

Abstract
SF3b1 is an essential component of the U2 snRNP implicated in branch site (BS) recognition and found to be frequently mutated in several human cancers. While recent structures of yeast and human SF3b1 have revealed its molecular architecture, the importance of specific RNA:protein contacts and conformational changes remains largely uncharacterized. Here, we performed mutational analysis of yeast SF3b1, guided by recent structures of the spliceosome. We find that conserved amino acids contacting the U2 snRNA backbone of the U2/BS duplex are nonessential, and that yeast can tolerate truncation of the HEAT repeats containing these amino acids. The pocket housing the branchpoint adenosine (BP-A) is also amenable to mutation despite strong conservation. However, mutations that support viability can still lead to defects in splicing pre-mRNAs with nonconsensus BS substitutions found at −3, −2, −1, and +1 positions relative to the BP-A or at the branchpoint position. Through the generation of yeast and human chimeric proteins, we further defined the functionally conserved regions of Hsh155 as well as identify changes in BS usage resulting from inclusion of human SF3b1 HEAT repeats. Moreover, these chimeric proteins confer a sensitivity to small molecule inhibition by pladienolide B to yeast splicing. Together, these data reveal the importance of individual contacts of Hsh155/SF3b1 to the U2/BS duplex and define their contribution to BS usage by the spliceosome.
Keywords: pladienolide B, spliceosome, snRNP, SF3b1, splicing
INTRODUCTION
Pre-mRNA splicing is an essential process in eukaryotic gene expression carried out by the spliceosome. The splicing machinery is composed of five small nuclear ribonucleoproteins (the U1, U2, U4, U5, and U6 snRNPs) and additional protein-only cofactors that assemble de novo on pre-mRNAs to catalyze intron removal and exon ligation (Wahl et al. 2009; Hoskins and Moore 2012). Spliceosome assembly occurs in a stepwise fashion from preassembled snRNP particles on nascent RNA during transcription to define the intron 5′ and 3′ splice sites (5′SS and 3′SS) and the branch site (BS) sequence used in catalysis (Hoskins and Moore 2012; Herzel et al. 2017). The 5′SS, 3′SS, and BS sequences are well conserved in yeast but variable in humans. For example, very few yeast introns deviate from the yeast consensus BS sequence (UACUAAC, branch point adenosine [BP-A] underlined), while human spliceosomes can use highly divergent BS sequences and have a more degenerate consensus sequence (CURAY) (Harris and Senapathy 1990; Mercer et al. 2015; Gould et al. 2016; Qin et al. 2016). However, in both yeast and humans the BS is recognized by base-pairing with the U2 snRNA GUAGUA sequence to form the U2/BS duplex containing a bulged adenosine that acts as the nucleophile in the first step of splicing (Query et al. 1994; Wahl et al. 2009).
Spliceosome assembly is a dynamic process involving the association and disassociation of numerous factors to ensure accurate and efficient intron removal (Will and Lührmann 2011; Chen and Moore 2014). During early stages of assembly, addition of the U1 and U2 snRNPs to the 5′SS and BS, respectively, mark the formation of the spliceosome A complex. The U4/U6.U5 tri-snRNP is then recruited to form the B complex spliceosome. Activation of the spliceosome converts B complex to Bact, wherein U1 and U4 have been ejected from the spliceosome and the protein-only, Prp19-containing complex (NTC) has joined. A number of rearrangements occur within the active site of the spliceosome to then generate a complex competent for catalysis (Brow 2002). Following exon ligation, the mRNA is released and the spliceosome disassembles to permit recycling of individual components for the next round of splicing (Wahl et al. 2009).
Defects in pre-mRNA splicing are implicated in a variety of cancers including myelodysplastic syndromes (MDS), chronic lymphocytic leukemia, breast cancer, and uveal melanoma (Dvinge et al. 2016). The splicing factor most frequently mutated in these diseases is the conserved U2 snRNP component, SF3b1 [Hsh155 in Saccharomyces cerevisiae (yeast)] (Dvinge et al. 2016; Agrawal et al. 2017; Jenkins and Kielkopf 2017). Mutation of SF3b1 results in activation of cryptic 3′SS and altered BS usage in mammalian cells (Darman et al. 2015; Alsafadi et al. 2016). MDS-associated mutations also change BS usage in yeast, as mutation of Hsh155 can either increase or decrease nonconsensus BS usage by the spliceosome (Tang et al. 2016; Carrocci et al. 2017). This suggests that the role and mechanism of SF3b1 at the BS is conserved between yeast and humans.
SF3b1 is a large protein containing an N-terminal region with multiple U2AF ligand motifs (ULM) and a C-terminal HEAT domain composed of 20 highly conserved HEAT repeats (Supplemental Fig. 1; Kielkopf 2017). The N terminus of human SF3b1 is heavily post-translationally modified (Wang et al. 1998; Boudrez et al. 2002) and contacts many other protein partners through its ULMs (Kielkopf 2017). SF3b1 associates with the pre-mRNA during assembly of the spliceosome and crosslinks directly to the intron (Gozani et al. 1998; McPheeters and Muhlenkamp 2003; Schneider et al. 2015). Structural studies of B and Bact spliceosomes have revealed that distinct regions of SF3b1 make contact with the U2/BS duplex as well as the region of the intron immediately downstream from the BS (Rauhut et al. 2016; Yan et al. 2016; Bertram et al. 2017; Plaschka et al. 2017). The N- and C-terminal HEAT repeats contact the U2/BS duplex, while HEAT repeats 3–8 bind downstream intronic RNA (Fig. 1). Interestingly, MDS-associated mutations that change BS usage are found in HEAT repeats 4–9 rather than in regions that directly contact the U2/BS duplex (Cretu et al. 2016; Rauhut et al. 2016; Yan et al. 2016). Cryo-EM structures revealed that the BP-A is flipped out from the U2/BS duplex and sequestered in a sequence-conserved pocket found between HEAT repeats 15 and 16 (Rauhut et al. 2016; Yan et al. 2016; Bertram et al. 2017; Plaschka et al. 2017). However, what role this pocket plays in BS recognition or activation of the spliceosome prior to catalysis is unclear.
