

Abstract
There is a fundamental gap in our understanding of how a eukaryotic cell apportions the limited space within its cell membrane. Upon infection, a cell competes with intracellular pathogens for control of this same precious resource. The struggle between pathogen and host provides us with an opportunity to uncover the mechanisms regulating subcel-lural space by understanding how pathogens modulate vesicular traffic and membrane fusion events to create a specialized compartment for replication. By comparing several important intracellular pathogens, we review the molecular mechanism and trafficking pathways that drive two space allocation strategies, the formation of tight and spacious pathogen-containing vacuoles. Additionally, we discuss the potential advantages of each pathogenic lifestyle, the broader implications these lifestyles might have for cellular biology and outline exciting opportunities for future investigation.
Keywords: Brucella, Chlamydia, Coxiella, intracellular pathogens, Legionella, membrane fusion, Mycobacterium, vesicular trafficking
Intracellular pathogens are restricted within the spatial confines of their host cell environments, and so they must implement strategies to gain sufficient real estate to support growth and replication. The quest for space thus constitutes a central challenge for intracellular parasites. To address this challenge, intracellular pathogens have evolved diverse strategies to secure valuable space at preferred locations inside host cells, which offers a selective advantage for survival and dissemination.
Intracellular pathogens have been reported to colonize two topologically distinct regions of the host cell. Some pathogens, including Listeria monocytogenes and Francisella tularensis, can gain access to and replicate within the cytoplasm (1). The cytoplasm constitutes an attractive niche for intracellular pathogens because most metabolic processes within host cells occur there, allowing pathogens access to available metabolic intermediates, nucleotides, amino acids and other essential nutrients. Moreover, in many uninfected cell types, the cytoplasm comprises the majority of the host cell volume and, thus, provides an expansive subcellular region in which pathogens can replicate. Finally, the cytoplasm offers the advantage of being physically separated from the extracellular environment, and thereby provides a clandestine hideout where pathogens can evade adaptive immune surveillance and killing.
Alternatively, intracellular pathogens can reside and replicate within the host endomembrane system. The endomembrane system of host cells, which is composed of an intricate network of membrane-bounded organelles and vesicular and tubular trafficking intermediates, coordinates the delivery of membrane and soluble proteins outside of the cell or to assorted intracellular compartments. In the biosynthetic secretory pathway, for example, newly synthesized proteins and membrane traffic in vesicles or tubules from the endoplasmic reticulum (ER) to the Golgi complex (2), where protein cargo is posttranslationally modified and sorted for targeting to diverse cellular destinations or for secretion. In the endocytic pathway, protein and membrane on the cell surface are internalized and recycled or targeted to early and late endosomes before being degraded in lysosomes.
Vacuolar pathogens can subvert trafficking pathways within the host endomembrane system to generate remodeled membrane-bounded compartments in which they persist and replicate. The specialized features of these compartments are largely determined in a pathogen-specific fashion. In most cases, vacuolar pathogens can tailor host membrane to form hybrid organelles that are biochemically and morphologically distinct from compartments found in uninfected cells (3). They may also modify the subcellular compartments in which they reside to aid nutrient acquisition, to promote trafficking to a replicative niche or to limit exposure to the cytosolic immune surveillance pathways (4). Moreover, vacuolar pathogens can often be transported between subcellular compartments as hitchhikers in the host endomembrane system. For example, Brucella initially transits through the endocytic pathway but ultimately fuses with the ER for replication (5). As such, vacuolar pathogens do not require motility machinery for intracellular transport.
Tight and spacious vacuoles represent two general classes of subcellular compartments that support the replication of intracellular vacuolar pathogens. In tight parasitophorous vacuoles (PVs), such as those inhabited by Brucella spp., the vacuolar membrane surrounds each bacterium, and the replication of the pathogen is accompanied by vacuolar membrane expansion and fission, resulting in the encapsulation of individual progeny (Figure 1A). In contrast, spacious vacuoles contain two or more bacteria bounded by a single vacuolar membrane. These remarkable structures can grow to occupy a large fraction of the total intracellular volume (Figure 1B) (7,8). Spacious vacuoles are known to interact and even fuse with myriad host vesicles and organelles (9–12), and this is likely an important source of membrane fueling their growth.
Figure 1: Tight and spacious represent two broad classes of PVs.

A) For intracellular bacteria that reside in tight vacuoles, the vacuolar membrane divides with each bacterium, resulting in a single cell within each PV Inset is a transmission electron micrograph of a Brucella abortus-infected J774.A1 macrophage (6). Copyright 2006, American Society for Microbiology. B) In contrast, spacious vacuoles expand as the bacteria divide, so that all individuals are housed within a single, large vacuole. Inset is a transmission electron micrograph of a Coxiella burnetii-infected L929 cell (7). Copyright 1993, American Society for Microbiology.
A diverse array of intracellular pathogens replicate within vacuoles (Table 1), each having pathogen-specific characteristics in their biochemical composition, biogenesis and acidification. For instance, Chlamydia spp. rapidly depart the endocytic pathway by modifying their PV (called the inclusion) to resemble a Golgi-derived exocytic vacuole (23), further customizing it with a family of chlamydial proteins called Incs that mediate many crucial interactions with host signaling molecules and organelles (24). In contrast, the Legionella-containing vacuole (LCV) more closely resembles the ER, although its association with many different host Rab GTPases that are often not found on the same host membrane makes the LCV a distinctive subcellular compartment (25). Coxiella burnetii is a unique bacterial pathogen that replicates in what is essentially a mature phagolysosome (26). In contrast, Mycobacterium tuberculosis (Mtb) stalls endocytic maturation of its vacuole at the early endosome stage and severely limits interactions with other endocytic vacuoles to avoid acidification (19,27). While the above are all examples of pathogens in spacious vacuoles, some pathogens, especially Brucella spp., replicate in tight vacuoles that are no less unique. Similarly to Legionella, Brucella replicates in an ER-derived vacuole but only after trafficking through early and late endocytic compartments, allowing transient acidification of their developing vacuole (28). In this review, we exploit a comparative approach presenting phylogenetically diverse intracellular bacterial pathogens to illustrate key features of vacuolar pathogens’ quest to secure space. Comparison of the tight and spacious vacuolar lifestyles can delineate mechanisms controlling vacuole formation and also provide a platform for analyzing how molecular machineries associated with the biogenesis of PVs secure precious space within infected host cells.
Table 1:
A diverse array of pathogens can interact with and/or replicate within spacious vacuoles, which contain two or more bacterial cells
| Replicative vacuole |
|||||
|---|---|---|---|---|---|
| Pathogen | Domain | Disease | Tight | Spacious | References |
| Brucella spp. | Prokarya | Brucellosis | Yes | No | (13,14) |
| Chlamydia spp. | Prokarya | Cervicitis, conjunctivitis | No | Yes | (8) |
| Coxiella burnetii | Prokarya | Q fever | No | Yes | (7) |
| Cryptococcus neoformans | Eukarya | Cryptococcosis | Yes | Yes | (15) |
| Francisella tularensis | Prokarya | Tularemia | No | No | (16) |
| Legionella pneumophila | Prokarya | Legionnaire’s disease, Pontiac fever | No | Yes | (17) |
| Leishmania amazonensis | Eukarya | Leishmaniasis | No | Yes | (18) |
| Mycobacterium tuberculosis | Prokarya | Tuberculosis | No | Yes | (19,20) |
| Salmonella enterica | Prokarya | Salmonellosis | Yes | Yes* | (21,22) |
indicates limited replication.
