

Abstract
Objective
Liquid biopsies are being rapidly used in adult cancers as new biomarkers of disease. Circulating tumor DNA (ctDNA) levels have been reported to be proportional to disease burden, correlate with treatment response, and predict relapse. However, little is known about how frequently ctDNA is detectable in pediatric patients with solid tumors. Therefore, we developed a next-generation sequencing approach to detect and quantify ctDNA in the blood of patients with the most common pediatric solid tumors.
Methods
Detection of ctDNA requires assays sensitive to somatic events typically observed in the cancer type being studied. In pediatric solid tumors, structural variants are more common than recurrent point mutations. We adapted an ultralow passage whole-genome sequencing approach to capture copy number variants and a hybrid capture sequencing assay to detect translocations in liquid biopsy samples from pediatric patients.
Results
Copy number changes seen by ultralow passage whole-genome sequencing enabled detection of ctDNA in patients with osteosarcoma, neuroblastoma, alveolar rhabdomyosarcoma, and Wilms tumor. In Ewing sarcoma, detection of the EWSR1 translocation was a more sensitive approach. For patients with samples collected at multiple time points, changes in ctDNA levels corresponded to treatment response. We also found that disease-specific genomic biomarkers of prognosis were detectable in ctDNA.
Conclusion
This study demonstrates that liquid biopsy approaches that detect somatic structural variants are well suited to pediatric solid tumors. We show that children with the most common solid tumor malignancies have detectable levels of ctDNA, which may be used to track disease response and identify genomic subclassifiers of disease. Efforts to profile larger collections of clinically annotated specimens are under way to validate the clinical use of these assays.
INTRODUCTION
Cancer remains one of the most common childhood causes of disease-related death in developed countries.1 Patients with pediatric solid tumor malignancies are commonly treated with regimens that combine intensive chemotherapy, surgery, and radiation therapy.2 This approach has led to tremendous improvements in outcome, with overall cure rates for pediatric solid tumors now > 80%.3 However, these cures come at the cost of exposing patients to a high risk of long-term toxicities resulting in significant morbidity.4 In some diseases, such as Wilms tumor, efforts to de-escalate therapy have resulted in stable cure rates for low-risk patients accompanied by reduced treatment toxicity.5,6 In other diseases, such as Ewing sarcoma, a lack of prognostic biomarkers limit the ability to risk stratify therapy.7,8 In both cases, the development of new and more precise assays for risk stratification could improve outcomes by facilitating patient-specific treatment modifications.
Advances in the development of liquid biopsy assays provide a new opportunity for prognostication in cancer. Numerous studies have now demonstrated that circulating tumor DNA (ctDNA) levels in adults with cancer correlate with disease burden and track with treatment responses over time.9-14 The development of ctDNA assays rely on identification and quantification of somatic mutations.9,15 Many ctDNA assays have been developed to detect highly recurrent hot-spot mutations that frequently drive adult malignancies.9,12,13,16-18 However, recent comprehensive sequencing efforts in pediatric cancers show that pediatric solid tumors are rarely driven by highly recurrent single-nucleotide variants.19 Instead, structural variants, including chromosomal copy number changes and DNA translocations, are common somatic events in these tumor types.20-26 To determine whether patients with childhood solid tumor malignancies express detectable levels of ctDNA, we adapted two next-generation sequencing strategies to detect and quantify structural variants in the plasma of these patients. We applied these assays to five of the most common types of pediatric solid tumors, excluding CNS tumors, to quantify ctDNA levels, identify genomic subtypes, and confirm the feasibility of using ctDNA assays to track disease response to therapy.
METHODS
Patients and Tissue Samples
Patients at the Dana-Farber Cancer Institute and Boston Children’s Hospital were consented to an institutional review board–approved protocol. Patients were included if an initial peripheral blood, pleural effusion, or bone marrow sample was collected within 3 days from the start of chemotherapy and before a surgical resection. A confirmed pathologic diagnosis of Ewing sarcoma, alveolar rhabdomyosarcoma, osteosarcoma, Wilms tumor, or neuroblastoma was required. Additional details are available in the Data Supplement.
Sequencing Library Preparation, Sequencing Alignment, and Ultralow Passage Whole-Genome Sequencing Coverage
For all experiments, sequencing library preparation and sequencing data alignment were performed using standard techniques. For ultralow passage whole-genome sequencing (ULP-WGS), whole-genome libraries were pooled and sequenced (without selection) to achieve an anticipated average coverage between 0.2× and 1×. Additional details are available in the Data Supplement.
Translocation-Specific Sarcoma Sequencing Assay Development and Sequencing
To detect pediatric sarcoma-specific translocations in cell-free DNA, we developed a unique hybrid capture sequencing assay. First, we reviewed the literature to identify the genomic introns involved in oncogenic translocations in the EWSR1, FUS, CIC, CCNB3, PAX3, PAX7, and TP53 genes.20,21,25-33 Second, we used the SureSelect Advanced Design Wizard (Agilent Technologies, Santa Clara, CA) to create capture probes targeting these regions and the coding regions of TP53 and STAG2 with the following design options: sense strand, 3× tiling density, least stringent masking, balanced boosting, and region extensions of 10 bases from the 3′ and 5′ ends (Data Supplement). The resulting hybrid capture bait set was named Translocation-Specific Sarcoma Sequencing assay (TranSS-Seq). Normalized and pooled barcoded sequencing libraries were enriched using the SureSelectXT Fast Target Enrichment System (Agilent Technologies) and the TranSS-Seq bait set. Postenrichment captures were sequenced with an intended coverage at target regions of 500×. The average measured coverage at enrichment sites for all samples tested by TranSS-Seq was 655× (range, 31× to 1,464×).
Sequencing Analysis
ULP-WGS analysis was performed using the ichorCNA algorithm (https://github.com/broadinstitute/ichorCNA), with manual curation of results.34 In brief, ULP-WGS uses relative sequencing coverage of whole-genome data and computational correction of sequencing bias to detect segmental copy number changes. Percentage of ctDNA was determined using established probabilistic modeling algorithms for estimating tumor allelic fractions.
