

SUMMARY
ER associated degradation (ERAD) removes misfolded proteins from the ER membrane and lumen by the ubiquitin-proteasome pathway. Retrotranslocation of ubiquitinated substrates to the cytosol is a universal feature of ERAD that requires the Cdc48 AAA-ATPase. Despite intense efforts, the mechanism of ER exit, particularly for integral membrane (ERAD-M) substrates, has remained unclear. Using a self-ubiquitinating substrate (SUS) that undergoes normal retrotranslocation independent of known ERAD factors, and the new SPOCK (Single Plate Orf Compendium Kit) micro library to query all yeast genes, we found the rhomboid derlin Dfm1 was required for retrotranslocation of both HRD and DOA ERAD pathway integral membrane substrates. Dfm1 recruited Cdc48 to the ER membrane with its unique SHP motifs, and catalyzed substrate extraction through its conserved rhomboid motifs. Surprisingly, dfm1Δ can undergo rapid suppression, restoring wild-type ERAD-M. This unexpected suppression explained earlier studies ruling out Dfm1, and revealed an ancillary ERAD-M retrotranslocation pathway requiring Hrd1.
Keywords: Dfm1, Retrotranslocation, ERAD, Endoplasmic Reticulum (ER), HRD, rhomboid, Derlins, DOA, SUS, Hmg2, HMG-CoA reductase, Cdc48
ETOC
ER-associated degradation (ERAD) is a conserved pathway of protein quality control. All ERAD pathways require the retrotranslocation of ubiquitinated ERAD substrates from the ER to the cytoplasm for degradation. Neal et al. show that Dfm1 protein is a critical component of a dedicated retrotranslocation pathway for integral membrane ERAD substrates.
INTRODUCTION
ERAD is an ER-localized, ubiquitin-mediated quality control pathway that degrades damaged or misfolded ER proteins (Hampton and Garza, 2009; Foresti et al., 2013). Substrate ubiquitination occurs through ERAD-specific E3 ubiquitin ligases, followed by substrate transfer to the 26S proteasome (Richly et al., 2005). ERAD degrades a variety of misfolded proteins including both fully luminal (ERAD-L) and integral membrane (ERAD-M) substrates (Plemper et al., 1998; Vashist and Ng, 2004). In this capacity, these degradation pathways play important roles in a variety of human maladies, including cystic fibrosis, retinal degeneration, aging, and type II diabetes (Zhao and Ackerman, 2006). ERAD pathways also degrade a number of normal proteins; the best studied being the essential enzyme HMGR (Hydroxy-3-Methyl-Glutaryl-CoA Reductase). In both yeast and mammals, HMGR undergoes regulated ERAD as part of cellular control of sterol synthesis. In mammals, the lone HMGR enzyme is subject to regulated ERAD, while in yeast the Hmg2 isozyme undergoes regulated degradation while the Hmg1 isozyme is stable in all conditions (Hampton and Garza, 2009; Jo and Debose-Boyd, 2010).
In S. cerevisiae, ERAD is mediated by the HRD (HMG-CoA Reductase Degradation) and DOA (Degradation of α 2) pathways, by the separate, conserved integral membrane E3 ligases Hrd1 and Doa10 (Carvalho et al., 2006; Chen et al., 2006; Foresti et al., 2013; Hampton and Garza, 2009). A unifying feature of all ERAD pathways is movement of ER substrates to the cytosol in a process known as retrotranslocation (Baldridge and Rapoport, 2016; Garza et al., 2009a; Hiller et al., 1996; Nakatsukasa et al., 2008). Retrotranslocation requires the Cdc48 ATPase (p97 in mammals) as an energy source for substrate extraction, that entails complete removal of integral membrane ERAD-M substrates from the ER membrane, and movement of lumenal ERAD-L substrates across the ER membrane (Braun et al., 2002; Meyer et al., 2000; Ye et al., 2001).
Retrotranslocation remains poorly understood, and in particular, the identity of a transmembrane channel for ERAD-M substrates is unclear. (Carvalho et al., 2010; Garza et al., 2009; Hampton and Sommer, 2012; Nakatsukasa et al., 2016; Plemper et al., 1997; Scott and Schekman, 2008; Wahlman et al., 2007). We employed a previously characterized self-ubiquitinating substrate SUS, that undergoes retrotranslocation independent of all known candidates, along with a new complete mutant array called SPOCK (Single Plate Orf Compendium Kit), to discover mutants with deficiencies in ERAD-M retrotranslocation (Jaeger, Ornelas et al., 2017). Our analysis revealed that the derlin Dfm1 had a broad and strong role in ERAD-M in both the HRD and DOA pathways. Dfm1 is a member of the rhomboid superfamily, and has a unique C-terminal “SHP box” motif that binds Cdc48 (Goder et al., 2008; Sato and Hampton, 2006; Stolz et al., 2010a). The conserved rhomboid sequences and the SHP box were each required for Dfm1 mediated retrotranslocation, and appeared to serve distinct functions. The SHP box allowed Cdc48 association with the membrane, and the rhomboid sequences were required for substrate extraction.
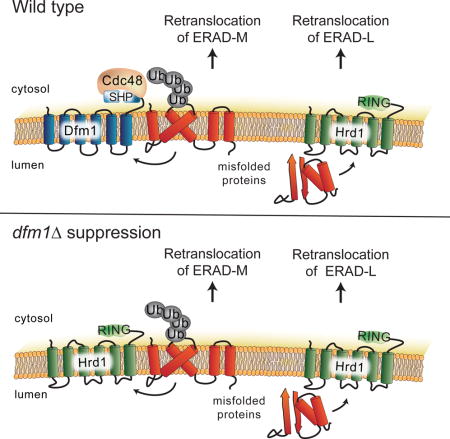
This strong, broad role for Dfm1 was perplexing because we had previously reported that Dfm1 was not involved in either HRD or DOA dependend ERAD (Sato and Hampton, 2006; Goder, Carvalho and Rapoport, 2008), while others have published a role for Dfm1 in only DOA-dependent branches of ERAD (Avci et al., 2014; Stolz et al., 2010a). We resolved this conundrum by showing dfm1Δ underwent rapid suppression during strong expression of ERAD-M substrates. dfm1Δ suppression required Hrd1, which was elevated in suppressees by a duplication of chromosome XV. Our results show that a dedicated ERAD-M retrotranslocation pathway mediated by Dfm1 exists, and that Hrd1 may provide an ancillary route in the absence of Dfm1.
RESULTS
SUS-GFP as an optical retrotranslocation reporter
We employed the well-characterized SUS (self-ubiquitinating substrate) to screen for genes required for late stages of ERAD. SUS consists of the catalytically active cytosolic RING domain of the Hrd1 E3 ligase fused to the transmembrane domain of the highly stable Hmg1 isoform of HMGR. SUS undergoes self-catalyzed degradation dependent on its Hrd1 RING domain, the E2 Ubc7, and Cdc48 (Garza et al., 2009). Full-length SUS is retrotranslocated to cytosol in a Cdc48-dependent manner, but does not require Hrd1 or Doa1 for degradation. We employed SUS in a screen biased towards the late stages of ERAD steps and particularly retrotranslocation.
To facilitate screening, we made a SUS-GFP optical reporter expressed from the strong TDH3 promoter (Fig. 1A). Because the Hmg1 included an added Myc epitope in the first lumenal loop, we confirmed that the GFP did not affect SUS topology by limited proteolysis with trypsin followed by blotting for the lumenal Myc or the cytosolic HA epitopes (Fig. 1B). SUS-GFP behaved identically to SUS in all assays used to characterize SUS initially (Garza et al., 2009): SUS-GFP degradation was rapid in WT or hrd1Δ strains, but drastically stabilized in ubc7Δ or cdc48-2 (Fig. 1C) strains. We examined retrotranslocation of SUS-GFP with our in vivo retrotranslocation assay (Neal et al., 2017). Proteasome inhibitor MG132- or vehicle-treated cells expressing SUS-GFP were lysed without detergent, and the lysate was ultracentrifuged to separate membrane-bound from retrotranslocated SUS-GFP. Retrotranslocated, ubiquitinated SUS-GFP was immunoprecipitated from the soluble (S) fraction using anti-GFP antibodies, and detected by ubiquitin immunoblotting (Fig. 1D, top panels). The pellet was solubilized and similarly analyzed (Fig. 1D, bottom panels). As expected, a fraction of the ubiquitinated SUS-GFP was soluble in untreated cells (Fig. 1D, lanes 1–2), indicating that SUS-GFP underwent retrotranslocation in normal conditions. Addition of MG132 increased ubiquitinated SUS-GFP in both the P and S fraction, as expected when the proteasome was blocked (Fig. 1D, lanes 3–4). Like SUS and Hmg2-GFP (Garza et al., 2009), full length SUS-GFP was retrotranslocated, as shown when enzymatic removal of the attached ubiquitin from the retrotranslocated material by protease Usp2 catalytic core (Usp2Core) restored full-length SUS-GFP (Fig. 1D, right panel).
Figure 1. SUS-GFP behaves as a HRD pathway substrate and is used as an optical retrotranslocation factor reporter in the yeast genomic screen.
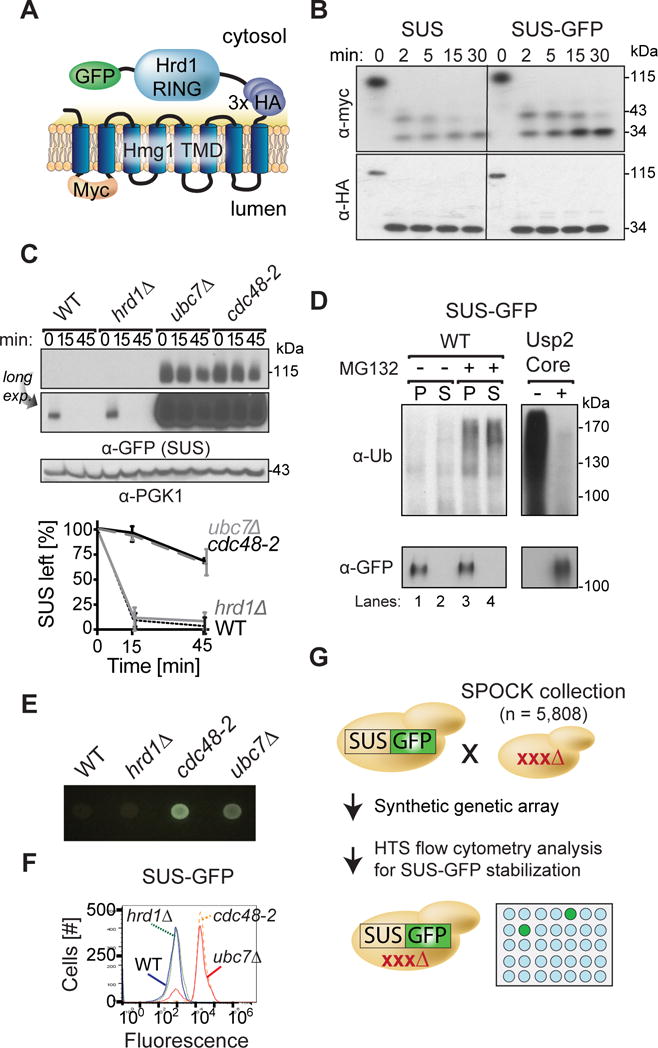
(A) Depiction of fusion protein, SUS-GFP. The transmembrane Hmg1 domain has a lumenal Myc epitope and the cytosolic domain has three HA epitopes followed by the HRD RING domain fused with the GFP epitope. (B) SUS-GFP is correctly inserted into microsomes. Microsomes prepared from strains expressing SUS or SUS-GFP were digested with trypsin for the indicated times and immunoblotted with α-Myc and α-HA. (C) Degradation of SUS-GFP depends on Ubc7 and cdc48-2. The indicated strains expressing SUS-GFP were grown into log phase and degradation was measured by a cycloheximide chase (CHX). After CHX addition, cells were lysed at the indicated times and analyzed by SDS-PAGE and immunoblotted for SUS-GFP with α-GFP. Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t=0 was taken as 100% and data is represented as mean ± SEM from at least three experiments. (D) Full-length SUS-GFP retrotranslocates in vivo. Left panel, in vivo retrotranslocation of SUS-GFP. WT strains were grown to log-phase and treated with MG132 (25 μg/mL). Crude lysate was prepared and ultracentrifuged to discern ubiquitinated SUS-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, SUS-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted for ubiquitin and SUS-GFP. Right panel, in vivo retrotranslocated SUS-GFP is full-length. Full-length SUS-GFP was immunoprecipitated and immunoblotted for SUS-GFP with α-GFP and α-Ubi. (E) Colony fluorescence shows increased steady-state levels of SUS-GFP in ubc7Δ and cdc48-2 strains. The indicated strains were grown to log phase and .03 OD of cells were spotted onto YPD plates and grown at 30°C for two days. Plates were imaged by a fluorescent imager. (F) Flow cytometry shows increased steady-state levels of SUS-GFP in ubc7Δ and cdc48-2 strains. The indicated strains were grown to log phase and were subjected to flow cytometry. Histograms of 10,000 cells are shown, with the number cells versus GFP fluorescence. (G) High throughput pipeline for identifying genes involved in SUS-GFP retrotranslocation. The SUS-GFP reporter was introduced into the SPOCK collection consisting of a 5,808 genome-wide library of yeast nulls and DaMP essential genes using SGA technology. The array was transferred to liquid YPD media in 384-well plates and SUS-GFP fluorescence was measured by a LS Fortessa high throughput flow cytometer.