Membrane Fusion Regulates Space
A defining characteristic of the spacious vacuoles of Coxiella and Chlamydia is their ability to interact and fuse with many different host cell compartments (23,26). Our working hypothesis is that vacuoles with broad fusogenic properties mature into spacious vacuoles. Put differently, we hypothesize that highly fusogenic vacuoles will become spacious, whereas vacuoles with limited fusogenicity will remain tight. In our discussion, we shall distinguish membrane fusion events involving similar subcellular compartments (homotypic fusion) from those between vesicles with different subcellular origins (heterotypic fusion) (Figure 2). For example, homotypic fusion generates spacious PVs as they combine with each other, allowing many bacteria to occupy the same compartment (7,11). Likewise, heterotypic fusion of a PV with host-derived vesicles can contribute to its growth. Consideration of the fusogenic properties of the Coxiella-containing vacuole (CCV) supports this hypothesis, as depletion of some host proteins required for vesicle fusion results in smaller CCVs (12,29). In contrast, after endocytic trafficking leading to transient interactions with lysosomes (28) followed by fusion with the ER, replicative Brucella-containing vacuoles (rBCVs) display limited fusogenicity. As a result, the size of the rBCV remains constant, accommodating only a single bacterium tightly wrapped by its vacuolar membrane, which divides with each daughter cell.
Figure 2: Fusogenic properties of tight versus spacious vacuoles.
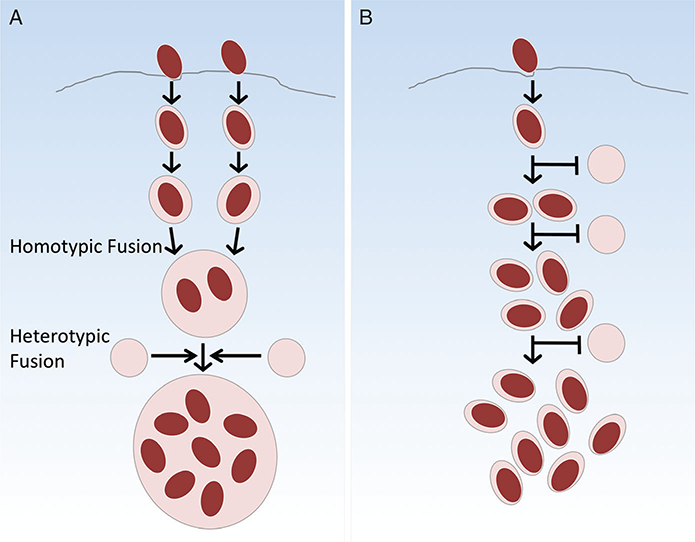
A) Spacious vacuoles allow membrane fusion to occur with each other (homotypic fusion), as well as host-derived vesicles (heterotypic fusion), fueling their growth. B) Tight vacuoles inhibit or avoid fusion with different host subcellular compartments and with each other, and as a result maintain their small size as they replicate.
Spacious Vacuoles Co-Opt Host Membrane Fusion Machinery
To build vacuoles within their eukaryotic host, intracellular pathogens subvert host membrane fusion proteins such as SNAREs and membrane-generating processes such as autophagy. In autophagy, cytosolic contents are sequestered within a double membrane and targeted for degradation by lysosomal fusion. Under conditions of host nutrient deprivation, or when any target cargo is identified, two multiprotein complexes, the ULK complex (containing ULK1, ULK2, ATG13, FIP200 and ATG101) and PI3 kinase complex (VPS34, p150, Beclin1 and ATG14L), initiate formation of the autophagic membrane, a process known as nucleation. Subsequently, the ATG12-ATG5-ATG16L1 complex, which is analogous to an E3 ligase, is recruited to the nascent membrane where it conjugates microtubule-associated protein 1A/1B light chain 3 (LC3) to phosphatidylethanolamine. Lipidated LC3 incorporates into both leaflets of the extending autophagic double membrane. LC3 has emerged as the sine qua non marker for canonical autophagosomes. However, an LC3-, ATG5/ATG7-independent autophagy pathway exists, in which nascent autophagosomes are supplied with membrane via Golgi-resident ATG9 (30). Once mature, autophagosomes fuse with lysosomes via SNAREs, including syntaxin 17, which has been implicated in homotypic vacuolar fusion of CCVs (29,31).
As a prime example of how autophagy supports intracellular pathogens, Coxiella utilizes its membrane fusion machinery to coordinate the homotypic fusion of CCVs. CCV acquisition of LC3 requires the Coxiella type IV secretion system (T4SS) and occurs within 5 min of entry (10,32,33). Interestingly, overexpression of LC3 accelerates CCV development (34), and inhibition of autophagy, or depletion of specific autophagic proteins, results in a multivacuolar phenotype, but no replication defects (29,33,35). Disruption of the gene encoding Coxiella T4SS effector Cig2/CvpB also abrogates homotypic CCV fusion and LC3 recruitment in HeLa cells (35). Remarkably, spacious vacuoles containing Brucella have also been described. A subset of intracellular Brucella populations co-opts the autophagic pathway to create a spacious vacuole after replication in ER-derived compartments (13). By 72 h postinfection, approximately 40% of infected HeLa cells, and 36% of macrophages, display large LAMP1-positive, calreticulin-negative vacuoles housing multiple bacteria. Formation of autophagic BCVs (aBCVs) requires ULK1, Beclin1 and ATG14L, but in contrast to Coxiella vacuole fusion, the autophagosome elongation factors ATG5, ATG7, ATG16L and LC3B are dispensable (13). Rab9 knockdown did not impact the formation of aBCVs, suggesting involvement of an uncharacterized Rab9-independent non-canonical autophagy pathway (30). Although the detailed mechanism remains enigmatic, future work will determine the role that these aBCVs play in the pathogenesis of Brucella in vivo as well as establish the relevance of a proposed cell-to-cell transmission model (13).
While homotypic fusion certainly contributes to spacious vacuole formation, heterotypic fusion of PVs with host membranes also drives vacuolar spaciousness. For example, the CCV is considered a promiscuous subcellular compartment, as fusion with host endosomal, lysosomal and secretory pathways has been observed (36–38). The core machinery that orchestrates docking and fusion of synaptic vesicles mediates membrane fusion events in a wide variety of membrane trafficking pathways (39). SNAREs coordinate the docking of host endomembranous vesicles and subsequent fusion with their target membranes. These proteins contain a highly conserved SNARE motif of about 65 amino acids that fold into characteristic coiled coils, and are categorized on the basis of the presence of a glutamine (Q) or arginine (R) residue therein. Tethering factors, which are often Rab effector proteins, help to bring compatible SNAREs together and regulate the specificity of membrane fusion. Bacterial pathogens that reside inside vacuoles use this host membrane fusion machinery to regulate their interactions with various subcellular compartments.