Identification of targeted translocations by TranSS-Seq was performed using BreaKmer.35 To quantify the number of translocations and wild-type reads detected in each sample, we developed a custom algorithm designed to realign all sequencing reads to either the reference human genome or the patient-specific translocation-positive reference sequence (https://github.com/vanallenlab/peds_ctDNA). The algorithm then reported the number of reads aligned at the patient-specific translocation breakpoint and the number of wild-type reads at the equivalent genomic base-pair location within the human reference genome. Because each cancer genome contains one translocated and one wild-type allele, whereas germline genomes contain two wild-type alleles, we used the following formula to calculate the percentage of ctDNA, where T equals the number of translocation reads and W equals the number of wild-type reads: % ctDNA = T/{[(W-T)/2]+T}.
Cell Lines and Digital Droplet Polymerase Chain Reaction
The EW8 Ewing sarcoma cell line dilution experiments, digital droplet polymerase chain reaction (ddPCR) methods, and ddPCR primers are listed in the Data Supplement.
RESULTS
Patients and Samples
A liquid biopsy sample was collected before the initiation of therapy from 45 pediatric patients with cancer with a confirmed diagnosis of Ewing sarcoma (n = 11), osteosarcoma (n = 10), neuroblastoma (n = 10), Wilms tumor (n = 8), or alveolar rhabdomyosarcoma (n = 7). Clinical characteristics were collected for each enrolled patient (Data Supplement). DNA from a tumor biopsy sample was available for eight of 11 Ewing sarcomas, four of 10 osteosarcomas, and six of seven rhabdomyosarcomas. DNA from tumor biopsy material from patients with neuroblastoma and Wilms tumor were not readily available.
Circulating Tumor DNA Is Detectable in Patients With Pediatric Solid Tumor Malignancies
Total cell-free DNA levels ranged from 2 ng to 3.5 µg of DNA per 1 mL of plasma, with the highest values originating from patients with neuroblastoma (Fig 1A). ULP-WGS was performed for each sample to detect and quantify ctDNA by measuring segmental copy number changes as recently described.34 ctDNA was detected in ≥ 50% of pretreatment plasma samples from patients with osteosarcoma, neuroblastoma, Wilms tumor, and alveolar rhabdomyosarcoma. ctDNA was detected in only four of 11 samples from patients with Ewing sarcoma (Fig 1B; Data Supplement). ULP-WGS was performed for all patients with available DNA from tumor biopsies (n = 18). In patients with detectable ctDNA, the pattern of segmental chromosomal copy number changes in cell-free DNA matched the copy number pattern observed in the tumor biopsy from the same patient (Figs 1C and 1D; Data Supplement). In four of the seven patients with Ewing sarcoma with no detectable ctDNA, ULP-WGS analysis of tumor biopsy DNA material demonstrated no discernable segmental copy number changes (Fig 2). This finding is consistent with previous reports that Ewing sarcoma tumors have few highly recurrent somatic events other than the EWSR1/ETS translocations and suggests that an alternative method for detection of ctDNA is necessary for this disease.21,26
Fig 1.
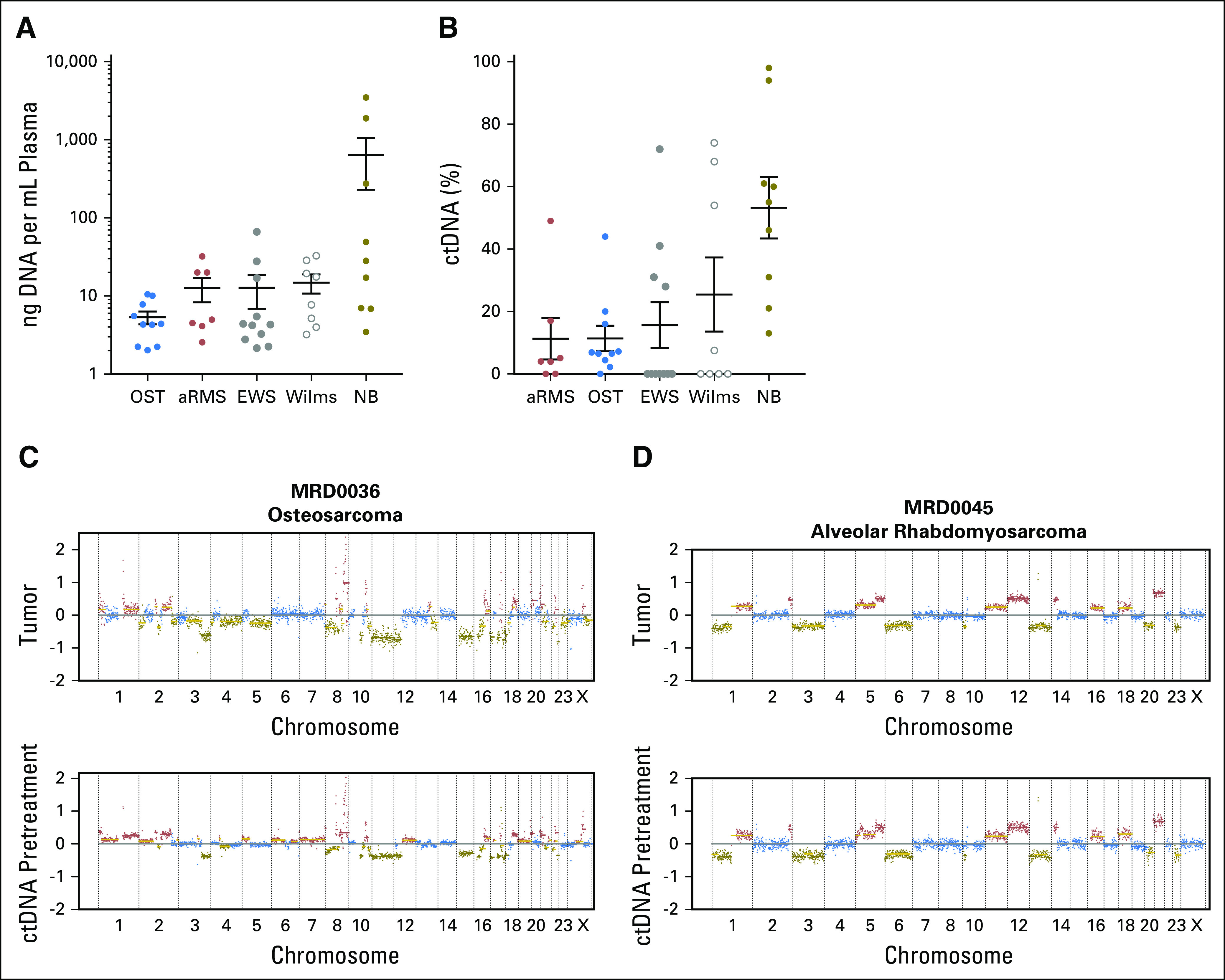
Circulating tumor DNA (ctDNA) is detectable in pediatric solid tumors. (A) Quantity of cell-free DNA extracted per mL of plasma or body fluid in patients with osteosarcoma (OST), alveolar rhabdomyosarcoma (aRMS), Ewing sarcoma (EWS), Wilms tumor (Wilms), and neuroblastoma (NB). (B) Percentage of ctDNA levels in cell-free DNA samples from panel A as determined by ultralow passage whole-genome sequencing. (C-D) Genome-wide plots represent the log2 ratio copy number for each data point determined by ultralow passage whole-genome sequencing. The color for each data point corresponds to the relative copy number change from baseline, with blue equal to two copies of the genomic location, green equal to copy number loss, and red equal to copy number gains. Chromosomal segmental medians are also plotted as horizontal lines, with gold lines representing likely subclonal events. The same pattern of copy number changes is observed in DNA sequenced from a tumor biopsy (top) and a pretreatment blood sample (bottom) in (C) a patient with osteosarcoma and (D) alveolar rhabdomyosarcoma.
