

Abstract
Targeting negative regulators downstream of the T cell receptor (TCR) represents a novel strategy to improve cancer immunotherapy. Two proteins that serve as critical inhibitory regulators downstream of the TCR are diacylglycerol kinase ζ (DGKζ), a regulator of Ras and PKC-θ signaling, and Casitas b-lineage proto-oncogene b (Cbl-b), an E3 ubiquitin ligase that predominantly regulates PI(3)K signaling. We sought to compare the signaling and functional effects that result from deletion of DGKζ, Cbl-b, or both (double knockout, DKO) in T cells, and to evaluate tumor responses generated in a clinically relevant orthotopic pancreatic tumor model. We found that whereas deletion of Cbl-b primarily served to enhance NF-κB signaling, deletion of DGKζ enhanced TCR-mediated signal transduction downstream of Ras/Erk and NF-κB. Deletion of DGKζ or Cbl-b comparably enhanced CD8+ T cell functional responses, such as proliferation, production of IFNγ, and generation of granzyme B when compared with WT T cells. DKO T cells demonstrated enhanced function above that observed with single knockout T cells after weak, but not strong, stimulation. Deletion of DGKζ, but not Cbl-b, however, resulted in significant increases in numbers of activated (CD44hi) CD8+ T cells in both non-treated and tumor-bearing mice. DGKζ-deficient mice also had enhanced control of pancreatic tumor cell growth compared to Cbl-b-deficient mice. This represents the first direct comparison between mice of these genotypes and suggests that T cell immunotherapies may be better improved by targeting TCR signaling molecules that are regulated by DGKζ as opposed to molecules regulated by Cbl-b.
Keywords: Diacylglycerol kinase zeta, Cbl-b, TCR signal transduction, T cell activation, tumor immunology
Introduction
Targeting inhibitory proteins within cytotoxic T cells as a means to improve T cell activity against cancer has led to impressive clinical successes, but only in a minority of treated patients (1). The primary current focus of cancer immunotherapy is to identify ways to increase the number of responding patients while limiting the induction of autoimmunity. This has resulted in accelerated research focused on understanding the mechanisms that tumors utilize to inactivate cytotoxic T cells and developing ways to overcome this inhibition (2, 3). Much of this work has centered on interfering with the activation of CTLA-4 and PD-1, two inhibitory cell surface receptors that antagonize TCR activation (4). While monoclonal antibodies that disrupt the interactions between inhibitory receptors and their cognate ligands have led to significant disease responses in a variety of cancers, only a minority of patients derive meaningful benefit from these therapies (5), and, in the case of PD-1, those who do benefit may develop tumor escape (6). Thus, current efforts have focused on finding novel targets to improve cytotoxic T cell activity that will augment existing therapies. These include therapies that target other immune checkpoint receptors such as Tim3 and Lag3 (2), or other inhibitory receptors including the adenosine 2A receptor, prostaglandin E2 (PGE2) receptor, and TGFβ receptor complex (7, 8). Recently, interest has developed regarding the possibility of targeting intracellular inhibitory proteins to improve T cell activity against cancer, including diacylglycerol kinase ζ (DGKζ) (9, 10), and Casitas b-lineage proto-oncogene b (Cbl-b) (11), which attenuate signal transduction events downstream of the T cell receptor (TCR) and CD28. Targeting these proteins may be superior to targeting cell surface proteins, since their elimination confers simultaneous insensitivity to multiple cell surface inhibitory receptors. For instance, we previously demonstrated that DGKζ-deficient T cells demonstrate partial insensitivity to several important mediators of immune inhibition within the tumor microenvironment including PGE2 (12), adenosine (12), TGFβ (13), and PD-L1 (10).
DGKζ is one of two DGK isoforms in T cells that phosphorylate diacylglycerol (DAG). DAG is a second messenger of TCR signaling that is generated by the cleavage of phophatidyl-inositol (4,5) bisphopshate (PIP2) by PLCγ1, and is required for activation of RasGRP1 and PKC-θ. (14). DAG phosphorylation results in production of phosphatidic acid (PA) and termination of DAG-mediated signaling (15). Deletion of DGKζ from T cells results in prolonged TCR signal transduction downstream of DAG, resulting in enhanced activation of Ras, increased effector T cell proliferation, and amplified cytokine production which leads to increased anti-tumor activity against subcutaneously implanted EL4 tumors or murine mesothelioma (12, 16, 17). DGKζ has been demonstrated to be the dominant isoform of DGK in T cells as assessed by functional changes in peripheral T cells and direct measurement of phosphatidic acid after TCR stimulation (18).
Cbl-b is an E3 ubiquitin ligase that targets multiple components of the TCR signal transduction pathway for degradation through the proteasome to facilitate termination of TCR signaling (19, 20). While several proteins have been demonstrated to be targets of Cbl-b, including the TCR itself, much of the inhibition attributable to Cbl-b results from ubiquitination of the p85α subunit of PI(3)K (21, 22). Similar to T cells deficient in DGKζ, Cbl-b-deficient T cells demonstrate enhanced proliferation, cytokine production, and anti-tumor activity in numerous tumor models (23, 24). Current phase I immunotherapy trials are underway to test the utility of inhibiting Cbl-b in peripheral blood mononuclear cells in the treatment of cancer patients (Clinical trials.gov: NCT02166255, NCT03087591).
Given the predominant role for DAG on regulating Ras/Erk and PKC-θ signaling and Cbl-b for regulating PI(3)K signaling, we hypothesized that T cells deficient in DGKζ or Cbl-b would alter TCR signal transduction through distinct mechanisms, such that DGKζ-deficient T cells would demonstrate strongly enhanced Ras/ERK activation, but only modestly enhanced NF-κB activation, and Cbl-b-deficient T cells would demonstrate modestly enhanced Ras/ERK activation and strongly enhanced NF-κB activation. We found that whereas DGKζ regulated Ras/ERK activation to a greater degree than Cbl-b, CD8+ T cells deficient in either DGKζ or Cbl-b exhibited similar enhancement of NF-κB signaling. We further determined that T cells deficient in DGKζ exhibit greater control of murine pancreatic tumor growth as compared with T cells deficient in Cbl-b. Moreover, we found that T cells from DKO mice did not demonstrate enhanced tumor activity above that observed in DGKζ single knockout mice. Additionally, we identified that combined deletion of DGKζ and Cbl-b resulted in similar impairment of antigen-specific memory CD8+ T cell generation and/or maintenance compared to single knockouts. Our data suggests that targeting DGKζ could prove to be a more useful clinical approach to augment cytotoxic T cell activity against tumor than targeting Cbl-b, perhaps resulting from enhanced Ras/ERK signaling.
Materials and Methods
Mice
Mice deficient in DGKζ or Cbl-b, backcrossed to C57BL/6 mice, were described previously (16, 25). Cbl-b−/− mice were generously provided by Richard Hodes, National Institutes of Health, Washington, DC. DGKζ−/− and Cbl-b−/− mice were crossed to create DKO mice. C57BL/6 mice were purchased from Jackson Laboratories. All experiments were performed in mice 6-13 weeks old. Animal housing and experimentation were done in accordance with the Institutional Animal Care and Use Committee at the Medical College of Wisconsin.
