data acquisition and analysis system (Biopac Systems Inc., CA, USA). The lead II electrocardiogram was continuously recorded by means of needle electrodes placed subcutaneously on the limbs.
A left thoracotomy was performed at the fourth intercostal space and the heart was exposed. By a surgical needle, a 5-0 silk ligature was placed under the left anterior descending coronary artery (LAD) and encircled with a suture. The LAD was ligated to induce ischemia for 30 min and then released to allow reperfusion for 120 min.
Experimental protocol
Sixty anesthetized rats with a verified diabetic condition were randomly divided into six groups: control group (C group), a 5-0 silk was placed under the LAD and encircled with a suture, and stay for 150 min; IRI group, the LAD was ligated for 30 min followed by opening LAD for reperfusion of 120 min; rapamycin group (R group), rapamycin 0.25 mg/kg was intraperitoneally injected 30 min before IRI;[22] wortmannin group (W group), wortmannin 0.6 mg/kg was intraperitoneally injected 30 min before IRI;[23] rapamycin + IPC group (R + IPC group), rapamycin 0.25 mg/kg was intraperitoneally injected and the IPC including three circles of a 5-min ischemia followed by a 5-min reperfusion was performed 30 min before IRI; and wortmannin + IPC group (W + IPC group), wortmannin (0.6 mg/kg) was intraperitoneally injected and the IPC procedure similar to the R + IPC group was carried out 30 min before IRI.
Measurements
Throughout the experiment, HR, SBP, and MAP were continuously monitored, and rate-pressure product (RPP) was calculated as the index of myocardial oxygen consumption. In all rats, ventricular arrhythmias, such as premature ventricular contraction (PVC), ventricular tachycardia (VT), and fibrillation (VF), were recorded from the initiation of ischemia to 30 min of reperfusion. The arrhythmia scores were graded as follows:[24] 0, without arrhythmias; 1, <3 PVCs per min; 2, ≥3 PVCs per min; 3, <3 episodes of VT per min; 4, >3 episodes of VT per min or transient VF; and 5, frequent or sustained VF or death.
At 120 min of reperfusion, a 4-ml blood sample was taken in the tube containing microscopic silica particles. After placing 30 min, the blood sample was centrifuged at 377.325 ×g for 15 min. The supernatant was collected and stored at −80°C condition until future analysis. The serum TnI and CK-MB levels were measured using the enzyme-linked immunosorbent assay according to the manufacturer's instructions.
After completion of the experiment, the rats in each group were further allocated into subgroups A and B. The subgroup A was used to evaluate myocardial infarct size. The LAD was reoccluded and 1 ml of 2% Evans blue dye was injected to stain the normally perfused region of the heart and delineate the area at risk (AAR). Then, rats were euthanized using an intravenous injection of 10% potassium chloride. After the heart was excised and rinsed of excess blue dye, the right ventricle and right and left atria were trimmed off. The remaining left ventricle was placed at −80°C condition for 10 min. The frozen left ventricle was then cut into approximately five sections from apex to base, and all tissues were incubated in a 2% solution of 2, 3, 5-triphenyltetrazolium chloride for 5 min at 37°C. The infarcted tissue stains a characteristic white color, whereas the viable tissue stains red. After overnight fixation in 4% formaldehyde, the slices were digitally photographed. Analysis of the sections using the Adobe Photoshop CS (Adobe Systems Inc., San Jose, CA, USA) by a blinded investigator allowed for assessment of the AAR and infarct size, which was expressed as a percentage of the AAR.[25]
The subgroup B was used to measure the myocardial expressions of LC3-II, beclin-1, mTOR, PI3K, and Akt by Western blotting. After myocardial tissues in the ischemic regions were taken, the proteins were extracted from myocardial tissue by suspension in radioimmunoprecipitation assay buffer, samples were centrifuged at 28,341.3 ×g at 4°C for 20 min, and the supernatants were retrieved for analysis. The protein concentrations were determined using the Bradford protein method and the bicinchoninic acid (BCA) protein assay kit (Beijing ComWin Biotech Co., Ltd., China). Protein (20 μg) was electrophoresed on a precast bis-Tris polyacrylamide gel (8–12%) for 20 min and then transferred onto a polyvinylidene difluoride (PVDF) membrane. Membranes were blotted with rabbit anti-LC3B (1:1000, Cell Signaling Technology, Inc., USA), rabbit anti-beclin-1 (1:1000, Cell Signaling Technology, Inc.), rabbit anti-PI3KP110a (1:1000, Cell Signaling Technology, Inc.), rabbit anti-mTOR (1:1000, Cell Signaling Technology, Inc.), rabbit anti-Akt (1:1000, Cell Signaling Technology, Inc.), rabbit anti-p-Akt (1:1000, Cell Signaling Technology, Inc.), and rabbit anti-GAPDH (1:1000, Cell Signaling Technology, Inc.), followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000, Jackson ImmunoResearch Laboratories, Inc., China). Immunoblots were visualized using enhanced chemiluminescence.
Statistical analysis
SPSS 13.0 statistical software (SPSS Inc., Chicago, IL, USA) was used for data analysis. The Kolmogorov–Smirnov test was used to test the normality of the distribution for all the parametric data. Furthermore, the Levene median test was adopted to test the homogeneity of variance for the parametric data. If the data were normally distributed and had homogeneous variance, they were expressed as a mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was used for intergroup comparisons. Repeated-measures ANOVA was applied for within-group comparisons. Tukey's multiple comparison tests were used for multiple post hoc comparisons. When the data were not normally distributed or had inhomogeneous variance, they were expressed as median (interquartile range) and were compared using the Kruskal-Wallis and Mann-Whitney U-tests. P < 0.05 was considered statistically significant.