To test the utility of SUS-GFP as an optical reporter, we examined SUS-GFP steady-state levels in ubc7Δ and cdc48-2 by colony fluorescence and flow cytometry. Colony fluorescence was assessed by plating early log phase cells onto YPD plates followed by two days outgrowth, followed by direct observation of colony fluorescence with appropriate illumination and filters (Hampton, 2005). Log-phase GFP fluorescence were also measured by flow cytometry. Colony fluorescence and flow cytometry each showed dramatic elevation of SUS-GFP steady state levels in ubc7Δ and cdc48-2 strains compared to WT or hrd1Δ, clearly seen by direct inspection of outgrown colonies (Fig. 1E), or a 12-fold increase in mean fluorescence in flow cytometry (Fig. 1F), validating each method for detection of stabilizing mutants.
Yeast genomic screen to identify retrotranslocation factors
We employed a high throughput yeast genomic screen to identify ERAD retrotranslocation. Using standard EMAP technology (Collins et al., 2010), SUS-GFP was introduced into the SPOCK (Single Plate ORF Compendium Kit) collection consisting of a 5,808 yeast strain array of non-essential gene deletion mutants and essential DaMP gene mutants (Jaeger, Ornelas et al). The resulting SUS-GFP array was transferred to 16×384-well plates containing 50 μL of YPD per well. Each resulting mutant was analyzed by high throughput flow cytometry to measure the corrected mean fluorescence intensity (Fig. 1G). Mutants exhibiting high fluorescence were validated for stabilization in SUS-GFP degradation by direct biochemical tests (full results in Table S3). Among these were a number of expected ERAD mutants, including ubc7Δ null a cdc48 hypomorph, both rad23 Δ and dsk2 Δ nulls, and several proteasome subunit hypomorphs. In addition, several unanticipated strong candidates emerged including the subject of this work.
Dfm1 was required for retrotranslocation
One of the leading candidates identified in the screen for SUS-GFP degradation factors was the derlin Dfm1 (Fig. 2A). Degradation assays showed strong stabilization of SUS-GFP or the bona fide HRD pathway substrate Hmg2-GFP (Fig. 2A & B) by the dfm1Δ null mutant. We directly tested Dfm1’s role in retrotranslocation with the in vivo retrotranslocation assay as described above (Neal et al., 2017), on both SUS-GFP (Fig. 2C) or Hmg2-GFP (Fig. 2D) as substrates. In each group, the hrd2-1 Rpn1 mutant of the 26S proteasome was included to show strong substrate retrotranslocation and the cdc48-2 allele of AAA-ATPase was included to show deficient substrate retrotranslocation (Garza et al., 2009). cdc48-2 caused a complete block of SUS-GFP or Hmg2-GFP retrotranslocation (Fig. 2C & D lanes 5,6), and the dfm1Δ null showed a similarly strong retrotranslocation defect with SUS-GFP or Hmg2-GFP, as indicated by the complete lack of ubiquitinated material in the S100 fraction (Fig. 2C & D lanes 7,8). In contrast, der1Δ or sec61-2 cells showed no effect on either substrate when compared to wild-type strains in the presence or absence of MG132, consistent with our earlier observation of the lack of involvement of these two candidates in Hmg2-GFP extraction (Fig. 2E & F)(Sato and Hampton, 2006).
Figure 2. Dfm1 is required for in vivo Hmg2-GFP retrotranslocation.
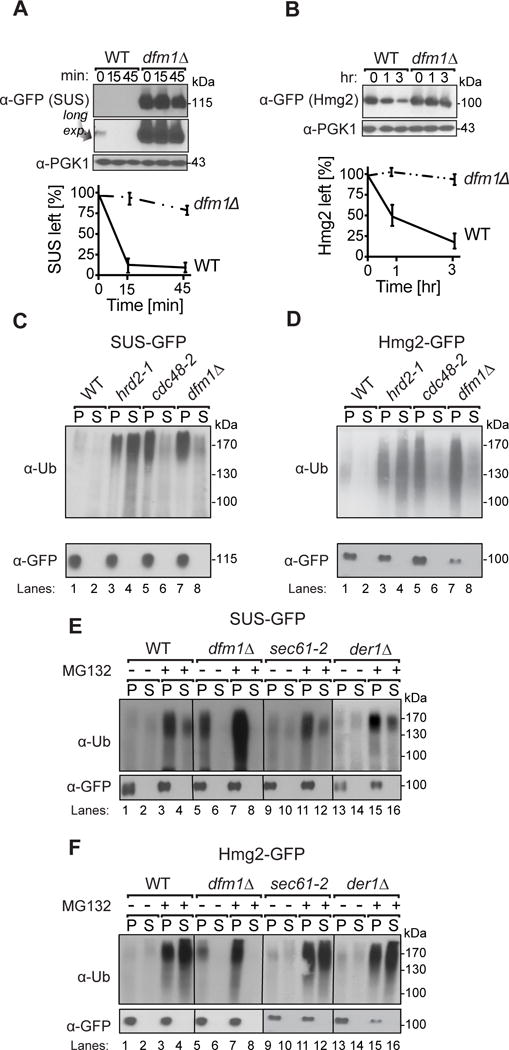
(A) Dfm1 is involved in degradation of SUS-GFP. WT and dfm1Δ strains were grown to log phase and degradation was measured by CHX. After CHX addition, cells were lysed at the indicated times and analyzed by SDS-PAGE and immunoblotted for SUS-GFP with α-GFP. (B) Dfm1 is involved in degradation of Hmg2-GFP. Same as (A), except CHX-chase assay was performed on Hmg2-GFP. (A–B) Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t=0 was taken as 100% and data is represented as mean ± SEM from at least three experiments. (C) Dfm1 is required for retrotranslocation of SUS-GFP. Crude lysate was prepared from each strain and ultracentrifuged to discern ubiquitinated SUS-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, SUS-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted with α-GFP and α-Ubi. (D) Dfm1 is required for retrotranslocation of Hmg2-GFP. Same as (C), except in vivo retrotranslocation assay was performed on Hmg2-GFP. (E–F) Der1 and Sec61 are not involved in retrotranslocation of SUS-GFP and Hmg2-GFP. Same as (C), except der1Δ and sec61-2 strains were grown to log-phase and treated with vehicle or MG132 prior to in vivo retrotranslocation assay.
Generality of Dfm1’s role in membrane protein ERAD
We next examined the extent of Dfm1 involvement in ERAD using various substrates (Hampton et al., 1996; Ravid et al., 2006; Swanson et al., 2001). Some studies of Dfm1 have found a role in DOA ERAD pathways (Avci et al., 2014; Stolz et al., 2010), while our earlier work found no involvement. Accordingly, we thoroughly and directly compared a variety of ERAD substrates in uniform strains. Test proteins included the additional integral membrane HRD substrates (ERAD-M) Pdr5* (Plemper et al., 1998) and Sec61-2; luminal HRD substrates (ERAD-L) CPY* and KHN; and the DOA pathway substrate (ERAD-C) Ste6*-GFP, with a misfolded cytoplasmic portion. The effect of a dfm1Δ was directly tested with CHX-chase assays on Pdr5*, Ste6* and Sec61-2 (Fig. 3A–C). In all three cases, the normally degraded substrates were stabilized by dfm1Δ. We confirmed that Ste6* retrotranslocation was strongly blocked in the dfm1Δ by directly examining a GFP fusion of this substrate, Ste6*-GFP, in the in vivo retrotranslocation assay (Fig. 3D) as used above. The extent of inhibition of Ste6*-GFP cause by dfm1Δ was again as strong as that caused by cdc48-2. In striking contrast, ERAD-L substrates, CPY* and KHN were unaffected by the dfm1Δ but strongly stabilized by der1Δ null (Fig. 3E–F). This was also true for the membrane-spanning Der1-dependent substrate KWW (Fig. 3G). Taken together, these data indicated that Dfm1 is generally involved in retrotranslocation integral membrane HRD and DOA substrates, but not any ERAD-L substrates examined.
Figure 3. Dfm1 is involved in degradation of integral membrane substrates.
