

Abstract
Terpene synthases comprise a family of enzymes that convert acyclic oligo-isoprenyl diphosphates to terpene natural products with complex, polycyclic carbon backbones via the generation and protection of carbocation intermediates. To accommodate this chemistry, terpene synthase active sites generally are lined with alkyl and aromatic, i.e., nonpolar, sidechains. Predicting the correct, mechanistically relevant binding modes for entire terpene synthase reaction pathways remains an unsolved challenge. Here we describe a method for identifying such modes: TerDockin, a series of protocols to predict the orientation of carbon skeletons of substrates and derived carbocations relative to the bound diphosphate group in terpene synthase active sites. Using this recipe for bornyl diphosphate synthase, we have predicted binding modes that are consistent with all current experimental observations, including the results of isotope labeling experiments and known stereoselectivity. In addition, the predicted binding modes recapitulate key findings of a seminal study involving more computationally demanding QM/MM molecular dynamics methods on part of this pathway. This work illustrates the value of the TerDockin approach as a starting point for more involved calculations and sets the stage for the rational engineering of this family of enzymes.
Keywords: docking, terpene, bornyl diphosphate, TerDockin, Rosetta, enzyme, Quantum Mechanics, QM
Introduction
Terpenes and their modified derivatives, terpenoids, comprise the largest class of natural products known.1–2 Made by organisms on every branch of the tree of life,3–5 these ubiquitous chemicals play important roles in chemical ecology.2–3, 6–11 Terpenes are often stereodense and polycyclic and they have been co-opted by humans for non-natural uses such as medicine, perfumery and food flavorings.12 Artemisinin13 and paclitaxel14 are two of the most famous examples of terpenoids that have had large impacts on human health, with the discovery of the former recently recognized with a Nobel prize.15 Many other terpenoids have been and are being investigated as antibiotics,16–19 anti-cancer therapies,20 enhancers of dermal penetration for other drugs,21 etc.
Terpenes are produced in secondary metabolism by a family of enzymes called terpene synthases (or cyclases).22 These enzymes convert simple, acyclic substrates into complex molecules via the intermediacy of carbocations. Terpene synthases can be separated into two groups on the basis of the two different ways in which they generate carbocations.22 Type 1 terpene synthases initiate carbocation formation by the magnesium-assisted removal of a diphosphate group, while type 2 terpene synthases generate carbocations by protonation of a C=C π-bond (or an epoxide). In this manuscript, we focus on type 1 terpene synthases.
In order to accommodate highly reactive carbocation intermediates, the active sites of terpene synthase enzymes are themselves largely nonpolar (greasy), usually lined with aromatics and alkyl side chains that (1) are unlikely to react with carbocations (or do so reversibly), (2) exclude water to prevent unintentional alcohol formation23 and (3) potentially promote formation of one product over another through weak interactions (e.g., CH–π and CH–O interactions).23–26 It has been demonstrated previously that the results of gas phase quantum chemical calculations on carbocation cyclization/rearrangement reactions are often consistent with product profiles observed for terpene synthase reactions, i.e., inherent carbocation reactivity is often expressed if the substrate adopts an appropriate conformation.27–34 These results are in accord with the long-standing hypothesis that one key role of terpene synthases is to pre-organize the substrate such that, once a carbocation is formed in a terpene synthase active site, it quickly reacts to give the known product.35–40
Several features of terpene synthases have made accurate molecular modeling of their roles in catalysis extraordinarily challenging. In general, most classical polar contacts between the substrate and terpene synthase enzyme involve the diphosphate moiety, which is removed in the first step of the mechanism. As a result, a largely greasy carbocation (while cationic, charge is substantially delocalized) is produced in a greasy, π-rich pocket, which makes predicting the bound conformation of the carbocation and its position relative to the diphosphate exceedingly difficult (described previously as akin to trying to identify a single piece of hay in a haystack).34 The difficulty arises in large part from the lack of specific intermolecular interactions with strong orientational preferences, such as the hydrogen bonds for which reasonable parameters exist in most force fields. Recently, we described preliminary work on a method for predicting the catalytically relevant ion pair (carbocation–diphosphate) orientation in the context of terpene synthase active sites that makes use of constraints derived from experimental observations on reactivity.34 This approach is referred to as TerDockin (short for terpene docking). Here, we apply the TerDockin workflow to the reaction catalyzed by bornyl diphosphate synthase (BPPS)35, 41 and describe a refined TerDockin recipe for carbocation docking, as well as docking of hydrocarbon diphosphate substrates, that can be used to generate experimentally testable hypotheses and starting points for more involved multiscale modeling efforts and enzyme engineering.
BPPS is an atypical type 1 terpene synthase, in that its reaction is not consummated by deprotonation, but rather by reattachment of the diphosphate moiety to give the primary product bornyl diphosphate (Figure 1). These features have led to a collection of experimental data that TerDockin should be able to reproduce—without this data being utilized during the TerDockin procedure. First, Croteau et al.35 found, via 18O labeling, that the oxygen atom that was attached directly to the hydrocarbon chain of the substrate also was the oxygen that was attached directly to the bornyl group in the product (Figure 1); does TerDockin predict an ion pair orientation consistent with this result? Second, there is an X-ray crystal structure of BPPS with the product bound (unusual for terpene synthases; PDB code 1N24);42 does TerDockin predict that the observed product orientation in the active site is preferred? Also of interest is whether this orientation reflects that formed during BPP formation or rather results from product rebinding in a thermodynamically preferred orientation. Third, the stereochemistry of the carbon in BPP to which the diphosphate group is attached is known;42 does TerDockin predict that this stereochemistry is preferred for the reattachment of the diphosphate? Fourth, BPPS selectively produces one enantiomer of BPP; does TerDockin discriminate against the incorrect enantiomers of the carbocation intermediates? In addition, computationally more demanding quantum mechanics/molecular mechanics (QM/MM) molecular dynamics (MD) studies were previously conducted by Major and Weitman;39, 43 are the TerDockin results consistent with these, i.e., could TerDockin be used as a reliable means of identifying correct carbocation–diphosphate ion pair orientations for subsequent use in multiscale modeling approaches, a problem identified by Major and others as a significant hurdle for generating meaningful results.36
Figure 1.
