

Abstract
Hepatoblastoma (HB) is the most common liver tumor in children. Despite recent improvements in treatment strategies, the survival of children with hepatoblastoma remains poor. In this study, we identified a novel role of microRNA‐26a‐5p (miR‐26a‐5p), lin‐28 homolog B (LIN28B), Ras‐related nuclear protein (RAN), and aurora kinase A (AURKA) in HB. The expression of LIN28B, RAN, and AURKA was significantly up‐regulated in human HB livers and cell lines. Knockdown of LIN28B and RAN by small interfering RNAs inhibited HB tumor cell proliferation and foci formation. We also elucidated miR‐26a‐5p‐mediated translational inhibition of LIN28B and AURKA in HB. Overexpression of miR‐26a‐5p markedly decreased LIN28B and AURKA 3′‐untranslated region activities and protein expression and repressed HB cell proliferation and colony formation. In contrast, re‐expression of LIN28B and AURKA rescued miR‐26a‐5p‐mediated suppression of HB cell growth and clonality. Importantly, a decreased miR‐26a‐5p expression correlated with the poor outcome of patients with HB. Conclusion: miR‐26a‐5p is a newly identified repressor of HB growth through its inhibition of the oncogenic LIN28B–RAN–AURKA pathway. (Hepatology Communications 2018;2:481‐491)
Abbreviations
- 3′‐UTR
3′‐untranslated region
- AURKA
aurora kinase A
- FFPE
formalin‐fixed paraffin‐embedded
- HB
hepatoblastoma
- LIN28B
lin‐28 homolog B
- miRNA
miR, microRNA
- mRNA
messenger RNA
- RAN
Ras‐related nuclear protein
- siRNA
small interfering RNA
Introduction
Hepatoblastoma (HB) is the most common liver tumor with an estimated incidence of 1 per 100,000 children. HB originates from liver progenitor cells that acquire malignant transformation during embryogenesis.1 Certain inherited conditions as well as congenital anomalies and low birth weight are believed to increase the risk of HB; however, the etiology of HB remains elusive due to the extreme rarity of this disease.2 Somatic mutations in genes encoding components of the Wnt/β‐catenin signaling pathwayare prevalent in sporadic HB cases. Recently, the interplay of Wnt/β‐catenin and Myc signaling has been shown to play a critical role in poorly differentiated aggressive HB.3 Overexpression of mutant β‐catenin in mice induces HBs that express high levels of c‐Myc.4 These findings suggest Myc as a potential therapeutic target for HB. However, MYC does not harbor any cavities into which small molecules can easily bind, indicating Myc oncoproteins are undruggable.5 In addition, small molecule drugs in combination with cytotoxic chemotherapy have not been developed for patients with HB. Because the event‐free survival rate is less than 50% in children with high‐risk HB6 and pediatric liver transplantation is not easily accessible, current major research efforts are directed toward identifying new regulators of HB.
MicroRNAs (miRNAs) are small, endogenous, noncoding RNAs that regulate gene expression by specifically binding to the 3′‐untranslated region (3′‐UTR) of target gene messenger RNAs (mRNAs).7 miRNAs are involved in many important processes, including development, proliferation, differentiation, apoptosis, and stress response.8, 9, 10 Because miRNAs regulate diverse physiological and developmental processes,11 their own expression and processing must be tightly controlled.12, 13, 14 Due to the rarity of HB and the limited availability of fresh‐frozen tumor samples, analyzing miRNA expression in HB has mostly been achieved from formalin‐fixed paraffin‐embedded (FFPE) samples. Several miRNAs have been reported aberrantly expressed in HB, including miRNA‐21 (miR‐21) and miR‐221.15 Despite these studies, how miRNAs control the development of HB remains little known.
Lin‐28 homolog B (LIN28B) is a highly conserved RNA binding protein that can regulate cell fate to control embryonic development.16 It sustains the proliferative and metabolic capacities of pluripotent stem cells and facilitates the transition from naive to primed pluripotency. Aberrantly expressed LIN28B plays an oncogenic role in various human cancers. Aurora kinase A (AURKA) is a serine–threonine mitotic kinase that plays an important role in regulating mitosis, cell division, and cell cycle progression.17 Overexpression of AURKA promotes cell cycle progression by disrupting cell cycle checkpoints, leading to aneuploidy and genomic instability; the latter is a hallmark of malignant transformation. Emerging results suggest that inhibiting AURKA may be a novel strategy for reducing the stability of Myc protein that has not been previously targetable. Ras‐related nuclear protein (RAN) is a well‐known nuclear trafficking protein that is a member of the Ras family.18 RAN is overexpressed in diverse malignancies and promotes AURKA phosphorylation. Recently, a study demonstrated that LIN28B–RAN–AURKA signaling drives neuroblastoma oncogenesis, providing a pathway that may be amenable to therapeutic targeting.19
In this study, we aimed to understand the function of LIN28B and AURKA in HB. We demonstrate that both genes are direct targets of miR‐26a‐5p. LIN28B and AURKA are up‐regulated in HB, and knockdown of both genes by small interfering RNAs (siRNAs) or by miR‐26a‐5p inhibits HB tumor cell proliferation and foci formation. Our results suggest that miR‐26a‐5p plays an important regulatory role in HB, providing new insights into developing treatment strategies for HB.