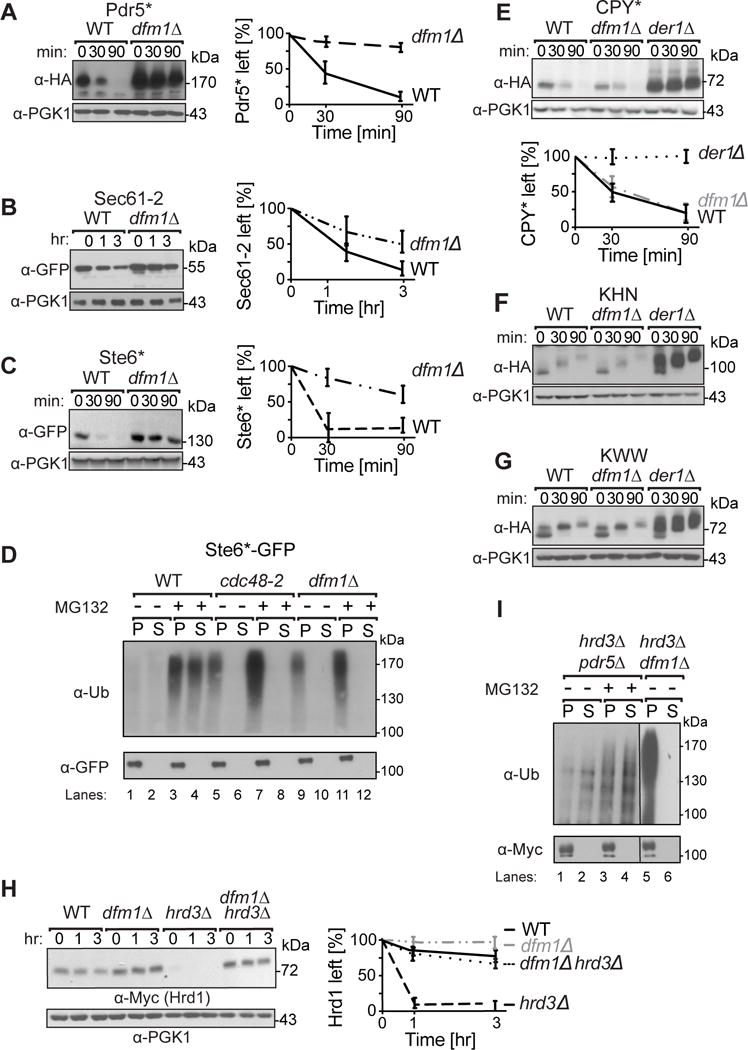
(A–C) Degradation of the indicated tagged ERAD-M and ERAD-C substrates were measured by CHX in isogenic strains. After CHX addition, cells were lysed at the indicated times, analyzed by SDS-PAGE and immunoblotted for each substrate. (D) Dfm1 is involved in retrotranslocation of Ste6*. The indicated strains were grown to log-phase and treated with MG132 (25 μg/mL). Crude lysate was prepared and ultracentrifuged to discern ubiquitinated Ste6*-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, Ste6*-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted for ubiquitin and Ste6*-GFP. (E–G) Dfm1 is not involved in degradation of ERAD-L substrates. Same as (A–C), except CHX-chase assay was performed on the indicated ERAD-L substrates. (H) Dfm1 is involved in degradation of HRD1. Same as above, except CHX-chase assay was performed on Hrd1. (I) Dfm1 is involved in retrotranslocation of Hrd1 in vivo. Crude lysate was prepared from each strain and ultracentrifuged to discern ubiquitinated Hrd1-5xmyc that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane fraction (P). Following fractionation, Hrd1-5xmyc was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted with α-Myc and α-Ubi. (A–C, E, & H) Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t=0 was taken as 100% and data is represented as mean ± SEM from at least three experiments.
Dfm1 was required for Hrd1 retrotranslocation
The Hrd1 E3 ligase can undergo self-ubiquitination and degradation in a proteasome-dependent manner (Carroll and Hampton, 2009; Vashistha et al., 2016). Recent studies have implicated Hrd1 as a channel for ERAD-L retrotranslocation (Baldridge and Rapoport, 2016). This begs the intriguing question of whether the multispanning Hrd1 protein meditates its own retrotranslocation during self-catalyzed ERAD, or rather, if it too requires Dfm1. In WT cells, Hrd1 was very stable and there was no effect on Hrd1 levels upon loss of Dfm1 (Fig. 3H). Conversely, when Hrd3 is absent, Hrd1 undergoes rapid self-ubiquitination and degradation (Carroll and Hampton, 2009; Gardner et al., 2000a; Vashistha et al., 2016). In a hrd3Δ, the levels of Hrd1 plummeted due to Hrd1 self-ubiquitination (Fig 3H). We examined the importance of Dfm1 in Hrd1 degradation in a hrd3Δ background. The dfm1Δ null completely stabilized Hrd1 in the hrd3Δ background. Furthermore, we observed a complete block of Hrd1 retrotranslocation when dfm1Δ was included in the hrd3Δ cells (Fig. 3I), resulting in a large buildup of ubiquitinated Hrd1 in the microsome (P) fraction, even in the absence of MG132 (Fig 3I, lanes 5–6). Thus, ironically, Hrd1 appears to require Dfm1 for its own retrotranslocation, despite its apparent ability to mediate that of other ERAD substrates.
Analysis of Dfm1 features important for retrotranslocation
The Dfm1 derlin is a member of the rhomboid protease family (depicted in Fig. 4A). The derlins are missing key rhomboid catalytic residues and are called inactive rhomboids (Lemberg, 2013; Lemberg and Adrain, 2016). These non-proteolytic rhomboids have a number of non-catalytic conserved residues important for rhomboid function. In addition, Dfm1 has a unique C-terminal cytoplasmic tail that contains SHP boxes that bind Cdc48 (Sato and Hampton, 2006) (Fig. 4A). This SHP motif is also present in mammalian Derlin 1, which functions in p97 binding and ERAD of several substrates (Greenblatt et al., 2011; Sun et al., 2006; You et al., 2017). We next studied the Dfm1 features needed for integral membrane retrotranslocation through use of chimeras and site-directed mutants.
Figure 4. Dfm1 SHP box is required, but not sufficient for retrotranslocation.
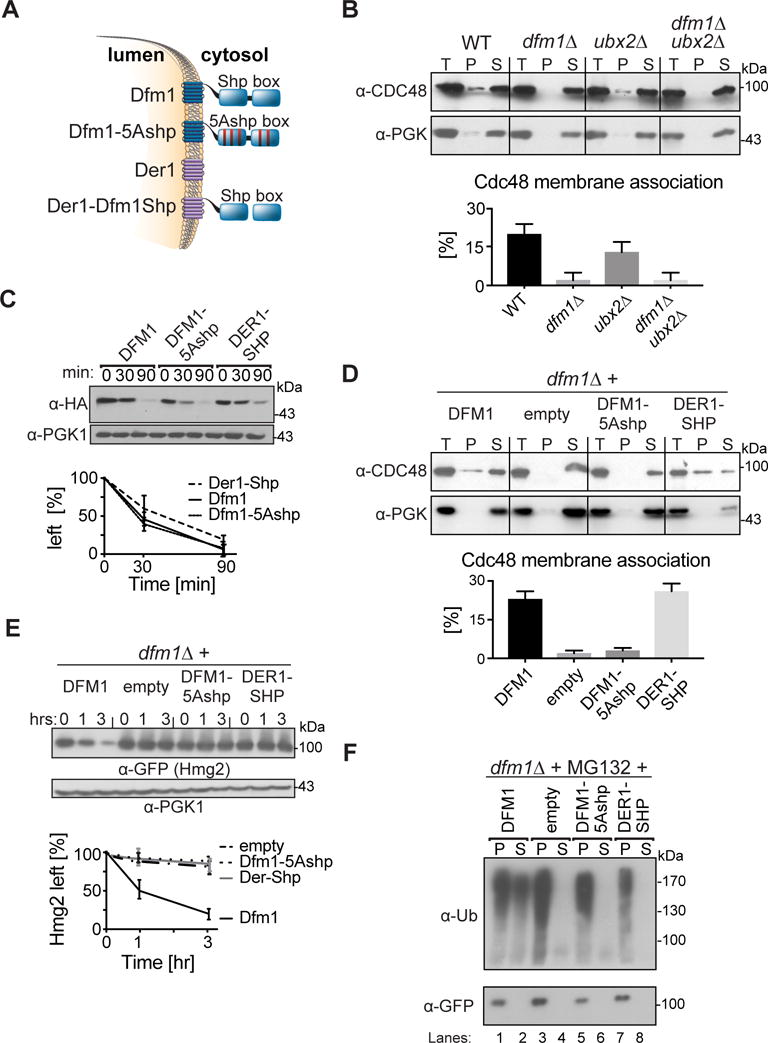
(A) Depiction of Dfm1, Der1, Dfm1-5Ashp, and Der1-Shp. Dfm1 and Der1 is an ER-localized membrane protein with six transmembrane domains (Greenblatt et al., 2011). Unlike Der1, Dfm1 has an extended cytoplasmic tail containing two SHP boxes. (B) Dfm1 is the major component for Cdc48 binding to the ER. Total cell lysate (T) from the indicated strains were separated into soluble cytosolic fraction (S) and pellet microsomal fraction (P) upon centrifugation at 14,000 × g. Each fraction was analyzed by SDS-PAGE and immunoblotted for Cdc48 with α-CDC48 and PGK1 with α-PGK1. (C) Stability of Dfm1 variants. Degradation of Dfm1, Dfm1-5Ashp, and Der1-Shp were measured by CHX-chase assay at the indicated times, cells were lysed, analyzed by SDS-PAGE and immunoblotted with α-HA. (D) SHP box is sufficient for Cdc48 recruitment. Same as (B), except strains expressing variants of Dfm1 were used. (E) Dfm1’s SHP box is required for degradation of Hmg2-GFP. In the indicated strains, degradation of Hmg2-GFP was measured by CHX-chase assay. Cells were analyzed by SDS-PAGE and immunoblotted for Hmg2-GFP with α-GFP. (F) in vivo retrotranslocation assay of Hmg2-GFP. After treatment with MG132, crude lysate was prepared from each strain and ultracentrifuged to discern ubiquitinated Hmg2-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, Hmg2-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted with α-GFP and α-Ubi. (B, D) The graph shows the quantification of Cdc48 in the pellet fractions of the respective cells as measured from ImageJ. Data is represented as percentage of Cdc48 that is bound to pellet fraction and is shown as mean ± SEM from three independent experiments. (C, E) Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t=0 was taken as 100% and data is represented as mean ± SEM from at least three experiments.
Dfm1 SHP box is required for retrotranslocation
We first tested the Cdc48-binding SHP box for a role in Dfm1 function (Fig. 4). Dfm1 recruitment of Cdc48 to the ER membrane was discerned with a direct assay of Cdc48 binding to microsomal membranes. The ubx2Δ null was also included since it has been reported that Ubx2 mediated Cdc48 recruitment to the ER membrane (Schuberth and Buchberger, 2005). Lysates from dfm1Δ, ubx2Δ and dfm1Δ ubx2Δ cells were centrifuged at 20,000 g to separate membrane pellet (P) from supernatant (S) (Fig. 4B). Fractions were analyzed on SDS-PAGE and immunoblotted for Cdc48 and the cytoplasmic enzyme PGK1. In all strains, cytosolic PGK1 remained in the supernatant and no PGK1 associated with the microsomal pellet. In contrast, about 15% of total Cdc48 was associated with the microsome pellet. Nearly all Cdc48 bound to the microsome was absent in the dfm1Δ strain, with no effect on the total pool. Curiously, in this assay, the ubx2Δ null had no effect on Cdc48 binding, either alone or in combination with the dfm1Δ. Thus, it appears that Dfm1 mediated Cdc48 recruitment to the ER surface, with no involvement of Ubx2. This was consistent with co-immunoprecipitation studies showing Ubx2 independence in the interaction of Dfm1 and Cdc48 (Stolz et al., 2010). We then tested the Dfm1 SHP motif for a role in Cdc48 membrane recruitment, using variants of Dfm1 or Der1 (Fig. 4A). We employed HA-tagged versions of each protein tested. It had been noted that HA tagging can cause destabilization of these proteins (Stolz et al., 2010), and it was clear that all of the proteins did undergo degradation (Fig. 4C). However, identical steady state levels of each were expressed in the strains used in our studies. Importantly, the HA tagged Dfm1 fully complemented the dfm1Δ null in both Cdc48 recruitment (Fig. 4D) and Hmg2-GFP degradation (Fig 2B vs 4E), indicating no effect on function caused by this modification.
Alteration of the 5 signature residues of the Dfm1 SHP box to alanine (Dfm1-5Ashp) removed its ability to recruit Cdc48 (Fig. 4A & D). Conversely, addition of the Dfm1 SHP motif to the normally SHP-less Der1 made this chimera able to promote Cdc48 recruitment comparable to Dfm1 (Fig. 4D). These results indicated that the Dfm1 SHP box was necessary and sufficient for Cdc48 binding, at least when part of a derlin. We then tested these SHP variants for ERAD of Hmg2-GFP. Neither the Der1-SHP chimera that recruited Cdc48, nor the Dfm1-5Ashp mutant, that failed to recruit Cdc48, could support Hmg2-GFP ERAD (Fig. 4E) or retrotranslocation (Fig. 4F). Thus, the SHP box, and presumably Cdc48 recruitment to the ER membrane, is necessary but not sufficient for Dfm1’s role in retrotranslocation.