Fig 2.
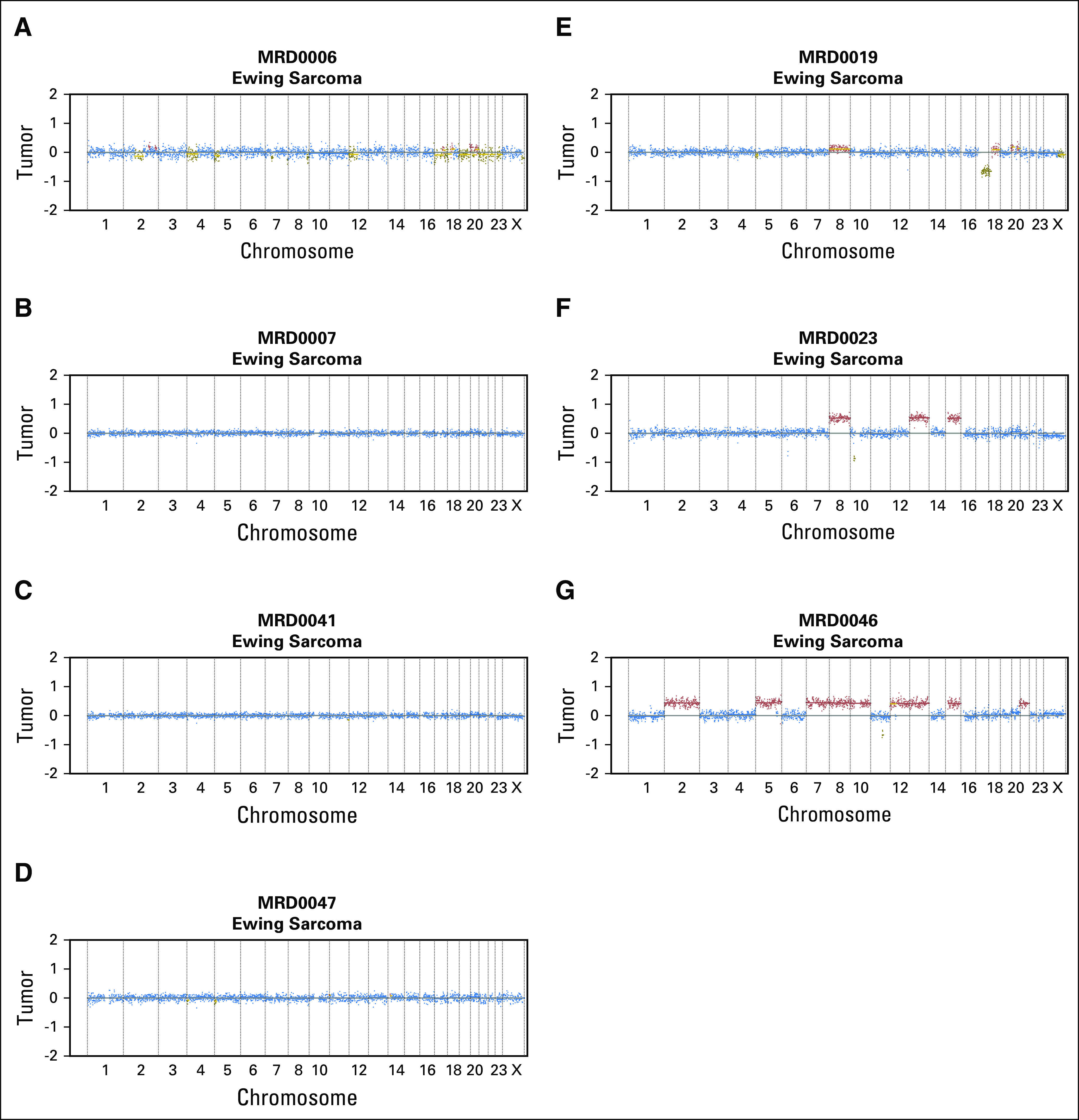
Ewing sarcoma tumors have few segmental copy number changes. Genome-wide copy number plots from seven Ewing sarcoma tumor biopsy samples detected by ultralow passage whole-genome sequencing in patients for which circulating tumor DNA could not be detected from peripheral blood. (A-D) Four Ewing sarcoma tumors that show no discernable segmental copy number changes. (E-G) Three Ewing sarcoma tumors with segmental copy number changes.