Materials and Methods
HB FFPE SAMPLES
Patient HB FFPE samples were collected retrospectively from the First Hospital of Jilin University, China. The study was approved by the institutional review board at the First Hospital of Jilin University.
CELL CULTURE
Human hepatocyte cell line HC‐04, human hepatoblastoma cell line HepG2 (generously provided by Dr. Yuzuru Shiio and Dr. Hongbing Wang20, 21), and cell line HUH6 (a generous gift from Dr. Thomas Pietschmann22) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% calf serum. Hep293TT cells (a generous gift from Dr. Yuzuru Shiio) were cultured in Roswell Park Memorial Institute 1640 supplemented with 10% fetal calf serum, 25 mM 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid, and 1 mM sodium pyruvate.20
OLIGONUCLEOTIDES, PLASMIDS, AND siRNAS
MiR‐26a‐5p mimics, miR‐29a‐3p mimics, and negative control oligonucleotides (miR‐CON) were purchased from Sigma (#HMI0415, #HMC0434, #HMC0002, respectively). Small interfering pool RNA of Lin28B, RAN, and control RNA (siLin28B, siRAN, siCon; five siRNAs per pool) were purchased from Santa Cruz Biotechnology (#SC‐105614, #SC‐36382, #SC‐37007, respectively). LIN28B and AURKA plasmids were purchased from Addgene (#51375 and #20427, respectively). Luciferase reporter gene plasmids, including wild‐type AURKA 3′‐UTR, wild‐type LIN28B 3′‐UTR, and their seed mutants, were synthesized and subcloned into pmirGLO vectors, which were confirmed by sequencing.
CELL TRANSFECTION
Cells were seeded in six‐well plates the day before the experiment and transfected using RNAiMAX transfection reagent (#13778075; Thermo Fisher Scientific), with microRNA mimics (miR‐26a‐5p or miR‐29a‐3p)/negative control at a final concentration of 50 nM or with siRNAs (siLin28B or siRAN)/negative control at a final concentration of 50 μM. After 48 hours, cells were harvested for RNA and protein isolation,23 respectively.
3′‐UTR LUCIFERASE REPORTER ASSAYS
HUH6 and HepG2 cells were seeded in 24‐well plates at a density of 5 × 104 cells/well. After 24 hours, wild‐type AURKA 3′‐UTR and LIN28B 3′‐UTR or their seed mutant luciferase reporter vectors with miR‐26a‐5p mimic or miR‐29a‐3p mimic were transfected into the cells using RNAiMAX transfection reagent. The dual‐luciferase reporter assay system (Promega, Madison, WI) was applied 48 hours after transfection to determine changes in relative luciferase units as described.24 All assays were performed in triplicate, and all values were normalized for transfection efficiency against Renilla luciferase activities.
CELL PROLIFERATION ASSAY
The 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) assay was performed to measure the proliferative ability of HUH6, Hep293TT, and HepG2 cells as described.25 In brief, 5 × 103 cells per well were seeded into 96‐well plates containing complete Dulbecco's modified Eagle's medium or Roswell Park Memorial Institute 1640 medium (100 μL) in triplicate for each condition and were maintained in an incubator at 37°C and 5% CO2. MTS solution (20 μL) was added to each well and incubated for 2 hours. Absorbance at 490 nM (A490) was read on a microplate reader (168‐1000 Model 680; Bio‐Rad Laboratories), and proliferation curves were plotted. The colony formation assay was conducted as described.8
QUANTITATIVE REAL‐TIME POLYMERASE CHAIN REACTION
Total RNA was extracted from tissues or cells with TRIzol reagent (#15596018; Thermo Fisher Scientific).26 Real‐time quantification of mRNA levels of the genes of interest was performed using Brilliant II SYBR Green QPCR Master Mix (#1725274; Bio‐Rad Laboratories, Munich, Germany) as described.27 Normalized cycle threshold (Ct) values were subjected to statistical analysis, and the fold difference was calculated by the ΔΔ Ct method. Human β‐actin and U6 mRNAs were also measured as internal controls for normalization. Specific primers for quantitative polymerase chain reaction are shown in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1185/full.
WESTERN BLOTTING
Tissue or cell lysate was harvested by radio immunoprecipitation assay lysis buffer with protease inhibitors (#78410; Thermo Fisher Scientific). Proteins (30 μg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes according to standard procedures.28 Membranes were blocked, immunostained with primary antibodies, and finally detected by horseradish peroxidase‐conjugated corresponding secondary antibody. Signals were visualized with SuperSignal West Pico Chemiluminescent Substrate (#34080; Thermo Fisher Scientific) according to the manufacturer's protocol. Equal loading of protein was verified with β‐actin.
RNA EXTRACTION FROM FFPE SAMPLES
Total RNAs from 14 FFPE tissues blocks were isolated using the commercially available miRNeasy FFPE kits according to the respective manufacturers' instructions (#217504; QIAGEN, Hilden, Germany).