Dfm1 WR and GxxxG rhomboid motifs were required for ERAD-M
Along with the unique SHP motifs, Dfm1 has rhomboid homologies. These include the highly conserved WR motif in the first cytoplasmic loop and a GxxxG motif in the sixth membrane span (Fig. 5A). Both are important for rhomboid intermembrane protease function (Fleig et al., 2012; Greenblatt et al., 2011). We created two mutants, Dfm1-AA and the Dfm1-Ax3A mutants, in which the conserved residues in the WR or the GxxxG motif were mutated to alanine (Fig. 5A). The AA mutation had the same stability as the HA tagged wild type protein, while the Ax3A mutant was more rapidly degraded (Fig. 5B). Nevertheless, the steady state levels of each mutant supported normal Cdc48 ER recruitment, and so appear to be at functionally effective levels at least for this action. Despite this, neither mutant could support Hmg2-GFP ERAD (Fig. 5D) or retrotranslocation (Fig. 5E). These results imply that additional functions are provided by the rhomboid motifs in Dfm1-mediated retrotranslocation.
Figure 5. WR and GxxxG motif is required for Hmg2-GFP retrotranslocation.
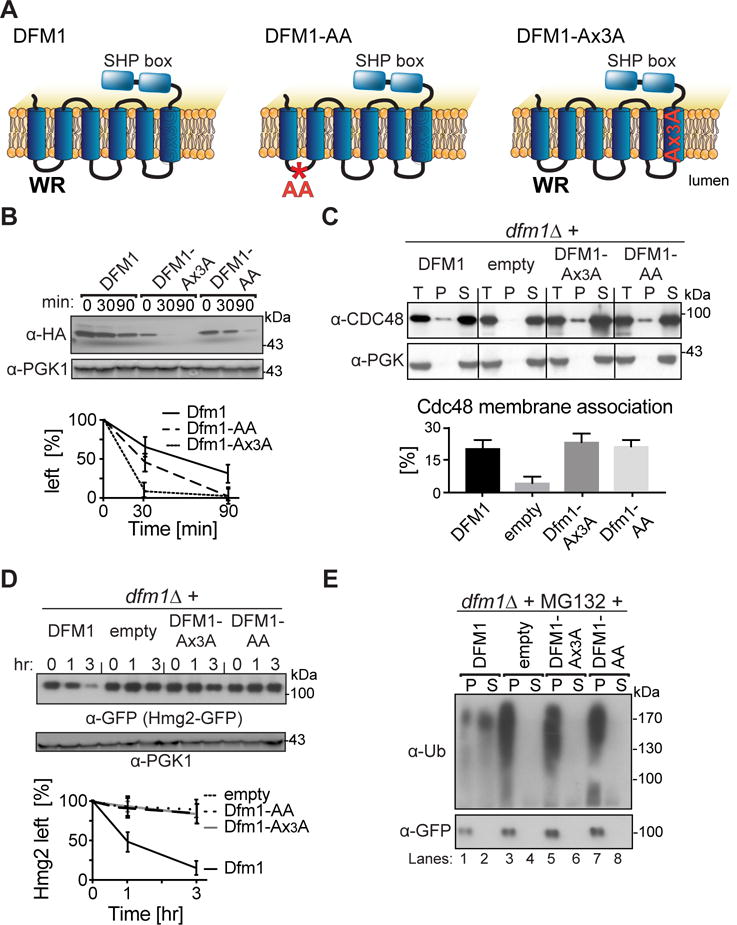
(A) Depiction of Dfm1, Dfm1-AA and Dfm1- Ax3A. Dfm1 has WR motif in the first lumenal loop and a GxxxG dimerization motif in the TMD. (B) Stability of Dfm1 variants. Degradation of Dfm1, Dfm1-AA, and Dfm1-Ax3A were measured by CHX-chase assay. At the indicated times, cells were lysed, analyzed by SDS-PAGE and immunoblotted with α-HA. (C) Dfm1-AA and Dfm1-Ax3A recruit Cdc48 to microsomes. Cell lysate (T) from the indicated strains were separated into soluble cytosolic fraction (S) and pellet microsomal fraction (P) upon centrifugation at 14,000 × g. Each fraction was analyzed by SDS-PAGE and immunoblotted for Cdc48 with α-CDC48 and PGK1 with α-PGK1. The graph shows the quantification of Cdc48 in the pellet fractions of the respective cells as measured from ImageJ. Data is represented as percentage of Cdc48 that is bound to pellet fraction and is shown as mean ± SEM from three independent experiments. (D) Dfm1’s WR and GxxxG motif is required for degradation of Hmg2-GFP. In the indicated strains, degradation of Hmg2-GFP was measured by CHX-chase assay. Cells were analyzed by SDS-PAGE and immunoblotted for Hmg2-GFP with α-GFP. (E) in vivo retrotranslocation assay of Hmg2-GFP. After treatment with MG132, crude lysate was prepared from each strain and ultracentrifuged to discern ubiquitinated Hmg2-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, Hmg2-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted with α-GFP and α-Ubi. (B, D) Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t=0 was taken as 100% and data is represented as mean ± SEM from at least three experiments.
Rapid suppression of dfm1Δ
The above studies show a strong and broad role for Dfm1 in ERAD-M. While exciting, this result was also initially perplexing. We and others had previously reported that Dfm1 had no role in ERAD (Goder et al., 2008; Sato and Hampton, 2006), while other investigators had observed a more specific involvement of Dfm1 in the DOA pathway (Stolz et al., 2010a), or in degradation of non-canonical ERAD-R (ERAD Regulatory) substrate Zrt1 (Avci et al., 2014). The strains we used in our earlier negative studies were the same background used herein, and the frozen stocks were reconfirmed to have the dfm1Δ allele and normal ERAD-M. Accordingly, we set out to resolve this conundrum. The results explain the discrepancy, suggest some intriguing ideas about ER retrotranslocation, and emphasize an important caveat in the study of null phenotypes.
Alerted to the possibilities of rapid suppression of null mutations recently described (Ryu et al., 2016; Szamecz et al., 2014; Teng et al., 2013), we explored suppression as a cause for the dichotomous results. We reasoned that the dfm1Δ null might be susceptible to rapid suppression, leading to phenotypic masking of dfm1Δs ERAD defect during normal strain curation. We developed a simple suppression assay to test this idea. We used a freshly generated dfm1Δ strain expressing SUS-GFP; the null strain was bright compared to a wild-type strain (mean fluorescence 23 K vs 1.5 K in WT), due to strong SUS-GFP stabilization (Fig. 6A). To test for suppression, a culture of the bright dfm1Δ SUS-GFP starting strain (P0) was repeatedly passaged into fresh minimal medium followed by growth to saturation (~ 24 hr). Remarkably, dark cells emerged rapidly: by the fourth passage (P4) approximately half of the cells were comparable to wild-type in mean fluorescence while the other half remained bright (Fig. 6A “P4”). By P11 the culture population was entirely composed of dark cells, with a histogram identical to that of wild-type strain (Fig. 6A “P11”). This suppression of ERAD is not typical: a phenotypically and optically identical cdc48-2 strain maintained the same brightness as initially observed after many passages (Fig. 6A, “cdc48-2”). Rapid suppression was also observed when Hmg2-GFP was the substrate expressed in the dfm1Δ strain: complete suppression was observed by passage nine (P9) (Fig. S1A). Again, suppression was not a typical observation for ERAD deficient strains. ERAD-deficient strains expressing Hmg2-GFP but lacking the E3 ligase Hrd1, remained bright and did not generate suppressors over time (Fig. S1A “hrd1Δ”). Thus, the suppression appeared to be a specific property of dfm1Δ; no other strongly ERAD deficient strain has so far shown this proclivity. Because of the speed of occurrence, suppressors can easily be accumulated during the course of normal strain maintenance. In fact, we demonstrate this in fact occurred in our own strains from our earlier work (see below).
Figure 6. dfm1Δ rapidly suppresses overtime.
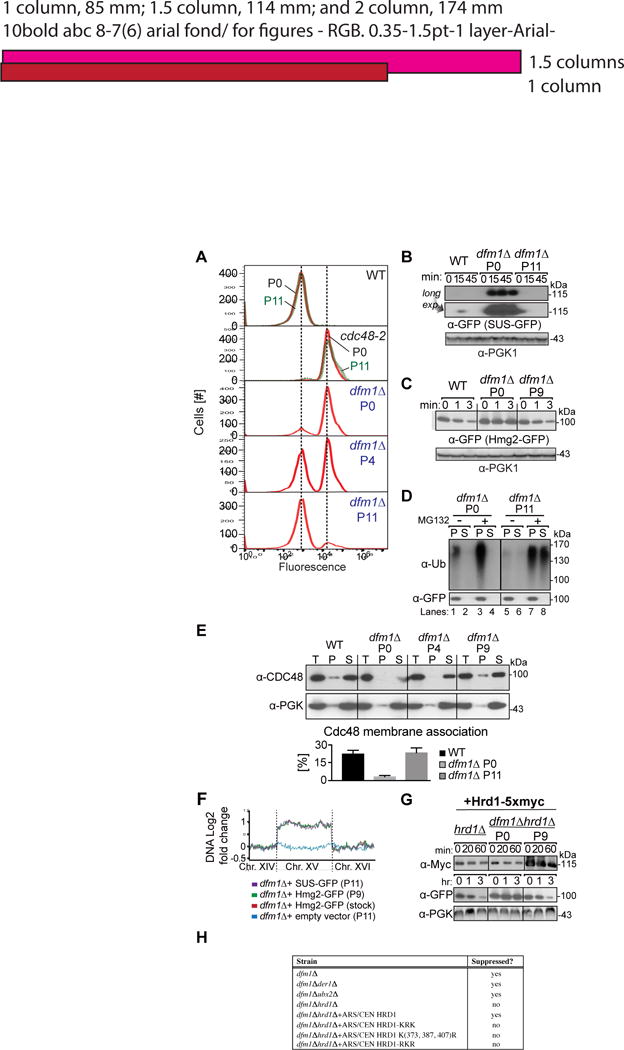
(A) dfm1Δ cells containing overexpressed SUS-GFP were passaged to suppression. The indicated cells with overexpressed SUS-GFP were passaged at the indicated number of times into fresh minimal media (P0, P4 and P11) and SUS-GFP levels were analyzed by flow cytometry. Histograms of 10,000 cells are shown, with the number of cells versus GFP fluorescence. Note: panels are aligned so all fluorescent histograms are comparable between panels (B) SUS-GFP is degraded to WT levels in dfm1Δ suppressed cells. Degradation of SUS-GFP was measured by CHX-chase assay in WT, dfm1Δ P0 and dfm1Δ P11 cells. After CHX addition, cells were lysed at the indicated times, analyzed by SDS-PAGE and immunoblotted for SUS-GFP with α-GFP. (C) Hmg2-GFP is degraded to WT levels in dfm1Δ suppressed cells. Same as (B) except degradation of Hmg2-GFP was measured by CHX-chase assay in WT, dfm1Δ P0 and dfm1Δ P9 cells. (D) in vivo Hmg2-GFP retrotranslocation completely restored dfm1Δ suppressed cells. Crude lysate was prepared from the indicated strains treated with vehicle or MG132 (25 μg/mL). Lysates were ultracentrifuged to discern ubiquitinated Hmg2-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, Hmg2-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted with α-GFP and α-Ubi. (E) Cdc48 recruitment to microsomes is restored in dfm1Δ suppressed cells. Total cell lysate (T) from the indicated strains were separated into soluble cytosolic fraction (S) and pellet microsomal fraction (P) upon centrifugation at 14,000 × g. Each fraction was analyzed by SDS-PAGE and immunoblotted for Cdc48 with α-CDC48 and PGK1 with α-PGK1. The graph shows the quantification of Cdc48 in the pellet fractions of the respective cells as measured from ImageJ. Data is represented as percentage of Cdc48 that is bound to pellet fraction and is shown as mean ± SEM from three independent experiments. (F) ChrXV duplication is substrate induced upon loss of Dfm1. Chromosome profiles of whole genome sequencing data mapped across ChrXV. Genomic levels through entire ChrXV are twice as high in suppressed dfm1Δ cells expressing Hmg2-GFP or SUS-GFP with respect to dfm1Δ cells containing empty vector. (G) Hrd1 levels are upregulated in dfm1Δ suppressees. Degradation of Hrd1-5xmyc was measured by CHX-chase assay in dfm1Δ P0 and dfm1Δ P11 overexpressing Hmg2-GFP. After CHX addition, cells were lysed at the indicated times, analyzed by SDS-PAGE and immunoblotted for Hrd1 with α-Myc and Hmg2 with α-GFP. (H) The indicated strains overexpressing SUS-GFP were passaged to suppression.
