

Summary
The spatiotemporal organization of chromatin plays central roles in cellular function. The ribosomal DNA (rDNA) chromatin undergoes dynamic structural changes during mitosis and stress. Here, we developed a CRISPR-based imaging system and tracked the condensation dynamics of rDNA chromatin in live yeast cells under glucose starvation. We found that acute glucose starvation triggers rapid condensation of rDNA. Time-lapse microscopy revealed two stages for rDNA condensation: a “primary stage,” when relaxed rDNA chromatin forms higher order loops or rings, and a “secondary stage,” when the rDNA rings further condense into compact clusters. Twisting of rDNA rings accompanies the secondary stage. The condensin complex, but not the cohesin complex, is required for efficient rDNA condensation in response to glucose starvation. Furthermore, we found that the DNA helicase Sgs1 is essential for the survival of cells expressing rDNA-bound dCas9, suggesting a role for helicases in facilitating DNA replication at dCas9-binding sites.
Subject Areas: Genetics, Techniques in Genetics, Molecular Biology, Chromosome Organization, Live Imaging
Graphical Abstract
Highlights
-
•
A CRISPR-based imaging system allows tracking of rDNA condensation in single cells
-
•
Glucose starvation triggers rDNA condensation in two prominent stages
-
•
Condensin contributes to efficient rDNA condensation caused by glucose starvation
-
•
Sgs1 helicase is required for normal rDNA replication at dCas9-binding sites
Genetics; Techniques in Genetics; Molecular Biology; Chromosome Organization; Live Imaging
Introduction
The spatial and temporal organization of chromatin plays an essential role in the regulation of cellular functions, such as transcription, DNA repair, and maintenance of genome stability (Dekker, 2008, Göndör and Ohlsson, 2009, Misteli and Soutoglou, 2012, Zhang et al., 2012). Our understanding of the 3D organization of chromatin has deepened remarkably owing to the recent developments in chromosome conformation capture and imaging technologies. For example, experiments using Hi-C, a chromosome conformation capture technique that analyzes genome-wide chromatin interactions, revealed that the interphase nucleus is organized into two multi-megabase compartments containing open and closed chromatin (Lieberman-aiden et al., 2009). Individual chromosomes are organized in topologically associating domains, which are arranged in a polarized manner in space (Wang et al., 2016). Super-resolution imaging revealed distinct packing behaviors for chromatin of different epigenetic states (Boettiger et al., 2016). Despite these insights, little is known about how chromatin is dynamically folded in 3D in real time.
In budding yeast Saccharomyces cerevisiae, the ribosomal DNA (rDNA) locus represents the largest repetitive chromosomal domain that is dedicated to the synthesis of ribosomal RNA (rRNA). The ∼150 copies of the rDNA genes are the most heavily transcribed region in the genome, producing ∼60% of the total transcription output in rapidly growing yeast cells (Warner, 1999). rDNA chromatin is highly dynamic and undergoes distinct conformational changes during the cell cycle (Lavoie et al., 2004). During G2 to metaphase transition, rDNA chromatin condenses and organizes into large “loop-like” structures containing paired sister chromatids (Lavoie et al., 2004, Miyazaki and Kobayashi, 2011). In addition, nitrogen starvation and exposure to the yeast mating hormone were also shown to effectively induce rDNA condensation (Stone et al., 2000, Tsang et al., 2003, Tsang et al., 2007). In both mitosis progression and nitrogen starvation conditions, condensation of yeast rDNA is dependent on the condensin complex (Freeman et al., 2000, Lavoie et al., 2003, Sullivan et al., 2004), a conserved protein complex that plays a pivotal role in the compaction and segregation of mitotic chromosomes (Hirano, 2016). Structurally, condensin consists of five protein subunits arranged in a large ring-like structure, which has been proposed to topologically entrap chromosomal fibers (Cuylen et al., 2011). Upon nitrogen starvation, condensin rapidly loads on rDNA arrays to help preserve rDNA stability (Tsang et al., 2003, Tsang et al., 2007). To date, tracking the dynamic process of rDNA condensation in live cells has not been possible using the conventional fluorescent in situ hybridization (FISH) method because FISH requires cell fixation.
Here, we present a CRISPR-based imaging system for visualizing the condensation of endogenous rDNA in live yeast cells. We found that glucose starvation induces rapid and robust rDNA condensation in a cell-cycle-independent manner. Our data reveal temporally biphasic dynamics of rDNA condensation: a primary phase in which relaxed chromatin remodels into higher order loop or ring structures and a secondary phase in which rDNA rings convert into highly compact clusters. The condensin complex, but not the cohesin complex or topoisomerase II, is required for efficient rDNA condensation in response to glucose starvation.
To date, the catalytically inactivated Cas9 (dCas9) proteins have been used for multiple purposes, including transcriptional regulation (Qi et al., 2013, Gilbert et al., 2014) and live imaging of DNA elements (Chen et al., 2013, Ma et al., 2015, Ochiai et al., 2015). However, little is known about how this bacterial protein interacts or interferes with essential cellular processes in eukaryotic organisms. In particular, it has not been examined whether transcription, replication, or chromatin condensation could affect dCas9 binding (and vice versa). Previous in vitro studies demonstrated that Cas9-guide RNA (gRNA) complex binds tightly to naked DNA targets and does not dissociate from DNA even under extremely harsh treatments (Sternberg et al., 2014). Such a stable interaction could potentially block the transcription and replication machinery, causing toxicity. A deeper understanding of dCas9-chromatin interactions in vivo is essential for the design of effective CRISPR-based gene regulation and live imaging experiments without toxicity. Our application of the CRISPR system for imaging chromatin in live yeast cells offers an opportunity to answer questions on how essential cellular processes affect dCas9 binding in eukaryotic cells. Our results unravel a role of DNA helicases in facilitating DNA replication near dCas9-binding sites. We also provide evidence that dCas9 binding on heavily transcribed genes is a highly dynamic process and depends on transcription activity. Moreover, dCas9 was found to access both nucleosomal and highly condensed chromatin compartments. These results have broad implications for experimental applications using CRISPR-based technologies in both basic science and clinical research.
Results
Development of a CRISPR-Based Imaging System for Visualizing rDNA in Live Cells
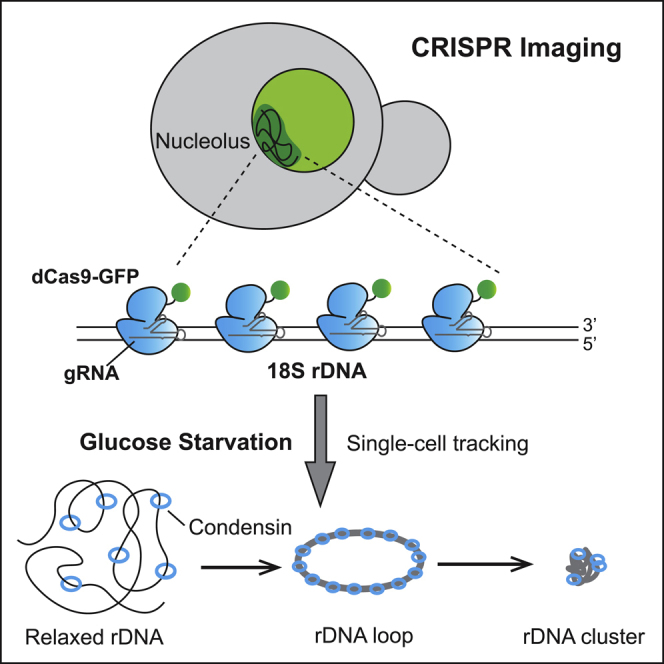
To visualize the rDNA chromatin in live budding yeast cells, we developed a CRISPR-based imaging system consisting of three components: a catalytically dead Cas9 (dCas9) from Streptococcus pyogenes, tagged with enhanced GFP (eGFP); a reverse trans-activator (rtTA); and tandem gRNA repeats with individual gRNAs whose transcription is regulated by the SNR52 promoter and the SUP4 terminator (Figure 1A). We initially constructed the system using the GalL promoter to drive dCas9-GFP expression, and later switched to the Tet promoter because the Tet promoter allowed for better fine-tuning of the dCas9-GFP protein levels. To ensure that all cells have similar dCas9 and gRNA expression levels, all components were stably integrated into the genome instead of being delivered on plasmids. This reduces the cell-to-cell heterogeneity of the dCas9-GFP signal, facilitating more reliable comparisons among yeast strains with different genetic backgrounds or grown under different media conditions.
Figure 1.

CRISPR Live Imaging System for Visualizing the Budding Yeast rDNA
(A) CRISPR imaging platform for the budding yeast. Doxycycline-inducible dCas9-GFP, rtTA, and nine gRNAs that target the 18S rDNA locus were stably integrated into the genome.
(B) A single ∼9.1-kb rDNA repeat unit of the budding yeast. The orientation toward telomere (TEL) and centromere (CEN) and the direction of rRNA transcription are indicated. Nine rDNA-targeting gRNAs were designed to bind the template strand of the 18S rDNA (red bars). The location of the replication fork barrier (RFB) and two non-transcribed spacer (NTS) regions are indicated.