STATISTICAL ANALYSIS
Statistical analysis and plots were performed and generated using GraphPad Prism software (version 6.0; GraphPad Software). Data are presented as mean values with SEM, and P < 0.05 was considered statistically significant.29 All experiments were performed independently at least 3 times. Statistical significance between two groups was measured using the Student t test.
Results
EXPRESSION OF LIN28B, RAN, AND AURKA IS UP‐REGULATED IN HUMAN HB LIVER
Hepatoblastoma results from the failure of cells to differentiate properly into their final cell types during fetal or postnatal development. Recently, LIN28B was identified as an oncogenic driver in high‐risk neuroblastoma.30 LIN28B–RAN–AURKA signaling was further revealed to drive neuroblastoma oncogenesis.19 We were interested in determining whether LIN28, RAN, and AURKA act as oncogenes in hepatoblastoma. We first examined their expression in human tissue biopsies from patients with HB by quantitative polymerase chain reaction and western blot. The mRNA levels of LIN28B and RAN were significantly higher in HB than in normal liver, whereas AURKA mRNAs showed greater variations among the samples (Fig. 1A). Interestingly, LIN28B protein exhibited larger variations among the samples, whereas RAN and AURKA proteins were notably up‐regulated in HB versus nontumors (Fig. 1B). The inconsistency between mRNA and protein expression of LIN28B and AURKA in HB suggests that both transcriptional and translational mechanisms may be involved.
Figure 1.
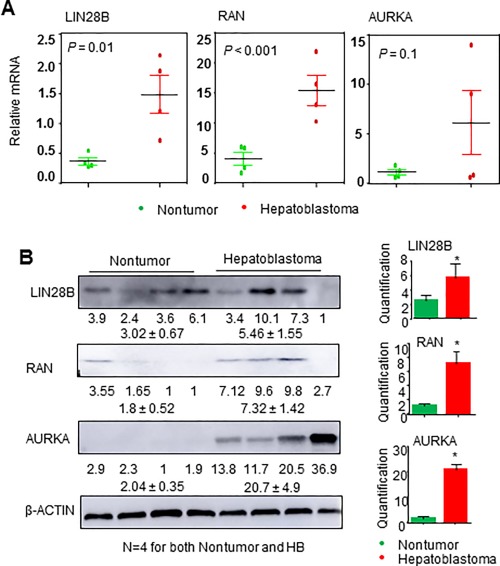
Lin28B, RAN, and AURKA are highly expressed in human HB. (A) Quantitative polymerase chain reaction analysis of LIN28B, RAN, and AURKA expression in nontumor livers and HB. (B) Western blot analysis of LIN28B, RAN, and AURKA protein expression in nontumor livers and HB, with β‐actin as a loading control (n = 4 samples/group). Quantification of the western blot analysis is presented on the right. Data are shown as mean ± SEM (triplicate assays). *P < 0.05 versus nontumor sample.
A MOLECULAR LINK EXISTS BETWEEN LIN28B, RAN, AND AURKA
We next assessed the expression levels of LIN28B, RAN, and AURKA in a panel of human HB cell lines versus normal hepatocyte cell line HC‐04. All three genes were highly expressed in HUH6, HepG2, and Hep293TT cells but with a low expression in HC‐04 (Fig. 2A). To further investigate the molecular link between LIN28B, RAN, and AURKA, we knocked down LIN28B using siLIN28B and examined mRNA expression of RAN and AURKA in all three HB cell lines. LIN28B depletion led to decreased RAN mRNA expression in HUH6, HepG2, and Hep293TT cells, whereas it decreased AURKA mRNA expression in HUH6 and HepG2 but not in Hep293TT cells (Fig. 2B). Interestingly, knockdown of RAN by siRNA significantly decreased AURKA mRNA but also had a modest effect in reducing LIN28B mRNA in all three HB cell lines. At the protein level, siLIN28B decreased RAN protein expression in HUH6 and HepG2 but not in Hep293TT cells, whereas RAN–siRNA decreased AURKA protein in all three lines but had a moderate effect on LIN28B protein expression (Fig. 2C). Taken together, the results suggest that LIN28B may act upstream of RAN and AURKA whereas RAN may be an activator of AURKA expression but may also regulate LIN28B in a feedback fashion. In addition, posttranscriptional regulation may be a mechanism contributing to the differential expression pattern between mRNA and protein.
Figure 2.