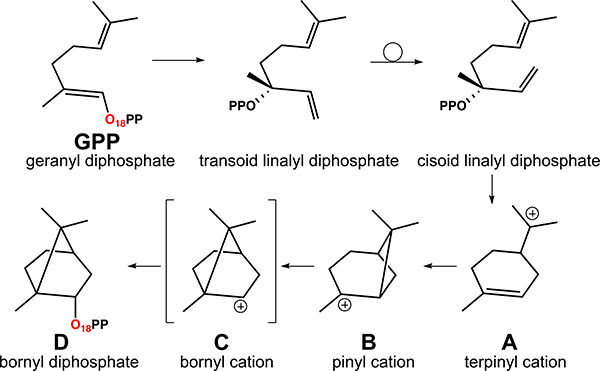
Putative mechanism for the formation of BPPS. Note that the bornyl cation (C) is not a discrete intermediate in the gas phase. The oxygen labeled by Croteau et al. is highlighted in red;35 the location of this oxygen in linalyl diphosphate (LPP) has not been definitively determined.
Methods
There are four essential steps required for a successful TerDockin analysis:
(1). Preparation of ligands.
In the TerDockin approach, two ligands are docked simultaneously: the diphosphate-magnesium complex and the hydrocarbon framework. For the particular reaction investigated here, carbocation intermediates were previously identified using quantum mechanics calculations (specifically, density functional theory [DFT] calculations).33, 43 We recommend that carbocation structures be determined in this manner, since many carbocations have delocalized structures that are not properly characterized by molecular mechanics. In this study, we use B3LYP44–48/6–31+G(d,p) geometries determined by Hong et al.33 Structures A, B, C and bornyl diphosphate from Figure 1 were docked into BPPS as described below. Note that neither Hong and Tantillo nor Major and Weitman identified the bornyl cation (C) as a potential energy surface minimum (i.e., an intermediate) in the gas phase. This structure was also not found to be a minimum in calculations with diphosphate models present,33 nor in the context of the active site using QM/MM calculations.35 A “C-like” structure described by Hong and Tantillo from an intrinsic reaction coordinate (IRC)49–51 calculation on a transition state structure connecting intermediate B to BPP was used here for the docking. The geometry of the diphosphate and three associated magnesium ions was taken from a BPPS crystal structure (PDB 1N23) and docked as a single ligand without relaxation.
Structures A-D lack conformational flexibility, allowing a single conformer to be docked for each. For most carbocations produced inside terpene synthases, however, multiple conformers must be located and docked. In contrast to our initial TerDockin study on epi-aristolochene synthase (TEAS),34 we here docked the oligoprenyl diphosphate reactant, GPP. Many conformations are energetically accessible for this substrate. Searching for these is a challenge, given the anionic nature, size and potential flexibility of the diphosphate moiety. In that the diphosphate group is bound in a relatively static orientation, held in place by counterions and enzymatic residues, we decided to replace the diphosphate group in GPP with an iodine for conformational searching (using Conflex)52 of the geranyl group. The resulting 18 conformers were then optimized using B3LYP/6–31+G(d,p) and, for any structure within 5 kcal/mol of the lowest energy optimized structure, the iodine was deleted and the resulting cations were combined and used as a library for the docking simulations described below. This OPP → I replacement approach is amenable to generating conformational libraries for oligoprenyl diphosphate reactants of any size for use in modeling terpene synthase reactions.
(2). Choosing a suitable crystal structure.
Many of the terpene synthase crystal structures available in the protein data bank (PDB)22, 53 have incomplete active sites or are apo structures. For modeling type 1 terpene synthases, a complete active site is required. In particular, a reliable structure of the C-terminal end of the protein sequence, which plays a role in capping the active site upon substrate binding (induced-fit),54 is essential for a catalytically relevant structure.55 Ideally, the crystal structure would also have a relevant ligand bound in a relevant conformation, such as a substrate or intermediate analog with the diphosphate moiety (either separate or together) and required magnesium ions.32 Also, it is ideal to have two (or more) crystal structures for the same enzyme that meet the outlined criteria, which allows one to dock into both and ensures that predictions are not the result of peculiarities in particular crystal structures or the artifacts of modeling errors. BPPS is an ideal terpene synthase for TerDockin analysis, with multiple complete crystal structures with a variety of ligands bound. For this study, BPPS structures with PDB codes 1N20 and 1N23 were used. For systems lacking “ideal structures,” one can resort to homology modeling, with or without MD relaxation (a challenge we will address in future studies).
(3). Definition of docking constraints.
The TerDockin approach involves the application of two sets of constraints, each serving a different purpose.56 First, a set of chemistry-based constraints is used to separate and sample possible ion-pair orientations. In BPPS, the diphosphate interacts, during the BPP-forming reaction, with three different carbons (Figure 1): initial attachment at C1, isomerization to a C3-attached structure (LPP; presumed) and reattachment at C2 to form BPP. In the crystal structure of BPPS (PDB code 1N23), there are two diphosphate oxygen atoms, (labeled A and B in Figure 2) pointed into the active site towards the hydrocarbon binding region. It is not known which of these two oxygens is attached to C1–3 during the reaction. This leads to 2×2×2 (i.e., 8) ion-pair orientations (Figure 2). For each, the relevant oxygen atom is constrained to be 3.0 ± 0.5 Å from a given carbon during docking, with no angle or dihedral constraints imposed for A, B or C These constraints were employed to be consistent with the hypothesis that, after a carbocation is generated, it is so reactive that it will progress to the final carbocation intermediate before the carbocation can move a significant distance away from the diphosphate group (GPP and BPP docking were treated differently, vide infra). The aforementioned labeling study conducted by Croteau et al. showed that the oxygen atom that starts attached to C1 in GPP becomes attached to C2 in BPP and if labeled LPP (label is attached to C3) is used as a substrate, the label also becomes attached to C2.35 Of the eight orientations sampled here, only two are consistent with this experimental result (highlighted in green in Figure 2).