Statistics
Statistical analysis was performed using ANOVA and unpaired two-tailed t-test with GraphPad Prism software (La Jolla, CA, USA). A p value <0.05 was considered to indicate statistical significance (*p<0.05, **p<0.01, ***p<0.001)
T cell phenotyping
Phenotypic analyses of cell surface markers in spleen, lymph node and thymus were performed on unchallenged mice at 6-8 weeks of age. Spleens were processed into cell suspensions through a 70μm strainer by a syringe plunger in RPMI with 10% FBS and then treated with ACK lysis buffer to remove red blood cells. Lymph nodes were processed using glass sides and the thymi were minced. The resulting cell suspensions were centrifuged and stained with antibodies specific for CD4 (RM4-5), CD8α (53-6.7), CD62L (MEL-14), CD44 (IM7), all BD Pharmingen (San Jose, CA), CD25 (PC61.5, ebioscience, San Diego CA), and a viability stain (Life Technologies, Carlsbad, CA). After staining for 25 minutes at 4°C cells were fixed in PBS containing 2% paraformaldehyde. All flow cytometry data was acquired on an LSRII cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Treestar, Ashland, Oregon).
Immunoblotting
T cells from mouse splenocytes were purified with magnetic naïve CD8+ T cell isolation kits using instructions provided by the manufacturer (STEMCELL Technologies, Vancouver, Canada). 4×105 purified CD8+ T cells in 100 μl serum free media were incubated with 5 μg/ml biotinylated anti-CD3 (2C11, BD Pharmingen San Jose, CA) and anti-CD28 antibodies (37.51, BD Pharmingen, San Jose, CA) for 1 min at 37° C followed by the addition of 25 μg/ml streptavidin (Thermo Scientific, Waltham, MA) for 0, 5, and 15 minutes. Reactions were terminated with 1 mL ice-cold phosphate-buffered saline, and cells were centrifuged and resuspended in 50 μl of cell lysis buffer containing 1% NP-40 and protease inhibitors. Lysates were subjected to high-speed centrifugation to remove nuclei and cell debris and total protein was quantified using Pierce BCA protein assay kit (BD Pharmingen, San Jose, CA). Normalized protein lysates were subjected to SDS-PAGE, transferred to nitrocellulose, and immunoblotted with primary antibodies from Cell Signaling (Danvers, MA) at concentrations recommended by the manufacturer.
Assessment of proliferation and cytokine production
Purified T cells from the spleen were labeled with CellTrace CFSE (carboxyfluorescein succinimidyl ester, Invitrogen, Carlsbad, CA) using protocols and reagents supplied by the manufacturer. 1×105 cells were placed in individual wells of a 96-well plate, pre-coated with 100 μl of varying concentrations of plate-bound anti-CD3 (2C11, BD Pharmingen, San Jose, CA) and 5 μg/ml anti-CD28 antibodies in PBS. Three days later, cells were surface-stained with antibodies against CD4 (RM4-5), CD8α (53-6.7), (Life Technologies, Carlsbad, CA), and subjected to flow cytometry for evaluation of CFSE dilution. Interferon gamma (IFNγ) and Interleukin-2 (IL-2) production from supernatants of individual wells was determined using an enzyme-linked immunosorbent assay (ELISA, Biolegend, San Diego, CA). To assess granzyme B production, CFSE-labeled cells were stimulated for 24 or 72 hours, surface stained as described above, permeabilized using Cytofix/Cytoperm kit (BD Pharmingen, San Jose, CA), and then stained with flurochrome- labeled granzyme B antibody (GB11, Life Technologies, Carlsbad, CA) at 4° C for 30 minutes prior to flow cytometric analyses. Naïve CD8+ T cell experiments were performed with 5×104 cells per stimulation. Cells were stimulated in the presence or absence of 100U/ml IL-2 (Peprotech, Rocky Hill, NJ) or 5 μg/ml anti-IL-2 (eBioscience, San Diego, CA).
Orthotopic injection of KPC1242 tumors
The KPC1242 cell line, derived from a spontaneous pancreatic tumor that arose in a KPC mouse, was a generous gift of David Tuveson (Cold Spring Harbor Laboratories, Cold Spring, New York). 1×106 KPC1242 tumor cells were injected orthotopically into the pancreas as previously described (26). 15 days later, mice were euthanized, tumor weight was measured, and T cells in the spleen, ascites and tumor were analyzed. Spleens and tumors were mashed through a 70 μm strainer by a syringe plunger in RPMI with 10% FBS. Spleens were than treated with ACK lysis buffer to remove red blood cells. Splenocytes, ascites and tumor cells were stained with a viability dye, antibodies against CD4, CD8α, CD62L (MEL-14), CD44 (IM7), and CD25 (PC61.5, eBioscience, San Diego, CA), and analyzed by flow cytometry. Additionally, an aliquot of 4×106 splenocytes was stained for Foxp3 expression using methods and reagents provided by the manufacturer (eBioscience, San Diego, CA), prior to flow cytometric analyses.
LCMV infection
Mice were infected intraperitoneally with 2×106 plaque forming units (PFU) LCMV (strain Armstrong) in PBS. Blood was collected on weeks 1, 2, 4, and 6 to assess virus-specific T cell expansion and contraction. Blood samples were underlayed with histopaque (Sigma, St. Louis, MO) and centrifuged at 2000 rpm for 15 minutes. Lymphocytes were harvested from the gradient interface and washed before further analysis. Cells were surface stained with antibodies against CD8α, CD62L, CD44, CD27 (LG.7F9, eBioscience, San Diego, CA), KLRG-1 (2F1), and CD127 (A7R34, Biolegend, San Diego, CA), along with viral epitope-specific GP33 tetramer for 25 min at 4° C. Cells were than analyzed by flow cytometry. Six weeks after infection, mice were euthanized, and spleens and inguinal lymph nodes isolated. Cells were stained with the same panel as described for blood samples with the addition of fluorochrome-labeled tetramers capable of binding to TCR clones GP276 or NP369, which were generated as previously described (27). For intracellular cytokine staining, lymphocytes were cultured in 96-well flat-bottom plates at 1×106 cells/well in 200 μl RPMI 1640 supplemented with 10% FBS in the presence or absence of GP33, GP276 and NP369 peptides. Stimulations were performed for 5 hours at 37° C in the presence of Golgi-plug (1:1000; BD Pharmingen, San Jose, CA), surface stained as described above, permeabilized, and stained with antibodies against IFNγ (XMG1.2, BD Pharmingen, San Jose, CA). Flow cytometry was performed and cells analyzed as described above.