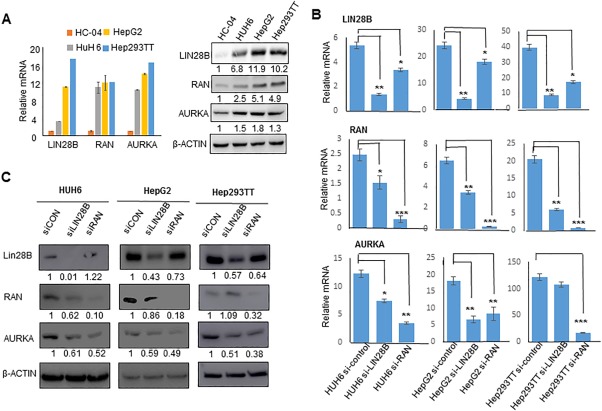
Lin28B directly regulates AURKA through RAN. (A) qPCR (left) and western blot (right) analysis of Lin28B, RAN, and AURKA expression in HB cell lines HUH6, HepG2, and Hep293TT and normal hepatocyte cell line HC‐04. (B) qPCR analysis of LIN28B, RAN, and AURKA mRNA levels in HUH6, HepG2, and Hep293TT cells transfected with control, siLIN28B, or siRAN after 48 hours. LIN28B, RAN, and AURKA mRNA levels are normalized to β‐actin levels. *P < 0.05, **P < 0.01, ***P < 0.001. Data are shown as mean ± SEM (triplicate assays). (C) Western blot analysis of LIN28B, RAN, and AURKA protein expression in HUH6, HepG2, and Hep293TT cells transfected with control, siLIN28B, or siRAN for 2 days, with β‐actin serving as a loading control. Pooled samples (n = 3 per group) were run in a single lane. Numbers indicate density relative to siCon. Abbreviations: qPCR, quantitative polymerase chain reaction; siCon, siControl.
KNOCKDOWN OF LIN28B AND RAN SUPPRESSES HB CELL PROLIFERATION AND COLONY FORMATION
To determine the functional roles of LIN28B and RAN in HB, we depleted LIN28B and RAN using their respective siRNAs in HUH6, HepG2, and Hep293TT cells. Knockdown of LIN28B or RAN markedly diminished HB cell proliferation (Fig. 3A). In addition, siLIN28B and siRAN notably suppressed colony formation of HB cells (Fig. 3B). The results suggest an oncogenic effect of LIN28B and RAN in HB.
Figure 3.
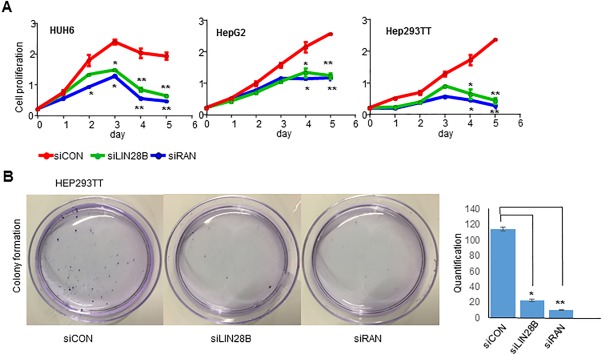
Knockdown of LIN28B and RAN inhibits HB cell growth. (A) Growth curves of control and HUH6 (left), HepG2 (middle), and Hep293TT (right) cells transfected with siLIN28B and siRAN for 5 days. Data are shown as mean ± SEM (triplicate assays). *P < 0.05, **P < 0.01. (B) Colony formation assay of Hep293TT cells transfected with siControl, siLIN28B, or siRNA for 14 days. Quantification of colony numbers is presented on the right. Abbreviation: siCon, siControl.
LIN28B AND AURKA ARE DIRECT TARGET GENES OF miR‐26a‐5p IN HB
The prediction of miRNA target regions by http://www.targetscan.org/ indicated that both LIN28B 3′‐UTR and AURKA 3′‐UTR contain a binding site for miR‐26a‐5p (Fig. 4A). Both target sequences are conserved in several different species. To further investigate whether LIN28B and AURKA were directly regulated by miR‐26a‐5p, luciferase reporter assays were performed in HUH6 and HepG2 cells. Cotransfection of miR‐26a‐5p mimics markedly decreased luciferase reporter activities of LIN28B 3′‐UTR and AURKA 3′‐UTR (Fig. 4B), which were relieved when the miR‐26a‐5p seed regions were mutated (Fig. 4C). MiR‐26a‐5p was significantly suppressed in HB cell lines HUH6, HepG2, and Hep293TT compared to HC‐04 cells (Fig. 4D). In addition, western blot showed decreased LIN28B and AURKA protein expression after transfection with miR‐26a‐5p mimics in HB cells (Fig. 4E). Interestingly, RAN protein was also decreased by miR‐26a‐5p mimics in HUH6 and HepG2 cells. We examined the microRNA target prediction databases http://www.targetscan.org/ and http://34.236.212.39/microrna/home.do but found no miR‐26a‐5p binding sites within RAN 3′‐UTR, which supports that RAN is not a canonical miR‐26a‐5p target. The results suggest that RAN down‐regulation likely resulted from LIN28B knockdown.
Figure 4.