RESULTS
Hemodynamics
Throughout the observed period, HR, MAP, and RPP kept the stable levels in the C group. At 1 min of ischemia, HR (392 ± 11 vs. 381 ± 12 beats/min, 394 ± 12 vs. 383 ± 13 beats/min, 388 ± 13 vs. 377 ± 11 beats/min, 389 ± 14 vs. 378 ± 12 beats/min, and 389 ± 15 vs. 379 ± 13 beats/min, P < 0.05) increased evidently and MAP (106 ± 7 mmHg [1 mmHg = 0.133 kPa] vs. 117 ± 7, 105 ± 6 vs. 116 ± 8 mmHg, 106 ± 7 vs. 114 ± 6 mmHg, 108 ± 8 vs. 116 ± 7 mmHg, and 108 ± 7 vs. 115 ± 8 mmHg, P < 0.05) decreased significantly compared to their baselines in the IRI, R, W, R + IPC, and W + IPC groups. At 60 min and 120 min of reperfusion, HR (364 ± 12 vs. 381 ± 12 beats/min, 364 ± 16 vs. 383 ± 13 beats/min, 377 ± 11 vs. 368 ± 18 beats/min, 360 ± 14 vs. 378 ± 12 beats/min, and 368 ± 12 vs. 379 ± 13 beats/min, P < 0.05; 363 ± 12 vs. 381 ± 12 beats/min, 358 ± 16 vs. 383 ± 13 beats/min, 360 ± 16 vs. 377 ± 11 beats/min, 355 ± 16 vs. 378 ± 12 beats/min, and 366 ± 13 vs. 379 ± 13 beats/min, P < 0.05), MAP (104 ± 6 vs. 117 ± 7 mmHg, 96 ± 8 vs. 116 ± 8 mmHg, 105 ± 7 vs. 114 ± 6 mmHg, 93 ± 8 vs. 116 ± 7 mmHg, and 106 ± 7 vs. 115 ± 8 mmHg, P < 0.05; 96 ± 6 vs. 117 ± 7 mmHg, 91 ± 6 vs. 116 ± 8 mmHg, 100 ± 8 vs. 114 ± 6 mmHg, 89 ± 8 vs. 116 ± 7 mmHg, and 102 ± 7 vs. 115 ± 8 mmHg, P < 0.05) significantly decreased compared to their baselines in the IRI, R, W, R + IPC, and W + IPC groups. At 60 min of reperfusion, RPP (40.5 ± 3.0 vs. 44.2 ± 4.3, 41.2 ± 5.4 vs. 44.6 ± 5.3, and 40.9 ± 2.9 vs. 43.9 ± 6.0, P < 0.05) significantly decreased compared to their baselines in the IRI, R, and W groups. At 120 min of reperfusion, RPP (38.6 ± 5.2 vs. 44.2 ± 4.3, 39.9 ± 6.1 vs. 44.6 ± 5.3, 37.7 ± 2.7 vs. 43.9 ± 6.0, 38.6 ± 3.1 vs. 43.8 ± 3.3, and 42.1 ± 3.2 vs. 44.0 ± 4.0, P < 0.05) significantly decreased compared to their baselines in the IRI, R, W, R + IPC, and W + IPC groups. At 60 min and 120 min of reperfusion, however, MAP (106 ± 7 vs. 104 ± 6 mmHg and 102 ± 7 vs. 96 ± 6 mmHg, P < 0.05) and RPP (42.1 ± 3.2 vs. 38.6 ± 5.2 and 41.6 ± 3.2 vs. 37.7 ± 5.2, P < 0.05) obviously increased in the W + IPC group compared to the IRI group [Table 1].
Table 1.