There are several examples of bacterial appropriation of host membrane fusion machinery. Coxiella requires the host endosomal SNARE Vamp7, which regulates fusion of late endosomes with lysosomes, to drive expansion and maturation of the CCV, and bacterial protein synthesis is necessary for the association (36). Growth of the CCV also requires fusion with early secretory vesicles, as siRNA knockdown of Rab1b, a regulator of traffic between the ER and Golgi, results in smaller PVs (37). Clathrin-coated vesicles may also supply membrane for the expanding CCV, as they appear to be enriched around the PV during infection (12). Other pathogens in spacious vacuoles stimulate membrane fusion events through the expression of membrane fusion machinery. For example, Chlamydiae express multiple SNARE-like proteins that localize to the inclusion membrane and mediate homotypic as well as heterotypic membrane fusion. The chlamydial SNARE mimic IncA regulates homotypic as well as heterotypic fusion, as it is involved in interactions with endocytic SNAREs Vamp7, Vamp8 and Vamp3 (40). Another chlamydial protein, CT813, shares some functional redundancy with IncA, as it also interacts with Vamp7 and Vamp8 (40). Legionella pneumophila also expresses SNARE-like proteins. Legionella SNARE effector A (LseA) specifically interacts in vitro with multiple host SNAREs (41). Based on its subcellular localization, LseA is likely involved in mediating interactions between LCVs and Golgi-derived vesicles. Another Legionella effector, SidM/DrrA, induces Rab1-dependent membrane fusion events between the plasma membrane-derived LCV and vesicles trafficking from the ER (42). Likewise, the effector RalF, an ARF1 guanine nucleotide exchange factor (GEF), recruits ARF1 to the LCV and promotes ER-derived vesicle fusion (43). Through expression of bacterial regulators of membrane fusion as well as their own SNARE-like proteins, pathogens that reside in spacious vacuoles encourage the fusion of PVs with multiple different host vesicles to gain membrane and space.
Pathogens in Tight Vacuoles Restrict Membrane Fusion
Brucella has evolved a very different intracellular lifestyle from the pathogens described thus far. The majority of replicating Brucella resides within tight replicative BCVs (rBCVs), in which the vacuolar membrane septates as the bacteria divide, yielding a single bacterium per vacuole (Figure 1A). The specific mechanisms by which Brucella maintains the tightness of rBCVs have not been fully elucidated. However, interactions between the endocytic BCV (eBCV) and lysosomes reveal that BCVs have the ability to interact with and acquire fluid-phase markers from lysosomes without complete membrane fusion (28). These limited interactions allow the necessary acidification of the eBCV and activation of T4SS apparently without expanding the vacuole through the acquisition of membrane. One can speculate that Brucella achieves this by expressing factors that exclude or inhibit membrane fusion machinery on the eBCV, but such proteins have yet to be identified. However, there certainly is precedence among other intracellular bacterial pathogens for the expression of virulence factors that inhibit membrane fusion events by interference with host SNARE and tethering factor function.
Mtb isan example of a pathogen that is capable of inhibiting vesicle fusion. Through the synthesis of the lipid lipoarabinomannan (LAM), Mtb specifically excludes EEA1, a Rab5 effector protein and membrane-tethering factor that recruits the endosomal and Golgi SNARE syntaxin 6, from its phagosomal membrane (27,44). By preventing syntaxin 6 recruitment, Mtb blocks delivery of V-ATPase and cathepsins to its PV. In addition to inhibitory lipids, Mtb expresses protein inhibitors of endosomal fusion. Ndk, a nucleoside diphosphate kinase, dephosphorylates cellular Rab7-GTP and Rab5-GTP, thereby preventing Rab7-dependent heterotypic fusion and phagosomal maturation (45). Another Mtb effector, PtpA, dephosphorylates VPS33b, a host protein involved in regulation of membrane fusion in the endocytic pathway (46). Much like Mtb, L. pneumophila expresses proteins to selectively inhibit adverse membrane fusion events. Legionella also evades endocytic maturation by depletion of EEA1 from its endosome. VipD localizes to early LCVs and catalyzes the removal of phosphatidylinositol 3-phosphate (PI(3)P) from the endosomal membrane through its phospholipase A1 activity (47). The net result is that EEA1, which binds to PI(3)P, no longer associates with the endosome. Legionella also expresses LegC3, a protein with a ‘SNARE-like’ N-terminal domain that blocks homotypic vacuole fusion events in yeast (48). It is not difficult to imagine that Brucella likewise expresses proteins that inhibit membrane fusion in order to maintain its preferred replicative niche and evade destruction within lysosomes.
Various factors may have driven the evolution of the tight vacuole lifestyle, including the adoption of strategies to evade destruction by complete lysosomal fusion. With that said, some pathogens have developed alternative strategies for limiting killing by acid hydrolases. In Salmonella-infected macrophages, intracellular survival correlates with the ability of the bacteria to induce the formation of spacious phagosomes shortly after uptake (49,50). It has been suggested that these initially spacious compartments dilute toxic compounds within the phagosome, allowing Salmonella to adapt to the intracellular environment prior to fusion with lysosomes, leading to shrinkage and acidification of the vacuole (50). It should be noted, however, that this strategy is not universally conserved, because the spacious CCV is known to harbor active hydrolytic enzymes capable of degrading Escherichia coli (51). This suggests that increased volume alone is not sufficient to inhibit the degradative capacity of lysosomes upon membrane fusion, and that C. burnetii must somehow resist these harsh conditions for survival.
Regulation of Space by Virulence Factor Secretion
A common theme among intracellular pathogens is the expression of one or more secretion systems that are required for modulating host membrane trafficking and fusion to tailor subcellular niches that enable replication and survival. Coxiella, Legionella and Brucella require active T4SS to build their replicative niches (52,53), and in the case of Coxiella, it is a prerequisite for vacuolar expansion. As mentioned above, the Coxiella protein Cig2/CvpB is a secreted effector that appears to be required for homotypic CCV fusion events (35). Whether the described phenotype of Cig2/CvpB mutation is a result of generally impaired intracellular growth (54,55) or a direct result of defective intracellular trafficking remains to be resolved. Disruption of the open reading frames encoding 17 other functionally uncharacterized T4SS effectors by Himar transposon insertion revealed that they are required for intracellular replication and vacuole expansion, as these mutants were confined to a tight CCV (55,56). In-frame deletion of the genes encoding T4SS effectors CvpA, CvpC, CvpD or CvpE, Coxiella proteins that are found on the CCV membrane, also results in a tight vacuole phenotype (12,54). Interestingly, CvpA has been implicated as having a role in the recruitment of clathrin-coated vesicles to the CCV (12), while CvpC colocalizes with transferrin receptor, suggesting it interacts with recycling endosomes (54). L. pneumophila also employs T4SS to create its spacious niche. As mentioned above, the effectors VipD and LegC3 have important roles to play in limiting membrane fusion events (47,48), while at the same time Legionella promotes favorable interactions via RalF, SidM/DrrA and LseA (41–43). These are just a few of the over 300 Legionella T4SS effectors, many of which play important roles in regulating host-pathogen interactions (reviewed in 25).