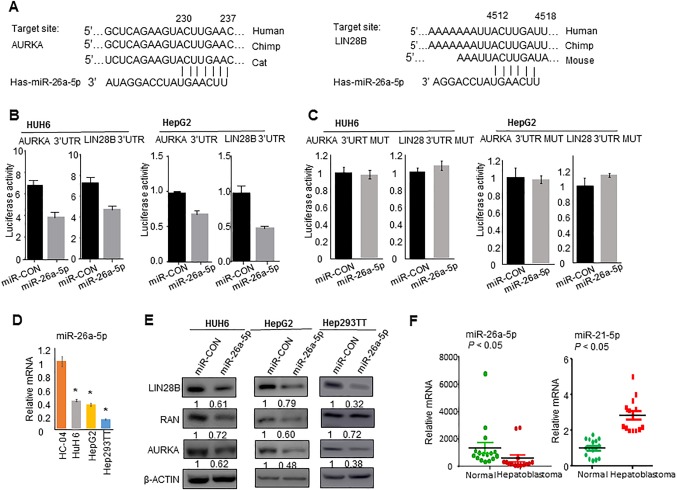
Lin28B and AURKA are novel target genes of miR‐26a‐5p. (A) Target prediction for miR‐26a‐5p using Targetscan identified miR‐26a‐5p binding sequences within LIN28B 3′‐UTR and AURKA 3′‐UTR. (B) Luciferase reporter assays of LIN28B 3′‐UTR and AURKA 3′‐UTR in HUH6 and HepG2 cells transfected with 100 nmol miR‐26a‐5p. (C) Luciferase reporter assays of LIN28B 3′‐UTR mutant and AURKA 3′‐UTR mutant in HUH6 and HepG2 cells transfected with 100 nmol miR‐26a‐5p. (D) qPCR analysis of miR‐26a‐5p expression in normal hepatocyte cells HC‐04 and HB cell lines HUH6, HepG2, and Hep293TT. (E) Western blot analysis of LIN28B, RAN, and AURKA protein expression in the indicated cells transfected with mimic control and miR‐26a‐5p mimic for 48 hours, with β‐actin serving as a loading control. Numbers indicate density relative to control. (F) qPCR analysis of miR‐26a‐5p and miR‐21‐5p expression in normal liver (n = 16) and FFPE HB tissues (n = 14). All data are shown as mean ± SEM (triplicate assays). *P < 0.05 versus HC‐04. Abbreviations: CON, control; MUT, mutant; qPCR, quantitative polymerase chain reaction.
DOWN‐REGULATION OF miR‐26a‐5p IS ASSOCIATED WITH POOR PATIENT OUTCOMES IN HB
To establish the clinical relevance of miR‐26a‐5p in HB, we determined its expression in 14 paraffin HB tissues (Table 1) and compared this with normal children liver specimens (Fig. 4F). Overall, the expression level of miR‐26a‐5p was much lower in HB tissues versus normal tissues. miR‐26a‐5p expression in stage one disease was compatible with normal liver; however, its level decreased drastically in stage four disease (Table 1). In addition, the expression level of miR‐26a‐5p in patients with poor outcomes (i.e., death of disease) was the lowest compared to other stages (P < 0.01). In contrast, miR‐21‐5p was expressed at higher levels in HB versus normal tissues.
Table 1.
Characteristics of FFPE HB Samples and the Relative Expression of miR‐26a‐5p
| Sex | Age | Histologic Type | PRETEXT Stagea | Outcome | Expression of miR‐26a‐5p | |
|---|---|---|---|---|---|---|
| 1 | Male | 4 months | fetal | I | Alive | 2,754.47 |
| 2 | Male | 1 year | fetal | I | Alive | 2,798.26 |
| 3 | Male | 1 year | embryonal | III | Alive | 327.425 |
| 4 | Male | 20 months | embryonal | III | Alive | 68.3725 |
| 5 | Male | 7 months | embryonal | IV | Alive | 576.186 |
| 6 | Male | 17 months | fetal | IV | Alive | 136.56 |
| 7 | Male | 2 years | fetal | IV | Alive | 167.30 |
| 8 | Female | 1 year | mixed | IV | Alive | 118.61 |
| 9 | Male | 2 months | mixed | IV | Alive | 276.85 |
| 10 | Male | 6 years | fetal | IV | Alive | 284.91 |
| 11 | Male | 3 years | embryonal | IV | Relapsed | 175.98 |
| 12 | Male | 10 years | unclear | IV | DOD | 49.46 |
| 13 | Female | 8 years | unclear | IV | DOD | 53.43 |
| 14 | Male | 15 months | mixed | IV | DOD | 53.79 |
PRETEXT = 2005 PRETEXT staging system.
Abbreviation: DOD, death of disease.
miR‐26a‐5p INHIBITS HB CELL PROLIFERATION AND COLONY FORMATION
To further investigate the potential function of miR‐26a‐5p in HB, we transfected miR‐26a‐5p mimics in Hep293TT, HUH6, and HepG2 cells. miR‐26a‐5p mimics but not the control mimics or miR‐29a‐3p mimics drastically attenuated the proliferation of these cells compared to the control cells (Fig. 5A). Because miR‐26a‐5p showed a similar effect on Hep293TT, HUH6, and HepG2 cells, we next examined the ability of miR‐26a‐5p to inhibit foci formation using Hep293TT cells. Overexpression of miR‐26a‐5p mimics also dramatically decreased colony formation (Fig. 5B); however, miR‐26a‐5p inhibitor increased colony formation (Fig. 5C). In addition, the inhibitory effect of miR‐26a‐5p on Hep293TT proliferation was rescued by miR‐26a‐5p inhibitor (Fig. 5C). The results suggest a tumor suppressor function of miR‐26a‐5p in HB.