(C) CRISPR imaging of the rDNA chromatin. Deconvolved, maximum projected z series are shown. Scale bar, 5 μm (overview) and 1 μm (inset). The control gRNA does not target the yeast genome.
(D) Classification of rDNA phenotypes by CRISPR imaging. For simplicity, only nuclei are shown. Scale bar, 1 μm.
(E) Distribution of rDNA phenotypic classes in cells grown to different densities. n = 3; error bars, mean ± SEM.
(F) Line scan analysis for different dCas9-GFP signals. Fluorescence intensity was normalized to the peak intensity value. Scale bar, 1 μm.
(G) Quantification of cells showing diffuse dCas9-GFP signal. dCas9-GFP-expressing cells carry genome-integrated gRNAs targeting either the 18S rDNA or the non-transcribed spacers NTS1 or NTS2. n = 3; error bars, mean ± SEM. p Values were calculated by unpaired Student's t test. *p < 0.05, **p < 0.01.
See also Figures S1–S3 and S7.
To image the yeast rDNA, we designed gRNAs that target the 18S region of the rDNA (Figure 1B). Each gRNA spacer sequence was 18–26 bp in length, and the average distance between adjacent gRNAs was 135 bp. We adopted a previously optimized gRNA design that harbored a stem loop extension and an A-U flip, because these features have been shown to improve imaging quality in mammalian cells (Chen et al., 2013). Initially, gRNAs were designed to bind both the template and non-template strands of the 18S gene. To test for potential toxicity caused by gRNAs, individual gRNAs were overexpressed on plasmids and cell growth was examined in the pGalL-dCas9-GFP system (Figure S1). We found that gRNAs binding to the non-template strand impaired growth, whereas gRNAs binding to the template strand did not (Figure S1). The growth defect was most likely due to blocking of RNA polymerase by dCas9 proteins targeted to the non-template strand, a phenomenon previously reported for bacteria and mammalian cells (Qi et al., 2013). Therefore, for live imaging, we selected nine gRNAs that bind solely on the template strand of rDNA (Figure 1B). The Golden Gate method (Engler et al., 2009) was used to assemble all gRNAs on a plasmid, which was subsequently integrated as a single copy in the yeast genome. No major growth defect was observed in the constructed strain, expressing both dCas9-GFP and 9x gRNAs when compared with wild-type (WT) cells in synthetic complete media (Figure S2A). However, we noticed that in YPD media, the integration of dCas9-GFP alone caused slight slowed growth compared with WT cells (Figure S2A). To test if rDNA-targeting dCas9 proteins affect rRNA transcription, we analyzed 18S rRNA transcript levels by quantitative RT-PCR. No significant change in 18S gene expression was detected in cells expressing both dCas9 and gRNAs (Figure S2B). In addition, using qPCR primers spanning the A0 or A1 cleavage sites within the 5′ external transcribed spacer region, we also quantified pre-rRNA levels whose half-life is on the order of minutes (Tollervey and Kos, 2010, Popov et al., 2017). This method allows precise measurement of newly synthesized rRNA levels. We found no significant change in the pre-rRNA levels in cells co-expressing dCas9 and gRNAs, suggesting that the chromatin-bound dCas9 proteins do not block rRNA transcription.
Having established the CRISPR imaging system, we performed live imaging on yeast cells carrying both dCas9-GFP and 9x gRNAs. In the absence of gRNA, we observed that dCas9-GFP distributed in the yeast nucleus in a “comet”-like pattern, with a major pool in the nucleolus (the crescent-shaped structure on the side of the nucleus) and a minor pool in the nucleoplasm (Figure 1C, left). This was consistent with the previous finding that excess dCas9 resided in the nucleolus in mammalian cells (Chen et al., 2013). In contrast, dCas9-GFP molecules co-expressed with a non-targeting control gRNA were homogenously distributed in the nucleus (Figure 1C, middle). In cells expressing rDNA-targeting gRNAs, dCas9-GFP partially localized to structures resembling chromatin fibers (Figure 1C, right). To further show how these elliptical structures are different from the ones shown for the nucleolus, we performed line scan analysis across the dCas9-GFP fluorescence signal. dCas9-GFP signal over the nucleolus showed one peak, whereas the one over the rDNA structures showed two separate peaks (Figure 1F). The pGalL-dCas9-GFP CRISPR imaging system produced imaging results similar to those of the pTet-dCas9-GFP system (Figure S3). No major cell-cycle-associated defects were observed in these cells during the long-term imaging conditions (Video S1).
Cells were grown to early log phase in synthetic complete media supplemented with 2% glucose and 0.5 μg/mL doxycycline. Then, cells were mounted on a freshly prepared 1.2% agarose pad made of the same media and imaged every 15 min at room temperature. Deconvolved, maximum projected z series are shown.
The dCas9-GFP signal on rDNA exhibited five distinct phenotypic classes within a population of cells: diffuse, spotted, ring-like, clustered, and linear (Figure 1D). The linear pattern was exclusively observed in anaphase cells. In early log phase cells (optical density [OD] = 0.2), the diffuse pattern of the dCas9-GFP signal was enriched in a population (Figure 1E), suggesting that this feature may be associated with the actively transcribed rDNA status. The fraction of cells with this pattern decreased as cells reached higher densities. In saturated culture, the dCas9-GFP signal on rDNA displayed ring-like and clustered patterns in the majority of cells (Figure 1E); these patterns likely reflected the condensed nature of rDNA chromatin in the saturated growth phase. To further examine if the diffuse dCas9 pattern was caused by fast on-and-off rate of dCas9 binding to the 18S rDNA during active transcription, we constructed yeast strains expressing gRNAs targeting the non-transcribed spacer (NTS) NTS1 or NTS2 regions. After growing cells to early log phase (OD600 = ∼0.2), we compared the fraction of cells showing diffuse dCas9 signal between the strains expressing 18S-targeting gRNAs and NTS1/2-targeting gRNAs (Figure 1G). The fraction of cells showing diffuse dCas9 pattern was significantly reduced, but not fully eliminated, in strains expressing NTS1- or NTS2-targeting gRNAs. Thus, we conclude that the diffuse dCas9 pattern can be attributed, at least in part, to the RNA transcription activity.
dCas9-GFP Specifically Binds to rDNA Chromatin in the Presence of gRNAs
To determine whether the dCas9-bound nuclear structures were rDNA chromatin, we performed co-localization experiments using a yeast strain expressing mRFPmars-tagged Net1. Net1 is a commonly used rDNA marker and a core member of the “regulator of nucleolar silencing and telophase exit” (RENT) complex (Straight et al., 1999). Previous chromatin immunoprecipitation (ChIP) experiments demonstrated that Net1 was specifically enriched on rDNA chromatin (Straight et al., 1999, Huang and Moazed, 2003). We detected strong co-localization of the dCas9-GFP signal with the Net1-mars signal using a widefield microscope (Figure 2A). To resolve finer details of the dCas9-bound structures, we applied structured illumination microscopy (SIM). SIM images revealed enrichment of dCas9-GFP signal on fiber-like structures resembling condensed chromatin in yeast nuclei (Figure 2B); the estimated width of these fiber structures was ∼129 ± 24 nm (n > 50).
Figure 2.

Specific Binding of dCas9-GFP to rDNA Chromatin in the Presence of rDNA-Targeting gRNAs
(A) dCas9-GFP signal co-localizes with the rDNA marker Net1. Cells were integrated with dCas9-GFP, 9x rDNA-targeting gRNAs, and Net1-mRFPmars. Deconvolved, maximum projected images are shown. Scale bar, 1 μm. Line scan data are shown to the right of the representative images. The fluorescence of each channel was normalized to the peak intensity value.
(B) Structured illumination microscopic (SIM) images of dCas9-bound chromatin structures in cells co-expressing dCas9-GFP and rDNA-targeting gRNAs. Brightness and contrast were adjusted for SIM images to highlight the dCas9-bound structures (right). Corresponding widefield images are shown (left). Scale bar, 1 μm.
(C) Chromatin immunoprecipitation experiments to detect dCas9-GFP binding at the 18S and the adjacent 5S and 25S rDNA regions. All cells were induced with 5 μg/mL doxycycline for 16 hr for dCas9 expression. ChIP enrichment values were calculated as (IP-secondary antibody)/Input*100%. n = 3; error bars, mean ± SEM.
Next, we performed ChIP experiments to probe for direct dCas9-GFP binding to the rDNA chromatin. In the presence of rDNA-targeting gRNAs, substantial dCas9-GFP enrichment was detected at the 18S rDNA locus (Figure 2C), whereas very limited dCas9-GFP enrichment was detected at the 5S or 25S regions (control loci). No enrichment was detected in cells expressing a non-targeting gRNA or in cells expressing dCas9-GFP alone. Taken together, these data demonstrated that dCas9-GFP proteins specifically bound to rDNA chromatin with the assistance of the gRNAs.