The T4SS can also be used to create a tight PV, as in the case of Brucella. RicA is a Brucella effector that specifically interacts with Rab2 in its GDP-bound state and is required for Rab2 recruitment to the BCV as well as for Brucella replication (57). SepA was recently described as a novel Brucella effector that is required for early trafficking to the LAMP 1-negative rBCV compartment (58). Myeni et al. have described a set of effectors, BspA, BspB and BspF, that affect host cell secretion and appear to be required for bacterial persistence in a mouse model of infection (59). These examples illustrate that while secreted effectors can modulate vacuolar space, they do so through the alteration of diverse host processes.
While many T4SS effectors have been described as having important roles in Coxiella, Legionella and Brucella infection, intracellular pathogens secrete virulence factors through several different secretion systems. Chlamydia secretes several virulence factors through its type III secretion system (T3SS), and disruption of T3SS function with pharmacologic inhibitors results in small PVs and can even be bactericidal (60,61). Mycobacterium spp. have multiple secretion systems that play a role in virulence, including two Sec-dependent pathways (SecA1 and SecA2) and five type VII secretion systems (T7SS or ESX) (reviewed in 62). PknG, an effector of the SecA2 system, is required to block endocytic maturation and create a replicative niche, as phagosomes of pknG and secA2 mutants acidify (63). The ESX-1 secretion system is also required for Mtb virulence, although the functions of its effectors are unknown (62). Taken together, these data indicate that secreted bacterial effector proteins are critical components of the biogenesis of both tight and spacious vacuoles. For a summary of bacterial effectors regulating vacuole fusion and biogenesis, see Table 2.
Table 2:
Selected pathogen and host-derived regulators of PV membrane fusion events
| Regulators of PV fusion |
||||
|---|---|---|---|---|
| Pathogen-derived |
Host-derived |
|||
| Pathogen | Homotypic | Heterotypic | Homotypic | Heterotypic |
| Brucella spp. | BMFPa (64,65) | RicA (57) | ULK1 (13) Beclin1 (13) ATG14L (13) |
Rab2 (66) GAPDH (66) Sar1 (67) Vamp3, 7, 8 (40) |
| Chlamydia trachomatis | IncA (68,69) | IncA (40) IncD (70) CT813 (40) |
||
| Coxiella burnetii | Cig2/CvpBb (35) | STX17 (29) ATG5, 12 (35) |
Vamp7 (36) Rab1b (37) Rab5, 7 (32) AP2 (12) ARF1 (43) Rab1 (42) Sec22b (42) |
|
| Legionella pneumophila | RalF (43) SidM/DrrA (42) LseA (41) VipD (47) LegC3 (48) LAM (27,44) Ndk (45) PtpA (46) |
|||
| Mycobacterium tuberculosis | ||||
Regulators in italics inhibit membrane fusion, and all others are known to positively regulate fusion of membranes.
BMFP induces membrane fusion in vitro, and its role during infection is undefined.
What Are the Advantages and Disadvantages of Spacious Versus Tight Vacuoles?
Many intracellular pathogens (Table 1) replicate within spacious vacuoles; yet, pathogens like Brucella also successfully replicate as individuals in tight PVs. This contrasting feature of their intracellular lifestyles leads to speculation about possible inherent advantages that spacious and tight vacuoles confer to invading pathogens.
Genetic exchange
By sharing the same intracellular compartment, bacteria in spacious vacuoles can exchange genetic information. Genetic recombination has been documented for Chlamydia. Comparative genomics provided the evidence for recombination among different chlamydial strains (71), and the phenomenon was recently observed in tissue culture (72,73). It was homotypic fusion that enabled the genetic exchange between chlamydiae that occupied the same inclusion. While natural competency and genetic recombination has not been documented for Coxiella, it is certainly an intriguing possibility. It is difficult to imagine a mechanism for genetic exchange between bacteria that inhabit tight vacuoles. Brucella also expresses a T4SS, but successful genetic transfer via the apparatus would require the bridging of two vacuolar membranes. Horizontal gene transfer is a powerful evolutionary driver, and among bacteria that are obligately intracellular, this may be the greatest benefit of occupying a shared vacuole.
Bacterial communication
In addition to the exchange of genetic information, spacious vacuoles provide a shared milieu through which individual bacteria may communicate through the exchange of soluble molecules. This may allow pathogen populations to optimize their collective behavior to promote growth and/or replication. For example, the lqs system of L. pneumophila produces the compound LAI-1 (Legionella autoinducer-1, 3-hydroxypentadecane-4-one) (74), which likely binds to and activates downstream cognate sensor kinases, ultimately activating LqsR (75), a protein that controls the switch from the stationary phase to the replicative phase (76), pathogen-phagocyte interactions and metabolic pathways (76–78). Therefore, the lqs system may enable the efficient coordination of pathogen activities in spacious vacuoles. Quorum sensing may also play a role during aBCV biogenesis in Brucella-infected cells. Current models suggest that aBCVs represent the terminal spacious vacuoles in which Brucella resides before egress from the cell. Interestingly, C12-HSL, a quorum sensing autoinducer, is produced in infected cells at times when the pathogen is predicted to reside in aBCVs. In the Brucella system, C12-HSL accumulation is associated with the suppression of pathogen gene transcription. These data suggest the hypothesis that aBCV-resident Brucella produces C12-HSL to suppress the expression of non-essential genes, thereby preparing the pathogen for pending egress (79). This model is supported by the observation that VirB expression decreases following in vitro exposure of Brucella melitensis to C12-HSL. Finally, despite the elegance of the hypothesis that quorum sensing facilitates coordinated action in spacious vacuoles, it is notable that quorum-sensing systems have not been identified in the majority of isolates of Coxiella (80,81) or Chlamydia (82,83), which replicate in spacious vacuoles. Therefore, quorum-sensing systems are not essential to the biogenesis of spacious vacuoles. If the need for intrabacterial communication contributes to the evolutionary drive for spacious vacuoles, then Coxiella and other intracellular pathogens must exploit as yet undescribed molecules for this purpose.