Figure 5.
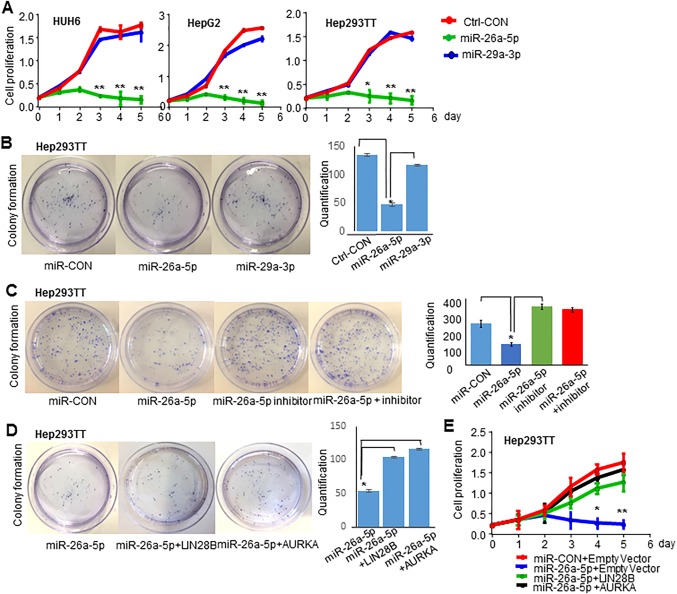
Re‐expression LIN28B and AURKA promotes HB cell growth. (A) Growth curves of control and HUH6 (left), HepG2 (middle), and Hep293TT (right) cells transfected with mimic control, 100 nmol miR‐26a‐5p mimic, and 100 nmol miR‐29a‐3p mimic. (B) Colony formation assay of Hep293TT cells transfected with mimic control, miR‐26a‐5p mimic, and miR‐29a‐3p mimic. (C) Colony formation assay of Hep293TT cells transfected with mimic control, miR‐26a‐5p mimic, miR‐26a‐5p inhibitor, and miR‐26a‐5p mimic with its inhibitor. (D) Colony formation assay of Hep293TT cells transfected with control, miR‐26a‐5p mimic, miR‐26a‐5p mimic plus LIN28B plasmid, and miR‐26a‐5p mimic plus AURKA plasmid. (E) Hep293TT cells were transfected with mimic control plus 2 μg empty vector control, miR‐26a‐5p mimic plus 2 μg empty vector control, miR‐26a‐5p mimic plus 2 μg LIN28B plasmid, and miR‐26a‐5p mimic plus 2 μg AURKA plasmid. The cell proliferation assay was performed from day 0 to day 5. All data are shown as mean ± SEM (triplicate assays). Abbreviation: CON, control.
LIN28B AND AURKA BLOCK THE INHIBITORY EFFECT of miR‐26a‐5p ON HB CELL GROWTH
Because LIN28B and AURKA are downstream targets of miR‐26a‐5p, as shown in Fig. 4, we conducted rescue experiments. As expected, re‐expression of LIN28B and AURKA blocked the effect of miR‐26a‐5p on the repression of colony formation (Fig. 5D) and cellular growth (Fig. 5E) of Hep293TT cells, suggesting that the effect of miR‐26a‐5p is mediated through both genes.
Discussion
Our work was motivated by a report that LIN28B and AURKA expressions were up‐regulated in a transcriptomic and genomic analysis of human HB.31 In addition, neuroblastoma tumorigenesis can be promoted by the LIN28B–RAN–AURKA signaling network. We propose a central regulatory role for LIN28B in HB by its effect on RAN and AURKA.
Our results show that malignant tissues have higher LIN28B, RAN, and AURKA expression than normal liver samples. LIN28B, RAN, and AURKA are also up‐regulated in a panel of malignant HB cells. We demonstrate RAN to be a key downstream component of LIN28B signaling, with LIN28B depletion decreasing RAN expression. As such, AURKA is a key downstream component of RAN signaling, with RAN depletion decreasing AURKA expression. We further show that inhibiting LIN28B by siRNAs significantly decreases HB cell proliferation and colony formation, suggesting that the LIN28B–RAN–AURKA pathway may play a critical role in HB development. Interestingly, knockdown of RAN expression using siRANs decreases LIN28B expression. Because RAN is a downstream signaling molecule of LIN28B, we hypothesize that there is a feedback regulatory loop between RAN and LIN28B.