Translocation Detection Outperforms Copy Number Detection for Ewing Sarcoma
To detect ctDNA in translocation-positive tumors, we developed a novel hybrid-capture assay, termed TranSS-Seq, designed to sequence intronic regions involved in genomic rearrangements in pediatric sarcomas. Coding regions of the TP53 and STAG2 genes were also targeted by this assay (Table 1; Data Supplement). First, we confirmed that the assay could detect the expected translocations in a panel of Ewing and Ewing-like sarcomas and alveolar rhabdomyosarcomas (Data Supplement). Second, DNA was extracted from the EW8 Ewing sarcoma cell line and mixed in decreasing concentrations with normal human cell line CEPH-1347-2. ddPCR in these samples confirmed the ability to detect and quantify the EW8-specific EWSR1/FLI translocation across the range of dilutions (Fig 3A; Data Supplement). Sequencing of the same DNA libraries with TranSS-Seq demonstrated a high correlation for this method with both the experimental dilution and ddPCR results and detected EW8 DNA at a concentration of 1.56% (Figs 3B and 3C; Data Supplement).
Table 1.
Gene Regions Targeted by Hybrid Capture Sequencing in the TranSS-Seq Assay

Fig 3.
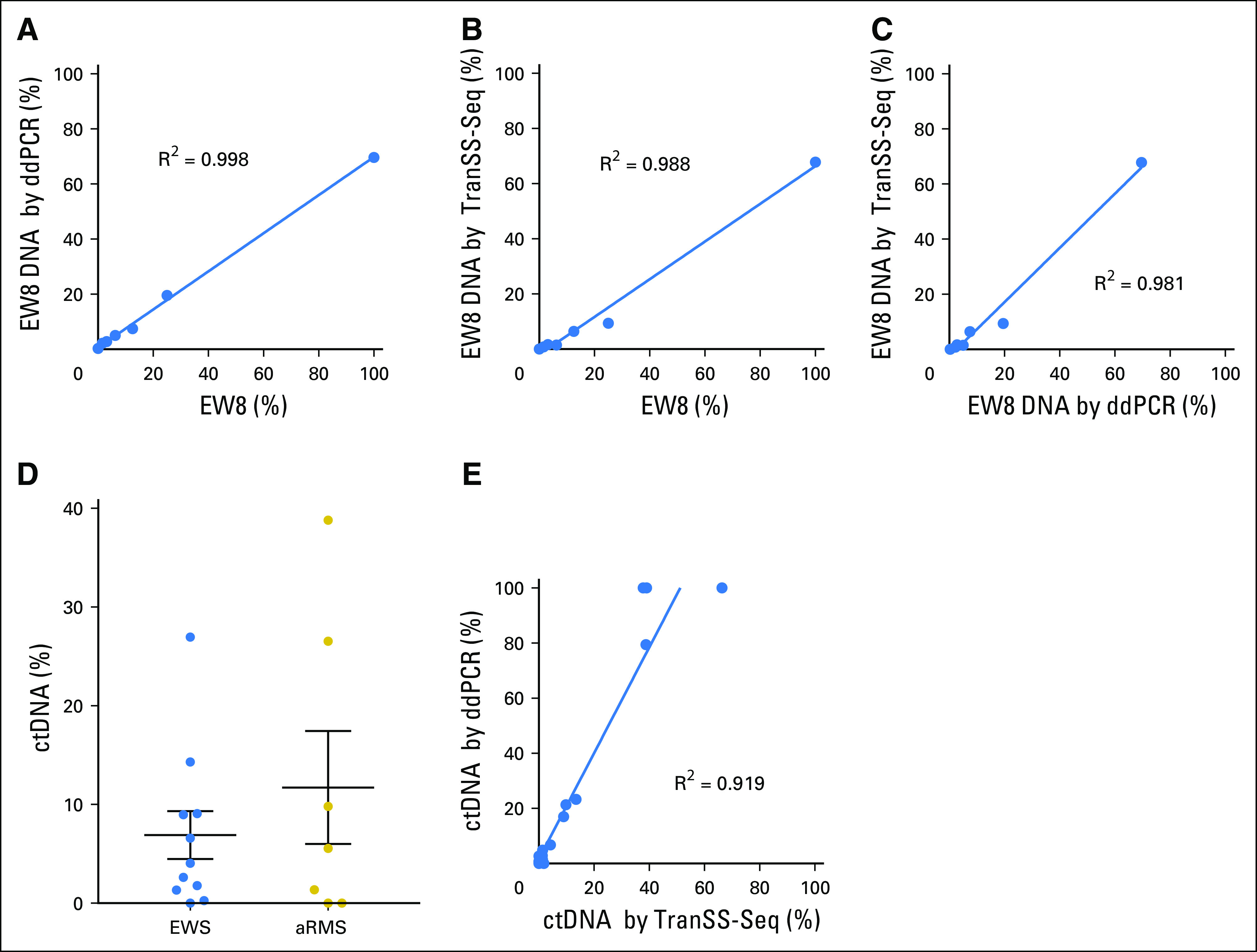
The Translocation-Specific Sarcoma Sequencing assay (TranSS-Seq) detects circulating tumor DNA (ctDNA) in Ewing sarcoma (EWS) and alveolar rhabdomyosarcoma (aRMS). (A) DNA extracted from the EW8 cell line was serially diluted with normal DNA extracted from CEPH1347-2. Sequencing libraries were made in duplicate and sequenced with TranSS-Seq to detect the EWSR1/FLI translocation. Samples are plotted on the x-axis by the percentage of EW8 by experimental dilution and on the y-axis by the percentage of EW8 DNA detected by digital droplet polymerase chain reaction (ddPCR). The line drawn on the plot is a linear regression curve of the data (slope, 0.66). (B) Percentage of EW8 DNA was determined from the same samples as in panel A by TranSS-Seq and plotted on the y-axis (slope, 0.6). (C) The same samples as in panels A and B with percentage of EW8 DNA by TranSS-Seq on the x-axis and ddPCR on the y-axis (slope, 0.83). (D) Percentage of ctDNA levels in cell-free DNA samples from patients with Ewing sarcoma and aRMS (same samples as in Figure 1B) determined by TranSS-Seq. (E) Comparison of percentage of ctDNA detected in Ewing sarcoma and alveolar rhabdomyosarcoma samples determined by TranSS-Seq on the x-axis and ddPCR on the y-axis (slope, 1.63).
TranSS-Seq was then applied to cell-free DNA samples from patients with Ewing sarcoma and alveolar rhabdomyosarcoma. ctDNA was detected by this method in 10 of 11 Ewing sarcoma samples and five of seven alveolar rhabdomyosarcoma samples (Fig 3D). For five patients with Ewing sarcoma and three patients with alveolar rhabdomyosarcoma, DNA from tumor biopsy samples was also profiled by TranSS-Seq (Data Supplement). In all cases, the unique genomic breakpoint in the ctDNA was identical to the breakpoint observed from the tumor biopsy sample.