Nutrient acquisition
The degree to which heterotypic fusion is allowed by a PV may affect a pathogen’s ability to acquire nutrients from its host. Coxiella and Legionella auxotrophs have been described, supporting the idea that highly fusogenic CCVs and LCVs are nutrient rich. For example, L. pneumophila, which uses amino acids as its primary carbon source, is an auxotroph for several amino acids including cysteine, arginine, isoleucine, leucine, threonine, valine and methionine. The observed auxotrophies correspond with observations that cysteine biosynthetic genes and other anabolic genes are absent in the genomes of L. pneumophila (84,85). At the same time, the genome sequences of several Legionella spp. as well as transcriptomic, proteomic and metabolic studies indicate that the bacteria possess broad catabolic abilities and are capable of utilizing carbohydrates such as glucose. Accordingly, L. pneumophila mutants lacking catabolic genes show intracellular growth defects, and thus, metabolism and virulence of the pathogen are intimately connected. Taken together, the data suggest that spacious vacuoles provide nutrient-rich niches. In contrast, most Brucella auxotrophs fail to replicate in host cells (86,87). Moreover, Brucella expresses many transporters for nutrient acquisition, and the loss of many of these transporters in Brucella ovis is suspected of contributing to its avirulence in humans (88). These observations suggest that the limited fusogenic capacities of Brucella tight vacuoles may restrict nutrient acquisition.
Coxiella and Brucella both usurp the autophagic membrane fusion machinery to create spacious vacuoles, but it may also serve to ramp up host nutrient availability(89) Lysosomal fusion enables digestion of autophagic cargo to constituent amino acids, carbohydrates and lipids, freeing these nutrients for reuse by the host cell. Microbial capitalization on autophagy for nutrient acquisition has been described for F. tularensis, an intracellular pathogen residing in the cytoplasm. The autophagy inhibitors 3-methyladenine, which prevents autophagosome formation, and Bafilomycin A (prevents lysosomal fusion) decreased Francisella uptake of amino acids from the host (90). In this case, Francisella nutrient uptake relied on ATG5-independent autophagy. The link between nutrient acquisition and autophagy is not as clear for Coxiella and Brucella; amino acid and serum starvation of host cells, which induces autophagy (among other stress responses), enhance replication of Brucella and infection with Coxiella (34,91,92). However, the details surrounding the subversion of autophagy by these intracellular bacteria remain an active area of investigation.
Membrane maintenance and vacuole integrity
Important benefits of the size of a vacuole may also be revealed when one considers the surface area-to-volume ratios of these compartments; the total membrane required to form a spacious vacuole is less than the corresponding amount for tight vacuoles. This fact holds true whether the bacterial number and/or size are varied. As illustrated by Figure 3, when comparing bacteria of the same size, those in tight vacuoles require much more total surface area (i.e. bounding membrane) than pathogens living in spacious vacuoles. This may provide the important benefit of increasing the host-pathogen interface by allowing each bacterium in tight vacuoles direct contact with the vacuolar membrane. However, pathogens in spacious vacuoles conserve the amount of membrane they require, putting less membrane biosynthetic stress on the host and possibly enhancing host survival, and evading immune surveillance. Conversely, bacteria in tight vacuoles, while requiring more total membrane, utilize less total volume within the host cell, and this could be seen as a benefit once replication begins.
Figure 3: Tight and spacious vacuoles require differing amounts of membrane and space.
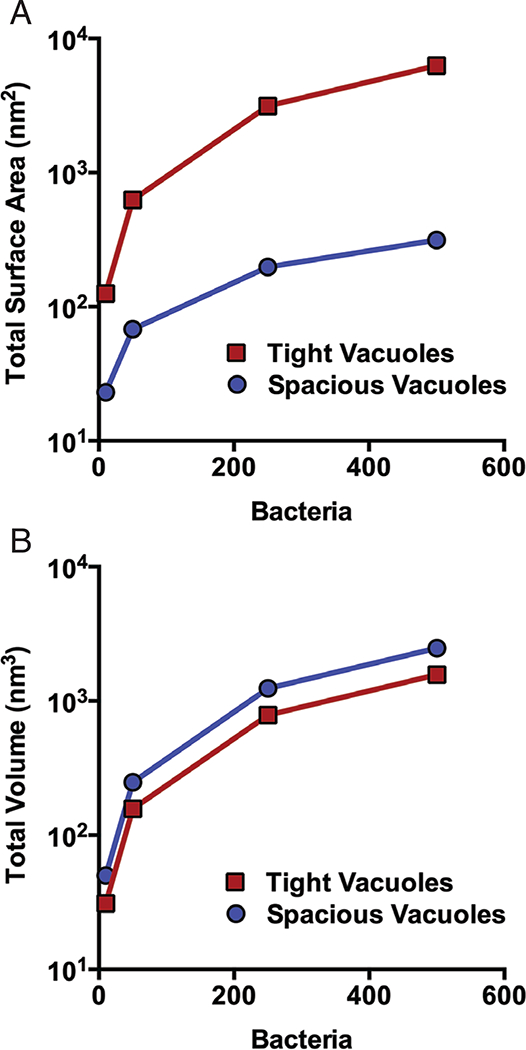
In each graph, the total surface area (A) or total volume (B) is shown for increasing numbers of bacteria of the same size. Surface area (SA) and volume (V) were calculated assuming spherical bacteria and vacuoles. Total surface area for tight vacuoles was calculated using the equation SAT = (4πr2)n, where SAT equals total surface area of all tight vacuoles, r is bacterial radius (1 μm) and n represents the number of bacteria. Total volume for tight vacuoles was calculated using the equation VT = πr3)n, where VT is the total volume of tight vacuoles, r is bacterial radius (1 μm) and n is the number of bacteria. For spacious vacuoles, we assume that bacteria are packed in an irregular configuration with a density limit that does not exceed 63.4% (93). Therefore, the volume of spacious vacuole containing n spheres (VS) is calculated as VS = VT/0.634. The surface area of the corresponding spacious vacuole (SAV) is calculated by first determining the radius of a sphere of volume VS, which is r = [(VS)(3/4π)]1/3. The surface area SAV can then be calculated as follows: SAV = (4πr2) = (4π)[(VS)(3/4π)]2/3.
Another important advantage of tight vacuoles is that the integrity or function of any one vacuole can be compromised without negatively impacting the other bacteria in the cell. The same cannot be said for spacious vacuoles, where the fate of every organism within is tied to the integrity of the single vacuole that envelops them. Experiments with the actin-destabilizing drugs latrunculin-A and -B demonstrated that chlamydial inclusion membrane integrity was dependent on F-actin. Once destabilized, the contents of the inclusion became accessible to the host cytosol, and IL-8 expression was upregulated (94), enhancing the vulnerability of all bacteria in the cell to innate immune responses. At early stages of Mycobacterium infection, expression of the essential ESX-1 secretion system increases the permeability of its phagosome, making the bacteria susceptible to innate immune sensors and the autophagy pathway (95,96). However, only about one third of infecting bacteria are targeted for killing by autophagy, leaving the remaining individuals in the cell, presumably living in less permeable phagosomes, to survive and replicate (96). Although Mycobacteria ultimately replicate in spacious PVs, this early infection dynamic illustrates how living in separate vacuoles is an advantage that limits the vulnerability of the infecting population. Finally, it should be noted that cells containing large lysosomal vacuoles can exhibit defects in MHC class II loading. This has been observed for Coxiella-infected cells and may contribute to immune deficiency in infected individuals (97).