Figure 2.
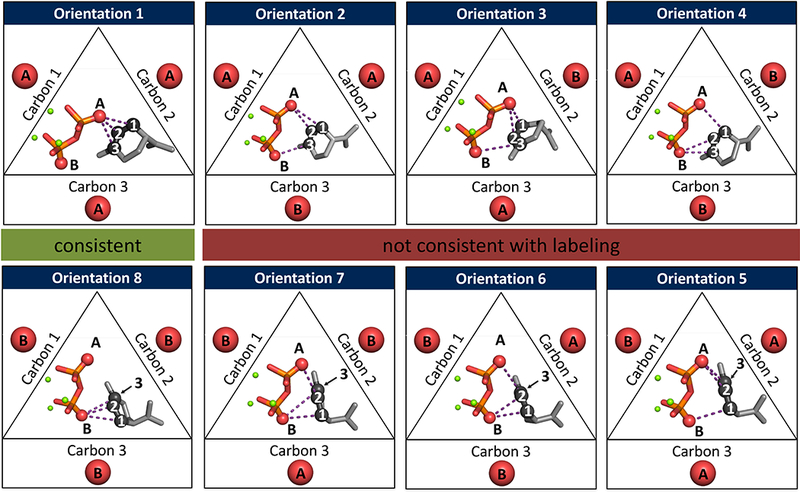
Depiction of the eight different ion-pair orientations that were explicitly tested by the implementation of constraints during docking calculations. Two oxygen atoms are pointed into the active site of BPPS, labelled A and B. There are three points of contact with the hydrocarbon skeleton during the BPP-forming reaction, labeled carbons 1, 2 or 3. Orientations 1 and 8 (above and below green highlighted box) are consistent with the labelling experiment conducted by Croteau et al.35 while the orientations that are not consistent with this experiment are above or below the red highlighted box.
Preliminary attempts to dock GPP and BPP as discrete ligands gave binding orientations not relevant to catalysis, due in large part to our inability to treat binding of magnesium ions appropriately. Consequently, a different approach using different constraints for the docking of GPP and BPP was employed in which the diphosphate and hydrocarbon chain were artificially separated but then constrained to be only 1.45 ± 0.5 Å apart (representing C–O distances from DFT minimizations; using C1 for GPP and C2 for BPP, but sampling oxygens A and B for both). This separation allows us to capture the C–O binding distance while docking the hydrocarbon group independently from the diphosphate moiety and its associated magnesium ions. The following constraints were also applied to capture key features of the C–O bonds: (1) C–C–O bond angles of 109 ± 5° were required to represent appropriately hybridized geometries and (2) the C4-C3-C2-O1 dihedral angle for GPP and the C3-C2-C1-O dihedral angle for BPP were constrained to 313 ± 5°, but with a periodicity of 180° to allow the explicit testing of both possible alignments of the C–O bond that breaks in GPP with respect to the adjacent C=C π-bond and the facial selectivity for diphosphate recapture leading to BPP (i.e., BPP versus its C2 epimer). For both the angle and dihedral constraints, values were derived from DFT minimizations of BPP. For GPP, only the C1–O constraints were utilized, which leads to only two orientations: C1 bound to oxygen A or oxygen B. For BPP, constraints to the all three carbons were used, but distance constraints to C1 and C3 were loosened to 2.5 ± 0.5 Å to avoid unreasonable crowding between the hydrocarbon and diphosphate groups. Thus, all eight possible ion-pair orientations described above were explicitly tested for BPP.
The second family of constraints applied during the TerDockin procedure, referred to as “coordination constraints”, are used to facilitate binding of the magnesium-diphosphate complex. Distances, bond angles and dihedral angles are constrained to their average values from the available crystal structures ± standard deviations. For BPP, these constraints were derived from all crystal structures having three magnesiums and either an individual diphosphate or a diphosphate-containing substrate analog bound (PDB codes 1N20, 1N22, 1N23 & 1N24) and are shown in Table S1.
(4). Docking and checking for convergence.
As described above, two BPPS crystal structures (PDB IDs 1N20 and 1N23) were used for docking. First, these structures were relaxed using a constrained FastRelax57 procedure from the Rosetta Modeling Suite.58–60 The diphosphate-magnesium complex and hydrocarbon (carbocation) structures were then docked into both relaxed crystal structures using Rosetta with the constraints described above. The protocol implemented for the docking employed a Monte Carlo simulation to sample binding orientations in a flexible active site; specific details are provided in the Supporting Information.
After docking runs were completed, the resulting structures were combined and then filtered by: (1) Constraint satisfaction – structures that did not satisfy all constraints were not considered further. (2) Total protein energy – only structures that were one standard deviation or lower than the mean in computed total protein energy were considered further; this allows solutions that greatly distort the overall protein structure in order to satisfy the constraints to be discarded. (3) Interface energy – only the top 10% of structures in terms of computed interface energy were carried forward from the structures in the low total protein energy population. These final filtered structures were then grouped by binding orientation to identify in which ion-pair orientation the low energy population resides. Details on the nature of total protein score and interface energy scores can be found in the Supporting Information.