Previous studies in Ewing sarcoma have used ddPCR assays that amplify patient-specific EWS/ETS translocations to measure ctDNA levels in patients.36-38 To compare TranSS-Seq with this previously validated approach, patient-specific polymerase chain reaction primers were developed for a subset of Ewing sarcoma and alveolar rhabdomyosarcoma samples (Data Supplement). The percentage of ctDNA levels identified by TranSS-Seq correlated with the percentage of ctDNA detected by ddPCR (Fig 3E). In three of the four patients with Ewing sarcoma for which no copy number change could be detected by ULP-WGS in the tumor (MRD0006, MRD0041, MRD0047), TranSS-Seq was able to detect an EWSR1-fusion in the tumor and cell-free DNA (Data Supplement). In the fourth patient (MRD0007), TranSS-Seq detected an EWSR1/FLI fusion in the tumor but not in cell-free DNA (Data Supplement). ddPCR could not be attempted on this plasma sample because there was no remaining cell-free DNA after sequencing. Finally, in all three patients with Ewing sarcoma for which the tumor biopsy specimen detected a segmental copy number change but ULP-WGS was unable to detect ctDNA from the same patient (MRD0019, MRD0023, MRD0046), TranSS-Seq detected low levels of ctDNA, suggesting that TranSS-Seq may have greater sensitivity for ctDNA than ULP-WGS in Ewing sarcoma (Data Supplement).
ctDNA Levels Track With Disease Burden in Pediatric Solid Tumors
When comparing ctDNA levels across cancer types, neuroblastoma demonstrated a significantly higher percentage of ctDNA (median, 55%; range, 13% to 98%) than other cancer types (Data Supplement). This may reflect the high disease burden typically present in patients with metastatic neuroblastoma. For a subset of patients, liquid biopsy samples were collected at multiple time points during clinical care (Data Supplement). ULP-WGS was applied to all serial samples, and TranSS-Seq was applied to Ewing sarcoma and alveolar rhabdomyosarcoma samples. Changes in ctDNA levels over time corresponded to response to therapy (Fig 4; Data Supplement). In patients with newly diagnosed Ewing sarcoma and alveolar rhabdomyosarcoma, ctDNA levels declined rapidly after initiation of chemotherapy, often becoming undetectable by the start of the second cycle (Figs 4A and 4B; Data Supplement). In osteosarcoma, studies have shown that a high percentage of tumor necrosis observed in the primary tumor after neoadjuvant chemotherapy is associated with a better prognosis.39,40 In five patients with newly diagnosed osteosarcoma, the percentage of tumor necrosis was available. Interestingly, for one patient with tumor necrosis of 90% (MRD0061) and one patient with 80% (MRD0053), ctDNA levels were undetectable after initiation of chemotherapy (Fig 4C; Data Supplement). However, four patients with tumor necrosis < 70% (MRD0031, MRD0036, MRD0040, MRD0054) had detectable ctDNA in at least one sample collected after initiation of chemotherapy (Fig 4D; Data Supplement). Finally, in patients whose ctDNA levels became undetectable with therapy but then experienced a clinical relapse or progression, ctDNA was again detectable before the initiation of relapsed therapy (Fig 4B; Data Supplement).
Fig 4.

Changes in circulating tumor DNA (ctDNA) levels over time in patients with pediatric solid tumors. Percentage of ctDNA levels from serial cell-free DNA samples collected from four patients with (A) Ewing sarcoma, (B) alveolar rhabdomyosarcoma, (C-D) osteosarcoma. Background shading indicates the relevant treatment course and clinical events (x-axis scale is arbitrary). Blue dots indicate peripheral blood samples, and the red dot (B) indicates a bone marrow sample collected simultaneously with a blood sample.
Genomic Markers of Poor Outcome Are Detectable in ctDNA
We also examined whether disease-specific genomic markers of prognosis could be detected in ctDNA. Recent studies demonstrate that mutations in STAG2 and TP53 may be associated with a worse outcome in Ewing sarcoma.21,26 A frame-shift mutation in STAG2 was detected by ctDNA from one patient (MRD0023), and a mutation in TP53 was detected in another patient (MRD0003; Data Supplement). In alveolar rhabdomyosarcoma, studies have demonstrated that patients with PAX3/FOXO1 have a worse prognosis than patients with PAX7/FOXO1 translocation.41 The FOXO1 fluorescent in situ hybridization probe, which is used in the diagnostic work-up of alveolar rhabdomyosarcoma, confirms a FOXO1 rearrangement but not the fusion partner. The TranSS-Seq assay distinguishes PAX3 from PAX7 translocations (Table 1). In our study, all patients with ctDNA were found to have a PAX3/FOXO1 translocation (Data Supplement). In osteosarcoma, copy number gains of chromosome arm 8q are associated with a poor outcome.42-44 ULP-WGS detected copy number gains in 8q in seven of nine osteosarcoma samples with detectable ctDNA (Fig 1C; Data Supplement). Amplification of MYCN is a well-established marker of poor prognosis in neuroblastoma.45,46 MYCN amplification was detectable in the ctDNA of two patients by ULP-WGS (Fig 5A; Data Supplement). These were the only two patients with neuroblastoma with fluorescent in situ hybridization–confirmed MYCN amplification in our cohort. Finally, in Wilms tumor, copy number gains of 1q were associated with poor prognosis in favorable histology tumors.47 ULP-WGS detected 1q gain in two of four Wilms tumor samples with detectable ctDNA (Figs 5B and 5C; Data Supplement).
Fig 5.