FIGURE 1.
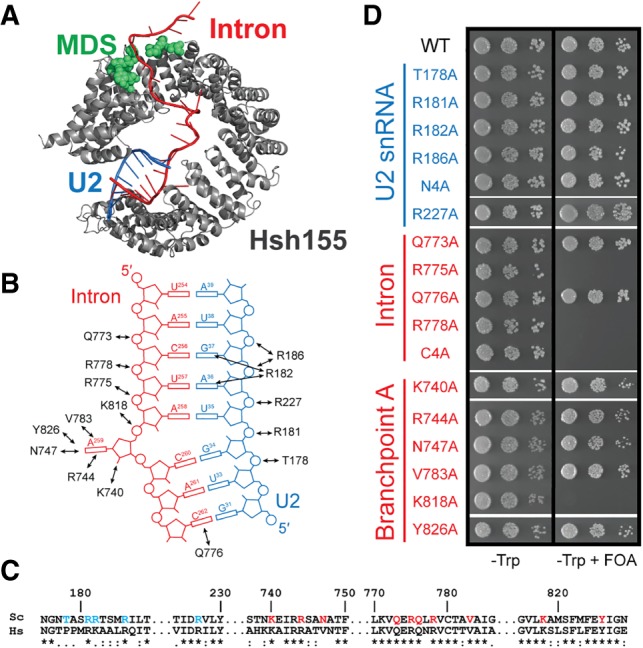
Alanine scan of Hsh155 contacts to the U2/BS duplex. (A) Cartoon representation of the HEAT domains of Hsh155 (gray) bound to the U2/BS duplex (blue/red) from a cryo-EM structure of an activated spliceosome (pdb 5GM6) (Yan et al. 2016). The region of human SF3b1 frequently mutated in MDS is colored in green. (B) Schematic illustration of Hsh155-U2/BS duplex contacts observed in the structure shown in A. (C) Sequence alignment of the U2/BS duplex binding site of Hsh155 from yeast (Sc) and human (Hs). (D) Growth of haploid yeast containing the indicated Hsh155 mutations on nonselective (−Trp) and selective (−Trp +FOA) media at 30°C.
SF3b1 has also been identified as the major target of several spliceosome inhibitors that block splicing in mammals including spliceostatin A, herboxidiene, and pladienolide B (PB) (Bonnal et al. 2012; Effenberger et al. 2017). These compounds halt the progression of spliceosome assembly, leading to widespread intron retention and changes in alternative splicing (Corrionero et al. 2011; Folco et al. 2011; Vigevani et al. 2017). The first identified small molecule inhibitor resistance mutation, SF3b1 R1074H, abolishes the activity of PB and its derivative E7107 and restores splicing (Yokoi et al. 2011; Teng et al. 2017). Multiple mutations in SF3b1 and another splicing factor PHF5A (Rds3 in yeast) confer resistance to spliceosome inhibitors, and this has led to a model wherein inhibitors bind to a composite binding site formed by the two proteins (Teng et al. 2017).
Despite the importance of SF3b1 to splicing, much remains unknown about how the protein functions. Here, we characterize structural features of Hsh155 and their functional impact on splicing in yeast. We performed structure-guided alanine scanning to identify important contacts between Hsh155 and RNA and show that many of these contacts are nonessential for yeast viability but influence BS usage. Furthermore, we demonstrate that the entire N terminus of the protein is dispensable for growth in yeast and deletion of N-terminal HEAT repeats that contact the U2 snRNA does not affect growth. Through the generation of yeast and human chimeric proteins, we show that human sequence can largely, but not fully, function in the context of the yeast spliceosome. Replacing Hsh155 HEAT repeats with their human SF3b1 counterparts renders yeast splicing susceptible to inhibition by PB, while the R1074H mutation confers resistance. These results further define the role SF3b1/Hsh155 plays in BS selection as well as provide a new strategy for chemically inhibiting yeast splicing.
RESULTS
Hsh155 contacts to the snRNA strand of the U2/BS duplex are not essential
Our laboratory and others have previously shown that MDS mutations in Hsh155 can alter splicing by modulating BS usage (Tang et al. 2016; Carrocci et al. 2017). However, the mechanism by which these mutations influence BS choice remains unclear. Spliceosome structures have revealed that Hsh155 contacts both the U2 snRNA and intron with three distinct regions of the protein: HEAT repeats 1–2 contact the snRNA strand of the U2/BS duplex, repeats 15–17 contact the intronic strand of the U2/BS duplex, and repeats 3–8 interact with the intronic RNA downstream from the BS (Fig. 1A; Rauhut et al. 2016; Yan et al. 2016; Bertram et al. 2017; Plaschka et al. 2017). MDS mutations must alter BS usage from a distance since the mutations are not found at the interface of Hsh155 and the U2/BS duplex (Cretu et al. 2016; Rauhut et al. 2016; Yan et al. 2016). Given this observation, we examined whether mutation of direct contacts to the U2/BS duplex would also influence splicing and BS selection.