Comparisons of hemodynamic changes during ischemia and reperfusion among different rat groups
| Groups | Baselines | Ischemia | Reperfusion | |||||
|---|---|---|---|---|---|---|---|---|
| 1 min | 5 min | 15 min | 30 min | 30 min | 60 min | 120 min | ||
| HR (beats/min) | ||||||||
| C | 379 ± 18 | 382 ± 14 | 380 ± 19 | 378 ± 15 | 379 ± 16 | 376 ± 16 | 378 ± 14 | 376 ± 19 |
| IRI | 381 ± 12 | 392 ± 11* | 383 ± 13 | 379 ± 18 | 374 ± 18 | 368 ± 12 | 364 ± 12* | 363 ± 12*,† |
| R | 383 ± 13 | 394 ± 12* | 384 ± 15 | 379 ± 13 | 376 ± 13 | 3684 ± 14 | 364 ± 16* | 358 ± 16*,† |
| W | 377 ± 11 | 388 ± 13* | 379 ± 13 | 377 ± 14 | 374 ± 12 | 373 ± 12* | 368 ± 18*,† | 360 ± 16*,† |
| R + IPC | 378 ± 12 | 389 ± 14* | 378 ± 16 | 373 ± 11 | 369 ± 15 | 364 ± 14* | 360 ± 14*,† | 355 ± 16*,† |
| W + IPC | 379 ± 13 | 389 ± 15* | 378 ± 12 | 375 ± 16 | 371 ± 12† | 371 ± 12*,† | 368 ± 12*,† | 366 ± 13* |
| MAP (mmHg) | ||||||||
| C | 118 ± 7 | 117 ± 8 | 116 ± 7 | 117 ± 7 | 116 ± 6 | 116 ± 7 | 115 ± 7 | 115 ± 7 |
| IRI | 117 ± 7 | 106 ± 7* | 115 ± 6 | 109 ± 7 | 112 ± 7 | 106 ± 8 | 104 ± 6* | 96 ± 6*,† |
| R | 116 ± 8 | 105 ± 6* | 114 ± 8 | 105 ± 8 | 103 ± 6* | 101 ± 7 | 96 ± 8* | 91 ± 6*,† |
| W | 114 ± 6 | 106 ± 7* | 115 ± 6 | 113 ± 6 | 110 ± 7 | 108 ± 6†,§ | 105 ± 7*,† | 100 ± 8*,† |
| R + IPC | 116 ± 7 | 108 ± 8* | 116 ± 6 | 110 ± 8 | 98 ± 7* | 95 ± 7* | 93 ± 8*,† | 89 ± 8*,† |
| W + IPC | 115 ± 8 | 108 ± 7* | 115 ± 8 | 113 ± 7 | 111 ± 8 | 109 ± 8‡,§ | 106 ± 7†,§ | 102 ± 7*,‡,§ |
| RPP (1000·min/mmHg) | ||||||||
| C | 43.9 ± 6.4 | 44.5 ± 5.3 | 44.0 ± 3.2 | 43.5 ± 3.4 | 43.1 ± 3.3 | 42.9 ± 3.6 | 42.6 ± 3.2 | 42.7 ± 4.2 |
| IRI | 44.2 ± 4.3 | 40.5 ± 3.0* | 42.9 ± 5.3 | 42.1 ± 3.3 | 41.3 ± 5.5 | 39.8 ± 3.2 | 38.6 ± 5.2* | 37.7 ± 5.2*,† |
| R | 44.6 ± 5.3 | 41.2 ± 5.4* | 44.9 ± 3.4 | 43.6 ± 4.4 | 42.1 ± 3.6 | 41.3 ± 5.3 | 39.9 ± 6.1*,† | 37.9 ± 6.8*,† |
| W | 43.9 ± 6.0 | 40.9 ± 2.9* | 43.7 ± 6.1 | 42.2 ± 2.9 | 40.4 ± 2.9 | 39.4 ± 3.0† | 37.7 ± 2.7*,† | 34.1 ± 2.7*,† |
| R + IPC | 43.8 ± 3.3 | 42.0 ± 3.5 | 44.0 ± 3.1 | 42.2 ± 5.2 | 40.0 ± 3.0 | 39.1 ± 6.1* | 38.6 ± 3.1*,† | 37.8 ± 3.2*,† |
| W + IPC | 44.0 ± 4.0 | 41.0 ± 3.3 | 43.3 ± 3.6 | 43.0 ± 3.7 | 42.9 ± 3.9 | 42.3 ± 3.5† | 42.1 ± 3.2*,†,‡,§ | 41.6 ± 3.2*,†,‡,§ |
Data are presented as mean ± SD, n = 10 in each group. 1 mmHg = 0.133 kPa. IPC: Ischemia preconditioning; C: Control group; IRI: Ischemia/reperfusion injury group; R: Rapamycin group; W: Wortmannin group; R + IPC: Rapamycin + IPC group; W + IPC: Wortmannin + IPC group. Repeated-measures analysis of variance for within-group comparisons. Tukey’s multiple comparison tests for multiple post hoc comparisons. *P<0.05 versus baseline values; †P<0.05 versus C group; ‡P<0.05 versus IRI group; §P<0.05 versus R + IPC group. HR: Heart rate; MAP: Mean arterial pressure; RPP: Rate-pressure product; SD: Standard deviation.
Arrhythmia
In the groups other than the C group, ventricular arrhythmias, including multi-derived VPC, a variable number of VTs with different durations and even VF, were observed during 6–15 min of ischemia. The scores of ventricular arrhythmia during the ischemia and initial reperfusion periods markedly decreased in the W group (3.0 [0.5] vs. 4.0 [0.0] and 3.0 [0.5] vs. 4.0 [0.5], P < 0.05; 2.0 [0.0] vs. 4.0 [1.0] and 2.0 [0.0] vs. 4.0 [1.0], P < 0.05) and W + IPC group (2.0 [0.5] vs. 4.0 [0.0] and 2.0 [0.5] vs. 4.0 [0.5], P < 0.05; 2.0 [1.0] vs. 4.0 [1.0] and 2.0 [1.0] vs. 4.0 [1.0], P < 0.05) compared with the IRI and R groups, but obviously increased in the R + IPC group (4.0 [1.0] vs. 3.0 [0.5] and 4.0 [0.5] vs. 2.0 [0.0], P < 0.05) compared to the W group. There were no significant differences in the incidence and score of ventricular arrhythmia during the ischemia and initial reperfusion among the IRI, R, and R + IPC groups [Table 2].
Table 2.