Results
T cells deficient in DGKζ and Cbl-b demonstrate differential enhancement of Erk and NF-κB signaling
Stimulation of the TCR results in cleavage of PI(4,5)P2 to form IP3, which induces intracellular calcium flux, and DAG, which binds to RasGRP1 and PKC-θ to contribute to activation of these proteins (28). DGKζ phosphorylates DAG terminating DAG-mediated protein activation. Cbl-b targets the p85α subunit of PI(3)K for degradation resulting in termination of substrate activation downstream of PI(1,4,5)P3 (21). Previous work demonstrated that loss of either DGKζ or Cbl-b results in enhanced activation of both Erk and NF-κB pathways in T cells after stimulation of the TCR (29); however, the relative intensity of signaling changes downstream of the TCR in T cells after loss of each protein has not been directly compared. To define the signaling changes resulting from each genotype, we isolated naïve CD8+ T cells from mice deficient in DGKζ, Cbl-b, or both (double knockout, DKO), stimulated the cells through their TCRs, and evaluated phosphorylation of Erk and phosphorylation and degradation of IκBα. We observed that Erk phosphorylation was modestly enhanced in Cbl-b−/− CD8+ T cells when compared with wild type (WT) T cells, but was greatly enhanced in DGKζ−/− and DKO CD8+ T cells when compared either with Cbl-b−/− or WT CD8+ T cells (Fig. 1a). This indicates that DGKζ plays a greater role in limiting Erk activation in CD8+ T cells than Cbl-b. We also observed that IκBα phosphorylation and degradation were enhanced in DGKζ−/−, Cbl-b−/− and DKO CD8+ T cells, indicating that DGKζ and Cbl-b are important regulators of NF-κB activation (Fig. 1b). These data indicate that DGKζ and Cbl-b differentially regulate TCR signal transduction.
Figure 1. DGKζ and Cbl-b deficient T Cells have enhanced ERK1/2 and IκBα phosphorylation.
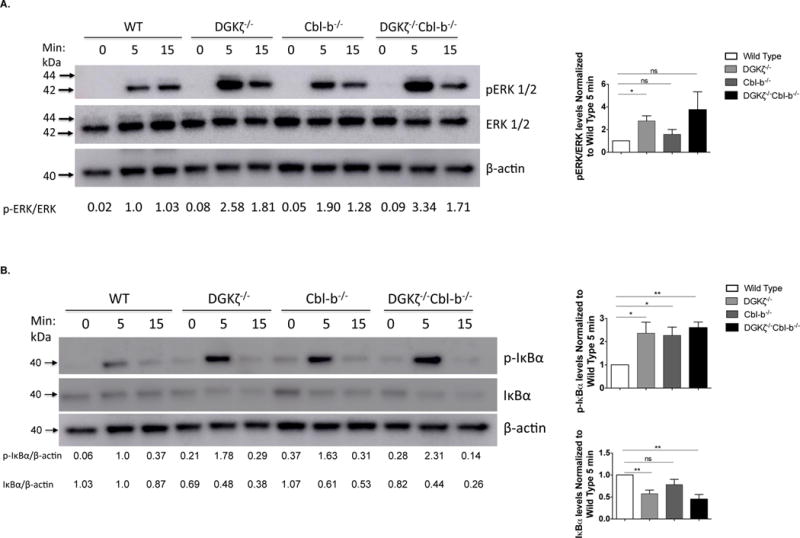
4×105 STEMCELL-purified WT, DGKζ−/−, Cbl-b−/−, or DKO naïve CD8+ T cells were incubated with biotinylated α–CD3 (5ug/ml) and α–CD28 (5ug/ml) and cross-linked with streptavidin (25ug/ml) for the indicated times. Lysates were immunoblotted for protein levels of (A) phosphorylated ERK (p-ERK), total ERK (tERK) and β-actin or (B) phosphorylated IκBα (p-IκBα), total IκBα (IκBα) and β-actin. Relative band intensities are indicated below each lane. Representative blots are depicted from one of three independent iterations and ImageJ quantification of the three iterations are displayed ± SEM in the graphs.
DGKζ, Cbl-b, and DKO mice exhibit normal T cell development and peripheral T cell numbers, but decreased naïve CD8+ T cells
Previous studies on DGKζ−/− and Cbl-b−/− transgenic mice have shown phenotypically normal thymic development, but decreased presence of naïve T cells (e.g., CD44loCD62Lhi) in peripheral immune organs (17). The mechanism underlying a decrease in naïve T cells is thought to result from several factors including increased non-TCR-mediated (e.g., homeostatic) T cell proliferation and increased TCR-mediated activation (12, 17). To compare T cell phenotypes between DGKζ−/− and Cbl-b−/− mice, and to evaluate DKO mice, we isolated thymi, lymph nodes (LN), and spleens from mice of each genotype, and determined expression of T cell surface markers. In the thymus, expression of CD4 and CD8 varies during T cell maturation, such that cells initially do not express CD4 or CD8 (double negative, DN) after arriving to the thymus, then express both CD4 and CD8 (double positive, DP) after TCR rearrangement, and finally downregulate either CD4 or CD8 to generate mature single positive (SP) cells that exit into the periphery. We observed no significant differences between percentages of DN and DP populations in the thymi from mice of each genotype (Fig. 2a), however some changes in absolute number of single positive populations were decreased in DKO and DGKζ−/− mice, of unclear significance. Overall, the results indicate that T cell development is grossly normal in mice that lack DGKζ, Cbl-b, or both.
Figure 2. Loss of DGKζ and Cbl-b results in a greater percentage of splenic CD8+ T cells with an activated phenotype.
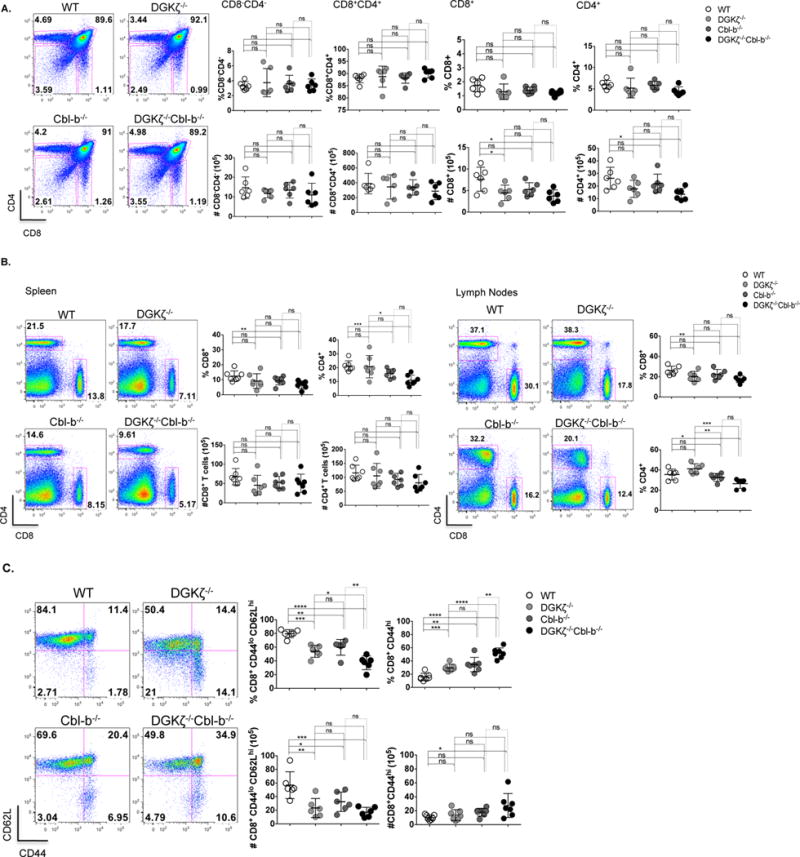
(A) Thymocytes, (B) splenocytes and lymph node cells from WT and knockout mice were stained for cell surface markers CD8 and CD4. Percentage and absolute number were determined for different cell populations. (C), Splenocytes were stained for cell surface markers CD8 and CD4, and CD62L and CD44 after gating for CD8+ cells, and the percentage and absolute number of CD8+CD44hi cells was determined. (n=7 for each group).