We performed alanine scanning of conserved amino acids nearby and potentially contacting the U2/BS duplex based on the yeast B and Bact spliceosome structures (pdb 5NRL and 5GM6, respectively; Fig. 1B; Rauhut et al. 2016; Yan et al. 2016; Plaschka et al. 2017). The majority of residues near RNA are conserved between yeast and humans, suggesting they play a similar role in the human spliceosome (Fig. 1C). We used a previously described hsh155Δ yeast strain to introduce point mutations in Hsh155 and assay them for function (Carrocci et al. 2017). We individually mutated five positions found in the N-terminal HEAT repeats that contact the U2 snRNA strand (T178, R181, R182, R186, and R227) to alanine. Each of these mutations supported yeast viability, with similar growth observed on FOA-containing media as the WT strain (Fig. 1D). A quadruple mutation of all of the contacts found in HEAT repeats 1 and 2 (N4A: T178, R181, R182, R186) also supported viability (Fig. 1D). These data argue that N-terminal contacts with the U2/BS duplex are not critical or may be functionally redundant with other components of the splicing machinery.
Yeast were much more sensitive to mutation of Hsh155 residues contacting the intron strand of the U2/BS duplex. We mutated four positions in C-terminal HEAT repeat 16 (Q773, R775, Q776, and R778) and assayed them for growth. While yeast were viable after mutation of Q773 and Q776, replacement of R775 or R778 with alanine was lethal as was simultaneous mutation of all four amino acids (C4A, Fig. 1D). Expression of lethal point mutations (R776 and R778A) was confirmed by western blot in merodiploid strains (Supplemental Fig. 2). It is possible that Hsh155 may be more sensitive to mutation at R775 and R778 compared to Q773 and Q776 due to arginine's greater ability to neutralize the negative charge of the phosphodiester backbone of the U2/BS duplex or due to the need for these particular contacts occurring more closely to the site of the bulged adenosine. While yeast can tolerate loss of nearly all of the contacts between the snRNA strand of the BS duplex and Hsh155, single point mutations on the complementary, intronic strand can be lethal.
The adenosine binding pocket of SF3b1 is amenable to mutation
The C terminus of SF3b1/Hsh155 engages the BP-A directly by encapsulating the bulged nucleotide in a pocket composed of residues R744, N747, V783, and Y826 (Fig. 1B). In addition, the BP-A ribose 2′ OH and 5′ phosphate are proximal to K740 and K818, respectively. Although many of the residues that form the pocket are well-conserved, yeast were surprisingly tolerant of alanine substitutions at K740, R744, N747, and V783 (Fig. 1D). Thus, while many amino acids involved in forming the binding site for the branchpoint adenosine are conserved, they are nonessential. An exception to this is the K818A mutation, which was lethal. This amino acid interacts with the 5′ phosphate of the BP-A, similar to how R775 and R778 (which also cannot be substituted for by alanine) interact with the RNA backbone of the two nucleotides that immediately precede the BP-A (Fig. 1B). Expression of epitope-tagged K818A Hsh155 was also confirmed by western blot (Supplemental Fig. 2), suggesting that the mutant protein is likely nonfunctional. Together, these data demonstrate that the only essential contacts formed between Hsh155 and the U2/BS duplex occur in the C terminus of the protein at positions immediately 5′ of the bulged BP-A.
The N terminus of Hsh155 is largely dispensable
The observation that Hsh155 could tolerate alanine substitution at most positions that contact the snRNA led us to define the regions of Hsh155 that are essential. To this end, we prepared 15 mutants of Hsh155 that contained successive truncations of either the N or C termini and assayed whether these proteins could support growth as the sole copy of Hsh155 (Fig. 2A). The N-terminus of Hsh155 contains one conserved U2AF ligand motif (ULM) and a second ULM-like sequence lacking the characteristic tryptophan (Thickman et al. 2006). We generated truncations of Hsh155 containing both ULMs but lacking the rest of the N terminus (residues 64–972), containing only the first ULM (residues 96–972) or lacking both ULMs (residues 103–972) and assayed them for function in yeast (Fig. 2A). Growth was observed for all of these truncations (Fig. 2B), consistent with this region of Hsh155 having a nonessential role in yeast splicing and previous results obtained for the Schizosaccharomyces pombe homolog (Habara et al. 1998). Further truncation of the protein to eliminate the entire N terminus while maintaining all of the HEAT repeats in the HEAT domain was nonviable (Hsh155 167–972, Fig. 2B). Surprisingly, additional truncation of the N terminus resulting in elimination of individual HEAT repeats restored growth. Hsh155 variants lacking HEAT repeats 1 (residues 197–972); 1 and 2 (residues 237–972); and 1, 2, and 3 (residues 275–972) could all support growth (Fig. 2B), whereas loss of HEAT repeat 4 or further truncation of the protein was lethal.
FIGURE 2.
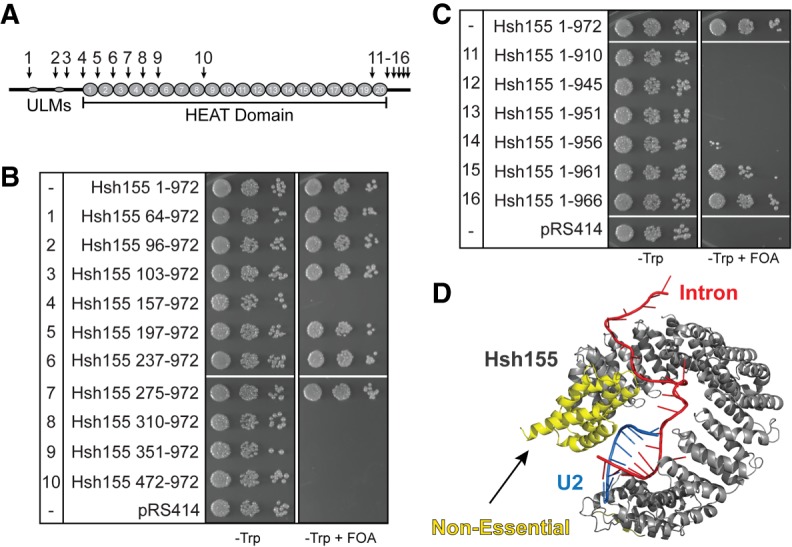
Identification of the essential regions of S. cerevisiae Hsh155. (A) Schematic illustration of Hsh155 domains. Numbers and arrows indicate the positions of truncation mutants shown in panels B and C. (B,C) Growth of haploid yeast containing the indicated Hsh155 N-terminal (panel B) and C-terminal (panel C) truncations on nonselective (−Trp) and selective (−Trp +FOA) media at 30°C. (D) Cartoon illustration of the HEAT domains of Hsh155 (gray) bound to the U2/BS duplex (blue/red) from a cryo-EM structure of an activated spliceosome (pdb 5GM6). The nonessential region is shown in yellow.