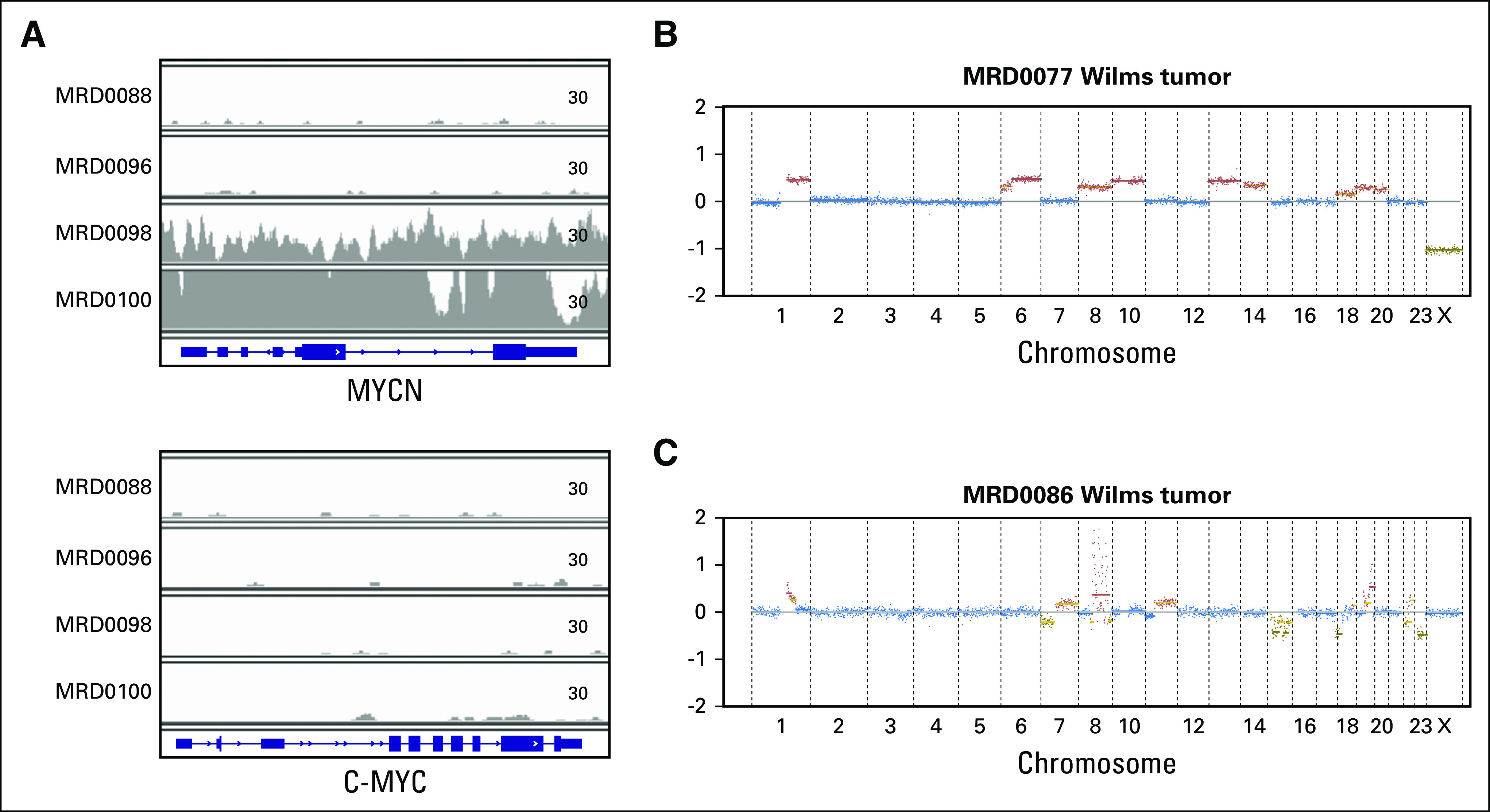
Genomic hallmarks of high-risk disease are detectable in circulating tumor DNA. (A) Sequencing coverage (read counts) across the MYCN gene (top) and c-MYC gene (bottom) from four cell-free DNA samples from patients with neuroblastoma. MRD0098 and MRD0100 correspond to patients with known MYCN amplification. (B-C) Genome-wide copy number plots of circulating tumor DNA detected by ultralow passage whole-genome sequencing from two patients with Wilms tumor. Plots show evidence of 1q copy number gain.
DISCUSSION
Liquid biopsy technologies are currently being adapted for prognostic purposes in many adult malignancies.11,48-50 It is critical that liquid biopsy strategies are also developed for childhood cancer. Because of the relative rarity of pediatric solid tumors, large prospective trials frequently rely on multi-institutional consortiums to reach adequate patient enrollment. In this setting, availability of tumor biopsy samples for correlative biology studies is inconsistent. In addition, it is preferable to have a liquid biopsy approach that can provide information about disease burden and prognosis before availability of molecular results from tumor. Therefore, we adapted liquid biopsy methods that could detect somatic structural variants without requiring access to tumor biopsy samples. In this article, we report our results from profiling cell-free DNA samples from 45 patients diagnosed with a wide range of pediatric solid tumors.
We first applied ULP-WGS to detect copy number changes in cell-free DNA and found that the majority of patients with pediatric solid tumors had detectable levels of ctDNA. Patients with neuroblastoma had the highest levels of ctDNA, including two patients with > 1 µg of cell-free DNA per mL of plasma, which was nearly 100% composed of tumor DNA. However, one obvious limitation to the use of ULP-WGS was in tumor types with low rates of copy number alterations, such as Ewing sarcoma.
Recent liquid biopsy studies in Ewing sarcoma have used the development of patient-specific ddPCR assays.36-38 Although this approach has proven to be highly sensitive and quantitative, the development of each assay requires genomic profiling of large amounts of tumor biopsy material to establish the patient-specific oncogenic translocation. One recent study used a combination of ddPCR and hybrid capture sequencing to detect EWSR1 translocations in Ewing sarcoma and desmoplastic small round-cell tumors.38 Results showed that it is feasible to detect EWSR1 translocations directly from the plasma of these patients without first sequencing the tumor sample. In our study, we developed a unique hybrid capture sequencing assay that targets a wider range of oncogenic translocations observed in pediatric sarcomas. Despite the larger genomic region targeted for hybrid capture, the TranSS-Seq assay detected ctDNA in 10 of 11 pretreatment samples obtained from Ewing sarcoma, similar to rates observed by other groups. Serial dilution studies demonstrated that our assay was sufficiently sensitive to detect oncogenic translocations diluted in germline DNA to as little as 1.56%. We also demonstrated that quantification of ctDNA levels was similar using either TranSS-Seq or ddPCR.