Alternative tRNA-gRNA Fusion System Allows Integration of a Larger Number of gRNAs
Due to the long repetitive sequence of each gRNA cassette (∼385bp), assembly of a large number of gRNAs for genomic integration is technically challenging. This limits the potential of applying CRISPR imaging for long-term visualization of single copy, non-repetitive loci in live cells. To increase the total number of gRNAs that can be stably integrated in the genome, we explored an alternative approach based on expressing gRNAs under various tRNA promoters (Mefferd et al., 2015) (Figures S4A and S4B). This approach reduced the size of each gRNA cassette to ∼200 bp and reduced the total repetitive sequences by half. In this method, each tRNA-gRNA fusion is expected to be cleaved by RNase Z at the fusion site after transcription (Mefferd et al., 2015), releasing functional gRNAs (Figure S4A). Using a CRISPR-based GFP silencing assay, we first tested a variety of human and yeast tRNA promoters for their ability to drive the expression of a single gRNA (Figure S4C). The assay showed that all tRNA-gRNA fusions were functional and tRNA promoters were capable of driving strong gRNA expression to a similar extent as the conventional SNR52 promoter (Figure S4D). We then fused nine tRNA promoters with three sets of gRNAs that target the template strand of the 18S and 25S rDNA genes. The Golden Gate assembly was used to further assemble these components into recipient plasmids to generate 9x, 18x, and 27x tRNA-gRNA fusions. The tRNA-gRNA fusions were integrated into the yeast genome. No growth or cell cycle defects were observed in any of the tRNA-gRNA fusion strains (data not shown), indicating that the tRNA-gRNA fusions were not toxic to the cells. We found that ∼30% of the cells expressing 9x tRNA-gRNA fusions showed the “comet”-like phenotype, possibly due to a lower combined expression level of gRNAs from the tandem tRNA-gRNA repeats (Figures S4E and S4F). However, cells expressing 18x and 27x tRNA-gRNA fusions showed no “comet”-like phenotype, and the dCas9 binding pattern to rDNA in these cells was similar to that in cells expressing 9x gRNAs from the conventional SNR52 promoter. For this reason, we used the 9x gRNA expression system in the remainder of this study.
CRISPR Imaging of rDNA Condensation in Single Cells
Conventional FISH methods are not compatible with imaging dynamic changes in rDNA structures in live cells. To address this issue, we applied CRISPR imaging to monitor stress-induced changes in rDNA. First, we assessed the impact of a variety of stress conditions on rDNA morphology by culturing cells in media deprived of glucose, amino acids, or vitamins (Figures 3A and 3B). In unstressed cells, dCas9 signal on rDNA showed mostly a diffuse pattern. In contrast, dCas9 signal appeared as short lines or clusters in cells grown for 1 hr in media deprived of glucose or amino acids, indicating that rDNA chromatin has condensed under these conditions. Exposure to 100 J/m2 of 254 nm UV light also triggered rDNA condensation. Interestingly, cells deprived of vitamins showed no change in the dCas9 signal on rDNA.
Figure 3.

CRISPR Imaging of rDNA Condensation
(A) rDNA condensation upon nutrient depletion or 100 J/m2 UV treatment. dCas9-GFP shows diffuse pattern on uncondensed rDNA but appears as short lines or clusters during condensation. Scale bar, 5 μm (overview) and 1 μm (inset).
(B) Fraction of nuclei showing the condensed rDNA lines and clusters.
(C) Kinetics of rDNA condensation in cells grown in glucose-free liquid culture versus in cells incubated on coverslips. Cells carrying both dCas9-GFP and 9x gRNAs were grown to early log phase in media containing 2% glucose. Glucose was removed at time 0, and cells were imaged at indicated time points. To slow down the condensation process, cells were mounted on a coverslip in a thin layer of media containing 2% glucose (see Transparent Methods for details).
(D) Time-lapse movies of individual yeast cells undergoing rDNA condensation. Cells carrying both dCas9-GFP and 9x gRNAs were mounted on a coverslip in a thin layer of media containing glucose. Widefield images were collected as z stacks, deconvolved, and max projected. For simplicity, only nuclei are shown. Scale bar, 1 μm.
(E) Late condensation process imaged by structured illumination microscopy (SIM). Experiments were set up as in (D), except that cells were incubated on a coverslip for 1 hr to induce rDNA condensation before imaging. Corresponding widefield images are shown (bottom panel). Scale bar, 1 μm.
To investigate the dynamics of rDNA condensation, we opted for the glucose-deprived condition, as it represents a more canonical phenomenon. For this, cells were grown to early log phase (OD = 0.2) in synthetic complete media supplemented with 2% glucose, washed twice with glucose-free media, and incubated in glucose-free media with shaking. Samples were harvested at multiple time points, and the fraction of nuclei with condensed rDNA was scored. Remarkably, the condensed rDNA phenotype was detectable as soon as 10 min after glucose removal in the majority (∼87%) of cells (Figure 3C). In addition, rDNA condensation was observed in all cell-cycle stages.
To better capture the dynamic changes of rDNA in single cells, we mounted cells on a coverslip and grew them in a thin layer (∼7 μL) of media containing 2% glucose so that cell growth would deplete glucose slowly, resulting in slow accumulation of stress. Indeed, we observed slower rDNA condensation dynamics in cells incubated on coverslips, when compared with cells exposed to a sudden drop in glucose concentration in the liquid culture (Figure 3C). The rDNA condensation process in individual cells was captured by taking z-stacked images every 10 min (Figure 3D). At time 0, the dCas9 signal on rDNA in the majority of cells displayed either a diffuse or a spotted pattern. At around 30–60 min, rDNA chromatin in ∼89% of cells aligned into higher order structures that resembled rings or loops. We termed this stage of the condensation process as the “primary stage.” The rDNA rings subsequently remodeled into short lines. In ∼70% cells, the rDNA lines further compacted into clusters during the 110-min experiment (Figure 3D, cells 1, 2, and 4). The remaining cells showed rDNA lines that did not undergo further compaction (Figure 3D, cells 3 and 5). We termed this stage of the condensation process as the “secondary stage.” Interestingly, the rDNA rings were observed in all cell-cycle stages.
To better understand the secondary stage of the rDNA condensation, we applied SIM microscopy to image cells that had already undergone slow glucose starvation on coverslips for 60 min. Following these cells between 60 and 90 min time points, we found that the rDNA rings underwent twisting, and this twisting continued until chromatin fibers became compacted into clusters (Figure 3E). At higher resolution, the rDNA clusters showed overlapping chromatin fibers.
Factors Responsible for rDNA Condensation in Response to Glucose Starvation
To identify the factors responsible for glucose-starvation-induced rDNA condensation, we generated temperature-sensitive (ts) or deletion mutants of condensin, cohesin, and topoisomerases. Time course experiments were performed at 37°C to examine the condensation dynamics of rDNA in response to glucose removal from liquid culture (Figure 4A). We found that the cohesin mutant mcd1-1 has a condensation dynamics profile indistinguishable from the one of WT, suggesting that the cohesin complex is not involved in rDNA condensation triggered by glucose starvation. In contrast, two condensin mutants, ycs4-1 and ycg1-2, showed slower condensation dynamics compared with WT (Figure 4A). Consistent with these results, individual ycs4-1 cells also showed impaired condensation phenotypes in time-lapse movies performed at 37°C (Figure 4B). rDNA loops were still able to form in ycs4-1 cells, but their further compaction into short lines and clusters was hindered or delayed in ycs4-1 cells compared with WT cells.
Figure 4.

Role of the Condensin and Cohesin Complexes in rDNA Condensation in Response to Glucose Starvation
(A) rDNA condensation dynamics of the temperature-sensitive condensin mutants in glucose-free liquid culture. Cells were grown overnight at 25°C until early log phase, and then were shifted to 37°C for 3 hr. Glucose was removed at time 0, and the samples were kept at 37°C and collected at indicated time points for live-cell imaging. Mean ± SEM of two to four independent experiments are presented.
(B) Time-lapse movies of individual ycs4-1 cells undergoing rDNA condensation. Cells were grown at 25°C overnight and shifted to 37°C for 3 hr. Then, cells were mounted on a coverslip in a thin layer of media containing glucose and imaged in a stage-top incubator at 37°C. Scale bar, 1 μm.
(C) rDNA morphologies during condensation in WT versus ycs4-1 cells. Scale bar, 1 μm.
(D) Quantification of rDNA condensation phenotypes at indicated time points in glucose-free media (n = 150–200).
(E) Comparison of rDNA area between ycs4-1 and WT cells in glucose-free liquid culture at 30 min time point. Mean and SD are plotted. p value was calculated by unpaired Student's t test. ***p < 0.001.
See also Figure S5.
To gain further insights into the defects caused by the ycs4-1 mutation, we examined rDNA condensation intermediates at different time points in liquid culture. At 30 min after glucose removal, 93% WT cells displayed uniform, hyper-condensed rDNA clusters and rDNA lines (Figures 4C and 4D). In contrast, only 44% ycs4-1 cells showed these phenotypes, whereas 33% cells showed branched, twisted, or irregular-shaped rDNA condensation intermediates (Figures 4C and 4D). Quantification of rDNA area at 30 min after glucose removal revealed that rDNA was significantly more condense in WT cells than in ycs4-1 mutants (Figure 4E). We also saw that the rDNA lines and loops in ycs4-1 cells were considerably longer than those in WT cells (Figure 4C) and that this phenotype was more striking in cells grown in YPD media (Figure S5), suggesting that axial compaction of chromatin may be impaired in ycs4-1 cells. Together, these results suggest that the ycs4-1 mutation causes a defect in rDNA condensation.