Incidence and scores of arrhythmia during ischemia and early reperfusion in different rat groups
| Groups | Ventricular arrhythmias (number of animals [%]) | Arrhythmia scores (median [interquartile range]) | ||
|---|---|---|---|---|
| Ischemia | Early reperfusion | Ischemia | Early reperfusion | |
| C | 0 | 0 | 0.0 (0.0) | 0.0 (0.0) |
| IRI | 10 (100) | 10 (100) | 4.0 (0.0) | 4.0 (1.0) |
| R | 10 (100) | 10 (100) | 4.0 (0.5) | 4.0 (1.0) |
| W | 6 (60)*,† | 5 (50)*,† | 3.0 (0.5)*,† | 2.0 (0.0)*,† |
| R + IPC | 10 (100)‡ | 9 (90)‡ | 4.0 (1.0)‡ | 4.0 (0.5)‡ |
| W + IPC | 5 (50)*,† | 4 (40)*,† | 2.0 (0.5)*,† | 2.0 (1.0)*,† |
IPC: Ischemia preconditioning; C: Control group; IRI: Ischemia/reperfusion injury group; R: Rapamycin group; W: Wortmannin group; R + IPC: Rapamycin + IPC group; W + IPC: Wortmannin + IPC group. Mann-Whitney U-tests for intergroup comparisons. n = 10 in each group. *P<0.05 versus IRI group; †P<0.05 versus R group; ‡P<0.05 versus W group.
Myocardial infarct sizes and serum cTnI and creatine kinase-myocardial isoenzyme MB levels
The infarct size (75.4% ± 7.1%, 77.6% ± 5.1%, 71.8% ± 6.0%, 77.8% ± 6.9%, and 56.1% ± 6.1% vs. 4.2% ± 1.0%, P < 0.05), serum cTnI (0.95 ± 0.26, 0.97 ± 0.25, 0.85 ± 0.22, 0.92 ± 0.25, and 0.61 ± 0.21 vs. 0.20 ± 0.06 ng/ml, P < 0.05), and CK-MB levels (11.51 ± 2.35, 12.11 ± 2.38, 9.70 ± 2.03, 11.32 ± 2.33, and 6.70 ± 1.25 vs. 2.98 ± 0.43 ng/ml, P < 0.05) significantly increased in the IRI, R, W, R + IPC, and W + IPC groups compared to the C group, but obviously decreased in the W + IPC group (56.10% ± 6.10% vs. 75.40% ± 7.10%, 0.61 ± 0.21 vs. 0.95 ± 0.26 ng/ml, and 6.70 ± 1.25 vs. 11.51 ± 2.35 ng/ml, P < 0.05) compared to the IRI group. Compared to the W and R + IPC groups, both infarct size (56.1% ± 6.1% vs. 71.8% ± 6.0% and 56.1% ± 6.1% vs. 77.8% ± 6.9%, P < 0.05) and serum CK-MB level (6.70 ± 1.25 vs. 9.70 ± 2.03 ng/ml and 6.70 ± 1.25 vs. 11.32 ± 2.33 ng/ml, P < 0.05) evidently decreased in the W + IPC group [Figure 1].
Figure 1.

Infarct size, serum cTnI, and CK-MB levels in different rat groups. (a) Infarct size determined by the Evans blue and 2, 3, 5-triphenyl tetrazolium chloride staining; (b) Serum cTnI levels measured by enzyme-linked immunosorbent assay; (c) Serum CK-MB levels measured by enzyme-linked immunosorbent assay. Data are presented as mean ± standard deviation, n = 5 in each group for infarct size measurement and n = 10 in each group for serum cTnI and CK-MB measurements. C: Control group; IRI: Ischemia/reperfusion injury group; R: Rapamycin group; W: Wortmannin group; R + IPC: Rapamycin + IPC group; W + IPC: Wortmannin + IPC group. One-way analysis of variance was used for intergroup comparisons. *P < 0.05 versus C group; †P < 0.05 versus IRI group; ‡P < 0.05 versus W group; §P < 0.05 versus R + IPC group. IS: Infarct size; cTnI: Cardiac troponin I; CK-MB: Creatine kinase isoenzyme MB.
Myocardial expressions of LC3-II, beclin-1, mammalian target of rapamycin, phosphoinositide 3-kinase, and P-Akt/Akt ratio
Compared to the C group, myocardial expressions of LC3-II (0.46 ± 0.04 and 0.56 ± 0.04 vs. 0.36 ± 0.04, P < 0.05) and beclin-1 (0.34 ± 0.08 and 0.38 ± 0.07 vs. 0.24 ± 0.03, P < 0.05) significantly increased, and myocardial expressions of mTOR (0.26 ± 0.08 and 0.25 ± 0.07 vs. 0.38 ± 0.06, P < 0.05), PI3K (0.29 ± 0.04 and 0.30 ± 0.03 vs. 0.38 ± 0.02, P < 0.05), and P-Akt/Akt ratio (0.49 ± 0.10 and 0.48 ± 0.06 vs. 0.72 ± 0.07, P < 0.05) evidently decreased in the IRI and R groups. Compared with the IRI group, myocardial expression of beclin-1 (0.26 ± 0.03 vs. 0.34 ± 0.08, P < 0.05) obviously decreased in the W + IPC group, and myocardial expressions of mTOR (0.36 ± 0.04 vs. 0.26 ± 0.08, P < 0.05), PI3K (0.37 ± 0.03 vs. 0.29 ± 0.04, P < 0.05), and P-Akt/Akt ratio (0.68 ± 0.05 vs. 0.49 ± 0.10, P < 0.05) evidently increased in the W + IPC group. Compared with the R + IPC group, myocardial expressions of LC3-II (0.40 ± 0.04 vs. 0.57 ± 0.03, P < 0.05) and beclin-1 (0.26 ± 0.03 vs. 0.37 ± 0.02, P < 0.05) markedly decreased, and myocardial expressions of mTOR (0.36 ± 0.04 vs. 0.25 ± 0.08, P < 0.05), PI3K (0.37 ± 0.03 vs. 0.24 ± 0.05, P < 0.05), and P-Akt/Akt ratio (0.68 ± 0.05 vs. 0.50 ± 0.07, P < 0.05) significantly increased in the W + IPC group [Figures 2 and 3].