We next evaluated T cell numbers in peripheral immune organs and found comparable numbers of CD8+ and CD4+ T cells in spleens of WT mice compared to single or DKO mice with some modest differences in CD8+ and CD4+ T cell percentages between genotypes (Fig. 2b). To test the activation state of peripheral T cells, we evaluated for the presence of two surface markers, CD44 and CD62L. Whereas naïve T cells express low levels of CD44 and high levels of CD62L, acutely activated CD8+ T cell express high levels of CD44 and low levels of CD62L, and memory CD8+ T cells express high levels of both CD44 and CD62L. Importantly, evaluation of activation markers on peripheral CD8+ T cells showed decreased numbers of naïve (CD44loCD62Lhi) T cells in splenocytes of single KO mice (Fig. 2c) as has been previously observed (17), that was enhanced in DKO mice. A similar trend was observed in activated CD4+ T cells (Sup. Fig. 1). Together, these data indicate that peripheral T cell numbers are relatively preserved in single or DKO mice compared with WT mice, but that less naïve CD8+ T cells are present in all genotypes when compared to WT.
T cell effector functions are enhanced in DKO mice compared with DGKζ−/− or Cbl-b−/− mice under sub-optimal stimulatory conditions
Deletion of DGKζ or Cbl-b is known to enhance the effector functions of CD8+ T cells (16, 17, 19, 25). To directly compare functional changes in DGKζ−/− and Cbl-b−/− mice and to determine if DKO CD8+ T cell demonstrated an enhanced functional phenotype relative to single knockouts, we stimulated purified T cells obtained from spleens of each genotype with limiting dilutions of anti-CD3 and/or anti-CD28, and measured CD8+ T functions such as proliferation, production of effector cytokine IFNγ, and generation of granzyme B. We found that, relative to WT T cells, single knockout T cells and DKO T cells demonstrated comparable enhancement of proliferation with high amounts (0.3 μg/mL) of anti-CD3 stimulation as assessed by dilution of CFSE (Fig. 3a, bottom panels), but proliferation occurred in DKO T cells only, after stimulation with low amounts (0.1 μg/mL) of anti-CD3 (Fig. 3a, middle panels). Further, while we could detect enhancement of granzyme B production in DKO T cells relative to WT, DGKζ−/−, or Cbl-b−/− T cells at early time points (Fig. 3b, upper panels), we did not observe enhanced granzyme B production in DKO relative to DGKζ−/−, or Cbl-b−/− T cells at later time points (Fig. 3b, lower panels), at higher or lower concentrations of anti-CD3 antibody, or with addition of anti-CD28 antibody (Sup. Fig. 2b). Lastly, we observed that production of IFNγ was generally greater in DKO and Cbl-b−/− T cells relative to DGKζ−/− T cells, which was increased above the level observed in WT T cells (Fig. 3c). Previous work with DGKζ−/− CD8+ T cells showed that increased TCR-mediated proliferation relative to WT T cells can be largely attributed to increased IL-2 production (17, 25). To determine if this was also true for Cbl-b−/− and DKO T cells, we isolated naïve CD8+ T cells from mice deficient in DGKζ, Cbl-b, or both prior to stimulation with anti-CD3 (Fig. 3d, e, f). We found that, similar to the responses observed after stimulation of mixed T cell populations, stimulated naïve CD8+ T cells from DGKζ−/−, Cbl-b−/− and DKO mice demonstrated enhanced proliferation compared to WT T cells (Fig. 3d). However, DGKζ−/− CD8+ T cells were less responsive to low amounts (0.3 μg/mL) of anti-CD3 stimulation than similarly stimulated Cbl-b−/− and DKO CD8+ T cells (Fig. 3d) or to DGKζ−/− CD8+ T cells present in a mixed T cell population (Fig. 3a). The addition of IL-2, as expected, enhanced the proliferation of WT T cells, but not completely to levels observed with DGKζ−/− CD8+ T cells + IL-2 or to Cbl-b−/− and DKO CD8+ T cells (Fig. 3d). Furthermore, an increased amount of IL-2 was present in the supernatants of anti-CD3 stimulated DKO and Cbl-b−/− stimulated CD8+ T cells relative to DGKζ−/− CD8+ T cells, which was increased relative to WT CD8+ T cells (Fig. 3e). Lastly, we determined the amount of IFNγ produced after administration of exogenous IL-2 or anti-IL-2 to naïve CD8+ T cells. As seen with stimulated T cells (Fig. 3c), Cbl-b−/− and DKO CD8+ T cells produced higher levels of IFNγ compared to DGKζ−/− or WT CD8+ T cells (Fig. 3f) in a manner that was enhanced by the presence of additional IL2 (Fig. 3f). In contrast, the addition of anti-IL-2 inhibited the production of IFNγ in all genotypes tested (Fig. 3f). Together, these data indicate that the deletion of DGKζ or Cbl-b confer different effects on CD8+ T cell functions, and that combined deletion of the proteins decrease the threshold for inducing CD8+ T cell functions after TCR activation.
Figure 3. Enhanced proliferation and cytokine production in DGKζ and Cbl-b deficient mice.