We tested the dark suppressees for restoration of ERAD-M and retrotranslocation. We compared the degradation rate of SUS-GFP degradation in suppressed (P11) vs. non-suppressed (P0) cultures of dfm1Δ. Cycloheximide chase assays on dfm1Δ P0 showed full stabilization of SUS-GFP degradation, while in P11 suppressed dfm1Δ cells, SUS-GFP was rapidly degraded, showing a return to the characteristic low WT levels of this rapidly degraded substrate (Fig. 6B); note that observing the 0 time point of SUS-GFP in the WT or P11 dfm1Δ required overexposure of the blots (Fig. 6B, lower panel). Similarly, Hmg2-GFP degradation was restored to wild-type rates in suppressed P9 dfm1Δ cultures (Fig. 6C). We next tested suppressed dfm1Δ strains for restored Hmg2-GFP retrotranslocation. Non-passaged dfm1Δ P0 showed the typical buildup of ubiquitinated Hmg2-GFP in the pellet fraction in both untreated and MG132 treated cells Fig. 6D lanes 1 & 3). In striking contrast, suppressed P11 dfm1Δ cells showed normal Hmg2-GFP retrotranslocation, that was enhanced by MG132 (Fig. 6D lanes 5–8). The P11 dfm1Δ suppressees also showed fully restored Cdc48 recruitment to the microsome (Fig. 6E). Finally, degradation and retrotranslocation of DOA pathway substrate Ste6*-GFP were also completely restored in suppressees (Fig. S1 B & C). Thus, by all criteria examined, the rapid suppression of dfm1Δ was complete, in terms of substrate degradation, Cdc48 recruitment, restored retrotranslocation, and substrate range.
Acquired specific aneuploidy underlies dfm1Δ suppression
The rapid suppression of the dfm1Δ led us to wonder if some type of DNA amplification was involved. Accordingly, we subjected suppressed and control strains to whole genome sequencing using positional bar codes of sufficient density to examine the entire genome for regions of amplification. We included a passaged dfm1Δ strain containing empty vector and a dfm1Δ strain expressing SUS-GFP or Hmg2-GFP that had been passaged to suppression. The suppressed strains showed a complete duplication of chromosome XV (Fig. 6F as indicated & Fig. S2A). Moreover, Ste6*-GFP expressing strain that had been passaged to suppression showed the same duplication of chromosome XV (Fig. S1D). We performed the same analysis on our frozen dfm1Δ strain stocks from the 2006 report that showed wild-type Hmg2-GFP degradation (Sato and Hampton, 2006), and they also had a full duplication of chromosome XV (Fig. 6F & Fig. S2A). Importantly, strains that were passaged with dfm1Δ but had no substrate-expressing plasmid did not duplicate any chromosome, indicating the combination of an expressed ERAD-M substrate and the presence of the dfm1Δ mutation imposes a stress that may promote selection of the suppressed state. We confirmed this need for strong substrate expression by using a GAL1 galactose-driven promoter to compare the effects of the presence and absence of Hmg2-GFP during a passaging experiment (Fig. S1E). A dfm1Δ strain bearing a plasmid with Hmg2-GFP expressed from the strong, galactose inducible GAL1 promoter was passaged either in glucose (promoter off) or minimal medium with galactose (promoter on). As expected from the above experiments, the brightness of the strain passaged in galactose, where the Hmg2-GFP is strongly expressed, rapidly declined in brightness and recovered Hmg2-GFP degradation due to suppression (Fig. S1E). In contrast, when the uninduced, glucose passaged cultures were subsequently placed in galactose after multiple passages to assess the dfm1Δ phenotype, the Hmg2-GFP was stable and the stains remained as bright as the P0 cultures even after 9 passages with the Hmg2-GFP promoter off (Fig. S1E). Thus, the suppression of dfm1Δ required strong ERAD-M substrate expression during passage.
Genes required for dfm1Δ suppression
To better understand the mechanisms at play, we tested several genes for involvement in dfm1Δ suppression (Fig. 6H & Fig. S3A), using the passaging-based suppression assay to evaluate which genes were required for restoration of ERAD and retrotranslocation in the dfm1Δ null. We first tested Der1, Dfm1’s homologue, and found Der1 was dispensable for dfm1Δ suppression (Fig. 6H & Fig. S3A). Because suppressees also showed full restoration of Cdc48 binding to the ER (Fig. 6E), we tested the importance of Ubx2 in dfm1Δ suppression since it had been implicated in Cdc48 recruitment to the ER membrane (Schuberth and Buchberger, 2005). Although we failed to see a role of Ubx2 in normal Cdc48 recruitment, it might have an ancillary role in suppression. Nevertheless, Ubx2 was not required for suppression, since the dfm1Δ ubx2Δ double null showed full restoration of SUS-GFP degradation and Cdc48 recruitment after passaging (Fig. 6H & Fig. S3B).
Hrd1 has a mulitspanning membrane domain that can serve as retrotranslocon for lumenal substrates (Baldridge and Rapoport, 2016; Sato et al., 2009; Stein et al., 2014). Thus, we tested if Hrd1 played a role in suppression of the dfm1Δ null mutation, and found this to be the case. In striking contrast to dfm1Δ, the dfm1Δ hrd1Δ double null never underwent growth dependent suppression: even after nine passages, the double null remained bright (Fig. 6H & Fig. S3A). This result is intriguing because Hrd1 is located on chromosome XV, which was duplicated during suppression of dfm1Δ, suggesting that elevation of Hrd1 creates an alternate route of retrotranslocation for ERAD-M substrates. In fact, we found that when Hrd1 was present on an autonomous ARS/CEN plasmid and expressed from its native promoter, suppression of dfm1Δ still occurred, but chromosome XV was no longer duplicated (Fig. 6F & S2B). Thus, it appears that the ability to suppress dfm1Δ required only elevated Hrd1, and that the duplication of chromosome XV is the most rapid way to achieve an increase in Hrd1 when expressed from its native locus. Importantly, it seems that simple duplication of Hrd1 was not sufficient to suppress the dfm1Δ null. dfm1Δ strains with an ARS/CEN expressing the native Hrd1 gene still required passaging of the strains (Fig. S3A), even though Hrd1 was then duplicated (one copy on the plasmid, one on the chromosome), indicating that either other factors or Hrd1 itself must be further induced for suppression. Consistent with this idea, we tested the levels of Hrd1 in normal and suppressed dfm1Δ strains using a 5myc-tagged Hrd1 expressed from the normal chromosomal locus. Suppression occurred with the same kinetics as strains expressing untagged Hrd1, and Myc blotting indicated that the Hrd1-5myc levels were elevated ~ 5 fold above wild-type in the suppressed strain, indicating that Hrd1-dependent suppression was more complex than simply doubling the normally expressed locus (Fig. 6G).
These suppression tests indicated that Hrd1 might function in ERAD-M retrotranslocation, although normally ERAD-M and ERAD-L retrotranslocation occur by separate routes. Recent elegant studies from the Rapoport group show that Hrd1 can catalyze ERAD-L substrate movement across a bilayer in vitro (Baldridge and Rapoport, 2016). This Hrd1-mediated retrotranslocation requires autoubiquitination of Hrd1 lysine residues. The authors scanned the Hrd1 cytoplasmic domain for lysines important in retrotranslocation, dividing the Hrd1 into three regions: lysines N-terminal to the RING domain (Kxx), lysines within the RING domain (xKx), and lysines C-terminal to the RING domain (xxK). The Ks in each region were converted en bloc to R, and the resulting Hrd1 mutants were tested for retrotranslocation. In that work, conversion of all lysines in the “middle” RING group to R was annotated Hrd1-KRK and conversion of all lysines N-terminal and C-terminal to the RING group to R was annotated Hrd1-RKR (Fig. S4A). The analysis showed that the RING domain lysines were uniquely important for Hrd1-mediated retrotranslocation, while the flanking lysines were not: the Hrd1-RKR mutant supports ERAD-L, while neither Hrd1-KRK, nor any other variant with the middle RING Ks changed to R (xRx), support ERAD-L. In fact, Hrd1 with only three RING region Ks modified, Hrd1 K(373, 387, 407)R, was also unable to support ERAD-L. (Baldridge and Rapoport, 2016). Because we could observe Hrd1-dependent suppression of dfm1Δ in strains harboring Hrd1 on an ARS/CEN plasmid, we were able to test those K-to-R Hrd1 mutants for their ability to restore ERAD-M to dfm1Δ. ARS/CEN-expressed Hrd1, and the variants Hrd1-RKR, Hrd1-KRK, or wild-type Hrd1-KKK, were each introduced into dfm1Δhrd1Δ strains expressing SUS-GFP, and subjected to the passaging suppression assay (Fig. 6H & Fig. S4B). Interestingly, while our own Hrd1 plasmid, and the provided wild type Hrd1-KKK plasmids supported dfm1Δ suppression, neither of the Hrd1 variants, including the ERAD-L competent Hrd1-RKR, allowed dfm1Δ suppression. We confirmed that in our strain background, CPY* degradation showed the appropriate response to the Baldridge Hrd1 mutants: Hrd1-KRK did not support CPY* degradation, but Hrd1-RKR and Hrd1-KKK did (Fig. S4C). Thus, if Hrd1 is indeed mediating retrotranslocation of ERAD-M substrates in the suppressed dfm1Δ strains, the mechanistic details might be distinct from those involved in ERAD-L. Nevertheless, it is intriguing that the suppressed retrotranslocation phenotype is sensitive to alterations of Hrd1 cytoplasmic lysines.
DISCUSSION
These studies revealed the derlin Dfm1 to be a key participant in ERAD-M retrotranslocation. Both HRD and DOA pathway integral membrane substrates showed complete dependence on Dfm1 for extraction from the ER membrane, but the dfm1Δ null had no effect on any ERAD-L substrate tested. This specificity extended to Hrd1 itself. In conditions where Hrd1 undergoes rapid self-degradation, it too showed a complete dependence on Dfm1 for ERAD and retrotranslocation. This is particularly interesting because Hrd1 has been strongly implicated as an ERAD-L retrotranslocation channel (Baldridge and Rapoport, 2016). Nevertheless, Hrd1 still required outside assistance in ER exit when it is a substrate. Perhaps in order for Hrd1 to serve in this capacity catalytically, it may be useful that Hrd1 is normally unable to bring about its own exit from the ER membrane.