Among the topoisomerase mutants that we examined, the Top2 ts mutant top2-4 showed a condensation dynamics profile considerably different from the WT cells (Figure 5A) and was further examined by time-lapse microscopy. Interestingly, 52% top2-4 cells were arrested at G2/M phase at 37°C with rDNA residing in the mother cell compartment at time 0 (Figure 5B). During the 110-min-long experiment, most of the G2/M phase top2-4 cells showed rDNA segregation defect, with rDNA being condensed in the mother compartment but not segregating into daughter cells (Figures 5C and 5D). In contrast, ∼90% G2/M phase WT cells under the same experimental conditions showed proper segregation of the rDNA followed by its further condensation in both mother and daughter cells (Figures 5C and 5D). Due to this rDNA segregation defect of top2-4 cells, many nuclei contained no rDNA signal in the experiment shown in Figure 5A, and this results in a considerably lower % value obtained for the top2-4 strain compared with that for the WT strain under glucose starvation. In the case when rDNA was able to segregate in top2-4 cells, we saw that rDNA segregation between mother and daughter cells was unequal, indicating that a DNA breakage might have occurred within the rDNA (Figure 5E). Measurements of rDNA area changes in top2-4 cells indicate that rDNA became more condensed over time (Figure 5F). Thus, we conclude that Top2 is not responsible for rDNA condensation induced by glucose starvation, but it is responsible for the resolution and segregation of rDNA between mother and daughter cells.
Figure 5.

Role of Topoisomerases in rDNA Condensation and Segregation in Response to Glucose Starvation
(A) rDNA condensation dynamics of topoisomerase mutants in glucose-free liquid culture. top4-2 cells were grown overnight at 25°C until early log phase, and then were shifted to 37°C for 3 hr. WT and top1Δ cells were cultured at 30°C. Glucose was removed at time 0, and samples were collected at indicated time points for live-cell imaging.
(B) Representative images of G2/M-arrested top2-4 mutant after 3 hr growth at 37°C. Scale bar, 1 μm. Inset image is shown on the right; scale bar, 1 μm.
(C) Time-lapse microscopy showing rDNA condensation and segregation in WT versus top2-4 cells. Cells were grown at 25°C overnight, shifted to 37°C for 3 hr, and imaged in a stage-top incubator at 37°C. Scale bar, 1 μm.
(D) Quantification of nuclear and rDNA segregation pattern in G2/M phase WT and top2-4 mutant in the experiment shown in (C).
(E) Unequal rDNA segregation between mother and daughter cells in top2-4 mutant in the experiment shown in (C). dCas9-GFP fluorescence images of WT and top2-4 cells before and after rDNA segregation are shown. Scale bar, 1 μm.
(F) Box-and-whisker plots of rDNA area changes in top2-4 mutant in the experiment shown in (C). Median, lower, and upper quartile, min to max values for whiskers are plotted (n = 25).
Neither Nucleosomes nor Condensation Affect the Accessibility of dCas9 to rDNA at the Steady State
Recent in vitro studies have demonstrated that nucleosomes can inhibit Cas9 binding and cleavage on artificial DNA sequences or chromatinized plasmids (Hinz et al., 2015, Horlbeck et al., 2016, Isaac et al., 2016). Therefore, we asked whether nucleosomes could affect dCas9 binding to the endogenous 18S rDNA genes in vivo. In exponentially growing yeast cells, approximately half of the rDNA genes are transcriptionally active and devoid of nucleosomes (“open”), whereas the other half is inactive and packaged in nucleosomes (“closed”) (Dammann et al., 1993, Merz et al., 2008). As cells approach stationary phase, all rDNA genes become nucleosome-associated (Dammann et al., 1993, Sandmeier et al., 2002). To test whether dCas9 has a preference for binding to “open” or “closed” rDNA genes, we performed ChIP of dCas9-GFP on cells grown to different densities. ChIP data showed no significant differences of dCas9 enrichment on the rDNA locus between cells in exponential and stationary phases (Figure 6A), indicating that nucleosomes did not inhibit dCas9 binding to the rDNA region at the steady state.
Figure 6.

Robust dCas9 Binding to rDNA In Vivo Irrespective of Nucleosomes or Condensation
(A) dCas9-GFP binding at the 18S rDNA locus in cells grown to different densities. Cells carrying both dCas9-GFP and 9x gRNAs were cultured for ∼16 hr in synthetic complete media containing 5 μg/mL doxycycline and grown to the indicated densities. An equal number of cells were harvested for ChIP experiments. Mean and SEM of four independent experiments are shown. NS, not significant (one-way ANOVA, p = 0.2809). No significant differences were found between samples with an OD600 of 0.2 and OD600 of 6 (unpaired t test, p = 0.1381).
(B) Newly synthesized dCas9 can access rDNA chromatin packaged into nucleosomes. Cells were first grown to stationary phase to trigger nucleosome packing of rDNA genes; dCas9-GFP expression was then induced by doxycycline. Deconvolved, max projected images are shown. Scale bar, 1 μm.
(C) dCas9 binding at the rDNA locus in unstressed cells versus in cells grown in glucose-free media for indicated time periods (n = 3; error bars, mean ± SEM). NS, not significant (one-way ANOVA, p = 0.8363).
To further validate that the nucleosomal rDNA is accessible for dCas9 binding, we first cultured cells to stationary phase (OD > 2) and then induced dCas9-GFP expression (Figure 6B). After ∼1.5 hr of induction, dCas9-GFP proteins were synthesized and transported into the nucleus. dCas9-GFP signal appeared as clusters or short lines that co-localized with the rDNA marker Net1, indicating that the nucleosomal rDNA was accessible for dCas9 binding.
Next, we asked whether condensation (i.e., increased density of chromatin meshwork) could inhibit dCas9 binding. For this, we induced rDNA condensation by treating cells with glucose-free media for different time periods. We also harvested cells grown in regular (2% glucose) media as an uncondensed rDNA control. Then, ChIP was performed to detect dCas9 binding to the 18S rDNA locus. We found that dCas9 binding on the condensed versus the uncondensed rDNA chromatin was not significantly different (Figure 6C), indicating that dCas9-gRNA complexes were able to find and bind to their DNA targets in a condensed chromatin environment.
Sgs1 Helicase Is Essential for Survival of Cells Expressing rDNA-Targeting dCas9
In cells overexpressing rDNA-bound dCas9, replication forks encounter numerous dCas9 molecules during every S phase, potentially blocking fork progression. However, we did not detect major growth or cell cycle defects in these cells (Figure S2A, Video S1), suggesting that the dCas9-gRNA complex was efficiently removed from targets during rDNA replication. Helicases are molecular motors that unwind DNA and play major roles in genome maintenance (Mackintosh and Raney, 2006, Brosh, 2013). Sgs1, a RecQ family DNA helicase, is required for genomic and rDNA replication, and the maintenance of genome stability (Gangloff et al., 1994, Versini et al., 2003). We therefore tested whether the Sgs1 is required for normal rDNA replication in the presence of rDNA-bound dCas9 proteins. When we deleted SGS1 from the cells carrying dCas9 and gRNAs, we interestingly observed two populations of sgs1Δ mutants, each exhibiting distinct phenotypes, as summarized in Figure 7A. Of the 29 sgs1Δ colonies 17 (group 1) grew relatively well when dCas9 expression was not induced, but displayed slower growth after induction. In contrast, the remaining 12 colonies (group 2) showed no sensitivity to dCas9 induction. Fluorescence-activated cell sorting (FACS) data revealed irregular size and increased granularity in group 1 sgs1Δ cells (Figure 7B). In addition, these cells had lower dCas9-GFP fluorescence than the SGS1+ cells, whereas their dCas9-GFP expression noise was higher (Figure S6). In contrast, group 2 sgs1Δ cells displayed normal size and shape and showed similar levels of dCas9-GFP fluorescence and expression noise as SGS1+ cells (Figures 7B and S6).
Figure 7.

Sgs1 Helicase Is Essential for Survival of Cells Expressing rDNA-Bound dCas9
(A) Summary of the phenotypes of sgs1Δ and rrm3Δ mutants. All strains were integrated with dCas9-GFP and 9x gRNAs. For doubling time estimation, cells were inoculated in synthetic complete media, transferred into media with or without 5 μg/mL doxycycline, and grown for 16 hr. Percentages of G2/M phase cells were scored in cells grown in the same conditions.
(B) Cellular morphology and dCas9 localization. All strains carry the dCas9-GFP and 9x gRNAs integrations. Cells were grown in synthetic complete media and induced with 5 μg/mL doxycycline for 16 hr for microscopy and FACS. Deconvolved, maximum projected images are shown. Scale bar, 5 μm (overview) and 1 μm (inset). Occasional cell death of sgs1Δ mutant is indicated (arrow). FSC, forward scatter, SSC, side scatter.
(C) qPCR quantification of gRNA copy number in sgs1Δ strains. All strains contain the dCas9-GFP integration, and a strain integrated with one copy of gRNA was used for normalization. Three independent sgs1Δ strains from each phenotypic group were examined (n = 3; error bars, mean ± SEM).