The C terminus of Hsh155 is involved in contacting both the U2/BS duplex and other components of the SF3 complex including Ysf3, Cus1, Prp11, and Rds3 (Supplemental Fig. 3). Consistent with the numerous contacts in this region, the C terminus of the protein was largely intolerant of truncation (Fig. 2C). Loss of just 15 amino acids from the C terminus results in a significant growth defect (Fig. 2C, residues 1–956) and further C-terminal truncations of the protein cannot support growth. This 15 amino acid region interacts extensively with both Rse1 and Ysf3, suggesting it may be important for SF3b complex stability. These data define the essential regions of Hsh155 as beginning with HEAT repeat 4, which is the region implicated in MDS and that contacts the downstream intronic RNA (Fig. 1A), and ending with amino acids very near the C terminus of the protein.
Contacts between Hsh155 and the U2/BS duplex can alter BS selection
MDS mutations in Hsh155 that result in no apparent growth phenotype can nonetheless alter BS usage (Tang et al. 2016; Carrocci et al. 2017). This is likely due to the highly specific impact of the MDS alleles on the splicing of pre-mRNAs containing certain nonconsensus BS substitutions at the −2, −1, and +1 positions relative to the BP-A adenosine. For this reason, we chose to examine viable mutations of the Hsh155 U2/BS duplex binding domains for defects in splicing using the ACT1-CUP1 reporter assay (Lesser and Guthrie 1993; Carrocci et al. 2017). This well-characterized assay relates reporter pre-mRNA splicing to copper tolerance in yeast, with the extent of copper resistance strongly correlating with the amount of spliced reporter (Fig. 3A; Carrocci et al. 2017).
FIGURE 3.

Mutation of BP-A pocket alters nonconsensus BS usage. (A) Schematic illustration of the ACT1-CUP1 reporter pre-mRNA and assay. The consensus 5′SS, 3′SS, and BS are shown in the intron. The position of A258 is noted and the branchpoint (BP-A; A259) is underlined. (B) Results from a Cu2+ growth assay of strains containing mutations to U2/BS duplex contacts and either the consensus ACT1-CUP1 reporter plasmid or one with a nonconsensus A258U BS substitution. (C) Results from a Cu2+ growth assay of strains carrying mutations to the BP-A pocket and containing a nonconsensus A258U BS reporter plasmid. (D) Heatmap summarizing ACT1-CUP1 reporter data for the indicated Hsh155 mutants and BS reporters. Plotted data represent the log2 transform of the ratio of the maximum [Cu2+] at which growth was observed for the indicated mutant to the maximum [Cu2+] at which growth was observed for WT Hsh155. Purple indicates decreased growth relative to WT, while orange indicates improved growth. In panels B and C, each bar represents the average of three independent experiments, and error bars represent the standard deviation.
We first assayed the viable Hsh155 N4A mutant, in which most of the contacts between Hsh155 and the snRNA strand of the U2/BS duplex have been mutated. As was previously observed with the MDS alleles, the N4A quadruple mutant did not appear to alter splicing of the consensus BS reporter (UACUAAC; Fig. 3B). However, this mutation slightly improved splicing of a reporter containing the A258U BS substitution (UACUuAC, 1.6 mM versus 1.8 mM [Cu2+]; Fig. 3B). These data do not allow us to distinguish if this improvement stemmed from the N4A mutation promoting spliceosome assembly at the A258U BS or if ablation of these contacts facilitates spliceosome activation during conversion from the Bact to B* complex.
Mutation of the adenosine pocket alters BS recognition
As discussed above, yeast are more sensitive to mutation of Hsh155 contacts to the intron strand of the U2/BS duplex than contacts to the snRNA strand with the exception of amino acids that form the pocket for the BP-A. We therefore focused on using the ACT1-CUP1 assay to determine if viable mutations to the BP-A pocket also alter splicing. In addition to the alanine mutations, we included a number of conservative mutations within the pocket and assayed each mutant (K740A/R, N747A/Q, V783A/L, and K818R) for splicing defects with the A258U ACT1-CUP1 reporter. Consistent with both the N4A mutant and the previously examined MDS alleles, yeast grew equally well as WT using the consensus ACT1-CUP1 reporters and any viable mutation of the BP-A pocket (Supplemental Fig. 4). However, each of the remaining BP-A pocket mutations altered growth on Cu2+-containing media when using the A258U BS mutant reporter (Fig. 3C). Several mutations improved Cu2+ tolerance of yeast expressing the A258U reporter, while others decreased tolerance. Intriguingly, alanine substitutions of K740, N747, and V783 all significantly improved Cu2+ tolerance in the presence of the A258U reporter while conservative mutations (K740R, V783L, and K818R) were inhibitory. It is possible that the greater steric bulk of these latter mutations disrupts splicing by preventing proper accommodation of the BP-A within Hsh155 when the U2/BS duplex is not properly paired. Conversely, alanine substitution may provide additional space and flexibility for accommodating the BP-A and improve spliceosome assembly when mismatches occur in the duplex. This may then lead to increased splicing of pre-mRNAs containing these BS.