Figure 2.

Expressions of myocardial LC3-II, beclin-1, mTOR, PI3K, and P-Akt/Akt ratio measured by Western blotting in different rat groups. Data are presented as mean ± standard deviation, n = 5 in each group. C: Control group; IRI: Ischemia/reperfusion injury group; R: Rapamycin group; W: Wortmannin group; R + IPC: Rapamycin + IPC group; W + IPC: Wortmannin + IPC group. One-way analysis of variance for intergroup comparisons. *P < 0.05 versus C group; †P < 0.05 versus IRI group; ‡P < 0.05 versus R + IPC group. PI3K: Phosphoinositide 3-kinase; mTOR: Mammalian target of rapamycin.
Figure 3.
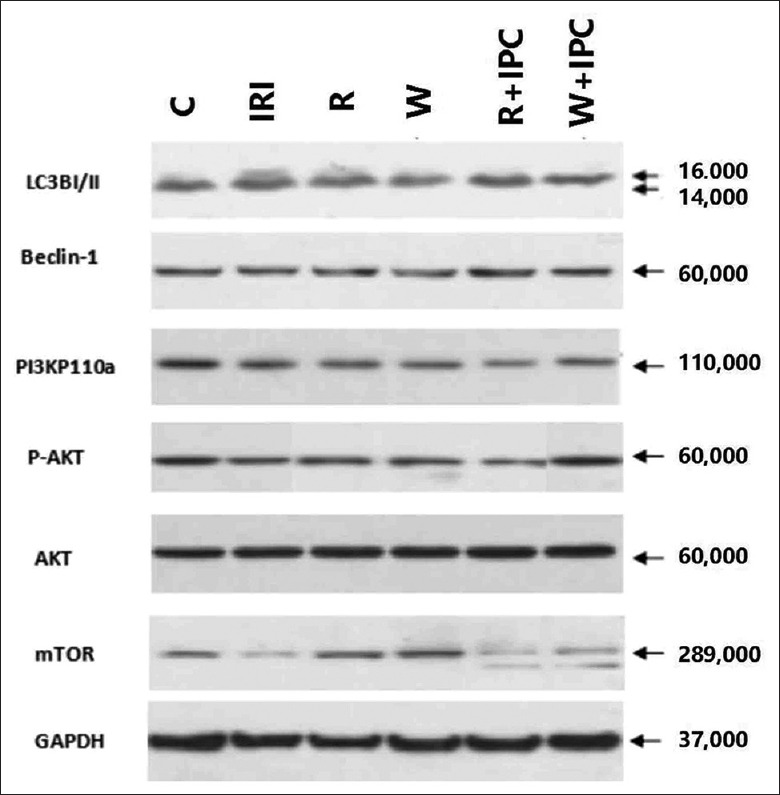
Western blotting for myocardial expressions of LC3-II, beclin-1, mTOR, PI3K, Akt, and P-Akt in different rat groups. C: Control group; IRI: Ischemia/reperfusion injury group; R: Rapamycin group; W: Wortmannin group; R + IPC: Rapamycin + IPC group; W + IPC: Wortmannin + IPC group. PI3K: Phosphoinositide 3 - kinase; mTOR: Mammalian target of rapamycin; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
DISCUSSION
Our study results showed that at 1 min of ischemia, HR significantly increased, and MAP and RPP evidently decreased compared to their baselines in the other groups except for C group. This suggests that the LAD is well occluded, and acute myocardial ischemia is successfully achieved. During 6–15 min of ischemia, the rats in the other groups except for C group also developed a large number of VPCs and VTs with different durations, even some developed VF. After reperfusion, infarct size, serum cTnI, and CK-MB levels significantly increased in the other groups compared to the C group. All of these results indicate that an in vivo model of myocardial IRI was successfully established.[26] Furthermore, the alone use of autophagy inhibitor wortmannin or activator rapamycin does not significantly affect the hemodynamic responses to IRI process.
There are no inconsistent results on the role of autophagy in myocardial IRI process; some report that an increased autophagy is beneficial,[27,28] but others show that an increased autophagy is harmful.[29,30,31] In our study, thus, protective effects of IPC against diabetic myocardial IRI with agonist and inhibitor of autophagy were assessed. The results showed that compared to the IRI group, infarct size, serum cTnI, and CK-MB levels evidently decreased in the W + IPC group. This indicates that with autophagy inhibition, IPC can produce a protection against diabetic myocardial IRI. However, infarct size, serum cTnI and CK-MB levels were not significantly different in the R and W groups compared with the IRI group, suggesting that alone activation or inhibition of autophagy does not obviously affect the occurrence and severity of diabetic myocardial IRI. Moreover, previous work had demonstrated that IRI increased the myocardial autophagy, as shown by increased conversion from LC3-I to LC3-II and the decreased expression of P62.[32] This is consistent with our results. Accordingly, we speculate that an increased autophagy in the diabetic myocardium may be attributable to decreased or disappeared protection of IPC against myocardial IRI.