(D) Western blot analysis of Rad53 phosphorylation. Whole-cell lysates were separated on a 7.5% Tris-Tricine gel and blotted with a Rad53 antibody. Phosphorylated Rad53 is indicated in bracket.
(E) Growth test of sgs1Δ and rrm3Δ strains. Serially diluted yeast cells were spotted on plates containing synthetic complete media supplemented with doxycycline and grown for 2–3 days at 30°C. All strains were integrated with dCas9-GFP and 9x gRNAs, except row 6, which did not contain gRNAs.
(F) Viability assay. ∼200 cells were seeded as single cells on synthetic complete media plates containing indicated concentrations of doxycycline. The number of viable colonies were scored after 2 days' growth at 30°C (n = 3; error bars, mean ± SEM).
(G) Role of homologous recombination in SGS1-mediated growth defect. Growth assay was done on plates supplemented with 1 μg/ml doxycycline. All strains were integrated with dCas9-GFP and 9x gRNAs.
See also Figure S6.
Because of the phenotypic heterogeneity caused by the SGS1 deletion, we carefully examined each sgs1Δ strain by microscopy. We observed that group 1 sgs1Δ cells had an elongated shape and larger size when induced (Figure 7B). A large fraction (51.7%) of these cells were found at the G2/M phase, and some of them died with large buds (Figure 7B, indicated by the arrow). dCas9-GFP still localized to the nucleoplasm and rDNA in the vast majority of these cells. In contrast, group 2 sgs1Δ cells showed normal size and shape under the microscope. Strikingly, in all group 2 cells analyzed, dCas9-GFP no longer targeted to rDNA, but accumulated in the nucleolus with a “comet”-like pattern, similar to that observed in cells that did not express gRNAs (Figure 1C). Therefore, we performed qPCR analysis to test if the gRNAs were still present. qPCR data revealed a complete loss of gRNAs in the group 2 sgs1Δ strains (Figure 7C), suggesting that the deletion of SGS1 has triggered hyper-recombination, leading to the excision of the repetitive gRNA sequences. The role of SGS1 in the maintenance of genome stability has been previously demonstrated (Watt et al., 1996). Therefore, we conclude that the group 2 sgs1Δ strains were survivors that showed early signs of genomic instability, and in these cells dCas9-gRNA was no longer functional. In contrast, group 1 sgs1Δ strains showed bona fide phenotypes.
Since the fraction of sgs1Δ cells found at the G2/M phase was unusually large, we next asked whether this was due to the activation of checkpoint pathways. Rad53 is a checkpoint kinase required for survival of yeast cells subjected to DNA damage or replication stress (Zhou and Elledge, 2000, Tercero and Diffley, 2001). Rad53 activation occurs through phosphorylation (Pellicioli et al., 1999) and phosphorylated Rad53 has slower electrophoretic mobility when characterized by western blot. Western blot analysis showed that Rad53 was not phosphorylated in cells co-expressing dCas9 and gRNAs (Figure 7D, lane2), or in group 2 sgs1Δ survivors (lane 6). However, Rad53 was phosphorylated in group1 sgs1Δ cells (lane 4), indicating that, in the absence of the helicase Sgs1, the rDNA-bound dCas9 activated DNA damage checkpoint pathways.
We found that group 1 sgs1Δ cells were sensitive to dCas9 induction even when the induction level was low (Figure 7E). To further confirm this result, we separately seeded an equal number of cells on plates containing 0–20 μg/mL doxycycline and scored the number of colonies that formed after 2 days. The fraction of viable colonies formed by group 1 sgs1Δ cells decreased to 27% on the 1 μg/mL doxycycline plates and further decreased at higher doxycycline concentrations. In contrast, no loss of viability was found in SGS1+ or group 2 sgs1Δ cells (Figure 7F). These results suggest that Sgs1 plays an essential role in the cells expressing rDNA-targeting dCas9. Importantly, deletion of SGS1 in cells expressing dCas9-GFP alone showed no growth defects (Figure 7E, lane 6), suggesting that the effect of Sgs1 on cell survival is dependent on the ability of dCas9 to bind to rDNA.
To test if the loss of viability of sgs1Δ mutants is due to toxic homologous recombination (HR) intermediates arising during rDNA replication, we deleted the HR-based repair pathway genes RAD51 or RAD52, and assayed cell growth on plates (Figure 7G). Deletion of either RAD51 or RAD52 rescued the growth defect of sgs1Δ mutants. These results suggest that a key function of Sgs1 in cells is to suppress inappropriate HR-based repair that leads to cellular toxicity.
The 5′-3′ DNA helicase Rrm3 is also involved in rDNA replication (Ivessa et al., 2003, Torres et al., 2004), therefore, we tested if Rrm3 had the same effect as Sgs1 in cells expressing rDNA-bound dCas9. Similar to group 1 sgs1Δ mutants, we found that rrm3Δ mutants displayed slower growth when induced (Figure 7A). A relatively high fraction (48.2%) of rrm3Δ cells was found at the G2/M phase, suggesting that the deletion of RRM3 led to checkpoint activation. Consistent with this interpretation, western blot analysis revealed Rad53 phosphorylation in rrm3Δ cells induced with doxycycline (Figure 7D, lane 8). Despite these defects, the rrm3Δ cells showed correct dCas9-GFP localization (Figure 7B) and no loss of viability when induced (Figures 7E and 7F). Therefore, we concluded that Rrm3 maintains normal growth in cells harboring rDNA-bound dCas9, but is not essential for survival of these cells.
Discussion
CRISPR Live Imaging System for the Yeast rDNA
Single cell live imaging is becoming an increasingly powerful tool for mechanistic dissection of cellular processes and behaviors (Spiller et al., 2010, Conlon et al., 2016). The CRISPR imaging system that we developed enables the visualization of the folding process of endogenous chromatin domains in real time in live cells. An important advantage of CRISPR-based imaging is that no genetic alteration of the DNA target is required. In comparison, imaging DNA using the LacO-LacI system requires the insertion of long repetitive sequences (∼10kb) in the DNA, which alters genetic structure with potential confounding effects on endogenous chromatin dynamics. Alternatively, the imaging method relying on GFP fusion proteins has limitations in terms of the selection of proteins that specifically bind to the genomic target; the fusion process itself may also alter the function and localization of the target protein, requiring further validation by immunofluorescence experiments. In contrast, the CRISPR-based imaging method is broadly applicable to many genomic targets. Recently, an improved transcription activator-like effector (TALE)-based system has been developed for live imaging of repetitive genomic sequences and has been applied to track rDNA changes during human aging (Ren et al., 2017). Although this method benefits from higher signal-to-noise ratio than the CRISPR-based imaging method, the construction of functional transcription activator-like effector nucleases (TALENs) with desired specificity remains a laborious and time-consuming process. Also, the TALE-based system currently relies on one type of effectors designed to bind short stretches (15–20 bp) of DNA targets and thus is only suitable for imaging repetitive sequences, whereas the CRISPR-based imaging system can be used to image much longer (>1 kb) regions, as well as non-repetitive regions, using different gRNAs tiling the DNA target.
The CRISPR-based imaging system developed in this study uses genomic integration of dCas9 and gRNAs, rather than delivering these components on plasmids. This facilitates imaging the same cells over many generations and also reduces copy-number-associated noise. Moreover, cells of different genetic backgrounds or cells grown in different conditions can be reliably cross compared. The tRNA-gRNA fusion system we developed allows stable expression of up to 27 gRNAs, thus paving the way for long-term imaging of single, non-repetitive genomic loci. Furthermore, this system would also be valuable for expanding the capacity of multiplexed CRISPR-gene targeting platforms.
Comparing the results from the CRISPR-based live imaging system with results obtained by traditional FISH (Guacci et al., 1994, Lavoie et al., 2003) and LacO-LacI tandem array methods (Miyazaki and Kobayashi, 2011), we observe several common features. For example, among the five phenotypic rDNA classes that we identified, three (ring-like, linear, and clustered patterns) have also been observed in previous FISH and LacO methods. However, by CRISPR imaging, we also frequently observed diffuse dCas9 signal on rDNA in early log phase cells. This pattern decreased as cells grew to higher densities. Based on our observations from cells expressing gRNAs targeting the NTS1 and NTS2 spacer regions, this diffuse signal was partly due to the dynamic binding and unbinding of dCas9 molecules on actively transcribed rDNA regions during rapid cell growth. This phenomenon could not be detected by the FISH or LacO methods, as the rDNA region is denatured for FISH experiments and the LacO repeats were inserted in the non-transcribed regions of the rDNA (Miyazaki and Kobayashi, 2011), hence the LacO system could not report how binding would be influenced by local transcription.
Glucose-Starvation-Induced rDNA Condensation Occurs in Two Prominent Stages
Combining CRISPR live imaging with time-lapse microscopy, we were able to capture native rDNA intermediates during glucose-starvation-induced condensation in individual cells. Our data reveal two stages of rDNA condensation upon glucose starvation, a “primary stage,” in which relaxed rDNA chromatin forms higher order rDNA loops or rings, and a “secondary stage,” in which the rDNA rings further compact into rDNA clusters (Figure 8). Consistent with this notion, a quantitative time-lapse microscopy study on C. elegans embryo also reported two discrete steps in the condensation of C. elegans chromosomes (Maddox et al., 2006). In this case, diffuse chromatin was initially converted into elongated, highly curved chromosomes, which were further compacted into shorter bar-shaped structures in a secondary phase. Thus, the biphasic dynamics of chromatin condensation may be a conserved feature in eukaryotic cells. It is also possible that the two phases of condensation are regulated by distinct mechanisms.