In this study, changes in ctDNA levels were observed during a patient’s clinical course and corresponded to changes in disease burden. This suggests that next-generation sequencing approaches could be used as a measure of disease burden in patients before the start of therapy and to track treatment response. Monitoring changes in ctDNA levels during therapy may facilitate new response-based risk-stratification approaches that allow refinement of treatment intensity for patients undergoing therapy. With next-generation sequencing becoming increasingly available for clinical decision making, we anticipate that our ctDNA assays could also be adapted to clinical laboratory testing if they demonstrate significant improvements to risk stratification for pediatric solid tumors. A similar approach to risk-stratified treatment by minimal residual disease testing in children with acute lymphoblastic leukemia has led to significant improvements in outcome.51 We believe that this study demonstrates the feasibility of measuring and tracking ctDNA across a wide range of pediatric solid tumors and justifies prospective efforts within each disease type to validate the use of ctDNA in prognostication. Importantly, our study was not optimized to determine which time points during a patient’s treatment course may be the most useful in measuring response to therapy. Therefore, prospective studies should be designed to obtain frequent samples throughout treatment so that the time points most predictive of outcome can be appropriately identified.
Finally, our study demonstrates that next-generation sequencing of ctDNA can detect existing genomic biomarkers of outcome and may provide another modality for genomic profiling of tumors in patients. Furthermore, it remains unclear whether these genomic biomarkers are primarily clonal or heterogeneous events within a tumor or across tumors and whether the clonality of these genomic events changes in patients who relapse after therapy. Recent studies demonstrate that ctDNA can detect intratumor and multitumor heterogeneity and detect complex patterns of treatment resistance.52-55 With the emergence of new sequencing modifications that improve sensitivity and decrease sequencing errors, we believe that ctDNA profiling by next-generation sequencing approaches will improve our understanding of tumor heterogeneity and patterns of somatic evolution in pediatric solid tumors.12,16 In addition, the assays described in this study could facilitate broader profiling of ctDNA, such as deep whole-exome sequencing, by providing a mechanism to screen samples for the presence of sufficiently abundant ctDNA, allowing selection of samples most likely to yield informative data.
In summary, our study demonstrates that patients with pediatric solid tumors have detectable levels of ctDNA, which can be measured with next-generation sequencing approaches that do not require profiling of tumor biopsy material or patient-specific assays. Disease-specific genomic hallmarks of pediatric solid tumors can also be identified in liquid biopsy samples, and ctDNA levels correlate with disease burden and response to therapy over time. Larger studies are needed to confirm the prognostic value of ctDNA detection and quantification in these patients. As more sensitive and less error-prone sequencing technologies become available, we believe that ctDNA studies may provide insight into tumor evolution and treatment resistance in pediatric solid tumors.
ACKNOWLEDGMENT
We thank the Boston Children’s Hospital Biorepository for processing a portion of the blood samples. We are also grateful to the patients who participated in this study.
Footnotes
Supported by Brielle’s Brigade Fund (K.A.J.); National Institutes of Health Grant No. K08 CA188073-01A1 (B.D.C.); Children’s Oncology Group Translational Pilot Studies Program for Solid Malignancies (B.D.C.); Boston Children’s Hospital Translational Research Program (B.D.C.); Pediatric Cancer Research Foundation (B.D.C.); Go 4 The Goal Foundation (B.D.C.); Fernando’s Fight (B.D.C.); Nate Cavallo Fund (B.D.C.).
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other funding agencies.
AUTHOR CONTRIBUTIONS
Conception and design: Kelly Klega, Catherine Clinton, Katherine Janeway, Matthew Meyerson, Brian D. Crompton
Financial support: Katherine Janeway, Brian D. Crompton
Administrative support: Catherine Clinton, Katherine Janeway
Provision of study materials or patients: Katherine Janeway, Brian D. Crompton
Collection and assembly of data: Andrea N. Clapp, Stephanie Meyer, Abigail Ward, Katherine Janeway, Brian D. Crompton
Data analysis and interpretation: Kelly Klega, Alma Imamovic-Tuco, Gavin Ha, Anwesha Nag, Eliezer Van Allen, Elizabeth Mullen, Steven G. DuBois, Aaron R. Thorner, Brian D. Crompton
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Kelly Klega
No relationship to disclose
Alma Imamovic-Tuco
No relationship to disclose
Gavin Ha
No relationship to disclose
Andrea N. Clapp
No relationship to disclose
Stephanie Meyer
No relationship to disclose
Abigail Ward
No relationship to disclose
Catherine Clinton
No relationship to disclose
Anwesha Nag
No relationship to disclose
Eliezer Van Allen
Stock and Other Ownership Interests: Syapse, Tango Therapeutics, Genome Medical
Consulting or Advisory Role: Syapse, Roche, Third Rock Ventures, Takeda, Novartis, Genome Medical, InVitae
Speakers' Bureau: Illumina
Research Funding: Bristol-Myers Squibb, Novartis
Elizabeth Mullen
No relationship to disclose
Steven G. DuBois
Honoraria: Loxo
Consulting or Advisory Role: Loxo
Research Funding: Millennium (Inst), Merck (Inst), Novartis (Inst), Roche/Genentech (Inst), Eli Lilly (Inst), Curis (Inst), Loxo (Inst)
Travel, Accommodations, Expenses: Loxo, Roche/Genentech
Katherine Janeway
No relationship to disclose
Matthew Meyerson
Stock and Other Ownership Interests: OrigiMed
Consulting or Advisory Role: OrigiMed
Research Funding: Bayer (Inst)
Patents, Royalties, Other Intellectual Property: Patent on EGFR mutations for lung cancer diagnosis
Aaron R. Thorner
No relationship to disclose
Brian D. Crompton
Employment: Mersana (I), Shire (I)
Stock and Other Ownership Interests: Mersana (I), Shire (I)
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Green DM, Kun LE, Matthay KK, et al. Relevance of historical therapeutic approaches to the contemporary treatment of pediatric solid tumors. Pediatr Blood Cancer. 2013;60:1083–1094. doi: 10.1002/pbc.24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlader N NA, Krapcho M, Miller D, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute; Bethesda, MD: 2016. [Google Scholar]