Conclusion and Perspectives
In considering the lifestyles of pathogens in either spacious or tight vacuoles, we are reminded of the specific adaptations they must make to generate a favorable intracellular environment for replication. For any intracellular pathogen, the quest to secure prime real estate involves optimal exploitation of the local cellular resources, including hijacking machinery for the manufacture and transport of raw materials for vacuolar membrane biosynthesis, scavenging for nutrients and energy by subverting autophagy machinery. By utilizing host membrane fusion machinery, as well as pathogen-specific fusogens, intracellular bacteria can promote vacuolar expansion. At the same time, by careful regulation of membrane fusion events, apathogenliving in a spacious vacuole can evade destruction via lysosomal fusion. While many intracellular pathogens opt for the spacious lifestyle, there are some likely benefits to replication in a tight vacuole. For example, as tight PVs take up less total volume, more bacteria can fit inside a host cell before it lyses (Figure 3B). It is also intriguing to consider the impact of regulating host-pathogen interactions at the level of a single bacterium, which is possible from within tight PVs.
In contemplating the regulation of pathogen-defined intracellular space, many interesting questions arise. It is evident that the tight vacuolar lifestyle adopted by Brucella spp. is quite unique among intracellular bacterial pathogens. What are the molecular mechanisms that coordinate fission of the rBCV membrane along with the bacteria? If rBCVs are autonomously replicating, i.e. separate from the ER, how do they acquire all the membrane that bounds them? Also, what causes some Brucella within tight rBCVs to transition to the spacious aBCV near the end of the infectious cycle? Are these events regulated by host or pathogen-derived factors? Questions also remain about the generation of spacious vacuoles. While many vacuolar bacteria live in spacious PVs, the execution and expansion dynamics vary widely among pathogens. For example, why does the Coxiella-containing vacuole expand rapidly after infection, prior to the replication of the majority of bacteria? For others, like Chlamydia, the PV appears to grow along with increases in bacterial numbers. The spaciousness of the inclusion also appears to be maintained through regulation of osmotic pressure (98), a strategy that has also been proposed for spacious Salmonella-containing phagosomes (49). It would be interesting to see if other pathogens use a similar strategy to regulate vacuolar space. There are also questions as to exit strategies for pathogens that replicate in spacious vacuoles. Many appear to exit via host cell lysis, which can induce proinflammatory responses and compromise pathogen stealth; how might this benefit the infection? There is still much we can learn about bacterial pathogenesis by investigating the structure and function of PVs. Understanding the pathogen-specific dynamics of vacuole generation, trafficking, an interactions at the vacuole-cytoplasm interface will significantly advance the development of pathogen-specific intervention strategies, which indeed is an important goal for studying any host-pathogen interactions at the cell biological level.
Acknowledgments
We are grateful to ASM Press for permission to reuse (with modification) previously published material (6,7). This review was supported by grants from the Bill & Melinda Gates Foundation, the Defense Threat Reduction Agency, the Texas A&M University-Weizmann Grant Program, the Texas A&M CAPES Program, Texas A&M Midas Program and Texas A&M AgriLife Research to P. d. F., NIH U54AI057156 and AI48496 to T. A. F., NIH R01AI073558 to J. S. and NIH A1090142, HDTRA1–13-1–0003 to J. S. The authors of this manuscript certify that they have no affiliations with or involvement in any organization or entity with any financial interest in the subject matter or materials discussed in this manuscript. Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the funding agencies.
References
- 1.Ray K, Marteyn B, Sansonetti PJ, Tang CM. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 2009;7:333–340. [DOI] [PubMed] [Google Scholar]
- 2.Bechler ME, de Figueiredo P, Brown WJ. A PLA1–2 punch regulates the Golgi complex. Trends Cell Biol 2012;22:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alix E, Mukherjee S, Roy CR. Subversion of membrane transport pathways by vacuolar pathogens. J Cell Biol 2011;195:943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herweg JA, Hansmeier N, Otto A, Geffken AC, Subbarayal P, Prusty BK, Becher D, Hensel M, Schaible UE, Rudel T, Hilbi H. Purification and proteomics of pathogen-modified vacuoles and membranes. Front Cell Infect Microbiol 2015;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Celli J The changing nature of the Brucella-containing vacuole. Cell Microbiol 2015;17:951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pei J, Turse JE, Wu Q, Ficht TA. Brucella abortus rough mutants induce macrophage oncosis that requires bacterial protein synthesis and direct interaction with the macrophage. Infect Immun 2006;74:2667–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hechemy KE, McKee M, Marko M, Samsonoff WA, Roman M, Baca O. Three-dimensional reconstruction of Coxiella burnetii-infected L929 cells by high-voltage electron microscopy. Infect Immun 1993;61:4485–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterson EM, de la Maza LM. Chlamydia parasitism: ultrastructural characterization of the interaction between the chlamydial cell envelope and the host cell. J Bacteriol 1988;170:1389–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson CG, Roy CR. Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell Microbiol 2006;8:793–805. [DOI] [PubMed] [Google Scholar]
- 10.Winchell CG, Graham JG, Kurten RC, Voth DE. Coxiellaburnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun 2014;82:2229–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsumoto A, Bessho H, Uehira K, Suda T. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J Electron Microsc (Tokyo) 1991;40:356–363. [PubMed] [Google Scholar]
- 12.Larson CL, Beare PA, Howe D, Heinzen RA. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci USA 2013;110:E4770–E4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, Lopez-Otin C, Virgin HW, Celli J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012;11:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Detilleux PG, Deyoe BL, Cheville NF. Entry and intracellular localization of Brucella spp. in Vero cells: fluorescence and electron microscopy. Vet Pathol 1990;27:317–328. [DOI] [PubMed] [Google Scholar]
- 15.Qin QM, Luo J, Lin X, Pei J, Li L, Ficht TA, de Figueiredo P. Functional analysis of host factors that mediate the intracellular lifestyle of Cryptococcus neoformans. PLoS Pathog 2011;7:e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci USA 2006;103:14578–14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sturgill-Koszycki S, Swanson MS. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J Exp Med 2000;192:1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antoine JC, Prina E, Jouanne C, Bongrand P. Parasitophorous vacuoles of Leishmania amazonensis-infected macrophages maintain an acidic pH. Infect Immun 1990;58:779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol 2004;20:367–394. [DOI] [PubMed] [Google Scholar]