Overexpression of LIN28B and AURKA has been observed in a variety of tumors. However, the mechanisms that control their expression in HB remain unknown. Recent emerging evidence suggests that miRNAs impact malignant behaviors of HB. Previous studies provided evidence for several prognostic miRNAs associated with HB, such as miR‐49232 and miR‐21.33 Here, we demonstrate that miR‐26a‐5p inhibits LIN28B and AURKA expression by binding to their 3′‐UTR regions. Interestingly, LIN28B regulates AURKA levels through RAN and both are target genes of miR‐26‐5p. Because HB is a rare disease, our research can only rely on FFPE samples. Recent research has demonstrated that miRNA expression is relatively stable and well preserved in FFPE samples.34 Accordingly, we extracted RNAs from 14 FFPE samples and obtained high‐quality miRNAs from these samples.
The miR‐26a‐5p level is significantly lower in HB tissues compared with normal liver. Although the sample size was relatively small, it is evident that miR‐26a‐5p expression shows a negative correlation with disease stage and prognosis. Stage one diseases have much higher miR‐26a‐5p expression and a better outcome compared to stage four diseases. Importantly, miR‐26a‐5p exhibits a remarkable ability to suppress HB cell proliferation and clone formation. Notably, miR‐26a‐5p‐mediated suppression of these events is subsequently impaired by re‐expression of LIN28B and AURKA, indicating that miR‐26a‐5p regulates HB development at least in part by restraining the LIN28B–RAN–AURKA pathway.
Taken together, our study established for the first time a crucial role of the LIN28B–RAN–AURKA signaling pathway in HB oncogenesis (Fig. 6). In addition, we revealed miR‐26a‐5p as a new repressor of LIN28B and AURKA expression. Further investigation of the potential of miR‐26a‐5p as a disease marker and therapeutic target for HB is warranted.
Figure 6.
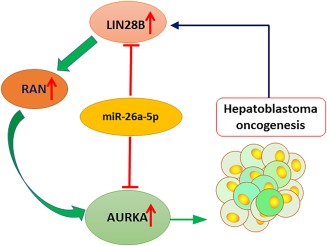
Working model of miR‐26a‐5p as a novel HB repressor. The LIN28B–RAN–AURKA signaling pathway plays a crucial role in hepatoblastoma oncogenesis. MiR‐26a‐5p translationally inhibits LIN28B and AURKA expression by targeting their 3′‐UTR, thereby suppressing HB cell growth.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1185/full.
Supporting Information Slide S1
Supporting Information Slide S2
Acknowledgment
We thank Dr. Yuzuru Shiio (University of Texas Health Science Center), Dr. Hongbing Wang (University of Maryland School of Pharmacy), and Dr. Thomas Pietschmann (TWINCORE Center for Experimental and Clinical Infection Research) for generously providing the cell lines. We thank our laboratory members Dr. Zhihong Yang, Dr. Melanie Tran, and Dr. Dong‐Ju Shin for their technical assistance.
Potential conflict of interest: Nothing to report.
Supported by National Institutes of Health grants R01DK104656, R01ES025909, R21AA022482, R21AA024935, and R01AA026322 (to L.W.); Veterans Affairs Merit Award 1I01BX002634 (to L.W.); and Yale Liver Center award P30DK34989 (to L.W.).
REFERENCES
- 1. Zhang YT, Feng LH, Zhong XD, Wang LZ, Chang J. Vincristine and irinotecan in children with relapsed hepatoblastoma: a single‐institution experience. Pediatr Hematol Oncol 2015;32:18‐25. [DOI] [PubMed] [Google Scholar]
- 2. Han ZG. Mutational landscape of hepatoblastoma goes beyond the Wnt‐beta‐catenin pathway. Hepatology 2014;60:1476‐1478. [DOI] [PubMed] [Google Scholar]
- 3. Cairo S, Wang Y, de Reynies A, Duroure K, Dahan J, Redon MJ, et al. Stem cell‐like micro‐RNA signature driven by Myc in aggressive liver cancer. Proc Natl Acad Sci U S A 2010;107:20471‐20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang H, Lu J, Edmunds LR, Kulkarni S, Dolezal J, Tao J, et al. Coordinated activities of multiple Myc‐dependent and Myc‐independent biosynthetic pathways in hepatoblastoma. J Biol chem 2016;291:26241‐26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW, Wuestefeld T, et al. A MYC‐aurora kinase A protein complex represents an actionable drug target in p53‐altered liver cancer. Nat Med 2016;22:744‐753. [DOI] [PubMed] [Google Scholar]
- 6. Katzenstein HM, Furman WL, Malogolowkin MH, Krailo MD, McCarville MB, Towbin AJ, et al. Upfront window vincristine/irinotecan treatment of high‐risk hepatoblastoma: a report from the Children's Oncology Group AHEP0731 study committee. Cancer 2017;123:2360‐2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song G, Zhang Y, Wang L. MicroRNA‐206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J Biol Chem 2009;284:31921‐31927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Z, Tsuchiya H, Zhang Y, Hartnett ME, Wang L. MicroRNA‐433 inhibits liver cancer cell migration by repressing the protein expression and function of cAMP response element‐binding protein. J Biol Chem 2013;288:28893‐28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer 2010;10:389‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Z, Zhang Y, Wang L. A feedback inhibition between miRNA‐127 and TGFbeta/c‐Jun cascade in HCC cell migration via MMP13. PLoS One 2013;8:e65256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang Z, Cappello T, Wang L. Emerging role of microRNAs in lipid metabolism. Acta Pharm Sin