- 4.Bhakta N, Liu Q, Ness KK, et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE) Lancet. 2017;390:2569–2582. doi: 10.1016/S0140-6736(17)31610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez CV, Perlman EJ, Mullen EA, et al. Clinical outcome and biological predictors of relapse after nephrectomy only for very low-risk Wilms tumor: A report from Children’s Oncology Group AREN0532. Ann Surg. 2017;265:835–840. doi: 10.1097/SLA.0000000000001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms’ tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1989;64:349–360. doi: 10.1002/1097-0142(19890715)64:2<349::aid-cncr2820640202>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 7.Shukla N. Ewing sarcoma: A tough road to clinically relevant biomarkers. Pediatr Blood Cancer. 2015;62:741–742. doi: 10.1002/pbc.25406. [DOI] [PubMed] [Google Scholar]
- 8.Shukla N, Schiffman J, Reed D, et al. Biomarkers in Ewing sarcoma: The promise and challenge of personalized medicine. A report from the Children’s Oncology Group. Front Oncol. 2013;3:141. doi: 10.3389/fonc.2013.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 11.Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015;6:42008–42018. doi: 10.18632/oncotarget.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698–1705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkinson CA, Gale D, Piskorz AM, et al. Exploratory analysis of TP53 mutations in circulating tumour DNA as biomarkers of treatment response for patients with relapsed high-grade serous ovarian carcinoma: A retrospective study. PLoS Med. 2016;13:e1002198. doi: 10.1371/journal.pmed.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 16.Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016;2:1014–1022. doi: 10.1001/jamaoncol.2016.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yung TK, Chan KC, Mok TS, et al. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009;15:2076–2084. doi: 10.1158/1078-0432.CCR-08-2622. [DOI] [PubMed] [Google Scholar]
- 19.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Bahrami A, Pappo A, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Reports. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4:1326–1341. doi: 10.1158/2159-8290.CD-13-1037. [DOI] [PubMed] [Google Scholar]
- 22.Gadd S, Huff V, Walz AL, et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet. 2017;49:1487–1494. doi: 10.1038/ng.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry JA, Kiezun A, Tonzi P, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci USA. 2014;111:E5564–E5573. doi: 10.1073/pnas.1419260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4:1342–1353. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15:2125–2137. doi: 10.1093/hmg/ddl136. [DOI] [PubMed] [Google Scholar]
- 28.Ng TL, O’Sullivan MJ, Pallen CJ, et al. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn. 2007;9:459–463. doi: 10.2353/jmoldx.2007.070009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters TL, Kumar V, Polikepahad S, et al. BCOR-CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod Pathol. 2015;28:575–586. doi: 10.1038/modpathol.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–466. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- 31.Shing DC, McMullan DJ, Roberts P, et al. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res. 2003;63:4568–4576. [PubMed] [Google Scholar]
- 32.Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: A genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol. 2014;38:1571–1576. doi: 10.1097/PAS.0000000000000286. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimoto M, Graham C, Chilton-MacNeill S, et al. Detailed cytogenetic and array analysis of pediatric primitive sarcomas reveals a recurrent CIC-DUX4 fusion gene event. Cancer Genet Cytogenet. 2009;195:1–11. doi: 10.1016/j.cancergencyto.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 34.Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8:1324. doi: 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abo RP, Ducar M, Garcia EP, et al. BreaKmer: Detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 2015;43:e19. doi: 10.1093/nar/gku1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayashi M, Chu D, Meyer CF, et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer. 2016;122:3015–3023. doi: 10.1002/cncr.30144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krumbholz M, Hellberg J, Steif B, et al. Genomic EWSR1 fusion sequence as highly sensitive and dynamic plasma tumor marker in Ewing sarcoma. Clin Cancer Res. 2016;22:4356–4365. doi: 10.1158/1078-0432.CCR-15-3028. [DOI] [PubMed] [Google Scholar]
- 38.Shukla NN, Patel JA, Magnan H, et al. Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO Precis Oncol. doi: 10.1200/PO.16.00028. 10.1200/PO.16.00028 [epub ahead of print on May 23, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyers PA, Heller G, Healey J, et al. Chemotherapy for nonmetastatic osteogenic sarcoma: The Memorial Sloan-Kettering experience. J Clin Oncol. 1992;10:5–15. doi: 10.1200/JCO.1992.10.1.5. [DOI] [PubMed] [Google Scholar]
- 40.Meyers PA, Schwartz CL, Krailo M, et al. Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol. 2005;23:2004–2011. doi: 10.1200/JCO.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 41.Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the Children’s Oncology Group. J Clin Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 42.Ozaki T, Schaefer KL, Wai D, et al. Genetic imbalances revealed by comparative genomic hybridization in osteosarcomas. Int J Cancer. 2002;102:355–365. doi: 10.1002/ijc.10709. [DOI] [PubMed] [Google Scholar]
- 43.Smida J, Baumhoer D, Rosemann M, et al. Genomic alterations and allelic imbalances are strong prognostic predictors in osteosarcoma. Clin Cancer Res. 2010;16:4256–4267. doi: 10.1158/1078-0432.CCR-10-0284. [DOI] [PubMed] [Google Scholar]
- 44.Tarkkanen M, Elomaa I, Blomqvist C, et al. DNA sequence copy number increase at 8q: A potential new prognostic marker in high-grade osteosarcoma. Int J Cancer. 1999;84:114–121. doi: 10.1002/(sici)1097-0215(19990420)84:2<114::aid-ijc4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 45.Brodeur GM, Seeger RC, Schwab M, et al. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 46.Seeger RC, Brodeur GM, Sather H, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313:1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- 47.Gratias EJ, Dome JS, Jennings LJ, et al. Association of chromosome 1q gain with inferior survival in favorable-histology Wilms tumor: A report from the Children’s Oncology Group. J Clin Oncol. 2016;34:3189–3194. doi: 10.1200/JCO.2015.66.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 49.Lecomte T, Berger A, Zinzindohoué F, et al. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer. 2002;100:542–548. doi: 10.1002/ijc.10526. [DOI] [PubMed] [Google Scholar]
- 50.Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vora A, Goulden N, Mitchell C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2014;15:809–818. doi: 10.1016/S1470-2045(14)70243-8. [DOI] [PubMed] [Google Scholar]
- 52.De Mattos-Arruda L, Weigelt B, Cortes J, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann Oncol. 2014;25:1729–1735. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murtaza M, Dawson SJ, Pogrebniak K, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760. doi: 10.1038/ncomms9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]