- 20.Moreira AL, Wang J, Tsenova-Berkova L, Hellmann W, Freedman VH, Kaplan G. Sequestration of Mycobacterium tuberculosis in tight vacuoles in vivo in lung macrophages of mice infected by the respiratory route. Infect Immun 1997;65:305–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eswarappa SM, Negi VD, Chakraborty S, Chandrasekhar Sagar BK, Chakravortty D. Division of the Salmonella-containing vacuole and depletion of acidic lysosomes in Salmonella-infected host cells are novel strategies of Salmonella enterica to avoid lysosomes. Infect Immun 2010;78:68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakowski MA, Braun V, Brumell JH. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 2008;9:2022–2031. [DOI] [PubMed] [Google Scholar]
- 23.Bastidas RJ, Elwell CA, Engel JN, Valdivia RH. Chlamydial intracellular survival strategies. Cold Spring Harb Perspect Med 2013;3:a010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirrashidi KM, Elwell CA, Verschueren E, Johnson JR, Frando A, Von Dollen J, Rosenberg O, Gulbahce N, Jang G, Johnson T, Jager S, Gopalakrishnan AM, Sherry J, Dunn JD, Olive A, et al. Global mapping of the Inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 2015;18:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.So EC, Mattheis C, Tate EW, Frankel G, Schroeder GN. Creating a customized intracellular niche: subversion of host cell signaling by Legionella type IV secretion system effectors. Can J Microbiol 2015;61:617–635. [DOI] [PubMed] [Google Scholar]
- 26.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol 2013;11:561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci USA 2003;100:5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starr T, Ng TW, Wehrly TD, Knodler LA, Celli J. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic 2008;9:678–694. [DOI] [PubMed] [Google Scholar]
- 29.McDonough JA, Newton HJ, Klum S, Swiss R, Agaisse H, Roy CR. Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. MBio 2013;4:e00606–e00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009;461:654–658. [DOI] [PubMed] [Google Scholar]
- 31.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012;151:1256–1269. [DOI] [PubMed] [Google Scholar]
- 32.Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 2007;9:891–909. [DOI] [PubMed] [Google Scholar]
- 33.Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun 2002;70:5816–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez MG, Vazquez CL, Munafo DB, Zoppino FC, Beron W, Rabinovitch M, Colombo MI. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol 2005;7:981–993. [DOI] [PubMed] [Google Scholar]
- 35.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 2014;10:e1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campoy EM, Mansilla ME, Colombo MI. Endocytic SNAREs are involved in optimal Coxiella burnetii vacuole development. Cell Microbiol 2013;15:922–941. [DOI] [PubMed] [Google Scholar]
- 37.Campoy EM, Zoppino FC, Colombo MI. The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infect Immun 2011;79:402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akporiaye ET, Rowatt JD, Aragon AA, Baca OG. Lysosomal response of a murine macrophage-like cell line persistently infected with Coxiella burnetii. Infect Immun 1983;40:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol 2008;15:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, Subtil A. SNARE protein mimicry by an intracellular bacterium. PLoS Pathog 2008;4:e1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King NP, Newton P, Schuelein R, Brown DL, Petru M, Zarsky V, Dolezal P, Luo L, Bugarcic A, Stanley AC, Murray RZ, Collins BM, Teasdale RD, Hartland EL, Stow JL. Soluble NSF attachment protein receptor molecular mimicry by a Legionella pneumophila Dot/Icm effector. Cell Microbiol 2015;17:767–784. [DOI] [PubMed] [Google Scholar]
- 42.Arasaki K, Toomre DK, Roy CR. The Legionella pneumophila effector DrrA is sufficient to stimulate SNARE-dependent membrane fusion. Cell Host Microbe 2012;1 1: 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 2002;295:679–682. [DOI] [PubMed] [Google Scholar]
- 44.Vergne I, Chua J, Deretic V. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med 2003;198:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS One 2010;5:e8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci USA 2011;108:19371–19376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaspar AH, Machner MP. VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci USA 2014;111:4560–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bennett TL, Kraft SM, Reaves BJ, Mima J, O’Brien KM, Starai VJ. LegC3, an effector protein from Legionella pneumophila, inhibits homotypic yeast vacuole fusion in vivo and in vitro. PLoS One 2013;8:e56798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alpuche-Aranda CM, Berthiaume EP, Mock B, Swanson JA, Miller SI. Spacious phagosome formation within mouse macrophages correlates with Salmonella serotype pathogenicity and host susceptibility. Infect Immun 1995;63:4456–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alpuche-Aranda CM, Racoosin EL, Swanson JA, Miller SI. Salmonella stimulate macrophage macropinocytosis and persist within spacious phagosomes. J Exp Med 1994;179:601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 2010;78:3465–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2011;2(4):e00175–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Comerci DJ, Martinez-Lorenzo MJ, Sieira R, Gorvel JP, Ugalde RA. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell Microbiol 2001;3:159–168. [DOI] [PubMed] [Google Scholar]
- 54.Larson CL, Beare PA, Voth DE, Howe D, Cockrell DC, Bastidas RJ, Valdivia RH, Heinzen RA. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect Immun 2015;83:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez E, Cantet F, Fava L, Norville I, Bonazzi M. Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog 2014;10:e1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber MM, Chen C, Rowin K, Mertens K, Galvan G, Zhi H, Dealing CM, Roman VA, Banga S, Tan Y, Luo ZQ, Samuel JE. Identification of Coxiella burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol 2013;195:3914–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Barsy M, Jamet A, Filopon D, Nicolas C, Laloux G, Rual JF, Muller A, Twizere JC, Nkengfac B, Vandenhaute J, Hill DE, Salcedo SP, Gorvel JP, Letesson JJ, De Bolle X. Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell Microbiol 2011;13:1044–1058. [DOI] [PubMed] [Google Scholar]
- 58.Dohmer PH, Valguarnera E, Czibener C, Ugalde JE. Identification of a type IV secretion substrate of Brucella abortus that participates in the early stages of intracellular survival. Cell Microbiol 2014;16:396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Myeni S, Child R, Ng TW, Kupko JJ III, Wehrly TD, Porcella SF, Knodler LA, Celli J. Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PLoS Pathog 2013;9:e1003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muschiol S, Bailey L, Gylfe A, Sundin C, Hultenby K, Bergstrom S, Elofsson M, Wolf-Watz H, Normark S, Henriques-Normark B. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc Natl Acad Sci USA 2006;103:14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Slepenkin A, Enquist PA, Hagglund U, de la Maza LM, Elofsson M, Peterson EM. Reversal of the antichlamydial activity of putative type III secretion inhibitors by iron. Infect Immun 2007;75:3478–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Woude A, Luirink J, Bitter W. Getting across the cell envelope: mycobacterial protein secretion In: Pieters J, McKinney JD, editors. Pathogenesis of Mycobacterium tuberculosis and Its Interaction with the Host Organism. Berlin/Heidelberg: Springer; 2013, pp. 109–134. [DOI] [PubMed] [Google Scholar]
- 63.van der Woude AD, Stoop EJ, Stiess M, Wang S, Ummels R, van Stempvoort G, Piersma SR, Cascioferro A, Jimenez CR, Houben EN, Luirink J, Pieters J, van der Sar AM, Bitter W. Analysis of SecA2-dependent substrates in Mycobacterium marinum identifies protein kinase G (PknG) as a virulence effector. Cell Microbiol 2014;16:280–295. [DOI] [PubMed] [Google Scholar]