Autophagy is regulated by autophagy-associated genes (Atg). The key protein complexes that are involved in the regulation of autophagy include mTOR, PI3K, Atg8 (LC3), p62, and Atg6 (beclin-1).[10,33] Of them, mTOR is a negative regulator which can inhibit the autophagy. LC3, a marker protein of autophagy expression, is involved in the formation of autophagosome together with the Atg5-Atg12-Atg16 conjugation system.[34] Furthermore, the levels of LC3 can represent the changes of autophagy expression. Beclin-1 can induce autophagy after separating from bcl-2,[35] and the expressions of beclin-1 and protein modifier of LC3 are commonly used as the indicators of autophagy activation.[36] In this experiment, thus, LC3-II, beclin-1, mTOR, PI3K, and Akt were used as measured variables assessing autophagy expression and associated signaling pathways.
Our results showed that compared with the C group, myocardial expressions of LC3-II and beclin-1 significantly increased, and myocardial expressions of mTOR, PI3K, and P-Akt/Akt ratio markedly decreased in the IRI and R groups. These results indicate that the IRI resulted in an obviously increased autophagy in diabetic myocardium. However, myocardial expression of LC3-II obviously increased and the P-Akt/Akt ratio evidently decreased in the W group compared to the C group. Compared with IRI group, myocardial expression of LC3-II significantly increased and myocardial expression of PI3K evidently decreased in the R group. The results that myocardial autophagy significantly increased in the R group and evidently reduced in the W group suggest that autophagy interventions with rapamycin and wortmannin are successful in this study. It was important to note that autophagy interventions evidently changed autophagy in the diabetic myocardium, but did not aggravate or alleviate severity of myocardial IRI assessed by infarct size, serum cTnI and CK-MB levels in the R and W groups. In contrast, myocardial expression of beclin-1 markedly decreased and myocardial expressions of mTOR, PI3K, and P-Akt/Akt ratio significantly increased in the W + IPC group compared to the IRI group. These results suggest that with autophagy inhibitor, IPC can provide a protection against diabetic myocardial IRI, with a reduced autophagy.
In this study, myocardial autophagy significantly increased in the IRI, R, and R + IPC groups compared to the C group. Furthermore, increased levels of autophagy were more significant in the R and R + IPC groups than in the IRI group. It suggests that autophagy plays an important role in the diabetic myocardial IRI, which is in accordance with the findings of the previous studies.[37,38] After autophagy was increased to a certain extent, however, severity of myocardial IRI was not further worsened. According to these results, we deduce that increased autophagy in the diabetic myocardium may have a “ceiling threshold” for the IRI. When this “ceiling threshold” is reached or exceeded, severity of diabetic myocardial IRI is no longer aggravated with increased autophagy. However, further researches are still needed to clarify whether this phenomenon really exists.
As an upstream signal subunit, PI3K is involved in the multiple signaling pathways. For example, the PI3K-SGK1-GSK3β signaling pathway plays an important role in protection of H2S against anoxia/reoxygenation injury of the neonatal rat cardiomyocytes by anti-autophagy, that is, anoxia/reoxygenation injury increases autophagy of cardiomyocytes, whereas H2S reduces autophagy of cardiomyocytes.[39] The use of LY294002 to block PI3K or knockout of SGK1 gene in the neonatal mice can lead to autophagy amplification, increased injury, and attenuated protective effect of H2S. However, the use of tws119 to block GSK3β expression produces the opposite effect.[39] With rapamycin intervention, moreover, anoxia preconditioning can inhibit the mTOR expression of cardiomyocytes, result in an increased autophagy and a decreased protection of anoxia preconditioning against anoxia/reoxygenation injury.[40] These findings are in line with our results, that is, autophagy can indeed affect the protection of preconditioning against myocardial IRI. In addition, Tsang et al.[41] showed that significant phosphorylation of Akt in the diabetic myocardium appeared only after 3 circles of IPC were performed and activation of the PI3K-Akt-mTOR signaling pathway is one of the most important mechanisms that IPC provides a protection against diabetic myocardial IRI. Based on our and previous findings, we think that autophagy inhibition by activating the PI3K-Akt-mTOR signaling pathway is attributable to improved protection of IPC against diabetic myocardial IRI.
In conclusion, this experiment determines that increased autophagy in the diabetic myocardium is attributable to decreased cardioprotection of IPC, and autophagy inhibited by activating the PI3K-Akt-mTOR signaling pathway can result in an improved protection of IPC against diabetic myocardial IRI.