Dfm1 has homology to both other derlins and rhomboid superfamily members, and a unique C-terminal motif known as a SHP box that allows interaction with Cdc48 (Greenblatt et al., 2011; Sato and Hampton, 2006). We showed that the binding of Cdc48 to ER microsomes was principally mediated by Dfm1 and required intact SHP motifs. However, recruitment of Cdc48 was not sufficient for ERAD-M retrotranslocation. Addition of the SHP box to Dfm1’s homologue Der1 produced a chimera that restored wild-type recruitment of Cdc48 to the dfm1Δ null but not ERAD-M. Similarly, the Dfm1 variants with altered rhomboid GxxxG or WR motifs showed normal Cdc48 recruitment, but also did not support ERAD-M.
In our hands, Cdc48 recruitment to the ER surface relied entirely on Dfm1. This was surprising in light of reports that Cdc48-binding factor Ubx2 was described as having this function (Neuber et al., 2005; Schuberth and Buchberger, 2005). In the assay we employed, we found no contribution of Ubx2 to Cdc48 recruitment under any circumstances. It is possible that Cdc48 recruitment can be attained through a number of mechanisms; this is clearly the case in the suppressed dfm1Δ null cells, which have restored Cdc48 binding but no Dfm1. However, even in the suppressed dfm1Δ null strains, Ubx2 was still not required for the restored binding of Cdc48 to the ER membrane (Fig S3B).
Dfm1 shares homology with the superfamily of rhomboid proteases, but it lacks key residues for proteolysis (Lemberg and Adrain, 2016). The two principle rhomboid sequences in Dfm1, WR and GxxxG were each required for Dfm1-mediated retrotranslocation of ERAD-M substrates, yet each inactive rhomboid fully retained its ability to mediate Cdc48 recruitment to the ER surface, indicating that the conserved rhomboid residues function in a different aspect of retrotranslocation. Rhomboid proteases typically cleave their substrates within the lipid bilayer. Perhaps the Dfm1 inactive rhomboid has retained membrane-perturbing properties used by the authentic proteases that are now used for channeling or moving substrates during retrotranslocation, as has been suggested (Greenblatt et al., 2011). Other mammalian rhomboid pseudoproteases, UBAC2 and Derlin-1, have been implicated for involvement in ERAD as well (Christianson et al., 2011; Greenblatt et al., 2011). In fact, the closest mammalian protein to Dfm1, RHBDL4, is an ER-resident membrane protein required for ERAD of a variety of mammalian substrates. RHBDL4 similarly requires its rhomboid identities to function in ERAD (Fleig et al., 2012). However, unlike Dfm1, RHBDL4 is an active rhomboid protease, and appears to require its proteolytic activity to function in ERAD. Thus, the details of how rhomboids participate in ERAD may vary in detail depending on the specific member and the presence or absence of protease activity. The ease of study and the breadth of Dfm1’s role in ERAD will allow us to unveil mechanistic features of this widely represented superfamily in ERAD and perhaps other functions mediated by these proteins.
Our functional studies suggest a model of at least two coordinated functions of Dfm1 in retrotranslocation. Through its SHP box, Dfm1 mediates Cdc48 recruitment to the ER surface and this activity appears to be tightly correlated with successful retrotranslocation. In addition, the Dfm1 rhomboid sequences function in a manner distinct from Cdc48 binding. Our recent studies on retrotranslocated ERAD-M substrates show that Cdc48 is required for the initial passage of ERAD-M substrates across the ER membrane (Neal et al., 2017), implying that the ATPase activity and the Dfm1 transmembrane domain execute the movement and energetics of ERAD-M retrotranslocation in a highly coordinated manner. It will be important to directly evaluate Dfm1’s ability to catalyzed movement of ERAD-M substrates across the lipid bilayer.
We were initially surprised and concerned when these studies showed Dfm1 to be required for ERAD-M retrotranslocation, since we had previously ruled out Dfm1 in our tests of candidates (Sato and Hampton, 2006). We resolved this discrepancy by showing that passage of dfm1Δ nulls strongly expressing ERAD-M substrates underwent rapid and complete suppression of the original ERAD and retrotranslocation defects. Suppression of dfm1Δ nulls was remarkably rapid, typically coming to completion within nine passages of a liquid culture. Suppression occurred though a duplication of chromosome XV, and analysis of the stocks of dfm1Δ null expressing Hmg2-GFP strains used in our early studies had the same complete duplication of chromosome XV. This suppression was highly atypical; no other ERAD mutants tested, including nulls in the HRD ligase components, or associated E2s, or strongly ERAD deficient cdc48-2 mutants showed any tendency for suppression even upon many passages with strongly expressed Hmg2-GFP or other substrates.
Rapid suppression of dfm1Δ absolutely required the presence of strongly expressed ERAD-M substrates. A number of substrates have been tested, including Hmg2-GFP, Ste6*, SUS-GFP, or Sec61-2 (Fig. 3), as well as 6myc-Hmg2 (data not shown), and the presence of any substrate expressed from the strong TDH3 promoter resulted in rapid suppression of dfm1Δ. Identical passage of dfm1Δ without a strongly expressed substrate did not result in suppression; this was best demonstrated by using a dfm1Δ strain with a plasmid that allowed galactose-driven expression of Hmg2-GFP. Growth of this strain in non-inducing conditions even for many passages had no effect on the dfm1Δ dependent stability of Hmg2-GFP (Fig. S1E). Conversely, passage of the identical strain in galactose, where Hmg2-GFP was strongly induced, lead to the expected rapid resumption of normal Hmg2-GFP degradation through suppression of dfm1Δ. Thus, production of a dfm1Δ null in an otherwise wild-type strain, followed by introduction of a substrate-expressing plasmid allows facile observation of the strong ERAD-M defect. However, curation of dfm1Δ strains with a constitutively expressing substrate plasmid would be expected to rapidly bring about the now-understood suppression, as appears to have occurred in our earlier studies (Sato and Hampton, 2006). This feature also explains why the SPOCK array allowed discovery of the dfm1Δ phenotype: the array mating used to place the integrated SUS-GFP expression plasmid into each candidate null proceeds such that substrate plasmid is first present in a heterozygous diploid with the null mutant of interest; robotic sporulation and selection for haploids occurs only at the end of the mating series, allowing expression of SUS-GFP in the presence of dfm1Δ without a protracted and suppressive growth period.
The ability to reproducibly observe dfm1Δ suppression also allowed us to study this process. In testing candidate genes, we found that Hrd1 was absolutely required for suppression of dfm1Δ. Appropriately, Hrd1 is found on chromosome XV, which was duplicated in dfm1Δ suppressees. Hrd1 appears to be the only chromosome XV gene that requires increased gene dosage to suppress dfm1Δ since providing Hrd1 on an ARS/CEN plasmid, which can stably exist in cells in varying copy numbers, allows suppression that no longer results in duplication of chromosome XV. Intriguingly, although chromosome XV is only duplicated in the suppressees, Hrd1 levels were elevated about 5-fold after suppression. This implies that there is upregulation of Hrd1 expression that occurs when the combination of elevated ERAD-M substrates and dfm1Δ are present. This will be an intriguing and fruitful avenue of future investigation. It is important to note that observing this ancillary Hrd1 ERAD-M retrotranslocation function was made possible by use of the self-ubiquitinating SUS-GFP substrate, since normal ERAD-M substrates require Hrd1 for both ubiquitination as well as possible extraction functions, obviating unambiguous study of the retrotranslocation functions of this E3 ligase.
Recent elegant work from the Rapoport group has shown that Hrd1 can catalyze reconstituted retrotranslocation of ERAD-L substrates through a mechanism that involves self-ubiquitination of specific Hrd1 lysine residues (Baldridge and Rapoport, 2016). This raises the interesting possibility that Hrd1 can be repurposed to perform ERAD-M retrotranslocation when Dfm1 is missing. In that work, specific K to R mutants of Hrd1 (xRx group) were shown to be deficient in the reconstituted ERAD-L function, and in vivo ERAD-L, whereas other K to R mutants (xKx groups) were without effect on Hrd1’s ERAD-L channeling function. In contrast, we found that none of the K to R mutants from that study could support dfm1Δ suppression, so the detailed rules of suppression are at least somewhat different from those that operate in Hrd1-mediated ERAD-L. Nevertheless, the connection to Hrd1 is compelling.
Very recently, this same group has shown that Hrd1’s structure by cryo EM appears to be that of a transmembrane channel (Schoebel et al., 2017), and perhaps this explains Hrd1’s abilities as an ancillary ERAD-M translocon. In contrast, structural modeling of Dfm1 indicates that it has a “rhomboid fold” without the expected structural features of a channel (Greenblatt et al., 2011; Lemberg and Adrain, 2016). This may mean that that Dfm1 can catalyze membrane substrate extraction in a distinct and perhaps entirely novel manner from a classic peptide pore. This will be an important and revealing issue for future studies.
It has been suggested that retrotranslocation’s recalcitrance to genetic discovery may be due to an “Escape from New York” effect, referring to the idea that there are several completely distinct ways to exit New York City that all provide the same outcome (Hampton and Sommer, 2012). The genetic analogy is that perhaps analogous but non-homologous factors, as distinct in features as the Holland Tunnel and the George Washington Bridge, play redundant roles in retrotranslocation, making genetic analysis difficult. And indeed, this may be the case with Hrd1’s ability to suppress the retrotranslocation function of the entirely distinct Dfm1. Nevertheless, in normal circumstances, it appears that Dfm1 is the principle route for both HRD and DOA pathway integral membrane substrates to escape from the ER membrane.
STARS METHOD
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GFP | Clontech Laboratories, Inc. | Cat#632381; RRID: AB_2313808 |
| Mouse monoclonal anti-HA | Thermo Fisher Scientific | Cat#32-6700; RRID: AB_2533092 |
| Rabbit polyclonal anti-Myc | Genscript | Cat#A00172; RRID: AB_914457 |
| Rabbit polyclonal anti-Cdc48 | Neal et al., 2016 | N/A |
| Mouse monoclonal anti-PGK | Thermo Fisher Scientific | Cat#459250; RRID: AB_2569747 |
| Mouse monoclonal anti-Ubiquitin | Richard Gardner: University of Washington | N/A |
| Bacterial and Virus Strains | ||
| Escherichia coli DH5 alpha Competent Cells | Thermo Fisher Scientific | Cat#18265017 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG132 (benzyloxycarbonyl-Leu-Leu-aldehyde) | Sigma-Aldrich | 474787; CAS: 133407-82-6 |
| Cycloheximide | Sigma-Aldrich | C7698; CAS: 66-819 |
| USP2core | LifeSensors | Cat#DB501 |
| Protein A Sepharose | GE Healthcare | 17-0780-01 |
| Critical Commercial Assays | ||
| GeneArt Site-Directed Mutagenesis PLUS kit | Life Technologies | Cat#A14604 |
| Nextera DNA Library Preparation Kit | Illumina | FC-121-1031 |
| Deposited Data | ||
| Raw Files | This study, Mendeley Data | doi:10.17632/ym9mt gmrwh.1 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae BY4741 | GE Dharmacon | Cat#YSC1048 |
| Saccharomyces cerevisiae S288C | This paper | N/A |
| Additional yeast strains used: refer to Table S2 | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Plasmids used: refer to Table S1 | ||
| Software and Algorithms | ||
| Prism 7 for Mac | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| FlowJo | Vashistha et al., 2016 | https://www.flowjo.com/solutions/flowjo |
| BD Accuri C6 | BD Accuri | Cat # 653122 |
| HOMER | Heinz et al., 2010 | N/A |
| Bowtie2 | Langmead and Salzberg, 2010 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Randolph Hampton (rhampthon@ucsd.edu).
EXPERIMENTAL MODELS
All experiments were carried out in Saccharomyces cerevisiae budding yeast in BY4741 and S288C background.