We next tested a collection of ACT1-CUP1 reporters sampling all positions in the BS in combination with several of the BP-A pocket mutants (Fig. 3D). Each of the tested mutations altered growth of multiple reporter-containing strains. In contrast with our previous results for the MDS alleles (Carrocci et al. 2017), mutations contacting the U2/BS duplex altered usage of introns containing an A to C substitution of the branchpoint (A259C reporter; UACUAcC) and showed mixed patterns of splicing changes by improving splicing of some reporters and inhibiting splicing of others. The K740A, N747A, and V783A mutations all decreased Cu2+ tolerance when using the A259C reporter, whereas N747Q improved growth. This latter observation is consistent with the hypothesis that a larger side chain could help stably accommodate a smaller nucleobase. Abolishing the K740 interaction with the ribose 2′ OH by introducing the K740A mutation likely leads to destabilization of the bulged nucleotide, and we observed a decrease in Cu2+ tolerance consistent with this hypothesis. K818R showed the strongest effect for most reporters excluding the BP-A mutant (A259C), consistent with K818 playing an important role in binding of the U2/BS duplex. This notion is further supported by the observation that K818A is lethal (Fig. 1D). It is less clear why N747A and V783A decrease Cu2+ tolerance in the presence of A259C, although it is possible that these substitutions in combination with a smaller nucleobase prevent proper alignment of the U2/BS duplex phosphodiester backbone with essential, positively charged Hsh155 amino acids (Fig. 1B).
A second interesting observation is that mutation of a single residue can either enhance or impair splicing depending on the BS sequence. For example, V783A improves A258U (UACUuAC) Cu2+ tolerance but reduces C260G (UACUAAg) growth on Cu2+-containing media. This result is in contrast to what has been described for MDS mutations where a single Hsh155 mutation will either enhance or decrease splicing of all affected BS mutants (Tang et al. 2016; Carrocci et al. 2017). For N747A and V783A, we observed an enhancement of Cu2+ tolerance when nonconsensus nucleotides were present at the −2 and −1 positions, while this was inhibited with A259C and +1 substitutions. This suggests additional functional asymmetry in how Hsh155 interacts with U2/BS duplex: Protein mutations are tolerated along the snRNA but not the intron phosphodiester backbone (Fig. 1), and usage of nonconsensus BS differs depending on whether mismatches are found 5′ or 3′ of the branchpoint.
We found no effect of mutating the −4 and −5 positions relative to the BP-A adenosine (UACUAAC; −4 and −5 positions in boldface) for any Hsh155 mutants, consistent with few observed contacts between this region of the duplex and the protein (Figs. 1B, 3D). However, most Hsh155 mutants showed changes in Cu2+ tolerance using reporters with substitutions at the −2, −1, and +1 positions. Changes in the splicing of reporters with substitutions at these same positions were previously observed upon incorporation of MDS mutations in Hsh155 (Tang et al. 2016; Carrocci et al. 2017). Unlike with MDS mutations, we also found changes upon substitution of the −3 position using the C256A (UAaUAAC) reporter in combination with N747Q, K740A/R, and K818R mutants. Interestingly, each of the 5′ phosphates of the −3, −2, −1, and +1 nt of the BS is in close proximity to a positively charged (and often essential) amino acid in Hsh155 (Fig. 1B). Together these data suggest a connection between binding of the BP-A within the pocket, interaction of the RNA backbone of the BS with conserved amino acids, and usage of a particular BS sequence by the spliceosome.
Genetic interactions between Hsh155 mutants and the Prp2 ATPase
Hsh155 mutations that impact how the protein interacts with the U2/BS duplex may also change how Hsh155 is ejected upon activation of the spliceosome. The ATPase Prp2 binds Hsh155 during spliceosome activation and triggers destabilization or restructuring of the SF3a, the SF3b, and the retention and splicing (RES) subcomplexes (Lardelli et al. 2010; Liu and Cheng 2012). Recently, it has been shown that the absence of the RES complex components can bypass the requirement for Prp2 (Bao et al. 2017b). A possible explanation for this may be that without RES, interactions between the U2/BS duplex and Hsh155 are less stable—eliminating the need for Prp2 remodeling. Given that we have previously shown that Hsh155 MDS alleles interact genetically with the cold-sensitive (cs) Prp2 Q548N mutation, we explored whether mutation of the Hsh155 U2/BS duplex binding site would also rescue the cs phenotype.
We constructed strains in which N-terminal Hsh155 contacts to the U2/BS duplex were either ablated (N4A) or removed (Hsh155275-972) in combination with WT or mutant Prp2. The N4A mutant grew similarly to WT Hsh155 in the presence of WT Prp2 at 16°C, and neither N4A nor Hsh155275−972 could rescue the cs phenotype of Prp2 Q548N (Supplemental Fig. 5). Interestingly, the Prp2 Q548N mutant was also synthetically sick with Hsh155275−972 at 30°C and 37°C. Together these data indicate that Prp2 activity can still be limiting for growth even if contacts between Hsh155 and the U2/BS duplex are significantly compromised.
We next tested for genetic interactions between Hsh155 BP-A pocket mutations and Prp2 by combining V783A and V783L with Prp2 Q548N. We reasoned that if BP-A pocket mutations changed how the U2/BS duplex was accommodated by Hsh155, they might also alter the requirement of Prp2 for activation. V783A and V783L grew similarly to WT Hsh155 regardless of the presence of WT Prp2 or Q548N at 30°C and 37°C (Supplemental Fig. 5). At 23°C, both Hsh155 V783 mutants grew similarly to WT Hsh155 in the presence of WT Prp2. However, V783A showed decreased growth and V783L showed increased growth relative to WT Hsh155 in the presence of Prp2 Q548N. The observed differences in growth were further exacerbated at 16°C, with the V783L mutant showing significantly better growth. These results are similar to those previously observed for the MDS-linked K335E and D450G Hsh155 mutations that inhibit and enhance nonconsensus splice site usage respectively (Carrocci et al. 2017). Thus, mutations in Hsh155 can genetically interact with a cs allele of Prp2 whether or not they are located within the U2/BS duplex binding site.