Financial support and sponsorship
This study was supported by a grant from the National Natural Science Foundation of China (No. 81170128).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lippi G, Franchini M, Cervellin G. Diagnosis and management of ischemic heart disease. Semin Thromb Hemost. 2013;39:202–13. doi: 10.1055/s-0032-1333543. doi: 10.1055/s-0032-1333543. [DOI] [PubMed] [Google Scholar]
- 3.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. doi: 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Dorado D, Barba I, Inserte J. Twenty-five years of preconditioning: Are we ready for ischaemia? From coronary occlusion to systems biology and back. Cardiovasc Res. 2011;91:378–81. doi: 10.1093/cvr/cvr140. doi: 10.1093/cvr/cvr140. [DOI] [PubMed] [Google Scholar]
- 5.Kiss A, Tratsiakovich Y, Gonon AT, Fedotovskaya O, Lanner JT, Andersson DC, et al. The role of arginase and rho kinase in cardioprotection from remote ischemic perconditioning in non-diabetic and diabetic rat in vivo. PLoS One. 2014;9:e104731. doi: 10.1371/journal.pone.0104731. doi: 10.1371/journal.pone.0104731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miki T, Itoh T, Sunaga D, Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol. 2012;11:67. doi: 10.1186/1475-2840-11-67. doi: 10.1186/1475-2840-11- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whittington HJ, Babu GG, Mocanu MM, Yellon DM, Hausenloy DJ. The diabetic heart: Too sweet for its own good? Cardiol Res Pract. 2012;2012:845698. doi: 10.1155/2012/845698. doi: 10.1155/2012/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baranyai T, Nagy CT, Koncsos G, Onódi Z, Károlyi-Szabó M, Makkos A, et al. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc Diabetol. 2015;14:151. doi: 10.1186/s12933-015-0313-1. doi: 10.1186/s12933-015-0313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charan K, Goyal A, Gupta JK, Yadav HN. Role of atrial natriuretic peptide in ischemic preconditioning-induced cardioprotection in the diabetic rat heart. J Surg Res. 2016;201:272–8. doi: 10.1016/j.jss.2015.10.045. doi: 10.1016/j.jss.2015.10.045. [DOI] [PubMed] [Google Scholar]
- 10.Anding AL, Baehrecke EH. Cleaning house: Selective autophagy of organelles. Dev Cell. 2017;41:10–22. doi: 10.1016/j.devcel.2017.02.016. doi: 10.1016/j.devcel.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Z, Han Z, Ye B, Dai Z, Shan P, Lu Z, et al. Berberine alleviates cardiac ischemia/reperfusion injury by inhibiting excessive autophagy in cardiomyocytes. Eur J Pharmacol. 2015;762:1–0. doi: 10.1016/j.ejphar.2015.05.028. doi: 10.1016/j.ejphar.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Ren J. Targeting autophagy for the therapeutic application of histone deacetylase inhibitors in ischemia/reperfusion heart injury. Circulation. 2014;129:1088–91. doi: 10.1161/CIRCULATIONAHA.113.008115. doi: 10.1161/CIRCULATIONAHA.113.008115. [DOI] [PubMed] [Google Scholar]
- 13.Ma S, Wang Y, Chen Y, Cao F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim Biophys Acta. 2015;1852:271–6. doi: 10.1016/j.bbadis.2014.05.010. doi: 10.1016/j.bbadis.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 14.Guo X, Jiang H, Yang J, Chen J, Yang J, Ding JW, et al. Spermine ameliorates ischemia/reperfusion injury in cardiomyocytes via regulation of autophagy. Int J Mol Med. 2016;38:885–93. doi: 10.3892/ijmm.2016.2686. [Google Scholar]
- 15.Han Z, Cao J, Song D, Tian L, Chen K, Wang Y, et al. Autophagy is involved in the cardioprotection effect of remote limb ischemic postconditioning on myocardial ischemia/reperfusion injury in normal mice, but not diabetic mice. PLoS One. 2014;9:e86838. doi: 10.1371/journal.pone.0086838. doi: 10.1371/journal.pone.0086838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou B, Lei S, Xue R, Leng Y, Xia Z, Xia ZY, et al. DJ-1 overexpression restores ischaemic post-conditioning-mediated cardioprotection in diabetic rats: Role of autophagy. Clin Sci (Lond) 2017;131:1161–78. doi: 10.1042/CS20170052. doi: 10.1042/CS20170052. [DOI] [PubMed] [Google Scholar]
- 17.Tsai JP, Lee CH, Ying TH, Lin CL, Lin CL, Hsueh JT, et al. Licochalcone A induces autophagy through PI3K/Akt/mTOR inactivation and autophagy suppression enhances Licochalcone A-induced apoptosis of human cervical cancer cells. Oncotarget. 2015;6:28851–66. doi: 10.18632/oncotarget.4767. doi: 10.18632/oncotarget.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Shreder KR, Gai W, Corral S, Ferris DK, Rosenblum JS, et al. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Mansor LS, Gonzalez ER, Cole MA, Tyler DJ, Beeson JH, Clarke K, et al. Cardiac metabolism in a new rat model of type 2 diabetes using high-fat diet with low dose streptozotocin. Cardiovasc Diabetol. 2013;12:136. doi: 10.1186/1475-2840-12-136. doi: 10.1186/1475-2840-12- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang SY, Cui XL, Xue FS, Duan R, Li RP, Liu GP, et al. Combined morphine and limb remote ischemic perconditioning provides an enhanced protection against myocardial ischemia/reperfusion injury by antiapoptosis. J Surg Res. 2016;202:13–25. doi: 10.1016/j.jss.2015.12.007. doi: 10.1016/j.jss.