Figure 8.

Model for rDNA Condensation Induced by Glucose Starvation
Upon glucose starvation, relaxed rDNA chromatin condenses to form higher order rDNA loops or rings (primary stage). Then, the rDNA loops twist and further condense into rDNA clusters (secondary stage). The condensin complex is loaded onto relaxed chromatin (possibly with other unknown factors) to catalyze rDNA condensation.
In the primary stage of condensation, the vast majority of nuclei formed rDNA rings regardless of the cell cycle stages. In contrast, in nutrient-rich conditions, the rDNA rings/loops have been reported to form only at the G2/M phase via associated sister chromatids (Lavoie et al., 2003, Miyazaki and Kobayashi, 2011). Hence, the ring structures may represent important intermediates that drive further compaction of rDNA chromatin into clustered structures. We noted that these ring structures often consisted of single DNA duplex, instead of sister chromatids. The high curvature of these structures is intriguing, and could be due to the repetitive DNA sequences in the rDNA. Our SIM data provide a step-by-step visualization of the condensation of rDNA rings to highly compact clusters. During this process, rDNA rings were regularly seen as twisted, indicating that torsional stress may be applied to these structures. The imaging data are consistent with earlier in vitro studies that showed that purified 13S condensin complex was able to introduce positive supercoiling on circular DNA in the presence of topoisomerase I and ATP (Kimura and Hirano, 1997). Another related study showed that 13S condensin could catalyze global, positive writhe on nicked, circular DNAs. As overlapping chromatin fibers were visible in the condensed rDNA clusters at the later stage, it is possible that these structures may be catenated; however, we do not have direct evidence for this possibility.
The primary stage observed in response to glucose starvation does not merely represent a situation in which increased dCas9 binding leads to ring-like rDNA structures as a simple consequence of halted rRNA transcription. This is because, under unstressed conditions, uncondensed rDNA can be visualized in a large fraction of the cells expressing gRNAs targeting the NTS1 and NTS2 spacer regions (Figure S7). The uncondensed rDNA typically exhibits extended fuzzy features (Figure S7A), and the ring-like rDNA is rarely observed in these cells (Figure S7B). In support of this view, traditional FISH studies also revealed that uncondensed rDNA exhibits disorganized, “puff-like” phenotype (Lavoie et al., 2004). Thus, the primary stage is a transitional step for rDNA from disordered chromatin fibers to higher order structures.
The condensed rDNA fibers in live budding yeast cells were estimated to be ∼129 nm in diameter in our study, which is in good agreement with earlier electron microscopic (EM) studies that showed in both Drosophila and human cells that mitotic chromosomes are organized in higher order structural domains with diameters of ∼130 nm. These structural domains appeared to be linear, fiber-like structures and were termed as chromonema (Belmont et al., 1989, Belmont and Bruce, 1994). On the other hand, contrary to our results, a recent study using EM tomography with DNA labeling showed that chromatin assembles into disordered chains with diameters of 5–24 nm (Ou et al., 2017). Several factors might have contributed to this discrepancy; for example, our study examined a specific genomic region (the rDNA) using GFP-labeled proteins in live cells, whereas the recent study (Ou et al., 2017) focused on imaging all chromatin non-specifically using DNA dyes in fixed cells. Although EM-based methods may be thought to be more suitable for detecting chromatin ultrastructure owing to the superior resolution limit of EM (∼50 pm) than SIM microscopy (∼80–120 nm XY resolution), SIM microcopy has the advantage of reporting chromatin structures in live cells.
Furthermore, our data suggest that dCas9 binding to rDNA does not significantly interfere with chromatin condensation. This is because, first, we did not observe cell-cycle defects in cells expressing rDNA-bound dCas9, suggesting that the mitotic chromatin condensation was not hindered. Second, rDNA condensation could be readily detected upon acute nutrient depletion or DNA damage (Figure 4). Since the large tripartite ring of condensin has a similar dimension and geometry as the cohesin complex (which has a diameter of ∼40 nm [Haering et al., 2002, Cuylen et al., 2011]), it is conceivable that the condensin rings could slide across numerous chromatin-bound dCas9 molecules that are estimated to be less than ∼10 nm in size.
Factors Required for rDNA Condensation Induced by Glucose Starvation
Apart from condensin, several other factors have been identified to be required for rDNA condensation during mitosis, including the cohesin complex (Guacci et al., 1997), Cdc14 (Sullivan et al., 2004), Fob1, Csm1, Irs4, and Tof2 (Waples et al., 2009). However, factors required for rDNA condensation induced by glucose starvation are not known. In this study, we found that the condensin complex plays an active role in rDNA condensation induced by glucose starvation. The secondary stage of rDNA condensation was absent or delayed in the condensin mutant ycs4-1 compared with WT, and condensation intermediates accumulated in ycs4-1 cells. The fact that rDNA rings were still able to form in ycs4-1 cells could mean that the primary stage of condensation did not require the condensin complex. Alternatively, it is also possible that ycs4-1 mutant cells at restrictive temperature contained a small amount of functional Ycs4 proteins that were sufficient to compact rDNA chromatin into ring-like structures, but not sufficient for further compaction.
We further found that Top2 is not responsible for rDNA condensation induced by glucose starvation, but that it plays a key role in rDNA segregation. Results from previous studies suggest that Top2 functions to resolve catenations between sister chromatids and its inactivation results in 4′,6-diamidino-2-phenylindole (DAPI)-stained anaphase bridges and broken chromosomes upon completion of cytokinesis (Dinardo et al., 1984, Shamu and Murray, 1992). Interestingly, we saw that the majority of the top2-4 cells exhibited a binucleated phenotype after ∼2 hr at 37°C, indicating that many chromosomal regions were capable of successful segregation; however, rDNA segregation was completely blocked. Thus, we conclude that rDNA is a primary target for Top2 action during anaphase. It is likely that the rDNA repeats are more prone to entanglement as a consequence of DNA replication than other genomic regions, and Top2 is essential for resolving these entanglements. These results highlight the importance of Top2 in maintaining stability in the rDNA region.
A previous study has shown that rDNA condenses in response to nitrogen starvation and rapamycin treatment (Tsang et al., 2007). Since both nitrogen depletion and rapamycin treatment inhibit the TOR pathway (Rødkær and Færgeman, 2014), it was concluded that rDNA condensation is mediated by the inhibition of the TOR pathway activity. In comparison, the rDNA condensation process reported in our study can be additionally regulated by glucose signaling pathways, such as the pathways regulated by the Snf1- or cyclic AMP (cAMP)-dependent kinases (Rødkær and Færgeman, 2014). The biological function of rDNA condensation under stressful conditions is not fully understood. It is known that rapamycin treatment in the absence of condensin causes fragmented nucleolus phenotype due to the formation of extrachromosomal rDNA circles (Tsang et al., 2007). Therefore, it is possible that rDNA condensation in response to glucose starvation serves a similar role by preserving rDNA stability. Alternatively, rDNA condensation may significantly affect ribosome biogenesis, which could in turn lead to a decreased translation of normal mRNAs and an increased translation of stress-response-specific mRNAs. Future studies are required to investigate these possibilities.
Effect of Transcription on dCas9 Binding to rDNA In Vivo
Using a long exposure time (∼5.8 s) on early log phase cells (OD600 = 0.2), we observed that dCas9 binding on rDNA had a predominantly diffuse pattern (Figure 1E), despite strong enrichment of dCas9 on the rDNA array (Figure 6A). Since rDNA is the heaviest transcribed region in the genome, we reason that this diffuse pattern was due to the dynamic binding and unbinding of dCas9 on rDNA. In support of this view, cells expressing dCas9 targeted to the non-transcribed NTS1 or NTS2 spacer regions showed significantly reduced levels of the diffuse dCas9 pattern (Figure 1G). It is likely that dCas9 molecules are frequently removed by RNA polymerase I during rRNA synthesis and they can rebind to target sites after the transcription bubble passes. This also leads to the expectation that, as cells grow to higher densities, dCas9 interaction with chromatin becomes more stable due to reduced Pol I activity.
How RNA polymerase removes dCas9 from its target remains an open question. It is possible that the intrinsic helicase activity of RNA polymerase is sufficient to displace dCas9. The strand specificity of the gRNA appears to be crucial in this process, since we and others (Qi et al., 2013) have noted that gRNAs must be designed on the template strand of the gene of interest to avoid blocking transcription.