Pladienolide B does not block splicing in S. cerevisiae
SF3b1 is the target of several splicing inhibitors such as PB that function in mammalian cells and these inhibitors are thought to bind the HEAT domain (Yokoi et al. 2011; Effenberger et al. 2016; Teng et al. 2017; Finci et al. 2018). The HEAT domain of SF3b1 is extremely well conserved between yeast and human, with several HEAT repeats possessing >60% identity (Supplemental Fig. 1). Previous work has also shown that S. pombe splicing is blocked upon incubation with inhibitors (Lo et al. 2007). Given these observations, we investigated whether PB would also block splicing in yeast splicing extract prepared from S. cerevisiae. While PB concentrations below 200 nM are sufficient to inhibit splicing in human nuclear extracts (Effenberger et al. 2014), PB concentrations 100-fold higher failed to inhibit yeast splicing (Fig. 4A). The surprising lack of PB efficacy suggests that the binding site of PB on Hsh155 is sufficiently different in yeast to prevent inhibition.
FIGURE 4.
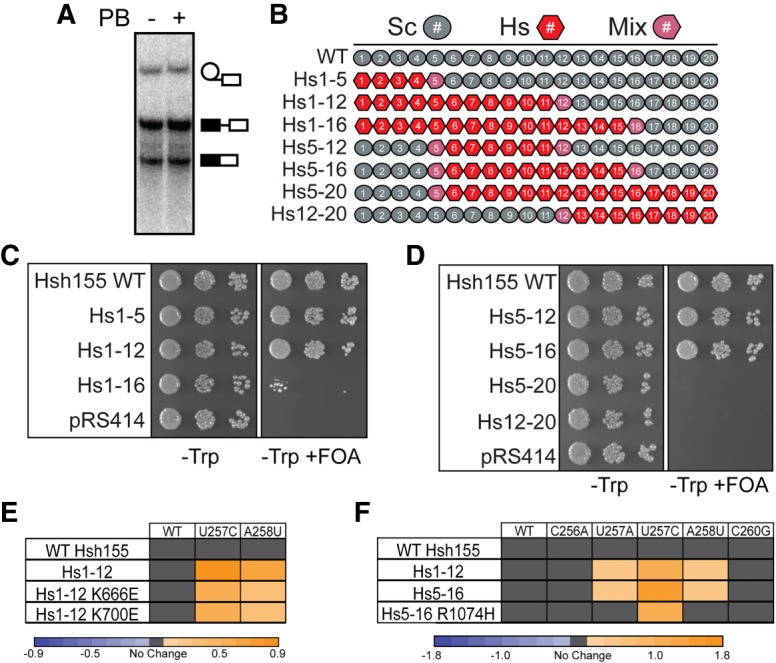
Humanized Hsh155 functions in yeast and alters nonconsensus BS usage. (A) In vitro splicing assay with WT Hsh155 in the presence or absence of PB (20 µM). PB does not inhibit yeast splicing at this concentration. (B) Schematic illustration of the humanized Hsh155 chimeric proteins. (C,D) Growth of haploid yeast containing the indicated humanized Hsh155 proteins on nonselective (−Trp) and selective (−Trp +FOA) media at 30°C. (E) Heatmap summarizing ACT1-CUP1 reporter data for the indicated humanized Hsh155 proteins and MDS mutants with several BS reporters. Plotted data represent the log2 transform of the ratio of the maximum [Cu2+] at which growth was observed for the indicated mutant to the maximum [Cu2+] at which growth was observed for WT Hsh155. Purple indicates decreased growth relative to WT, and orange indicates improved growth. (F) Heatmap of ACT1-CUP1 reporter data as in panel E but including the Hs5-16 humanized protein with and without the R1074H splicing inhibitor resistance mutation (note that numbering refers to human SF3b1).
Human SF3b1 HEAT repeats support yeast growth
To explore the differences between yeast Hsh155 and human SF3b1, we generated chimeric constructs and assayed them for function in yeast (Fig. 4B). We began by replacing yeast HEAT repeats 1–5, 1–12, and 1–16 with the human sequence to generate the chimeric proteins Hs1-5, Hs1-12 and Hs1-16. While not essential for growth (Fig. 2B), the N-terminal extension was not altered in these constructs and consists entirely of the native yeast sequence. Hs1-5 and Hs1-12 grow equally well as WT on FOA, but further exchange of sequence in Hs1-16 results in a significant growth defect (Fig. 4C). This result demonstrates that the region of SF3b1 mutated in MDS (HEAT repeats 4–9) in humans can functionally complement for this same region in yeast Hsh155. This same region has also been shown to interact with the spliceosomal ATPases Prp2 and Prp5 (Rauhut et al. 2016; Tang et al. 2016; Yan et al. 2016; Carrocci et al. 2017), and so it is likely that the human HEAT repeats also conserve these interactions to some extent.
We also exchanged sequence internal to the protein starting with HEAT repeat 5. Consistent with the above data, Hs5-12 was well tolerated and, surprisingly, this now permitted inclusion of additional HEAT repeats. A chimeric protein containing human HEAT repeats 5–16 (Hs5-16) supported yeast growth (Fig. 4D). From this, we propose that the Hs1-16 mutant was likely very sick due to the exchange of a large number of repeats and not due to incompatibility of HEAT repeats 12–16. Whereas the N-terminal HEAT repeats appear to be functionally interchangeable, exchange of the C-terminal HEAT repeats are not tolerated (Fig. 4D). This observation is consistent with C-terminal truncations of Hsh155 being lethal and the general importance of the region to SF3b function (Fig. 2C).