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Durrant D, Koka S, Salloum FN, Xi L, Kukreja RC, et al. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem. 2014;289:4145–60. doi: 10.1074/jbc.M113.521062. doi: 10.1074/jbc.M113.521062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jian J, Xuan F, Qin F, Huang R. Bauhinia championii flavone inhibits apoptosis and autophagy via the PI3K/Akt pathway in myocardial ischemia/reperfusion injury in rats. Drug Des Devel Ther. 2015;9:5933–45. doi: 10.2147/DDDT.S92549. doi: 10.2147/DDDT.S92549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtis MJ, Walker MJ. Quantification of arrhythmias using scoring systems: An examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc Res. 1988;22:656–65. doi: 10.1093/cvr/22.9.656. doi: 10.1093/cvr/22.9.656. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Liu GP, Xue FS, Wang SY, Cui XL, Li RP, et al. Combined vagal stimulation and limb remote ischemic perconditioning enhances cardioprotection via an anti-inflammatory pathway. Inflammation. 2015;38:1748–60. doi: 10.1007/s10753-015-0152-y. doi: 10.1007/s10753-015-0152-y. [DOI] [PubMed] [Google Scholar]
- 26.Yang XM, Cui L, White J, Kuck J, Ruchko MV, Wilson GL, et al. Mitochondrially targeted endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res Cardiol. 2015;110:3. doi: 10.1007/s00395-014-0459-0. doi: 10.1007/s00395-014-0459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang JL, Lu JK, Chen D, Cai Q, Li TX, Wu LS, et al. Myocardial autophagy variation during acute myocardial infarction in rats: The effects of carvedilol. Chin Med J. 2009;122:2372–9. doi: 10.3760/cma.j.issn.0366-6999.2009.19.033. [PubMed] [Google Scholar]
- 28.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–88. doi: 10.1038/nm.3322. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao X, Chen A, Yang P, Song X, Liu Y, Li Z, et al. Alpha-lipoic acid protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Biochem Biophys Res Commun. 2013;441:935–40. doi: 10.1016/j.bbrc.2013.10.166. doi: 10.1016/j.bbrc.2013.10.166. [DOI] [PubMed] [Google Scholar]
- 30.Zhao G, Wang S, Wang Z, Sun A, Yang X, Qiu Z, et al. CXCR6 deficiency ameliorated myocardial ischemia/reperfusion injury by inhibiting infiltration of monocytes and IFN-γ-dependent autophagy. Int J Cardiol. 2013;168:853–62. doi: 10.1016/j.ijcard.2012.10.022. doi: 10.1016/j.ijcard.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Li R, Luo X, Zhu Y, Zhao L, Li L, Peng Q, et al. ATM signals to AMPK to promote autophagy and positively regulate DNA damage in response to cadmium-induced ROS in mouse spermatocytes. Environ Pollut. 2017;231:1560–8. doi: 10.1016/j.envpol.2017.09.044. doi: 10.1016/j.envpol.2017.09.044. [DOI] [PubMed] [Google Scholar]
- 32.Xie H, Xu Q, Jia J, Ao G, Sun Y, Hu L, et al. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Commun. 2015;458:632–8. doi: 10.1016/j.bbrc.2015.02.017. doi: 10.1016/j.bbrc.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Obara K, Noda T, Niimi K, Ohsumi Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells. 2008;13:537–47. doi: 10.1111/j.1365-2443.2008.01188.x. doi: 10.1111/j.1365-2443.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- 34.Barth S, Glick D, Macleod KF. Autophagy: Assays and artifacts. J Pathol. 2010;221:117–24. doi: 10.1002/path.2694. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nopparat C, Porter JE, Ebadi M, Govitrapong P. 1-methyl-4-phenylpyridinium-induced cell death via autophagy through a Bcl-2/Beclin 1 complex-dependent pathway. Neurochem Res. 2014;39:225–32. doi: 10.1007/s11064-013-1208-8. doi: 10.1007/s11064-013-1208-8. [DOI] [PubMed] [Google Scholar]
- 36.Ghavami S, Gupta S, Ambrose E, Hnatowich M, Freed DH, Dixon IM, et al. Autophagy and heart disease: Implications for cardiac ischemia-reperfusion damage. Curr Mol Med. 2014;14:616–29. doi: 10.2174/1566524014666140603101520. doi: 10.2174/1566524014666140603101520. [DOI] [PubMed] [Google Scholar]
- 37.Mellor KM, Reichelt ME, Delbridge LM. Autophagic predisposition in the insulin resistant diabetic heart. Life Sci. 2013;92:616–20. doi: 10.1016/j.lfs.2012.03.042. doi: 10.1016/j.lfs.2012.03.042. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–92. doi: 10.1074/jbc.M113.474650. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, Xiao J, Kang B, Zhu X, Xin N, Wang Z, et al. PI3K/SGK1/GSK3β signaling pathway is involved in inhibition of autophagy in neonatal rat cardiomyocytes exposed to hypoxia/reoxygenation by hydrogen sulfide. Exp Cell Res. 2016;345:134–40. doi: 10.1016/j.yexcr.2015.07.005. doi: 10.1016/j.yexcr.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Zheng H, Maimaitili Y, Yu J, Yu J, Guo H, Ma HP, et al. The influence of rapamycin on the early cardioprotective effect of hypoxic preconditioning on cardiomyocytes. Arch Med Sci. 2014;20:145–75. doi: 10.5114/aoms.2016.59712. doi: 10.5114/aoms.2016.59712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the diabetic heart: The importance of Akt phosphorylation. Diabetes. 2005;54:2360–4. doi: 10.2337/diabetes.54.8.2360. doi: 10.2337/diabetes.54.8.2360. [DOI] [PubMed] [Google Scholar]