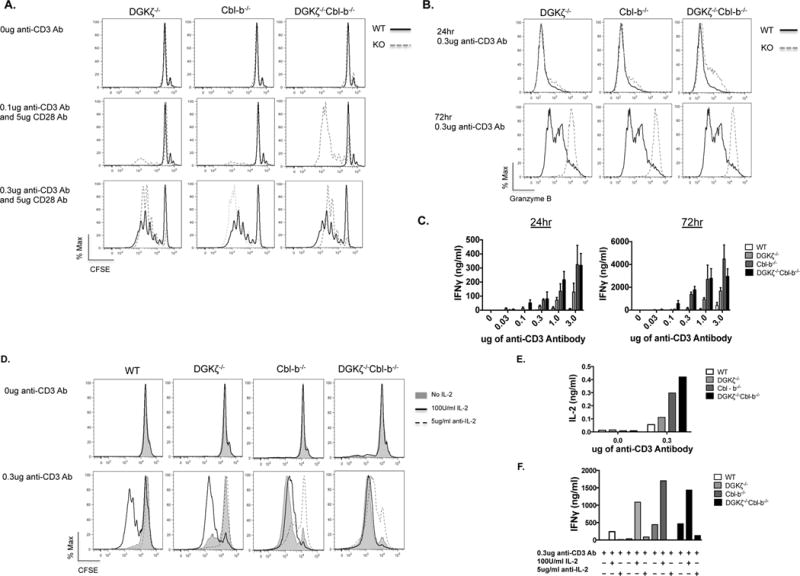
(A) 1×105 MACS-purified T cells were CFSE-labeled and incubated with varying concentrations of plate-bound anti–CD3 and 5ug/ml anti–CD28 antibodies at 37°C for 72 hours. Cells were surface-stained for viability, CD8 and CD4 and evaluated for proliferation as assessed by dilution of CFSE within CD8+–gated cells. (B) Using ELISA, IFNγ was measured from culture supernatants of (A) after 24 or 72 hours of stimulation. (C) Intracellular granzyme B was measured from purified T cells gated from CD8+ populations after 24 and 72 hours of stimulation with 0.3 μg/ml anti–CD3 stimulation. Representative of three iterations. For (D), (E), and (F), 5×104 STEMCELL-purified naïve CD8+ T cells were incubated with 0.3ug/ml anti-CD3 antibody at 37°C for 72hrs. Cells were surface-stained for viability and evaluated for proliferation as assessed by dilution of CFSE within CD8+–gated cells (D). (E, F) Using ELISA, IL-2 and IFNγ production were measured from culture supernatants of (D) after 72 hours of stimulation. Representative of two iterations.
Enhanced control of KPC1242 pancreatic tumor in DGKζ−/− and DKO mice relative to Cbl-b−/− or WT mice
DGKζ−/− mice and T cells deficient in DGKζ are known to demonstrate enhanced clearance of tumors in subcutaneous models (12, 17, 30, 31). Similarly, Cbl-b−/− mice and T cells lacking Cbl-b demonstrate improved control of subcutaneously implanted tumors (23) and disseminated leukemia (32), along with decreased spontaneous tumor formation in ATM−/− mice (23) and ultraviolet B-treated mice (24). However, tumor phenotypes in DGKζ−/− or Cbl-b−/− mice have not been directly compared. To evaluate tumor growth in mice of each genotype, we used an orthotopic implant model derived from a spontaneous pancreatic tumor arising in a C57BL/6 KPC mouse (33), which express mutant forms of K-Ras and p53 selectively in pancreatic tissue. We chose this model because (i) it more closely recapitulates features of human pancreatic cancer than do subcutaneously implanted tumors, (ii) it can be used in immunocompetent mice to permit assessment of immune responses, and (iii) the cells grow in vivo with predictable kinetics (34). 15 days after orthotopic implantation, we detected a trend toward smaller tumors in Cbl-b−/− mice when compared with WT mice, and significantly smaller tumors in DGKζ−/− and DKO mice, including a complete absence of tumors in several DGKζ−/− and DKO mice (Fig. 4a). To assess whether changes in T cell numbers within tumor could be responsible for the observed differences in tumor size, we processed spleen and tumor from mice and calculated percentages of CD4+ and CD8+ T cells. We found that the percentages of T cells were similar in tumors among all genotypes, however DKO mice were incidentally noted to have decreased amounts of splenic total CD8+ T cells (Fig 4b). Note that tumor could not be evaluated in all DGKζ−/−or DKO mice, as tumor was not present in approximately 50% of animals. We then assessed activation of status of T cells. We observed an increased percentage of activated (CD44hi) CD8+ T cells in tumors of DGKζ-deficient mice relative to WT mice (Fig. 4c). Further, consistent with T cell phenotypes in non-tumor bearing mice (Fig. 2), we observed an increased percentage of spleen-derived CD8+ T cells expressing high levels of the activation marker CD44 in DGKζ−/− or DKO mice that had been inoculated with tumor when compared with Cbl-b−/− or WT mice (Fig. 4c). In a reciprocal manner, tumor-inoculated DGKζ−/− or DKO mice demonstrated a decrease in percentages of naïve (CD44hiCD62Llo) CD8+ T cells within the spleen when compared with WT mice and, in the case of DGKζ−/− mice, when compared with Cbl-b−/− mice (Fig. 4c). We also evaluated the presence of CD4+ regulatory T cells (Tregs) within the spleen since regulatory T cells are known to play an important role in limiting anti-tumor immunity, and because an increase in natural Tregs has been reported in DGKζ−/− mice (29). Consistent with prior reports, an increase in percentages of splenic Tregs was observed in DGKζ−/− and DKO mice in comparison to WT or Cbl-b−/− mice (Fig. 4d). Collectively, these data indicate that DGKζ−/− mice exert improved control of orthotopically implanted KPC1242 tumors than WT mice, in a manner that may result from changes in the number of intra-tumoral activated CD8+ T cells in DGKζ−/− mice.
Figure 4. Deletion of DGKζ and Cbl-b does not result in a greater tumor response than deletion of DGKζ alone.
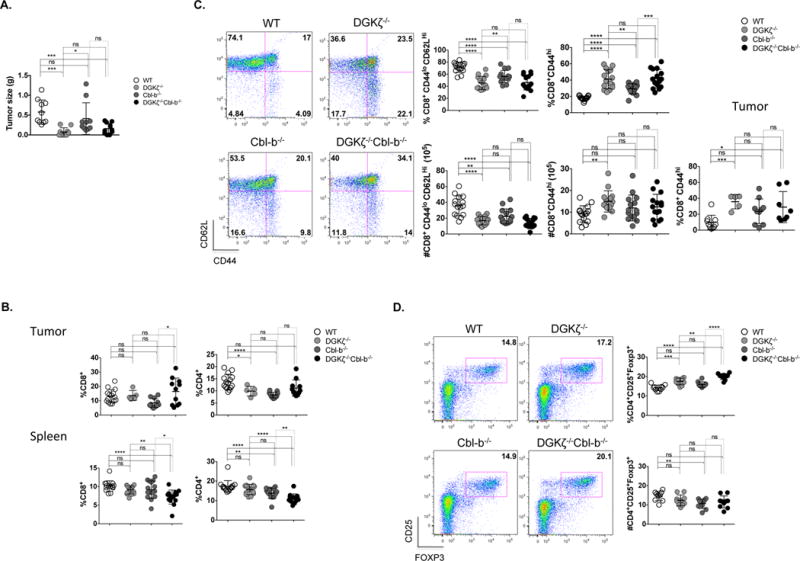
1×106 KPC1242 tumor cells were injected orthotopically into the pancreas. 15 days later, mice were euthanized; tumor presence was assessed and measured (A). Tumors were evaluated for the presence of infiltrating T cells and the spleen was analyzed for CD4+ and CD8+ T cell percentages (B). The activation phenotype (CD44hi) of CD8+ T cells was examined in spleen and tumor (C) along with the presence of regulatory T cells in the spleen (D). (A), Data from two pooled experiments, and (B, C, and D), data from 3 pooled experiments. (C), Tumor data from two pooled experiments, n=5 in each group).