Given the stochastic sampling method used and the large number of degrees of freedom to be sampled, care is necessary to ensure adequate sampling has been performed. To verify the amount of sampling required for BPPS, docking runs were conducted with different numbers of total structures (Supporting Figure S2) for intermediate A. In general, we recommend checking for convergence in this way using the most flexible carbocation structure involved in the mechanism. For BPPS, it appears that even a low number of structures leads to a qualitative answer, as all the runs, ranging in size from 50 to 10000 structures, predict the same preferred ion-pair orientation. However, it is possible that false positives can occasionally occur. In addition, when the number of structures that pass the filtering (see below for details on the filtering) are analyzed for the smaller number of runs (see nstruc = 50 in Supporting Figure S2), a single false positive would greatly affect the confidence associated with a binding mode prediction. In general, one would like to have more than a 10-fold enrichment for one orientation over another to make a confident prediction.
Results and Discussion
Predicted binding orientations from TerDockin.
GPP, carbocations A-C and BPP (D) were each docked into both relaxed crystal structures (PDB codes 1N20 and 1N23) and filtered using the TerDockin procedure described above. The results for 1N20 are compiled into the heat map shown in Figure 3 (the heat map for 1N23, which is very similar, is shown in the Supporting Information). Based on these results, only a single ion-pair orientation connects all docked structures: Orientation 1, as defined in Figure 2. This orientation is consistent with the 18O labeling experiment described above.35 For GPP and carbocations A-C, hundreds of low energy structures were found, but for the final product, D, the number of low energy structures that survived filtering is much smaller for each structure modeled. This is primarily a result of the additional constraints applied to the product.
Figure 3.
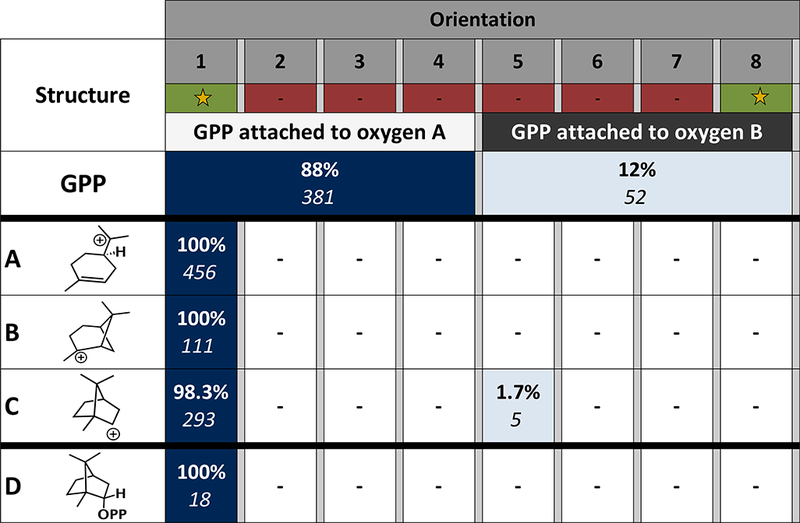
Heat map of the docking results. For GPP, only two discreet orientations were tested: attachment to oxygen A or B. Ion-pair orientations for other structures are depicted in Figure 2. The orientations highlighted with a star are consistent with the labeling experiment conducted by Croteau et al.35 The darker the color filling each cell of the table, the more low energy solutions were found for that orientation. The percentage of low energy structures identified for each docked structure are shown in bold, the number of low energy structures remaining after filtering is shown in italics. The thick black lines separate structures docked with different constraints.
Assuring that the predicted pathway is continuous.
Since a large amount of structural space was sampled during the docking simulations, it is possible that the low energy solutions for any one intermediate are not structurally similar to the low energy solutions for the next intermediate. Given that carbocations are extremely reactive,28 we hypothesized that large movements as one carbocation morphs into the next are unlikely (i.e., while vibration is reasonable, translation and rotation are less so). Therefore, we identified a pathway of least movement for Orientation 1. A pair-wise RMSD (carbon atoms only) was calculated for all the GPP structures and all the A structures that passed filtering. The structure of A that aligned most well with GPP was then used to calculate the RMSD with B, C and D. The results of these comparisons are plotted in Figure 4A. The lowest RMSD for the GPP to A transition was 0.84 Å, for the A to B transition was 1.64 Å, for A to C transition was 0.79 Å and for the A to D transition was 2.34 Å (Figure 4B – E). When the best matching structures are overlaid (Figure 4F), it is clear that they converge to a single region of the active site and it is clear how one structure can proceed to the next without large movements The orientation and the stereochemistry of the product, bornyl diphosphate (D), arrived at in this manner is consistent with the orientation found in the crystal structure of BPPS (PDB code 1N24) as shown in Figure 4G (the same orientation used in Major and Weitman’s more intensive QM/MM MD simulations).39, 43 It is not guaranteed that the orientation found in a crystal structure for a product complex is catalytically relevant, but, based on our TerDockin results, such an orientation does appear to be relevant for the BPPS reaction.
FIGURE 4.