Robust dCas9 Binding to rDNA In Vivo Irrespective of Nucleosomes or Condensation
Our ChIP and imaging data indicate that, at the steady state, the association of dCas9 proteins with the rDNA chromatin was not inhibited by nucleosomes, and that the “closed” rDNA compartment was accessible for dCas9 binding. In agreement with this view, single molecule studies have shown that, despite reduced sampling efficiency, HaloTag-dCas9 proteins could bypass chromatin obstacle and target the pericentromeric heterochromatin (Knight et al., 2015). dCas9-GFP proteins were also able to target the telomeric heterochromatin in live-cell imaging experiments (Chen et al., 2013). Several possibilities could account for the lack of apparent inhibitory effect of nucleosomes on dCas9 binding in vivo. First, the endogenous 18S rDNA chromatin may exhibit high breathing property, a phenomenon referring to the spontaneous unwrapping and rewrapping of nucleosomal DNA from the histone octamer (Polach and Widom, 1995). This would allow dCas9 to gain access to DNA targets in the exposed state. This hypothesis is supported by the recent in vitro studies showing that Cas9 cleavage was enhanced on the 5S rDNA sequences that had ∼100-fold higher breathing dynamics compared with the artificial Widom 601 sequence (Isaac et al., 2016). Second, chromatin remodeling factors such as the SWI/SNF and RSC complexes may cause sliding or eviction of nucleosomes and therefore exposure of the PAM sites for dCas9 recognition, a process that has also been demonstrated in vitro (Horlbeck et al., 2016, Isaac et al., 2016). It will be interesting to test whether chromatin-bound dCas9 can effectively compete with nucleosomes and inhibit their incorporation on DNA sequences in live cells.
Furthermore, our results show that dCas9 binding to rDNA did not significantly change during rDNA condensation in response to acute glucose starvation. This suggests that dCas9 accessibility is not hindered by the folded topology of chromatin. However, it is important to note that the ChIP method is unable to provide information on whether dCas9 proteins stably associate with the same targets, or dynamically bind and unbind to targets during condensation.
Role of Helicases in Facilitating DNA Replication Near dCas9-Binding Sites
Our results show that Sgs1, the yeast homolog of the Werner and Bloom syndrome helicase in humans, plays an essential role in cells expressing rDNA-bound dCas9. In the absence of Sgs1, the dCas9-gRNA complexes activate DNA damage checkpoint pathways, leading to cell-cycle arrest at the G2/M phase and loss of viability. Deletion of genes from the HR machinery rescues the growth defect of sgs1Δ mutants. Based on these results, we reasoned that the rDNA-bound dCas9 may impede the progression of replication forks at the rDNA region, possibly causing replication forks to stall. Studies in both E. coli (Possoz et al., 2006) and S. cerevisiae (Greenfedert and Newlon, 1992) have shown that replication forks can stall when they encounter tightly bound protein-DNA complexes, so we reasoned that the dCas9-gRNA complex can form similar “roadblocks” for replication due to its strong binding to DNA (Sternberg et al., 2014) (Figure 9). Stalled replication forks can regress to form “chicken foot” structures; these structures have been observed in hydroxyurea-treated cells deficient in DNA damage checkpoint kinase Rad53 (Sogo et al., 2002). Sgs1 may be important for resolving these aberrant structures and re-starting replication forks. In the absence of Sgs1, the chicken foot structures may be further processed by the HR repair pathway, which can generate toxic repair intermediates or broken replication forks (Figure 9). It has been shown that the deletion of SGS1 causes hyper-recombination in the rDNA locus (Gangloff et al., 1994, Versini et al., 2003). Furthermore, activation of HR repair pathway was shown to be the major cause of premature cell death in the sgs1Δsrs2Δ double mutant (Gangloff et al., 2000). Taken together, our results suggest that Sgs1 suppresses inappropriate HR-based DNA repair that takes place after replication fork stalling around dCas9 binding sites.
Figure 9.

Potential Roles for DNA Helicases in Cells Expressing rDNA-Bound dCas9
dCas9 binding to the 18S rDNA region may cause replication fork stalling. Stalled replication forks can reverse to form “chicken foot” structures. Sgs1 helps to restart replication fork at the dCas9 binding sites by preventing or resolving these “chicken foot” structures. Rrm3 plays an auxiliary role in displacing dCas9-gRNA from DNA template. In sgs1Δ cells, stalled replication forks are processed by the homologous recombination pathway, which can generate repair intermediates causing cellular toxicity.
Similar to the sgs1Δ mutant, rrm3Δ mutant also showed growth defect and activated DNA damage checkpoint response. Rrm3 has been shown to promote fork progression through non-nucleosomal protein-DNA complexes, and its absence leads to increased fork stalling and breakage at many genomic loci including the rDNA (Torres et al., 2004). Thus, our data support the role of Rrm3 in facilitating the clearance of dCas9 from DNA target, and this allows the replication fork to pass the dCas9-gRNA complexes.
Our results suggest that dCas9 proteins may form transient, site-specific replication roadblocks that can be rapidly removed by DNA helicases. Helicase genes are abundant in higher eukaryotic organisms (Umate et al., 2011), thus it is likely that other helicases can function redundantly with Sgs1 and Rrm3 to facilitate DNA replication around dCas9 binding sites. The recently developed CRISPR interference (CRISPRi) technology relies on the persistent binding of dCas9 to DNA to achieve gene activation or silencing (Qi et al., 2013, Gilbert et al., 2014). Thus, the ability of cells to efficiently restart replication forks and suppress inappropriate repair mechanisms may be particularly important in CRISPRi experiments. Our results highlight the need to exercise caution when applying CRISPRi in animal models or patient tissues if essential helicase functions are compromised.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are very grateful to Dr. Malaiyalam Mariappan for providing western blot facilities, Dr. Vladimir Polejaev (Yale West Campus Imaging Core) for microscopy training, Dr. Patrina Pellett (GE Healthcare) for assistance with SIM imaging, Dr. Charlie Boone (University of Toronto) for providing ycg1-2 and ycs4-1 strains, Dr. Vincent Guacci (University of California, Berkeley) for providing sequence information for the mcd1-1 mutation, Ruijie Song for image analysis discussions, Pamela Carley and Liese Klein for editorial advice, and Tanya Berbasova, Jessica Ye, Gregory Elison, and Ethan Sarnoski for critical reading of the manuscript. M.A. acknowledges funding from the U.S. National Institutes of Health (1DP2AG050461-01 and 1U54CA209992-01).
Author Contributions
Y.X. and M.A. planned the project. Y.X. designed and performed the experiments and analyzed the data. Y.X. and M.A. interpreted the data and results and wrote and approved the manuscript. M.A. supervised the project.
Declaration of Interests
The authors declare no competing interests.
Published: June 29, 2018
Footnotes
Supplemental Information includes Transparent Methods, seven figures, four tables, and one video and can be found with this article online at https://doi.org/10.1016/j.isci.2018.06.001.