METHOD DETAILS
Yeast and Bacteria Growth Media
Standard yeast Saccharomyces cerevisiae growth media were used as previously described (Hampton and Rine, 1994), including yeast extract-peptone-dextrose (YPD) medium and ammonia-based synthetic complete dextrose (SC) and ammonia-based synthetic minimal dextrose (SD) medium supplemented with 2% dextrose and amino acids to enable growth of auxotrophic strains at 30°C. Escherichia coli DH5 were grown in standard LB media with ampicillin at 37°C as previously described (Gardner et al., 1998).
Plasmids and Strains
Plasmids used in this study are listed in Table S1. Plasmids for this work were generated using standard molecular biological techniques as previously described (Sato et al., 2009) and verified by sequencing (Eton Bioscience, Inc.). Primer information is available upon request. The KHN (pRH1958), KWW (pRH1960) and CPY* (pRH1377) plasmids were a gift from Davis Ng (National University of Singapore, Singapore). The Ste6* plasmid (pRH2058) was a gift from S. Michaelis (Johns Hopkins School of Medicine, MD). The Pdr5* plasmid was a gift from Dieter H. Wolf (University of Stuttgart, Stuttgart, Germany). The Hrd1, Hrd1-RKR, Hrd1-KRK, Hrd1 K(373, 387, 404)R plasmids were a gift from T. Rapoport (Harvard Medical School). A complete list of yeast strains and their corresponding genotypes are listed in Table S2. All strains used in this work were derived from S288C or Resgen. The Y7092 query strain was a gift from Carla Koehler (University of CA, Los Angeles, CA). Yeast strains were transformed with DNA or PCR fragments using the standard LiOAc method (Ito et al., 1983). Null alleles were generated by using PCR to amplify a selection marker flanked by 50 base pairs of the 5′ and 3′ regions, which are immediately adjacent to the coding region of the gene to be deleted. The selectable markers used for making null alleles were genes encoding resistance to G418 or CloNat/nourseothricin. After transformation, strains with drug markers were plated onto YPD followed by replica-plating onto YPD plates containing (500 μg/mL G418 or 200 μg/mL nourseothricin). All gene deletions were confirmed by PCR.
For construction of the SUS-GFP query strain for Synthetic Genetic Array (SGA) analysis, the TRP1 coding region was replaced in situ with the fusion coding region for TDH3pro-SUS-GFP-PGK1term by homologous recombination with two cotransformed PCR products. These products were amplified from plasmid pRH2900, which contains TDH3pro-SUS-GFP-PGK1term. The first PCR product consisted of the 5′ flanking region of the TRP locus, the TDH3pro-SUS-GFP-PGK1term region, and the 5′ promoter region of URA3. The second PCR product consisted of the complete URA3 coding region followed by the 3′ flanking region of the TRP1 locus. Co-transformation of a native yeast strain with both products resulted in replacement of the TRP1 coding region with that of TDH3pro-SUS-GFP-PGK1term, followed by the URA3 marker allowing selection for successful replacement. Cotransformed cells were selected for growth on SC-Ura plates, confirmed by PCR and tested by immunoblotting for SUS-GFP. Primer sequences are available upon request.
dfm1Δ strain handling
To observe the phenotypic effect of dfm1Δ null strains, freshly transformed dfm1Δ null cells with the respective ERAD-M substrates was used in every assay.
Protease Protection Assay
Protease protection assay was adapted and modified from (Gardner et al., 2000). Cells expressing SUS and SUS-GFP were grown to log-phase (OD600 0.2–0.3) and 30 ODs of cells were pelleted. Cells were resuspended in H2O, centrifuged and lysed with the addition of 0.5 mM glass beads and 400 μL of diluted XL buffer (.24 M sorbitol, 1 mM EDTA, 20 mM KH2PO4, final pH 7.5) with protease inhibitors (PIs) followed by vortexing in 1 minute intervals for 6–8 min at 4°C. Lysates were combined and clarified by 5 second pulses at 16,000 × g in 1.5 ml eppendorf tubes until no pellet was formed (typically 4 pulses required). The resulting clarified supernatant was pelleted for 5 min at 14,000 × g at 4°C. The supernatant was aspirated, and the microsome pellet overlaid with fresh XL buffer followed by a spin at 14,000 × g at 4°C to ensure pellet cohesion. Microsomes were resuspended in XL buffer (1.2 M sorbitol, 5 mM EDTA, 0.1 M KH2PO4, final pH 7.5) and incubated with 100 μg/mL trypsin (Sigma) for 0, 2, 5,15 and 30 minutes. An equal volume of 2× USB buffer was added to stop the reactions. The samples were analyzed on SDS-PAGE and immunoblotted for SUS-GFP with α-Myc and α-HA.
In Vivo Retrotranslocation Assay
in vivo retrotranslocation assay was adapted and modified from (Jarosch et al., 2002). Cells in log phase (OD600 0.2–0.3) were treated with MG132 (benzyloxycarbonyl-Leu-Leu-aldehyde, Sigma) at a final concentration of 25 μg/mL (25 mg/mL stock dissolved in DMSO) for 2 hours at 30°C and GGPP (Geranylgeranyl pyrophosphate ammonium salt, Sigma) at a final concentration of 11 M for 1 hour at 30°C and 15 ODs of cells were pelleted. Cells were resuspended in H2O, centrifuged and lysed with the addition of 0.5 mM glass beads and 400 μL of XL buffer (1.2 M sorbitol, 5 mM EDTA, 0.1 M KH2PO4, final pH 7.5) with PIs, followed by vortexing in 1 minute intervals for 6–8 min at 4°C. Lysates were combined and clarified by centrifugation at 2,500 g for 5 min. Clarified lysate was ultracentrifuged at 100,000 g for 15 min to separate pellet (P100) and supernatant fraction (S100). P100 pellet was resuspended in 200 μL SUME (1% SDS, 8 M Urea, 10 mM MOPS, pH 6.8, 10 mM EDTA) with PIs and 5 mM N-ethyl maleimide (NEM, Sigma) followed by addition of 600 μL immunoprecipitation buffer (IPB) with PIs and NEM. S100 supernatant was added directly to IPB with PIs and NEM. 15 μL of rabbit polyclonal anti-GFP antisera (C. Zuker, University of California, San Diego) was added to P100 and S100 fractions for immunoprecipitation (IP) of Hmg2-GFP. Samples were incubated on ice for 5 minutes, clarified at 14,000 g for 5 min and removed to a new eppendorf tube and incubated overnight at 4°C. 100 μL of equilibrated Protein A-Sepharose in IPB (50% w/v) (Amersham Biosciences) was added and incubated for 2 h at 4°C. Proteins A beads were washed twice with IPB and washed once more with IP wash buffer (50 mM NaCl, 10 mM Tris), aspirated to dryness, resuspended in 2× Urea sample buffer (8 M urea, 4% SDS, 1mM DTT, 125 mM Tris, pH 6.8), and incubated at 55°C for 10 min. IPs were resolved by 8% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with monoclonal anti-ubiquitin (Fred Hutchinson Cancer Center, Seattle) and anti-GFP (Clontech, Mountain View, CA). Goat anti-mouse (Jackson ImmunoResearch, West Grove, PA) and goat anti-rabbit (Bio-Rad) conjugated with horseradish peroxidase (HRP) recognized the primary antibodies. Western Lightning® Plus (Perkin Elmer, Watham, MA) chemiluminescence reagents were used for immunodetection.
Proteolytic Removal of Ubiquitin from Retrotranslocated Hmg2-GFP
Ubiquitin removal was accomplished with the broadly active Usp2 ubiquitin protease as previously described (Garza et al., 2009b), except that human recombinant Usp2Core (LifeSensors Inc., Malvern, PA) was used, and leupeptin and NEM were excluded from all buffers. Briefly, 100 μL of S100 supernatant containing in vivo retrotranslocated Hmg2-GFP was incubated with 20 μL of Usp2Core (5 μg) for 1 hr at 37°C. The reaction was quenched with 200 μl of SUME (1% SDS, 8 M Urea, 10 mM MOPS, pH 6.8, 10 mM EDTA) with PIs and retrotranslocated Hmg2-GFP was immunoprecipitated as described above. 20 μL of IP was used for detection of Hmg2-GFP with α -GFP.
Cycloheximide-Chase Assay
Cycloheximide chase assays were performed as previously described (Sato et al., 2009). Cells were grown to log-phase (OD600 0.2-.03) and cycloheximide was added to a final concentration of 50 μg/mL. At each time point, a constant volume of culture was removed and lysed. Lysis was initiated with addition of 100 μl SUME with PIs and glass beads, followed by vortexing for 4 min. 100 μl of 2xUSB was added followed by incubation at 55°C for 10 min. Samples were clarified by centrifugation and analyzed by SDS-PAGE and immunoblotting.
High-throughput Retrotranslocation Factor Screen
The SUS-GFP query strain was crossed with the SPOCK collection, consisting of 5,808 yeast non-essential null and essential DaMP gene mutants (Jaeger, Ornelas et al., 2017). All library manipulations including selection of diploids, sporulation, and selection of haploids were done using standard EMAP technology (Collins et al., 2010) using a RoToR pinning robot (Singer Instruments, Taunton, UK) and Uracil and G418 selection. Once SUS-GFP was introduced into every yeast viable null and hypomorphic allele, the yeast array was grown for 24 hrs at 30°C. The resulting array was then transferred to liquid YPD media (50 μL/well) using the RoToR. After further incubation at 30°C for 2 days, we analyzed SUS-GFP stabilization with an LSR Fortessa with High Throughput Sampler (BD Biosciences, San Jose) set to collect 10,000 events per sample. GFP and autofluorescence were excited at 488 nm and 405 nm respectively and detected with 510/25 and 450/50 bandpass filters. Data was analyzed in FlowJo version 9. GFP+ events were gated by plotting autofluorescence versus GFP fluorescence.
Cdc48 Microsome Association Assay
Yeast strains were grown to log phase (OD600 0.2–0.3) and 15 ODs of cells were pelleted. Cells were resuspended in H2O, centrifuged and lysed with the addition of 0.5 mM glass beads and 400 μL of XL buffer with PIs and vortexed in 1 minute intervals for 6–8 min at 4°C. Lysates were combined and clarified by centrifugation at 2,500 g for 5 min. 50 μL of lysate was transferred to another tube and designated as total fraction (T). The rest of clarified lysate was centrifuged at 20,000 × g for 5 min to separate microsome pellet (P) and cytosolic supernatant fraction (S). An equivalent volume of 2xUSB was added to T, P and S fractions followed by solubilization at 55°C for 10 min. Samples were clarified by centrifugation, analyzed by SDS-PAGE and immunoblotted for Cdc48 and PGK1 with α-CDC48 and α-PGK1 respectively.
Flow Cytometry
Flow cytometry for GFP was performed as previously described (Garza et al., 2009c). Cells were grown to OD600 .15–.2. Data was obtained through using an Accuri machine (BD Biosciences) and Flowjo software.
Yeast genome resequencing and analysis
Genomic DNA was isolated with the MasterPure Yeast DNA purification kit (Epicentre). Approximately 50 ng of gDNA was tagmented using the Nextera® DNA Sample Preparation Kit (Illumina) with 0.5 μl Tn5 (Tagment DNA Enzyme 1) in a total volume of 20 μl and 5 minute incubation time at 55°C. Reaction was purified using the ChIP DNA Clean and Concentrate kit (Zymo Research) and amplified and barcoded for 9 PCR cycles. Libraries were size selected for 200–250bp by gel isolation and sequenced SE75 on a NextSeq 2500 (Illumina). Adapter sequences were trimmed from the 3′ ends and the reads were then aligned using bowtie 2 (version 2.3; default parameters) (Langmead and Salzberg, 2012) to the S. cerevisiae genome (sacCer3). HOMER (Heinz et al., 2010) was used to tile the genome in 10 kb regions and generate normalized read densities per 10kb region using the annotatePeaks.pl command.