- 64.de Souza Filho JA, de Paulo Martins V, Campos PC, Alves-Silva J, Santos NV, de Oliveira FS, Menezes GB, Azevedo V, Cravero SL, Oliveira SC. Mutant Brucella abortus membrane fusogenic protein induces protection against challenge infection in mice. Infect Immun 2015;83:1458–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carrica Mdel C, Craig PO, Alonso Sdel V, Goldbaum FA, Cravero SL. Brucella abortus MFP: a trimeric coiled-coil protein with membrane fusogenic activity. Biochemistry 2008;47:8165–8175. [DOI] [PubMed] [Google Scholar]
- 66.Fugier E, Salcedo SP, de Chastellier C, Pophillat M, Muller A, Arce-Gorvel V, Fourquet P, Gorvel JP. The glyceraldehyde-3-phosphate dehydrogenase and the small GTPase Rab 2 are crucial for Brucella replication. PLoS Pathog 2009;5:e1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Celli J, Salcedo SP, Gorvel JP. Brucella coopts the small GTPase Sar1 for intracellular replication. Proc Natl Acad Sci USA 2005;102:1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fields KA, Fischer E, Hackstadt T. Inhibition of fusion of Chlamydia trachomatis inclusions at 32 degrees C correlates with restricted export of IncA. Infect Immun 2002;70:3816–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell Microbiol 1999;1:119–130. [DOI] [PubMed] [Google Scholar]
- 70.Agaisse H, Derre I. Expression of the effector protein IncD in Chlamydia trachomatis mediates recruitment of the lipid transfer protein CERT and the endoplasmic reticulum-resident protein VAPB to the inclusion membrane. Infect Immun 2014;82:2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Joseph SJ, Didelot X, Rothschild J, de Vries HJ, Morre SA, Read TD, Dean D. Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol Biol Evol 2012;29:3933–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jeffrey BM, Suchland RJ, Eriksen SG, Sandoz KM, Rockey DD. Genomic and phenotypic characterization of in vitro-generated Chlamydia trachomatis recombinants. BMC Microbiol 2013;13:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suchland RJ, Sandoz KM, Jeffrey BM, Stamm WE, Rockey DD. Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob Agents Chemother 2009;53:4604–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spirig T, Tiaden A, Kiefer P, Buchrieser C, Vorholt JA, Hilbi H. The Legionella autoinducer synthase LqsA produces an alpha-hydroxyketone signaling molecule. J Biol Chem 2008;283:18113–18123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schell U, Kessler A, Hilbi H. Phosphorylation signalling through the Legionella quorum sensing histidine kinases LqsS and LqsT converges on the response regulator LqsR. Mol Microbiol 2014;92:1039–1055. [DOI] [PubMed] [Google Scholar]
- 76.Tiaden A, Spirig T, Weber SS, Bruggemann H, Bosshard R, Buchrieser C, Hilbi H. The Legionella pneumophila response regulator LqsR promotes host cell interactions as an element of the virulence regulatory network controlled by RpoS and LetA. Cell Microbiol 2007;9:2903–2920. [DOI] [PubMed] [Google Scholar]
- 77.Kessler A, Schell U, Sahr T, Tiaden A, Harrison C, Buchrieser C, Hilbi H. The Legionella pneumophila orphan sensor kinase LqsT regulates competence and pathogen-host interactions as a component of the LAI-1 circuit. Environ Microbiol 2013;15:646–662. [DOI] [PubMed] [Google Scholar]
- 78.Tiaden A, Spirig T, Carranza P, Bruggemann H, Riedel K, Eberl L, Buchrieser C, Hilbi H. Synergistic contribution of the Legionella pneumophila lqs genes to pathogen-host interactions. J Bacteriol 2008;190:7532–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weeks JN, Galindo CL, Drake KL, Adams GL, Garner HR, Ficht TA. Brucella melitensis VjbR and C12-HSL regulons: contributions of the N-dodecanoyl homoserine lactone signaling molecule and LuxR homologue VjbR to gene expression. BMC Microbiol 2010;10:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, Ward NL, Tettelin H, Davidsen TM, Beanan MJ, Deboy RT, Daugherty SC, Brinkac LM, Madupu R, Dodson RJ, et al. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci USA 2003;100:5455–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beare PA, Unsworth N, Andoh M, Voth DE, Omsland A, Gilk SD, Williams KP, Sobral BW, Kupko JJ 3rd, Porcella SF, Samuel JE, Heinzen RA. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect Immun 2009;77:642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998;282:754–759. [DOI] [PubMed] [Google Scholar]
- 83.Collingro A, Tischler P, Weinmaier T, Penz T, Heinz E, Brunham RC, Read TD, Bavoil PM, Sachse K, Kahane S, Friedman MG, Rattei T, Myers GS, Horn M. Unity in variety - the pan-genome of the Chlamydiae. Mol Biol Evol 2011;28:3253–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chien M, Morozova I, Shi S, Sheng H, Chen J, Gomez SM, Asamani G, Hill K, Nuara J, Feder M, Rineer J, Greenberg JJ, Steshenko V, Park SH, Zhao B, et al. The genomic sequence of the accidental pathogen Legionella pneumophila. Science 2004;305:1966–1968. [DOI] [PubMed] [Google Scholar]
- 85.D’Auria G, Jimenez-Hernandez N, Peris-Bondia F, Moya A, Latorre A. Legionella pneumophila pangenome reveals strain-specific virulence factors. BMC Genomics 2010;11:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Drazek ES, Houng HS, Crawford RM, Hadfield TL, Hoover DL, Warren RL. Deletion of purE attenuates Brucella melitensis 16M for growth in human monocyte-derived macrophages. Infect Immun 1995;63:3297–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alcantara RB, Read RD, Valderas MW, Brown TD, Roop RM 2nd.. Intact purine biosynthesis pathways are required for wild-type virulence of Brucella abortus 2308 in the BALB/c mouse model. Infect Immun 2004;72:4911–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsolis RM, Seshadri R, Santos RL, Sangari FJ, Lobo JM, de Jong MF, Ren Q, Myers G, Brinkac LM, Nelson WC, Deboy RT, Angiuoli S, Khouri H, Dimitrov G, Robinson JR, et al. Genome degradation in Brucella ovis corresponds with narrowing of its host range and tissue tropism. PLoS One 2009;4:e5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013;14:759–774. [DOI] [PubMed] [Google Scholar]
- 90.Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, Kawula T. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog 2013;9:e1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pizarro-Cerda J, Meresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goni I, Moreno E, Gorvel JP. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun 1998;66:5711–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pizarro-Cerda J, Moreno E, Sanguedolce V, Mege JL, Gorvel JP. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infect Immun 1998;66:2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song C, Wang P, Makse HA. A phase diagram for jammed matter. Nature 2008;453:629–632. [DOI] [PubMed] [Google Scholar]
- 94.Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 2008;4:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao LY. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun 2008;76:5478–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012;150:803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lem L, Riethof DA, Scidmore-Carlson M, Griffiths GM, Hackstadt T, Brodsky FM. Enhanced interaction of HLA-DM with HLA-DR in enlarged vacuoles of hereditary and infectious lysosomal diseases. J Immunol 1999;162:523–532. [PubMed] [Google Scholar]
- 98.Grieshaber S, Swanson JA, Hackstadt T. Determination of the physical environment within the Chlamydia trachomatis inclusion using ion-selective ratiometric probes. Cell Microbiol 2002;4:273–283. [DOI] [PubMed] [Google Scholar]