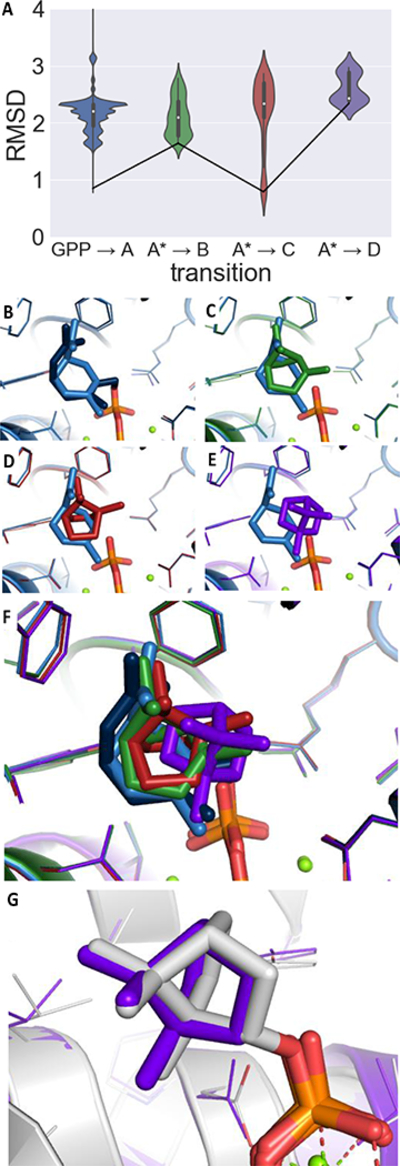
RMSD analysis of the structures identified in the docking results. (A) Violin plot of the RMSD for each transition. The mean value is indicated as a white dot and the interquartile range as a thick black box around the mean, the 95% confidence interval are shown as a black line. The population at any given RMSD score is mirrored on both sides. A line connecting the lowest RMSD structure identified in the search has been drawn in black. (B) Overlay of GPP (navy) and intermediate A (blue), (C) overlay of intermediate A and intermediate B (green), (D) overlay of intermediate A and intermediate C (red), (E) overlay of the intermediate A and the product, D in purple. (F) Overlay of all identified structures together. (G) The orientation of the product shown in purple overlaid with the crystal structure with the product bound in white (PDB 1N24).
Enantiomer docking.
As a test of TerDockin’s ability to predict absolute stereochemistry, enantiomers of all structures were docked using the same parameters as described above. As shown in Figure 5, there is no orientation which links the substrate, intermediates and the product, which is required to make a prediction of the catalytically relevant binding orientation. This result is consistent with ent-BPP not being a known product of this enzyme from GPP.61 The lack of a pathway linking all the structures docked for this enzyme is due primarily to structure ent-D, for which no structures passed the filtering criteria outlined in the materials section above (e.g., an inability to meet stringent distance constraint which is enforcing the covalent nature of the O-C bond in the product; see Supporting Information Figures S7 and S8 for additional details on the effects of alternate filtering criteria). Primarily, ent-D could not satisfy the constraints, indicating that, while enantiomers of the natural carbocations can bind, ent-C cannot effectively be trapped by OPP. In addition, a very low number of structures pass the filtering for ent-A, nine (Figure 5) rather than >450 for A (Figure 4), suggesting the active site contour can preorganize the substrate into one enantiomeric conformation. These results provide evidence that TerDockin can indeed recapitulate the stereochemical preference of the enzyme.
FIGURE 5.
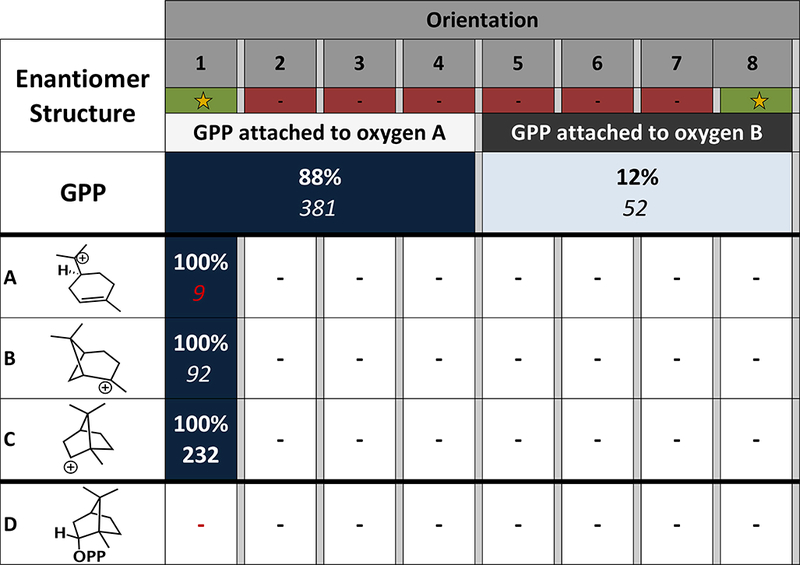
Heatmap of the results for docking the enantiomer of the carbon skeleton into the active site of BPPS (PDB 1N20). Each orientation is described in Figure 2. The orientations highlighted with a star are consistent with the labeling experiment conducted by Croteau et al.35 The darker the color the more low energy solutions are in that orientation. The percentage of low energy structures after filtering are in bold and the number of structures are underneath in italics. The number of structures for intermediate ent-A is in red to contrast it with the number of structures identifiedfor A in Figure 3.
Comparison to QM/MM studies.
When comparing the results from the docking simulations to the more expensive computations conducted by Weitmen et al. a similar prediction arises.36, 43 The Major group used the orientation of azabornane in the crystal structure of BPPS (PDB code 1N23) as the starting orientation for their QM/MM studies. Indeed, this orientation is consistent with the crystal structure with the product bound (PDB 1N24) and is consistent with the prediction made by TerDockin for the orientation of the bound product (Figure 5D, above) and with the orientation of GPP and intermediate A. Thus, the prediction made by TerDockin is consistent with the more elaborate QM/MM calculations run on this enzyme.
Conclusions
Predicting the catalytically relevant orientation of the substrate and reactive species derived from it in a terpene synthase active site is exceedingly challenging. Here we describe an approach to tackling this problem that can provide binding modes for all reaction intermediates. These high-resolution simulations provide a starting point for future multiscale modeling efforts, but also set the stage for rational engineering of terpene synthases.