Inefficient CD8+ T cell memory development in mice deficient in DGKζ or Cbl-b
Whereas acute CD8+ T cell effector responses are enhanced in DGKζ deficient mice after infection with intracellular pathogens, persistent memory formation is impaired, most dramatically when both DGKζ and DGKα, the other isoform of DGK that metabolizes DAG downstream of the TCR in T cells, are deleted (35, 36). To determine the impact of Cbl-b deletion on memory formation in conjuction with DGKζ-deficiency, we inoculated WT, DGKζ−/−, Cbl-b−/−, and DKO mice with LCMV (strain Armstrong). Consistent with prior reports on effector cells (35), we observed that, among CD8+ T cells specific for the dominant epitope of LCMV (GP33), the percentages of short lived effector cells (KLRG1+CD127-) were increased, and the percentages of memory precursor cells (KLRG1+CD127+) were decreased in the peripheral blood of DGKζ−/−, Cbl-b−/−, and DKO mice relative to WT mice after acute infection (Fig. 5a). While similar numbers of total splenic gp33-LCMV-specific T cells were observed between genotypes (Fig. 5b), the distribution of gp33-LCMV-specific CD8+ T cell effector and memory subsets was altered, such that there were significantly fewer percentage of short-term effector T cells and reciprocal changes in memory precursor cells among gp33-specifc CD8+ T cells in WT relative to DKO mice (Fig. 5c), with a trend in changes in absolute cell numbers, consistent with temporal data from peripheral blood (Fig. 5a). As has been noted by others (35), T cells from infected DGKζ−/− or DKO mice demonstrated enhanced IFNγ production after stimulation with LCMV-specific peptides gp33 and np396 (Fig. 5d), and T cells from infected DGKζ−/− mice demonstrated enhanced IFNγ production after stimulation with LCMV-specific peptide gp276 relative to T cells from WT mice (Fig. 5d). Peptide stimulation of T cells from Cbl-b−/− infected mice were similar in their ability to produce IFNγ relative to WT controls (Fig. 5d), as previously seen in mice injected with a low dose of LCMV Docile (37). These data indicate that enhanced TCR signaling resulting either from deletion of DGKζ or Cbl-b results in skewed effector versus memory development after infection with LCMV. Deletion of DGKζ or Cbl-b also results in differential effects on memory CD8+ T cell expansion and effector cytokine production.
Figure 5. DKO CD8+ T cells demonstrate a strong effector response, but altered memory differentiation and maintenance after LCMV infection.
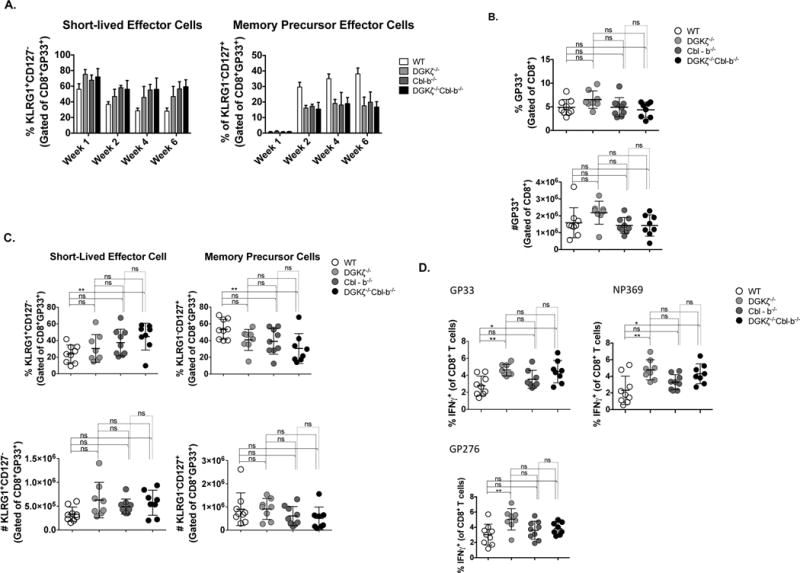
WT and knockout mice were infected with LCMV Armstrong. (A) Short lived effector (KLRG1+CD127−) and memory precursor (KLRG1−CD127+) cells were examined from blood at 1, 2, 4, and 6 weeks after infection. 6 weeks after infection splenocytes were analyzed for gp33-LCMV-specific total CD8+ T cells (B) or short-lived effector cells and memory precursor effector cells (C). (D) Splenocytes were stimulated in vitro with GP33, NP369, and GP276 peptides. Graphs show the frequency of IFNγ effector CD8+ T cells. (Data from two pooled experiments, n=8-9 in each group).
Discussion
Recently, there has been progress in the treatment of patients with malignancy through targeting receptors on the surface of CD8+ T cells that inhibit their activity within the highly suppressive tumor microenvironment. Clinically, these efforts have been most successful when targeting the inhibitory receptor PD-1, or its ligand PD-L1, with overall response rates between 20-35% for malignancies with favorable characteristics including high expression of PD-L1 on tumor cells, high mutational burden within tumor cells, or robust CD8+ T cell infiltration within a tumor prior to treatment. A current focus of immuno-oncology research is to identify additional T cell targets that are able to improve anti-tumor efficacy while maintaining a side effect profile suitable for clinical development. Whereas the vast majority of investigations in the field have focused on inhibitory cell surface proteins such as CTLA4, Tim3, Lag-3, TGFβ-R and others, much less is known about intracellular regulators that could be targeted to enhance T cell-mediated immune responses. The studies described herein directly compared the functional characteristics of T cells deficient in two intracellular negative regulators of TCR signal transduction, DGKζ and Cbl-b. We chose these two proteins since they regulate TCR signaling through distinct mechanisms. DGKζ, a lipid kinase, phosphorylates the second messenger DAG, terminating DAG-mediated activation of RasGRP1 and PKC-θ, whereas Cbl-b, an E3 ubiquitin ligase, facilitates ubiquitination and subsequent degradation of the p85 subunit of PI(3)K. Previous studies have evaluated numerous aspects of T cell biology in the two models (16–18, 25, 38), but the signaling and functional effects of DKGζ deficiency and Cbl-b deficiency in T cells had not been directly compared.
In the studies presented herein, we sought to directly compare signaling downstream of the TCR resulting from deletion of DGKζ or Cbl-b. Although other reports suggest that T cells deficient in either DGKζ or Cbl-b demonstrate enhanced signal transduction downstream of the TCR (14, 16, 17, 25, 39), we show that DGKζ is more important than Cbl-b in limiting Erk activation in T cells. However, DGKζ and Cbl-b both play a role in the negative regulation of NF-κB. We also show that deletion of both DGKζ or Cbl-b results in enhanced signaling events reflective of each individual deletion, but that little synergy is apparent among downstream pathways. In order to evaluate physiological alterations resulting from deletion of DGKζ or Cbl-b, we first sought to evaluate T cell development among wt mice and each of the three genetic deletion models. As previously reported, we observed no changes in T cell development in DGKζ−/− and Cbl-b−/− mice (16, 25), and found that DKO mice also displayed grossly normal T cell development. These characterizations were requisite since work by others demonstrated that expression of a constitutively active form of another DGK allele important in regulating DAG levels in T cells, DGKα, resulted in developmental blockade of T cells between progression from DN to DP (40). Furthermore, although T cell development in the thymus appeared normal, the distribution of T cells expressing markers consistent with a naïve versus activated state was altered resulting in decreased numbers of naïve T cells in peripheral lymphoid tissues of DGKζ−/−, Cbl-b−/− and DKO mice compared to WT mice. This alteration likely can be attributed to a combination of decreased persistence of memory T cells (41) and enhanced homeostatic proliferation resulting from increased strength of signal downstream of the TCR (16).