Exchanging human and yeast sequences alters splicing
We next examined how inclusion of human HEAT repeats altered BS usage in yeast and how this was further changed by two common MDS alleles (K666E and K700E; human SF3b1 numbering). The Hsh155 chimeras containing Hs1-12 showed no growth defects on Cu2+ when assayed with the consensus ACT1-CUP1 reporter. However, these human sequences increased Cu2+ tolerance using reporters containing the nonconsensus BS U257C and A258U relative to the yeast protein even without inclusion of MDS-linked mutations (Fig. 4E). Not all nonconsensus BS were affected, as no changes relative to Hsh155 were observed for any of the chimeras with the C256A reporter. While both yeast and human HEAT repeats promote splicing of pre-mRNAs with consensus BS equally well, human HEAT repeats alone are sufficient to promote usage of nonconsensus BS in yeast. This suggests that features of human SF3b1 may contribute to use of weak BS.
The K666E and K700E MDS alleles in the Hs1-12 chimera both impaired growth in the presence of a nonconsensus BS reporter relative to the WT Hs1-12 chimera (Fig. 4E). Reduced Cu2+ tolerance was observed using both the U257C and A258U reporters but not with the consensus BS. This indicates that the MDS alleles altered splicing in ways consistent with prior work in which homologous mutations were introduced in Hsh155 (Tang et al. 2016; Carrocci et al. 2017). These results support the notions that HEAT repeats 4–9 function similarly in yeast and humans and that MDS alleles in human or yeast may alter splicing through similar mechanisms.
We carried out similar experiments with the Hs5-16 chimera to test how a PB resistance mutation (R1074H) in SF3b1 alters BS usage. As with the Hs1-12 chimeras (Fig. 4E), we observed increased Cu2+ tolerance using reporters containing U257C and A258U BS with Hs5-16 and no change in growth using the WT reporter (Fig. 4F). Interestingly, inclusion of the R1074H PB resistance mutation alters this behavior. While growth using the WT reporter was unaffected, Hs5-16 R1074H Cu2+ tolerance more closely resembled that of WT Hsh155 with nonconsensus U257A and A258U reporters and decreased Cu2+ tolerance using the U257C reporter relative to Hs5-16 (Fig. 4F). Like MDS and BP-A pocket mutations, the PB-resistance R1074H mutation can also alter usage of nonconsensus BS.
PB inhibits splicing in chimeric Hsh155 mutants
Since yeast were able to tolerate substitution of the majority of the Hsh155 HEAT repeats with those from human SF3b1, we next tested whether PB would inhibit in vitro splicing in extracts containing chimeric SF3b1. We generated splicing extracts from cells expressing Hs1-12, Hs5-16, or Hs5-16 R1074H. Extracts were incubated with PB or a DMSO control prior to the addition of radiolabeled pre-mRNA to initiate splicing (Fig. 5A). In the absence of PB, all extracts containing the chimeras spliced in vitro, albeit to different extents and with somewhat reduced activity compared to WT. This was observed in multiple extract preps (data not shown) and likely indicates reduced function of these mutant proteins in vitro. Addition of 500 nM PB did not inhibit the formation of the lariat intron-3′ exon intermediate or mRNA product from extracts containing Hsh155 WT or the Hs1-12 chimera (Fig. 5A,B). However, product formation was strongly inhibited in Hs5-16 extract upon addition of PB (Fig. 5A,B). When the R1074H PB resistance mutation was introduced in the Hs5-16 chimera, generating Hs5-16 R1074H, splicing product formation in the presence of PB was restored.
FIGURE 5.
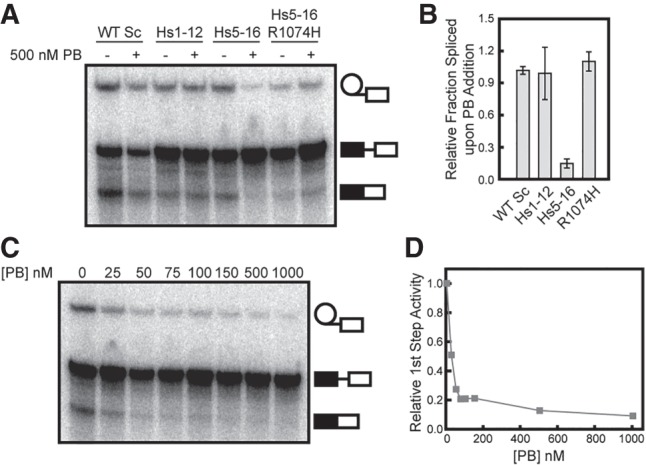
PB inhibits yeast splicing with humanized Hsh155. (A) In vitro splicing assay with WT Hsh155 or the indicated humanized Hsh155 variants in the presence or absence of PB (500 nM). (B) Quantification of in vitro splicing data represented by panel A. Each bar represents the average of three independent experiments, and error bars represent the standard deviation. Data were normalized to the extent of splicing observed for each individual strain in the absence of PB. (C) Titration of PB inhibition of in vitro splicing of extracts containing Hs5-16. (D) Quantification of the data shown in panel C. Approximately 50% inhibition (∼IC50) is observed at 25 nM PB.