The application of TerDockin specifically to BPPS allowed us to: (1) recapitulate the outcome of a key 18O labelling experiment, (2) predict an orientation of the product that is consistent with that found in the crystal structure of the BPPS-product complex (and that is similar to that used in previous QM/MM MD simulations), (3) correctly predict the diastereoselectivity for reincorporation of the diphosphate to form the final product, and (4) correctly predict the enantioselectivity of the BPPS reaction. As stated by Major, “the promiscuous binding in terpene synthases is one of the greatest challenges to theory due to weak interactions between the substrate and the hydrophobic pocket in the active site,”40 The TerDockin method addresses this challenge.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the NIH (GM076324). D.J.T. gratefully acknowledges support from the National Science Foundation (CHE-0957416, CHE-1565933 and CHE030089 for supercomputing resources via XSEDE). J.B.S. acknowledges the Sloan Foundation (#BR2014–012) and UC Davis for their support. T.E.O. was supported by the United States Department of Education for the Graduate Assistance in Areas of National Need (GAANN), the Alfred P. Sloan foundation and UC Davis’ Bradford Borge fellowship. S.J.B. gratefully acknowledges the Chemistry Graduate Program Fellowship (2014) from UC Davis for support. T.E.O thanks Prof. D. E. Cane (Brown) for his input on this project.
ABBREVIATIONS
- BPPS
bornyl diphosphate synthase
- GPP
geranyl diphosphate
- LPP
linolyl diphosphate
- BPP
bornyl diphosphate
- RMSD
root mean square deviation
- QM
quantum mechanics
- MM
molecular mechanics
- MD
molecular dynamics
- TEAS
tobacco epi-aristolochene synthase
- IRC
intrinsic reaction coordinate
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript.
Supporting Information1
The Supporting Information is available free of charge on the ACS Publications website.
Docking results from other crystal structure.
Values for coordination constraints used in Mg/PPi modeling.
Explanation on checking for sufficient sampling when using a monte-carlo based simulation.
The structures identified in the RMSD search in Figure 4 as mol2 files.
Sample Rosetta input files used in the modeling.
Energies and structures of the conformational search conducted on the iodo replacement of GPP.
Heatmap of results conducted with the recently updated Rosetta score function.
REFERENCES
- 1.McGarvey DJ; Croteau R, Terpenoid Metabolism. Plant Cell 1995, 7, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gershenzon J; Dudareva N, The Function of Terpene Natural Products in the Natural World. Nat. Chem. Biol 2007, 33, 408. [DOI] [PubMed] [Google Scholar]
- 3.Chen F; Tholl D; Bohlmann J; Pichersky E, The Family of Terpene Synthases in Plants: a Mid-Size Family of Genes for Specialized Metabolism that is Highly Diversified Throughout the Kingdom. The Plant J 2011, 66, 212–229. [DOI] [PubMed] [Google Scholar]
- 4.De Rosa M; Gambacorta A; Nicolaus B, Regularity of Isoprenoid Biosynthesis in the Ether Lipids of Archaebacteria. Phytochemistry 1980, 19, 791–793. [Google Scholar]
- 5.Yamada Y; Kuzuyama T; Komatsu M; Shin-ya K; Omura S; Cane DE; Ikeda H, Terpene Synthases are Widely Distributed in Bacteria. Proc. Natl. Acad. of Sci. USA 2015, 112, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byers KJRP; Vela JP; Peng F; Riffell JA; Bradshaw HD, Floral Volatile Alleles Can Contribute to Pollinator-Mediated Reproductive Isolation in Monkeyflowers (Mimulus). Plant J. 2014, 80, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisner T; Meinwald J, Chemical Ecology. Proc. Natl. Acad. Sci. USA 1995, 92, 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grayer RJ; Kokubun T, Plant–Fungal Interactions: the Search for Phytoalexins and Other Antifungal Compounds from Higher Plants. Phytochemistry 2001, 56, 253–263. [DOI] [PubMed] [Google Scholar]
- 9.Kessler A; Baldwin IT, Defensive Function of Herbivore-Induced Plant Volatile Emissions in Nature. Science 2001, 291, 2141. [DOI] [PubMed] [Google Scholar]
- 10.Umehara M; Hanada A; Yoshida S; Akiyama K; Arite T; Takeda-Kamiya N; Magome H; Kamiya Y; Shirasu K; Yoneyama K; Kyozuka J; Yamaguchi S, Inhibition of Shoot Branching by New Terpenoid Plant Hormones. Nature 2008, 455, 195–200. [DOI] [PubMed] [Google Scholar]
- 11.Wicker-Thomas C, Pheromonal Communication Involved in Courtship Behavior in Diptera. J. Insect Physiol 2007, 53, 1089–1100. [DOI] [PubMed] [Google Scholar]
- 12.Bicas JL; Dionísio AP; Pastore GM, Bio-oxidation of Terpenes: An Approach for the Flavor Industry. Chem. Rev 2009, 109, 4518–4531. [DOI] [PubMed] [Google Scholar]
- 13.Klayman DL, Qinghaosu (Artemisinin): An Antimalarial Drug from China. Science 1985, 228, 1049–1055. [DOI] [PubMed] [Google Scholar]
- 14.Wani MC; Taylor HL; Wall ME; Coggon P; McPhail AT, Plant Antitumor Agents. VI. Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus brevifolia. J. Am. Chem. Soc 1971, 93, 2325–2327. [DOI] [PubMed] [Google Scholar]