A major goal of these studies was to compare the functional consequences of enhanced signaling through Ras-Erk/AP-1 versus NF-κB downstream of TCR stimulation. As previously seen (16, 19, 42), CD8+ T cells deficient in DGKζ or Cbl-b demonstrated comparable increases in proliferation compared to WT T cells. More specifically, we found that T cells from DGKζ−/− and Cbl-b−/− mice exhibit similar levels of enhanced proliferation at low levels of TCR stimulation. However, stimulated naïve DGKζ−/− CD8+ T cells are less responsive to low levels of anti-CD3 stimulation than Cbl-b−/− CD8+ T cells. This is likely due to enhanced production of IL-2 and possibly related to a decreased dependence on CD28 engagement for full stimulation in Cbl-b deficient CD8+ T cells (25). In contrast, proliferation differences seen between DGKζ−/− or Cbl-b−/− CD8+ T cells mostly dissipate upon administration of exogenous IL-2, which indicates that the enhanced production of IL-2 in stimulated Cbl-b−/− CD8+ T cells is the primary factor responsible for the observed differences between the two genotypes. Consistent with previously reported analyses, we also observed enhanced IFNγ and granzyme B production in DGKζ and Cbl-b-deficient T cells. Further, DKO T cells demonstrated enhanced production of IFNγ compared with WT T cells, in a manner quantitatively similar to Cbl-b-deficient T cells. When we isolated naïve CD8+ T cells and stimulated with low level of anti-CD3, we also observed enhanced IFNγ production in Cbl-b−/− and DKO T cells relative to DGKζ−/− or wt T cells. This is likely due to differences in cell number resulting from dissimilar proliferation among genotypes (Fig. 3d,e,f). After addition of exogenous IL-2, the levels of IFNγ production in culture media of stimulated CD8+ T cells increased in all genotypes, again likely attributable to cell number (Fig. 3f). However, the absolute amount of IFNγ did not completely normalize between DGKζ−/− and Cbl-b−/− CD8+ T cells. Since cell number was similar between the two genotypes (Fig. 3d), this indicates that at least some intrinsic difference in cytokine production is present between DGKζ−/− and Cbl-b−/− CD8+ T cells. In contrast to addition of exogenous IL-2, addition of anti-IL2 resulted in relatively little proliferation and thus little production of IFNγ in any of the genotypes (Fig. 3f). As an additional finding, we observed little stimulation of purified naïve DGKζ-deficient CD8+ T cells, relative to Cbl-b-deficient or DKO cells (compare Fig. 3a with Fig. 3d), at limiting dilutions of anti-CD3 (0.3 μg/mL), when compared with DGKζ-deficient T cells in a mixed population. This suggests that DGKζ-deficient CD8+ T cells are more dependent on help from CD4+ T cells at limiting dilutions of anti-CD3 stimulation. Together, these data indicate that DGKζ and Cbl-b may differentially regulate the threshold of cytokine production in CD8+ T cells.
To evaluate the functional impact of deleting of DGKζ, Cbl-b or both molecules (DKO) on T cell immune responses, we evaluated two model systems: tumor growth in a model of pancreatic cancer, and memory T cell responses in LCMV infection. First, we tested T cell responses to tumor in the setting of DGKζ deficiency, Cbl-b deficiency or both. We observed enhanced tumor rejection in DGKζ-deficient mice and DKO mice relative to WT or Cbl-b-deficient mice. The decreased tumor size and enhanced tumor rejection in DGKζ-deficient mice indirectly suggests that enhanced Erk signaling may be superior to enhanced NF-κB activation in facilitating T cell activity against tumor, especially since DKO mice did not exhibit improved tumor control relative to DGKζ−/− mice. This provides evidence that targeting DGKζ may prove superior to targeting Cbl-b in cancer immunotherapies, and that combined targeting may not be beneficial. In our second model, we tested CD8+ T cell responses to LCMV infection. Results demonstrated that mice deficient in DGKζ, Cbl-b or both molecules generate impaired memory T cell responses relative to WT mice, confirming previous reports with mice deficient in DGKζ (41) or Cbl-b (19). This implies that TCR signal strength is titrated to balance generation of effector responses with sustained memory (43, 44), such that decreased TCR signaling results in poor acute effector memory expansion, and enhanced TCR signaling results in diminished establishment of memory. The mechanistic basis of decreased memory establishment secondary to enhanced TCR signaling is an area of ongoing investigation. DKO mice however, have a decreased percentage of memory precursor cells and an increase in short lived effector cells compared with WT mice. These data are consistent with previous work in DGKζ−/− mice, where others identified decreased percentages and total numbers of splenic CD8TetG+ (tetramer of H-2Dbgp33-41) cells four months post infection (35).
Together our data support the notion that targeting DGKζ, a negative regulator of T cell activation, or other regulators of Erk activation, could be useful in the treatment of human malignancy, and that additional preclinical studies of intracellular “brakes” on T cell activation are warranted.
Supplementary Material
Acknowledgments
The authors wish to thank Joseph Barbieri, Thomas Zahrt, and Carol Williams for their suggestions and Sandra Holzhauer for technical assistance.
Funding sources:
National Institutes of Health (Grant K08-CA151893) (MJR)
Institutional Award from the American Cancer Society (MJR)
Footnotes
Conflict of Interest: The authors report no conflict of interest with the data presented in this manuscript.