Supplemental Information
References
- Belmont A.S., Braunfeld M.B., Sedat J.W., Agard D.A. Large-scale chromatin structural domains within mitotic and interphase chromosomes in vivo and in vitro. Chromosoma. 1989;98:129–143. doi: 10.1007/BF00291049. [DOI] [PubMed] [Google Scholar]
- Belmont A.S., Bruce K. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J. Cell Biol. 1994;127:287–302. doi: 10.1083/jcb.127.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger A.N., Bintu B., Moffitt J.R., Wang S., Beliveau B.J., Fudenberg G., Imakaev M., Mirny L.A., Wu C.T., Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529:418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer. 2013;13:542–558. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon P., Gelin-Licht R., Ganesan A., Zhang J., Levchenko A. Single-cell dynamics and variability of MAPK activity in a yeast differentiation pathway. Proc. Natl. Acad. Sci. USA. 2016;113:E5896–E5905. doi: 10.1073/pnas.1610081113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuylen S., Metz J., Haering C.H. Condensin structures chromosomal DNA through topological links. Nat. Struct. Mol. Biol. 2011;18:894–901. doi: 10.1038/nsmb.2087. [DOI] [PubMed] [Google Scholar]
- Dammann R., Lucchini R., Koller T., Sogo J.M. Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:2331–2338. doi: 10.1093/nar/21.10.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. Gene regulation in the third dimension. Science. 2008;319:1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinardo S., Voelkel K., Sternglanzt R. DNA topoisomerase II mutant of Saccharomyces cerevisiae: topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc. Natl. Acad. Sci. USA. 1984;81:2616–2620. doi: 10.1073/pnas.81.9.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C., Gruetzner R., Kandzia R., Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman L., Aragon-Alcaide L., Strunnikov A. The condensin complex governs chromosome condensation and mitotic transmission of rDNA. J. Cell Biol. 2000;149:811–824. doi: 10.1083/jcb.149.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S., McDonald J.P., Bendixen C., Arthur L., Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S., Soustelle C., Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C., Qi L.S. Genome-Scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göndör A., Ohlsson R. Chromosome crosstalk in three dimensions. Nature. 2009;461:212–217. doi: 10.1038/nature08453. [DOI] [PubMed] [Google Scholar]
- Greenfedert S.A., Newlon C.S. Replication forks pause at yeast centromeres. Mol. Cell. Biol. 1992;12:4056–4066. doi: 10.1128/mcb.12.9.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V., Hogan E., Koshland D. Chromosome condensation and sister chromatid pairing in budding yeast. J. Cell Biol. 1994;125:517–530. doi: 10.1083/jcb.125.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V., Koshland D., Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haering C.H., Löwe J., Hochwagen A., Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell. 2002;9:773–788. doi: 10.1016/s1097-2765(02)00515-4. [DOI] [PubMed] [Google Scholar]
- Hinz J.M., Laughery M.F., Wyrick J.J. Nucleosomes inhibit Cas9 endonuclease activity in vitro. Biochemistry. 2015;54:7063–7066. doi: 10.1021/acs.biochem.5b01108. [DOI] [PubMed] [Google Scholar]
- Hirano T. Condensin-based chromosome organization from bacteria to vertebrates. Cell. 2016;164:847–857. doi: 10.1016/j.cell.2016.01.033. [DOI] [PubMed] [Google Scholar]
- Horlbeck M.A., Witkowsky L.B., Guglielmi B., Replogle J.M., Gilbert L.A., Villalta J.E., Torigoe S.E., Tjian R., Weissman J.S. Nucleosomes impede cas9 access to DNA in vivo and in vitro. Elife. 2016;5:1–21. doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Moazed D. Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev. 2003;17:2162–2176. doi: 10.1101/gad.1108403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac R.S., Jiang F., Doudna J.A., Lim W.A., Narlikar G.J., Almeida R. Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. Elife. 2016;5:1–14. doi: 10.7554/eLife.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivessa A.S., Lenzmeier B.A., Bessler J.B., Goudsouzian L.K., Schnakenberg S.L., Zakian V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell. 2003;12:1525–1536. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- Kimura K., Hirano T. ATP-dependent positive supercoiling of DNA by 13S condensin: a biochemical implication for chromosome condensation. Cell. 1997;90:625–634. doi: 10.1016/s0092-8674(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Knight S.C., Xie L., Deng W., Guglielmi B., Witkowsky L.B., Bosanac L., Zhang E.T., El Beheiry M., Masson J.B., Dahan M. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015;350:823–826. doi: 10.1126/science.aac6572. [DOI] [PubMed] [Google Scholar]
- Lavoie B.D., Hogan E., Koshland D. In vivo intermediates in condensation reveal dual pathways for mitotic chromosome folding. Genes Dev. 2003;18:76–87. doi: 10.1101/gad.1150404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie B.D., Hogan E., Koshland D. In vivo requirements for rDNA chromosome condensation reveal two cell-cycle-regulated pathways for mitotic chromosome folding. Genes Dev. 2004;18:76–87. doi: 10.1101/gad.1150404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Naseri A., Reyes-Gutierrez P., Wolfe S.A., Zhang S., Pederson T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA. 2015;112:3002–3007. doi: 10.1073/pnas.1420024112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackintosh S.G., Raney K.D. DNA unwinding and protein displacement by superfamily 1 and superfamily 2 helicases. Nucleic Acids Res. 2006;34:4154–4159. doi: 10.1093/nar/gkl501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox P.S., Portier N., Desai A., Oegema K. Molecular analysis of mitotic chromosome condensation using a quantitative time-resolved fluorescence microscopy assay. Proc. Natl. Acad. Sci. USA. 2006;103:15097–15102. doi: 10.1073/pnas.0606993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefferd A.L., Kornepati A.V., Bogerd H.P., Kennedy E.M., Cullen B.R. Expression of CRISPR/Cas single guide RNAs using small tRNA promoters. RNA. 2015;21:1683–1689. doi: 10.1261/rna.051631.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz K., Hondele M., Goetze H., Gmelch K., Stoeckl U., Griesenbeck J. Actively transcribed rRNA genes in S. cerevisiae are organized in a specialized chromatin associated with the high-mobility group protein Hmo1 and are largely devoid of histone molecules. Genes Dev. 2008;22:1190–1204. doi: 10.1101/gad.466908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T., Soutoglou E. The Emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2012;10:243–254. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T., Kobayashi T. Visualization of the dynamic behavior of ribosomal RNA gene repeats in living yeast cells. Genes Cells. 2011;16:491–502. doi: 10.1111/j.1365-2443.2011.01506.x. [DOI] [PubMed] [Google Scholar]
- Ochiai H., Sugawara T., Yamamoto T. Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res. 2015;43:e127. doi: 10.1093/nar/gkv624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O'Shea C.C. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017;357 doi: 10.1126/science.aag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A., Lucca C., Liberi G., Marini F., Lopes M., Plevani P., Romano A., Di Fiore P.P., Foiani M. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 1999;18:6561–6572. doi: 10.1093/emboj/18.22.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach K.J., Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- Popov A., Smirnov E., Kováčik L., Raška O., Hagen G., Stixová L., Raška I. Duration of the first steps of the human rRNA processing Duration of the first steps of the human rRNA processing. Nucleus. 2017;4:134–141. doi: 10.4161/nucl.23985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possoz C., Filipe S.R., Grainge I., Sherratt D.J. Tracking of controlled Escherichia coli replication fork stalling and restart at repressor-bound DNA in vivo. EMBO J. 2006;25:2596–2604. doi: 10.1038/sj.emboj.7601155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R., Deng L., Xue Y., Suzuki K., Zhang W., Yu Y., Wu J., Sun L., Gong X., Luan H., Yang F. Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 2017;27:483–504. doi: 10.1038/cr.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rødkær S.V., Færgeman N.J. Glucose- and nitrogen sensing and regulatory mechanisms in Saccharomyces cerevisiae. FEMS Yeast Res. 2014;14:683–696. doi: 10.1111/1567-1364.12157. [DOI] [PubMed] [Google Scholar]
- Sandmeier J.J., French S., Osheim Y., Cheung W.L., Gallo C.M., Beyer A.L., Smith J.S. RPD3 is required for the inactivation of yeast ribosomal DNA genes in stationary phase. EMBO J. 2002;21:4959–4968. doi: 10.1093/emboj/cdf498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu C.E., Murray A.W. Sister chromatid separation in frog egg extracts requires DNA topoisomerase II activity during anaphase. J. Cell Biol. 1992;117:921–934. doi: 10.1083/jcb.117.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo J.M., Lopes M., Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–603. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Spiller D.G., Wood C.D., Rand D.A., White M.R. Measurement of single-cell dynamics. Nature. 2010;465:736–745. doi: 10.1038/nature09232. [DOI] [PubMed] [Google Scholar]
- Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone E.M., Heun P., Laroche T., Pillus L., Gasser S.M. MAP kinase signaling induces nuclear reorganization in budding yeast. Curr. Biol. 2000;10:373–382. doi: 10.1016/s0960-9822(00)00413-9. [DOI] [PubMed] [Google Scholar]
- Straight A.F., Shou W., Dowd G.J., Turck C.W., Deshaies R.J., Johnson A.D., Moazed D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999;97:245–256. doi: 10.1016/s0092-8674(00)80734-5. [DOI] [PubMed] [Google Scholar]
- Sullivan M., Higuchi T., Katis V.L., Uhlmann F. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell. 2004;117:471–482. doi: 10.1016/s0092-8674(04)00415-5. [DOI] [PubMed] [Google Scholar]
- Tercero J.A., Diffley J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- Tollervey D., Kos M. Article yeast Pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell. 2010;37:809–820. doi: 10.1016/j.molcel.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres J.Z., Schnakenberg S.L., Zakian V.A. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol. Cell. Biol. 2004;24:3198–3212. doi: 10.1128/MCB.24.8.3198-3212.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang C.K., Bertram P.G., Ai W., Drenan R., Zheng X.F. Chromatin-mediated regulation of nucleolar structure and RNA Pol I localization by TOR. EMBO J. 2003;22:6045–6056. doi: 10.1093/emboj/cdg578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang C.K., Li H., Zheng X.S. Nutrient starvation promotes condensin loading to maintain rDNA stability. EMBO J. 2007;26:448–458. doi: 10.1038/sj.emboj.7601488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umate P., Tuteja N., Tuteja R. Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol. 2011;4:1–20. doi: 10.4161/cib.4.1.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versini G., Comet I., Wu M., Hoopes L., Schwob E., Pasero P. The yeast Sgs1 helicase is differentially required for genomic and ribosomal DNA replication. EMBO J. 2003;22:1939–1949. doi: 10.1093/emboj/cdg180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Su J.H., Beliveau B.J., Bintu B., Moffitt J.R., Wu C.T., Zhuang X. Spatial organization of chromatin domains and compartments in single chromosomes. Science. 2016;544:2955–2960. doi: 10.1126/science.aaf8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples W.G., Chahwan C., Ciechonska M., Lavoie B.D. Putting the brake on FEAR: Tof2 promotes the biphasic release of Cdc14 phosphatase during mitotic exit. Mol. Biol. Cell. 2009;20:245–255. doi: 10.1091/mbc.E08-08-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Watt P.M., Hickson I.D., Borts R.H., Louis E.J. SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., McCord R.P., Ho Y.J., Lajoie B.R., Hildebrand D.G., Simon A.C., Becker M.S., Alt F.W., Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.S., Elledge S.J. Checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were grown to early log phase in synthetic complete media supplemented with 2% glucose and 0.5 μg/mL doxycycline. Then, cells were mounted on a freshly prepared 1.2% agarose pad made of the same media and imaged every 15 min at room temperature. Deconvolved, maximum projected z series are shown.