- 15.Su X.-z.; Miller LH, The Discovery of Artemisinin and Nobel Prize in Physiology or Medicine. Science China. Life Sci 2015, 58, 1175–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dairi T; Hamano Y; Kuzuyama T; Itoh N; Furihata K; Seto H, Eubacterial Diterpene Cyclase Genes Essential for Production of the Isoprenoid Antibiotic Terpentecin. J. Bacteriol 2001, 183, 6085–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamano Y; Kuzuyama T; Itoh N; Furihata K; Seto H; Dairi T, Functional Analysis of Eubacterial Diterpene Cyclases Responsible for Biosynthesis of a Diterpene Antibiotic, Terpentecin. J. Biol. Chem 2002, 277, 37098–37104. [DOI] [PubMed] [Google Scholar]
- 18.He Y; Hu Z; Sun W; Li Q; Li X-N; Zhu H; Huang J; Liu J; Wang J; Xue Y; Zhang Y, Spiroaspertrione A, a Bridged Spirocyclic Meroterpenoid, as a Potent Potentiator of Oxacillin against Methicillin-Resistant Staphylococcus aureus from Aspergillus sp. TJ23. J. Org. Chem 2017, 82, 3125–3131. [DOI] [PubMed] [Google Scholar]
- 19.Tamamura T; Sawa T; Isshiki K; Masuda T; Homma Y; Iinuma H; Naganawa H; Hamada M; Takeuchi T; Umezawa H, Isolation and Characterization of Terpentecin, a New Antitumor Antibiotic. JPN. J. Antibiot 1985, 38, 1664–1669. [DOI] [PubMed] [Google Scholar]
- 20.Tan W; Lu J; Huang M; Li Y; Chen M; Wu G; Gong J; Zhong Z; Xu Z; Dang Y; Guo J; Chen X; Wang Y, Anti-cancer Natural Products Isolated from Chinese Medicinal Herbs. Chin. Med-UK 2011, 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J; Dong C; Song Z; Zhang W; He X; Zhang R; Guo C; Zhang C; Li F; Wang C; Yuan C, Monocyclic Monoterpenes as Penetration Enhancers of Ligustrazine Hydrochloride for Dermal Delivery. Pharm. Dev. Technol 2017, 22, 571–577. [DOI] [PubMed] [Google Scholar]
- 22.Christianson DW, Structural Biology and Chemistry of the Terpenoid Cyclases. Chem. Rev 2006, 106, 3412–3442. [DOI] [PubMed] [Google Scholar]
- 23.Hong YJ; Tantillo DJ, Tension between Internal and External Modes of Stabilization in Carbocations Relevant to Terpene Biosynthesis: Modulating Minima Depth via C–H···π Interactions. Org. Lett 2015, 17, 5388–5391. [DOI] [PubMed] [Google Scholar]
- 24.Hong YJ; Tantillo DJ, Feasibility of Intramolecular Proton Transfers in Terpene Biosynthesis – Guiding Principles. J. Am. Chem. Soc 2015, 137, 4134–4140. [DOI] [PubMed] [Google Scholar]
- 25.Hong YJ; Tantillo DJ, C-H···π Interactions as Modulators of Carbocation Structure - Implications for Terpene Biosynthesis. Chem. Sci 2013, 4, 2512–2518. [Google Scholar]
- 26.Bojin MD; Tantillo DJ, Nonclassical Carbocations as C−H Hydrogen Bond Donors. J. Phys. Chem. A 2006, 110, 4810–4816. [DOI] [PubMed] [Google Scholar]
- 27.Zu L; Xu M; Lodewyk MW; Cane DE; Peters RJ; Tantillo DJ, Effect of Isotopically Sensitive Branching on Product Distribution for Pentalenene Synthase: Support for a Mechanism Predicted by Quantum Chemistry. J. Am. Chem. Soc 2012, 134, 11369–11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tantillo DJ, Importance of Inherent Substrate Reactivity in Enzyme-Promoted Carbocation Cyclization/Rearrangements. Angew. Chem. Int. Edit 2017, 56, 10040–10045. [DOI] [PubMed] [Google Scholar]
- 29.Lodewyk MW; Willenbring D; Tantillo DJ, Pentalenene Formation Mechanisms Redux. Org. Biomol. Chem 2014, 12, 887–894. [DOI] [PubMed] [Google Scholar]
- 30.Hong YJ; Tantillo DJ, Biosynthetic Consequences of Multiple Sequential Post-transition-state Bifurcations. Nat. Chem 2014, 6, 104–111. [DOI] [PubMed] [Google Scholar]
- 31.Hong YJ; Tantillo DJ, Branching Out from the Bisabolyl Cation. Unifying Mechanistic Pathways to Barbatene, Bazzanene, Chamigrene, Chamipinene, Cumacrene, Cuprenene, Dunniene, Isobazzanene, Iso-γ-bisabolene, Isochamigrene, Laurene, Microbiotene, Sesquithujene, Sesquisabinene, Thujopsene, Trichodiene, and Widdradiene Sesquiterpenes. J. Am. Chem. Soc 2014, 136, 2450–2463. [DOI] [PubMed] [Google Scholar]
- 32.Hong YJ; Tantillo DJ, The Taxadiene-Forming Carbocation Cascade. J. Am. Chem. Soc 2011, 133, 18249–18256. [DOI] [PubMed] [Google Scholar]
- 33.Hong YJ; Tantillo DJ, Quantum Chemical Dissection of the Classic Terpinyl/Pinyl/Bornyl/Camphyl Cation Conundrum - the Role of Pyrophosphate in Manipulating Pathways to Monoterpenes. Org. Biomol. Chem 2010, 8, 4589–4600. [DOI] [PubMed] [Google Scholar]
- 34.O’Brien TE; Bertolani SJ; Tantillo DJ; Siegel JB, Mechanistically Informed Predictions of Binding Modes for Carbocation Intermediates of a Sesquiterpene Synthase Reaction. Chem. Sci 2016, 7, 4009–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Croteau RB; Shaskus JJ; Renstrom B; Felton NM; Cane DE; Saito A; Chang C, Mechanism of the Pyrophosphate Migration in the Enzymic Cyclization of Geranyl and Linalyl Pyrophosphates to (+)- and (−)-Bornyl Pyrophosphates. Biochemistry 1985, 24, 7077–7085. [DOI] [PubMed] [Google Scholar]
- 36.Major DT; Freud Y; Weitman M, Catalytic Control in Terpenoid Cyclases: Multiscale Modeling of Thermodynamic, Kinetic, and Dynamic Effects. Curr. Opin. Chem. Biol 2014, 21, 25–33. [DOI] [PubMed] [Google Scholar]