References
- 1.Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125(9):3335–3337. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what“s here, what”s next? Curr Opin Immunol. 2015;33:23–35. doi: 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen J, Sznol M. Therapeutic combinations of immune-modulating antibodies in melanoma and beyond. Semin Oncol. 2015;42:488–494. doi: 10.1053/j.seminoncol.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 6.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Moreno B Homet, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TNM, Lo RS, Ribas A. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. 2012;61:917–926. doi: 10.1007/s00262-011-1155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antonia SJ, Vansteenkiste JF, Moon E. Immunotherapy: Beyond Anti-PD-1 and Anti-PD-L1 Therapies. Am Soc Clin Oncol Educ Book. 2016;35:e450–8. doi: 10.1200/EDBK_158712. [DOI] [PubMed] [Google Scholar]
- 9.Riese MJ, Moon EK, Johnson BD, Albelda SM. Diacylglycerol Kinases (DGKs): Novel Targets for Improving T Cell Activity in Cancer. Front Cell Dev Biol. 2016;4:108. doi: 10.3389/fcell.2016.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jing W, Gershan JA, Holzhauer S, Weber J, Palen K, McOlash L, Pulakanti K, Wesley E, Rao S, Johnson BD, Riese MJ. T cells deficient in diacylglycerol kinase zeta are resistant to PD-1 inhibition and help create persistent host immunity to leukemia. Cancer Res. 2017;77(20):5676–5686. doi: 10.1158/0008-5472.CAN-17-1309. [DOI] [PubMed] [Google Scholar]
- 11.Wallner S, Gruber T, Baier G, Wolf D. Releasing the brake: targeting Cbl-b to enhance lymphocyte effector functions. Clin Dev Immunol. 2012;2012:692639. doi: 10.1155/2012/692639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riese MJ, Wang LCS, Moon EK, Joshi RP, Ranganathan A, June CH, Koretzky GA, Albelda SM. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73(12):3566–77. doi: 10.1158/0008-5472.CAN-12-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arumugam V, Bluemn T, Wesley E, Schmidt AM, Kambayashi T, Subramaniam M, Riese MJ. TCR signaling intensity controls CD8+ T cell responsiveness to TGF-beta. J Leukoc Biology. 2015;98(5):703–712. doi: 10.1189/jlb.2HIMA1214-578R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi RP, Koretzky GA. Diacylglycerol kinases: regulated controllers of T cell activation, function, and development. Int J Mol Sci. 2013;14:6649–6673. doi: 10.3390/ijms14046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shulga YV, Loukov D, Ivanova PT, Milne SB, Myers DS, Hatch GM, Umeh G, Jalan D, Fullerton MD, Steinberg GR, Topham MK, Brown HA, Epand RM. Diacylglycerol kinase delta promotes lipogenesis. Biochemistry. 2013;52:7766–7776. doi: 10.1021/bi401178y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong XP, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, Shen H, Koretzky GA. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003;4:882–890. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 17.Riese MJ, Grewal J, Das J, Zou T, Patil V, Chakraborty AK, Koretzky GA. Decreased diacylglycerol metabolism enhances ERK activation and augments CD8+ T cell functional responses. J Biol Chem. 2011;286:5254–5265. doi: 10.1074/jbc.M110.171884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi RP, Schmidt AM, Das J, Pytel D, Riese MJ, Lester M, Diehl JA, Behrens EM, Kambayashi T, Koretzky GA. The ζ isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Science Signaling. 2013;6(303):ra102. doi: 10.1126/scisignal.2004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, Le J, Ohashi PS, Sarosi I, Nishina H, Lipkowitz S, Penninger JM. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 20.Gruber T, Hermann-Kleiter N, Hinterleitner R, Fresser F, Schneider R, Gastl G, Penninger JM, Baier G. PKC-theta modulates the strength of T cell responses by targeting Cbl-b for ubiquitination and degradation. Science Signaling. 2009;2(76):ra30. doi: 10.1126/scisignal.2000046. [DOI] [PubMed] [Google Scholar]
- 21.Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem. 2001;276:4872–4878. doi: 10.1074/jbc.M008901200. [DOI] [PubMed] [Google Scholar]
- 22.Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol. 2001;2:870–875. doi: 10.1038/ni0901-870. [DOI] [PubMed] [Google Scholar]
- 23.Chiang JY, I, Jang K, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007;117:1029–1036. doi: 10.1172/JCI29472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, Beissert S, Melief CJM, Penninger JM. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007;204:879–891. doi: 10.1084/jem.20061699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 26.Roy I, McAllister DM, Gorse E, Dixon K, Piper CT, Zimmerman NP, Getschman AE, Tsai S, Engle DD, Evans DB, Volkman BF, Kalyanaraman B, Dwinell MB. Pancreatic Cancer Cell Migration and Metastasis Is Regulated by Chemokine-Biased Agonism and Bioenergetic Signaling. Cancer Res. 2015;75:3529–3542. doi: 10.1158/0008-5472.CAN-14-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt AM, Zou T, Joshi RP, Leichner TM, Pimentel MA, Sommers CL, Kambayashi T. Diacylglycerol kinase ζ limits the generation of natural regulatory T cells. Science Signaling. 2013;6(303):ra101. doi: 10.1126/scisignal.2004411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang E, Singh BK, Paustian AMS, Kambayashi T. Diacylglycerol Kinase ζ Is a Target To Enhance NK Cell Function. J Immunol. 2016;197:934–941. doi: 10.4049/jimmunol.1600581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrada E, Liébana R, Mérida I. Diacylglycerol Kinase ζ Limits Cytokine-dependent Expansion of CD8(+) T Cells with Broad Antitumor Capacity. EBioMedicine. 2017;19:39–48. doi: 10.1016/j.ebiom.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest. 2010;120:3722–3734. doi: 10.1172/JCI41991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 34.Tseng WW, Winer D, Kenkel JA, Choi O, Shain AH, Pollack JR, French R, Lowy AM, Engleman EG. Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin Cancer Res. 2010;16:3684–3695. doi: 10.1158/1078-0432.CCR-09-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin J, O’Brien TF, Grayson JM, Zhong X-P. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J Immunol. 2012;188:2111–2117. doi: 10.4049/jimmunol.1102265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Zhang P, Krishna S, Wang J, Lin X, Huang H, Xie D, Gorentla B, Huang R, Gao J, Li QJ, Zhong XP. Unexpected positive control of NFκB and miR-155 by DGKα and ζ ensures effector and memory CD8+ T Cell differentiation. Oncotarget. 2016;7(23):33744–64. doi: 10.18632/oncotarget.8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ou R, Zhang M, Huang L, Moskophidis D. Control of virus-specific CD8+ T-cell exhaustion and immune-mediated pathology by E3 ubiquitin ligase Cbl-b during chronic viral infection. J Virol. 2008;82:3353–3368. doi: 10.1128/JVI.01350-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shamim M, Nanjappa SG, Singh A, Plisch EH, LeBlanc SE, Walent J, Svaren J, Seroogy C, Suresh M. Cbl-b regulates antigen-induced TCR down-regulation and IFN-gamma production by effector CD8 T cells without affecting functional avidity. J Immunol. 2007;179:7233–7243. doi: 10.4049/jimmunol.179.11.7233. [DOI] [PubMed] [Google Scholar]
- 39.Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, Penninger JM, Zhang J. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. 2008;28:2470–2480. doi: 10.1128/MCB.01505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almena M, Andrada E, Liebana R, Mérida I. Diacylglycerol metabolism attenuates T-cell receptor signaling and alters thymocyte differentiation. Cell Death Dis. 2013;4:e912. doi: 10.1038/cddis.2013.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo R, Wan CK, Carpenter JH, Mousallem T, Boustany RMN, Kuan CT, Burks AW, Zhong XP. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc Natl Acad Sci U S A. 2008;105:11909–11914. doi: 10.1073/pnas.0711856105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 43.Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, Palmer E. Different T cell receptor signals determine CD8+ memory versus effector development. Science. 2009;323:502–505. doi: 10.1126/science.1163612. [DOI] [PubMed] [Google Scholar]
- 44.Smith-Garvin JE, Burns JC, Gohil M, Zou T, Kim JS, Maltzman JS, Wherry EJ, Koretzky GA, Jordan MS. T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood. 2010;116:5548–5559. doi: 10.1182/blood-2010-06-292748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.