Code
getGenomeTilingPeaks -res 10000 -d Exp -TagDir/> regions.txt annotatePeaks.pl regions.txt sacCer3 –d Exp1 -TagDir/Exp2-TagDir/Exp3-TagDir/> coverage.txt
QUANTIFICATION AND STATISTICAL ANALYSIS
ImageJ (NIH) was used for all western blot quantifications. Band intensities were measured directly from films scanned in high resolution (600 dpi) in TIFF file format. “Mean gray value” was set for band intensity measurements. In such experiments, a representative western blot was shown and band intensities were normalized to PGK1 loading control and quantified. t=0 was taken as 100% and data is represented as mean ± SEM from at least three experiments.
Supplementary Material
Table S2: Yeast strains used in this study, Related to STAR Methods.
Table S3: Identification of gene deletions stabilizing SUS-GFP in the retrotranslocation factor screen, Related to Figure 1
Figure 7. Model of Hrd1-mediated suppression in the dfm1Δ null.
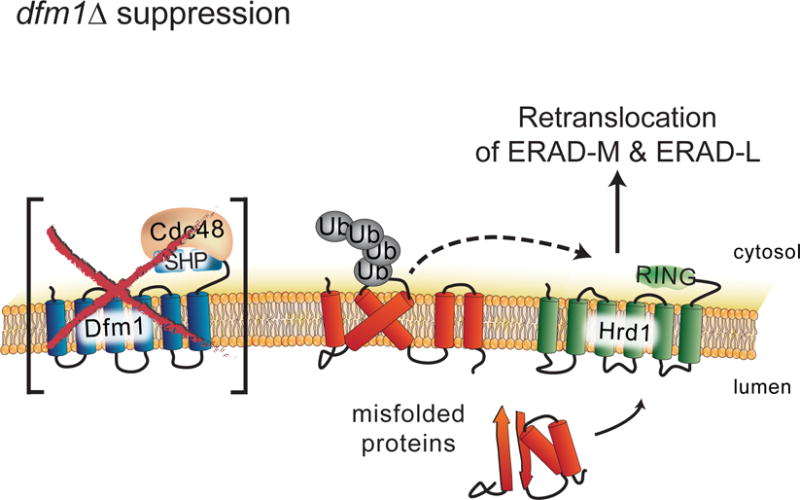
dfm1Δ null mutants undergo rapid suppression when ERAD-M substrates are strongly expressed, revealing an ancillary Dfm1-independent retrotranslocation route for ERAD-M substrates, which mediated by Hrd1.
HIGHLIGHTS.
-
-
Retrotranslocation of integral membrane ERAD (ERAD-M) substrates requires Dfm1
-
-
Dfm1 provides this function in both the Hrd and Doa pathways
-
-
Dfm1’s binds Cdc48 at the ER surface and moves ERAD-M substrates to the cytosol
-
-
dfm1Δ null mutants are rapidly suppressed when ERAD-M substrates are strongly expressed
Acknowledgments
We thank Tom Rapoport (Harvard Medical School), Davis Ng (National University of Singapore), Randy Schekman (University of California, Berkeley), and Susan Michaelis (John Hopkins University) for providing plasmids and antibodies. We thank Amy Cortez and Yoav Altman from the Flow Cytometry Shared Resource (Sanford Burnham Prebys Medical Discovery Institute) for the high throughput FACs screening and analysis. We also thank the Hampton lab members for in depth discussions and technical assistance. These studies were supported by NIH grant 5R37DK051996-18 (to R.Y.H.), R41 TR001908 (to P.A.J.), R01 ES014811 and R01 GM084279 (to T.I.), HL088083 (to C.G.). S.E.N was funded by the postdoctoral NIH grant 1F32GM111024-01 and Burroughs Wellcome Fund 1013987. S.D. was supported from funding by the CRI Irvington Postdoctoral Fellowship. S.E.N wishes to dedicate this work to the memory of her late brother, Michael Neal, who was a remarkable athlete, father, son, husband and friend. You will be missed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS: S.E.N. and R.Y.H. designed research; S.E.N., P.A.J., and S.D. performed research; S.E.N, R.Y.H., P.A.J., T.I., S.D., C.G., and C.B. analyzed data; S.E.N. and R.Y.H. wrote the paper; and S.D. designed illustrations for figures. All authors reviewed the results and approved the final version of the manuscript.
DECLARATION OF INTERESTS: The authors declare they have no competing interests within the contents of this article.
References
- Avci D, Fuchs S, Schrul B, Fukumori A, Breker M, Frumkin I, Chen C, Biniossek M, Kremmer E, Schilling O, et al. The Yeast ER-Intramembrane Protease Ypf1 Refines Nutrient Sensing by Regulating Transporter Abundance. Mol Cell. 2014;56:630–640. doi: 10.1016/j.molcel.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Baldridge RD, Rapoport TA. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell. 2016 doi: 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S, Bays N, Gardner R, Seelig L, Joazeiro C, Hampton R, et al. Role of the ubiquitin-selective CDC48(UFD1/NPL4) chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SM, Hampton RY. Usa1p Is Required for Optimal Function and Regulation of the Hrd1p Endoplasmic Reticulum-associated Degradation Ubiquitin Ligase. J Biol Chem. 2009;285(8):5146–5156. doi: 10.1074/jbc.M109.067876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci U S A. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2011;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Roguev A, Krogan NJ. Quantitative genetic interaction mapping using the E-MAP approach. Methods Enzymol. 2010;470:205–231. doi: 10.1016/S0076-6879(10)70009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, Lemberg MK. Ubiquitin-Dependent Intramembrane Rhomboid Protease Promotes ERAD of Membrane Proteins. Mol Cell. 2012;47:558–569. doi: 10.1016/j.molcel.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Foresti O, Ruggiano A, Hannibal-Bach HK, Ejsing CS, Carvalho P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife. 2013;2:e00953. doi: 10.7554/eLife.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R, Cronin S, Leader B, Rine J, Hampton R, Leder B. Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1998;9:2611–2626. doi: 10.1091/mbc.9.9.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multSUSispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Tran PN, Hampton RY. Geranylgeranyl pyrophosphate (GGPP) is a potent regulator of HRD- dependent HMG-CoA reductase degradation in yeast. J Biol Chem. 2009;284:35368–80. doi: 10.1074/jbc.M109.023994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V, Carvalho P, Rapoport TA. The ER-associated degradation component Der1p and its homolog Dfm1p are contained in complexes with distinct cofactors of the ATPase Cdc48p. FEBS Lett. 2008;582:1575–1580. doi: 10.1016/j.febslet.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant α-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY. Fusion-based strategies to identify genes involved in degradation of a specific substrate. Methods Enzymol. 2005;399:310–323. doi: 10.1016/S0076-6879(05)99021-6. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Garza RM. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev. 2009;109:1561–1574. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Rine J. Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol. 1994;125:299–312. doi: 10.1083/jcb.125.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol. 2012;24:460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger PA, Ornelas L, McElfresh C, Wong L, Hampton RY, Ideker T. Systematic Gene-to-Phenotype Arrays, SGPA: A High-Throughput Technique for Molecular Phenotyping. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.12.016. Manuscript under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Jo Y, Debose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010;45:185–198. doi: 10.3109/10409238.2010.485605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK. Sampling the membrane: function of rhomboid-family proteins. Trends Cell Biol. 2013;23:210–217. doi: 10.1016/j.tcb.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Lemberg MK, Adrain C. Inactive rhomboid proteins: New mechanisms with implications in health and disease. Semin Cell Dev Biol. 2016 doi: 10.1016/j.semcdb.2016.06.022. [DOI] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Kamura T, Bagola K, Mehnert M, Jarosch E, Sommer T, Xie W, Ng D, Hampton R, Sommer T, et al. Subcellular Fractionation Analysis of the Extraction of Ubiquitinated Polytopic Membrane Substrate during ER-Associated Degradation. PLoS One. 2016;11:e0148327. doi: 10.1371/journal.pone.0148327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Mak R, Bennett EJ, Hampton R. A Cdc48 ? Retrochaperone? Function Is Required for the Solubility of Retrotranslocated, Integral Membrane Endoplasmic Reticulum-associated Degradation (ERAD-M) Substrates. J Biol Chem. 2017;292:3112–3128. doi: 10.1074/jbc.M116.770610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Böhmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Egner R, Kuchler K, Wolf DH. Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J Biol Chem. 1998;273:32848–32856. doi: 10.1074/jbc.273.49.32848. [DOI] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Ryu HY, Wilson NR, Mehta S, Hwang SS, Hochstrasser M. Loss of the SUMO protease Ulp2 triggers a specific multichromosome aneuploidy. Genes Dev. 2016;30:1881–1894. doi: 10.1101/gad.282194.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato BK, Hampton RY. Yeast Derlin Dfm1 interacts with Cdc48 and functions in ER homeostasis. Yeast. 2006:1053–1064. doi: 10.1002/yea.1407. [DOI] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoebel S, Mi W, Stein A, Ovchinnikov S, Pavlovicz R, DiMaio F, Baker D, Chambers MG, Su H, Li D, et al. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature. 2017;548:352–355. doi: 10.1038/nature23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- Scott DC, Schekman R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol. 2008;181:1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Schweizer RS, Schäfer A, Wolf DH. Dfm1 Forms Distinct Complexes with Cdc48 and the ER Ubiquitin Ligases and Is Required for ERAD. Traffic. 2010a;11:1363–1369. doi: 10.1111/j.1600-0854.2010.01093.x. [DOI] [PubMed] [Google Scholar]
- Stolz A, Schweizer RS, Schäfer A, Wolf DH. Dfm1 forms distinct complexes with Cdc48 and the ER ubiquitin ligases and is required for ERAD. Traffic. 2010b;11:1363–1369. doi: 10.1111/j.1600-0854.2010.01093.x. [DOI] [PubMed] [Google Scholar]
- Sun F, Zhang R, Gong X, Geng X, Drain PF, Frizzell RA. Derlin-1 Promotes the Efficient Degradation of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) and CFTR Folding Mutants. J Biol Chem. 2006;281:36856–36863. doi: 10.1074/jbc.M607085200. [DOI] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szamecz B, Boross G, Kalapis D, Kovács K, Fekete G, Farkas Z, Lázár V, Hrtyan M, Kemmeren P, Groot Koerkamp MJA, et al. The Genomic Landscape of Compensatory Evolution. PLoS Biol. 2014;12:e1001935. doi: 10.1371/journal.pbio.1001935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng X, Dayhoff-Brannigan M, Cheng WC, Gilbert C, Sing C, Diny N, Wheelan S, Dunham M, Boeke J, Pineda F, et al. Genome-wide Consequences of Deleting Any Single Gene. Mol Cell. 2013;52:485–494. doi: 10.1016/j.molcel.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DTW. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashistha N, Neal SE, Singh A, Carroll SM, Hampton RY. Direct and essential function for Hrd3 in ER-associated degradation. Proc Natl Acad Sci. 2016;113:5934–5939. doi: 10.1073/pnas.1603079113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlman J, DeMartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell. 2007;129:943–955. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- You H, Ge Y, Zhang J, Cao Y, Xing J, Su D, Huang Y, Li M, Qu S, Sun F, et al. Derlin-1 promotes ubiquitylation and degradation of the epithelial Na+ channel. ENaC J Cell Sci. 2017;130 doi: 10.1242/jcs.198242. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: Yeast strains used in this study, Related to STAR Methods.
Table S3: Identification of gene deletions stabilizing SUS-GFP in the retrotranslocation factor screen, Related to Figure 1