- 37.Abe I; Rohmer M; Prestwich GD, Enzymatic Cyclization of Squalene and Oxidosqualene to Sterols and Triterpenes. Chem. Rev 1993, 93, 2189–2206. [Google Scholar]
- 38.Stadler PA; Eschenmoser A; Schinz H; Stork G, Untersuchungen über den Sterischen Verlauf Säurekatalysierter Cyclisationen bei Terpenoiden Polyenverbindungen. 3. Mitteilung. Zur Stereochemie der Bicyclofarnesylsäuren. Helv. Chim. Acta 1957, 40, 2191–2198. [Google Scholar]
- 39.Major DT; Weitman M, Electrostatically Guided Dynamics—The Root of Fidelity in a Promiscuous Terpene Synthase? J. Am. Chem. Soc 2012, 134, 19454–19462. [DOI] [PubMed] [Google Scholar]
- 40.Dixit M; Weitman M; Gao J; Major DT, Comment on “Substrate Folding Modes in Trichodiene Synthase: A Determinant of Chemo- and Stereoselectivity”. ACS Catalysis 2017, 8, 1371–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croteau R; Karp F, Biosynthesis of Monoterpenes: Preliminary Characterization of Bornyl Pyrophosphate Synthetase from Sage (Salvia officinalis) and Demonstration that Geranyl Pyrophosphate is the Preferred Substrate for Cyclization. Arch. Biochem. Biophys 1979, 198, 512–522. [DOI] [PubMed] [Google Scholar]
- 42.Whittington DA; Wise ML; Urbansky M; Coates RM; Croteau RB; Christianson DW, Bornyl Diphosphate Synthase: Structure and Strategy for Carbocation Manipulation by a Terpenoid Cyclase. Proc. Natl. Acad. Sci 2002, 99, 15375–15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weitman M; Major DT, Challenges Posed to Bornyl Diphosphate Synthase: Diverging Reaction Mechanisms in Monoterpenes. J. Am. Chem. Soc 2010, 132, 6349–6360. [DOI] [PubMed] [Google Scholar]
- 44.Becke AD, Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. US 1993, 98, 5648–5652. [Google Scholar]
- 45.Becke AD, A New Mixing of Hartree–Fock and Local Density‐Functional Theories. J. Chem. Phys. US 1993, 98, 1372–1377. [Google Scholar]
- 46.Lee C; Yang W; Parr RG, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 47.Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Chem. Phys. US 1994, 98, 11623–11627. [Google Scholar]
- 48.Tirado-Rives J; Jorgensen WL, Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory and Comput 2008, 4, 297–306. [DOI] [PubMed] [Google Scholar]
- 49.Fukui K, The Path of Chemical Reactions - the IRC Approach. Accounts Chem. Res 1981, 14, 363–368. [Google Scholar]
- 50.Gonzalez C; Schlegel HB, Reaction Path Following in Mass-Weighted Internal Coordinates. J. Chem. Phys. US 1990, 94, 5523–5527. [Google Scholar]
- 51.Maeda S; Harabuchi Y; Ono Y; Taketsugu T; Morokuma K, Intrinsic Reaction Coordinate: Calculation, Bifurcation, and Automated Search. Int. J. Quantum Chem 2015, 115, 258–269. [Google Scholar]
- 52.Goto H; Osawa E, An Efficient Algorithm for Searching Low-Energy Conformers of Cyclic and Acyclic Molecules. J. Chem. Soc. Perk. T 2 1993, 0, 187–198. [Google Scholar]
- 53.Christianson DW, Structural and Chemical Biology of Terpenoid Cyclases. Chem.l Rev 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baer P; Rabe P; Fischer K; Citron CA; Klapschinski TA; Groll M; Dickschat JS, Induced-Fit Mechanism in Class 1 Terpene Cyclases. Angew. Chem. Int. Edit 2014, 53, 7652–7656. [DOI] [PubMed] [Google Scholar]
- 55.Pechous SW; Whitaker BD, Cloning and Functional Expression of an (E,E)-α-Farnesene Synthase cDNA from Peel Tissue of Apple Fruit. Planta 2004, 219, 84–94. [DOI] [PubMed] [Google Scholar]
- 56.Fu DY; Meiler J, The Predictive Power of Different Types of Experimental Restraints in Small Molecule Docking: A Review. J. Chem. Inf. Model 2017, 58, 225–233. [DOI] [PubMed] [Google Scholar]
- 57.Conway P; Tyka MD; DiMaio F; Konerding DE; Baker D, Relaxation of Backbone Bond Geometry Improves Protein Energy Landscape Modeling. Protein Sci 2014, 23, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman KW; Renfrew PD; Smith CA; Sheffler W; Davis IW; Cooper S; Treuille A; Mandell DJ; Richter F; Ban Y-EA; Fleishman SJ; Corn JE; Kim DE; Lyskov S; Berrondo M; Mentzer S; Popović Z; Havranek JJ; Karanicolas J; Das R; Meiler J; Kortemme T; Gray JJ; Kuhlman B; Baker D; Bradley P, Chapter nineteen - Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules In Method Enzymol, Michael LJ.; Ludwig B, Eds. Academic Press: USA; San Diego, CA, 2011; Vol. 487, pp 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meiler J; Baker D, ROSETTALIGAND: Protein–Small Molecule Docking with Full Side-Chain Flexibility. Proteins: Struct., Funct., Bioinf 2006, 65, 538–548. [DOI] [PubMed] [Google Scholar]
- 60.Richter F; Leaver-Fay A; Khare SD; Bjelic S; Baker D, De Novo Enzyme Design Using Rosetta3. PLoS ONE 2011, 6, e19230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wise ML; Savage TJ; Katahira E; Croteau R, Monoterpene Synthases from Common Sage (Salvia officinalis) : cDNA Isolation, Characterization, and Functional Expression of (+)-Sabinene Synthase, 1,8-Cineole Synthase, and (+)-Bornyl Diphosphate Synthase. J. Biol.l Chem 1998, 273, 14891–14899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.