B 2015;5:145‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Yang Z, Whitby R, Wang L. Regulation of miR‐200c by nuclear receptors PPARalpha, LRH‐1 and SHP. Biochem Biophys Res Commun 2011;416:135‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song G, Wang L. A conserved gene structure and expression regulation of miR‐433 and miR‐127 in mammals. PLoS One 2009;4:e7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song G, Wang L. Nuclear receptor SHP activates miR‐206 expression via a cascade dual inhibitory mechanism. PLoS One 2009;4:e6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leichter AL, Purcell RV, Sullivan MJ, Eccles MR, Chatterjee A. Multi‐platform microRNA profiling of hepatoblastoma patients using formalin fixed paraffin embedded archival samples. GigaScience 2015;4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsanov KM, Pearson DS, Wu Z, Han A, Triboulet R, Seligson MT, et al. LIN28 phosphorylation by MAPK/ERK couples signalling to the post‐transcriptional control of pluripotency. Nature cell biology 2017;19:60‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu Q, Yu X, Zhou Z‐W, Zhou C, Chen X‐W, Zhou S‐F. Inhibition of aurora A kinase by alisertib induces autophagy and cell cycle arrest and increases chemosensitivity in human hepatocellular carcinoma HepG2 cells. Curr Cancer Drug Targets 2017;17:386‐401. [DOI] [PubMed] [Google Scholar]
- 18. de Boor S , Knyphausen P, Kuhlmann N, Wroblowski S, Brenig J, Scislowski L, et al. Small GTP‐binding protein Ran is regulated by posttranslational lysine acetylation. Proc Natl Acad Sci U S A 2015;112:E3679‐E3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schnepp RW, Khurana P, Attiyeh EF, Raman P, Chodosh SE, Oldridge DA, et al. A LIN28B‐RAN‐AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer Cell 2015;28:599‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elzi DJ, Song M, Blackman B, Weintraub ST, López‐Terrada D, Chen Y, et al. FGF19 functions as autocrine growth factor for hepatoblastoma. Genes Cancer 2016;7:125‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mackowiak B, Li L, Welch MA, Li D, Jones JW, Heyward S, et al. Molecular basis of metabolism‐mediated conversion of PK11195 from an antagonist to an agonist of the constitutive androstane receptor. Mol Pharmacol 2017;92:75‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haid S, Windisch MP, Bartenschlager R, Pietschmann T. Mouse‐specific residues of claudin‐1 limit hepatitis C virus genotype 2a infection in a human hepatocyte cell line. J Virol 2010;84:964‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo GL, et al. E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr‐1/SHP/EID1 network. Hepatology 2014;60:919‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou T, Zhang Y, Macchiarulo A, Yang Z, Cellanetti M, Coto E, et al. Novel polymorphisms of nuclear receptor SHP associated with functional and structural changes. J Biol Chem 2010;285:24871‐24881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Z, Koehler AN, Wang L. A novel small molecule activator of nuclear receptor SHP inhibits HCC cell migration via suppressing Ccl2. Mol Cancer Ther 2016;15:2294‐2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee SM, Zhang Y, Tsuchiya H, Smalling R, Jetten AM, Wang L. Small heterodimer partner/neuronal PAS domain protein 2 axis regulates the oscillation of liver lipid metabolism. Hepatology 2015;61:497‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choiniere J, Wu J, Wang L. Pyruvate dehydrogenase kinase 4 deficiency results in expedited cellular proliferation through E2F1‐mediated increase of cyclins. Mol Pharmacol 2017;91:189‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsuchiya H, da Costa KA, Lee S, Renga B, Jaeschke H, Yang Z, et al. Interactions between nuclear receptor SHP and FOXA1 maintain oscillatory homocysteine homeostasis in mice. Gastroenterology 2015;148:1012‐1023.e1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tran M, Lee SM, Shin DJ, Wang L. Loss of miR‐141/200c ameliorates hepatic steatosis and inflammation by reprogramming multiple signaling pathways in NASH. JCI Insight 2017;2 pii: 96094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Diskin SJ, Capasso M, Schnepp RW, Cole KA, Attiyeh EF, Hou C, et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet 2012;44:1126‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luo JH, Ren B, Keryanov S, Tseng GC, Rao UN, Monga SP, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006;44:1012‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. von Frowein J, Pagel P, Kappler R, von Schweinitz D, Roscher A, Schmid I. MicroRNA‐492 is processed from the keratin 19 gene and up‐regulated in metastatic hepatoblastoma. Hepatology 2011;53:833‐842. [DOI] [PubMed] [Google Scholar]
- 33. Leichter AL, Sullivan MJ, Eccles MR, Chatterjee A. MicroRNA expression patterns and signalling pathways in the development and progression of childhood solid tumours. Mol Cancer 2017;16:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet 2012;13:358‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1185/full.
Supporting Information Slide S1
Supporting Information Slide S2