The Hs5-16 chimera was very sensitive to PB and titration of the extract revealed an IC50 of ∼25 nM (Fig. 5C,D), comparable to values reported for inhibition of human splicing in vitro (Effenberger et al. 2014). These data demonstrate that exchange of HEAT repeats 5–16 is sufficient to render the yeast spliceosome susceptible to small molecule inhibition by a human SF3b1 inhibitor. This strongly suggests that the binding site for PB resides in these HEAT repeats. Recent work suggests that PB binds close to the BP-A pocket, at a site formed by SF3b1 HEAT repeats 15 and 16 as well as the SF3b protein PHF5A (yeast Rds3) (Cretu et al. 2016; Teng et al. 2017). Since we did not modify Rds3 in these assays, it is likely that it can compensate for PHF5A interactions with PB. Consistent with this hypothesis, the only known residue in PHF5A/Rds3 that can confer resistance to PB upon mutation, Y36, is conserved in yeast. The failure of PB to inhibit yeast splicing likely then arises from differences in the Hsh155 portion of the composite binding site.
DISCUSSION
We have used recently determined cryo-EM structures of SF3b1 and Hsh155 to analyze how the observed protein/RNA contacts promote splicing in yeast. Site-directed mutagenesis of the RNA binding site and the BP-A pocket in particular link SF3b1/RNA interactions with use of nonconsensus BS. The high sequence conservation of SF3b1 among eukaryotes allowed us to replace yeast Hsh155 HEAT repeats with their human counterparts. Yeast containing humanized Hsh155 showed increased usage of nonconsensus BS, in agreement with the notion that SF3b1/RNA interactions are an important determinant in BS selection. Furthermore, splicing in extracts prepared from yeast expressing humanized Hsh155 was strongly inhibited by addition of PB. Together, these data reveal new insights into SF3b1 function in splicing.
The essential domains and BS duplex interactions of SF3b1/Hsh155
SF3b1/Hsh155 has now been resolved in a number of structures of the human and yeast spliceosome at varying resolutions (Rauhut et al. 2016; Yan et al. 2016; Bertram et al. 2017; Plaschka et al. 2017). While specific amino acid/RNA contacts are not well-defined in every structure, they all identify the N- and C-terminal HEAT repeats of SF3b1 as the key interaction sites for the U2/BS duplex. Both RNA strands of the U2/BS duplex are similarly engaged by Hsh155 via interactions between conserved, positively charged lysine and arginine sidechains found in the N- and C-terminal HEAT repeats and the RNA phosphodiester backbone (Fig. 1). However, only contacts made with the intronic RNA of the U2/BS duplex are essential. Thus, despite structural similarities in how both strands of the U2/BS duplex are recognized by SF3b1/Hsh155, recognition of the BS strand is more critical for function. BP-A pocket mutations can also alter the usage of nonconsensus BS. Substitution with bulkier amino acids decreases nonconsensus BS usage, while substitution with smaller amino acids increases usage (Fig. 3). We hypothesize that for SF3b1/Hsh155 to bind U2/BS duplexes containing mismatches additional space or flexibility within the BP-A pocket may be required to properly accommodate the bulged nucleotide. In other words, deformation of the U2/BS duplex due to mismatches from weak BS may place additional constraints or requirements on SF3b1 for duplex interaction. While the BP-A pocket is highly conserved between yeast and humans (Supplemental Figs. 1, 6), humans contain a smaller, hydrophobic valine amino acid in place of N747 in yeast (Fig. 1C). It would be interesting to determine the contribution of this particular amino acid to usage of weak BS by the human splicing machinery.
In agreement with the above mutational data, we also found that large portions of the Hsh155 N terminus, but not the C terminus, are dispensable for growth (Fig. 2). The N-terminal ULM region as well as HEAT repeats 1–3 (which are responsible for contacts to the snRNA strand of the U2/BS duplex) are nonessential in S. cerevisiae, consistent with earlier work on the S. pombe homolog, Prp10 (Habara et al. 1998). Further truncation of the protein beyond HEAT repeat 3 was lethal. Interestingly, biochemical data from our laboratory and others previously indicated that this region is important for Prp5 ATPase binding in addition to binding of the intronic RNA downstream from the BS (Tang et al. 2016; Carrocci et al. 2017). It is possible that additional N-terminal truncations beyond HEAT repeat 3 cannot be tolerated due to disruption of necessary interactions with Prp5, that intronic RNA binding to this region is essential, or both. In contrast, very little of the C-terminal region of SF3b1 could be removed without impacting yeast growth. Each of the C-terminal HEAT repeats interacts extensively with multiple other proteins (Supplemental Fig. 3), and we speculate that their removal is too destabilizing to SF3b structure for yeast to tolerate.
While many of the HEAT repeats of Hsh155 can be replaced or deleted, it does not mean that these same regions do not influence the splicing reaction. Recently, a number of Hsh155 mutations have been isolated in HEAT repeat 3 that improve splicing of pre-mRNAs with nonconsensus BS (Tang et al. 2016). We have also shown that simultaneous alanine substitution of several amino acids within HEAT repeats 1 and 2 improves yeast growth when tested in combination with the nonconsensus A258U ACT1-CUP1 reporter (N4A, Fig. 3B). Whether or not these changes are the direct result of altered interactions between the N-terminal HEAT repeats and the snRNA strand of the U2/BS duplex is unclear. It is possible that perturbation of interactions between the N-terminal HEAT repeats and the snRNA strand also change interactions between the C-terminal HEAT repeats and the intronic strand of the duplex. Additional structural data of SF3b1 or Hsh155 bound to U2/BS duplexes containing mismatches could help clarify how the protein accommodates these structures.
Multiple features of SF3b1 impact BS usage
Cryo-EM structures of precatalytic spliceosomes have shown that binding of the U2/BS duplex is carried out predominantly by Hsh155 with a few additional interactions made between the duplex and Rds3 and Prp11 (PHF5A and SF3a2 in humans, respectively) (Rauhut et al. 2016; Yan et al. 2016; Plaschka et al. 2017). SF3b1/Hsh155 contacts to the U2/BS duplex do not appear to involve specific recognition of the RNA sequence. Instead, its interaction mode is predominated by contacts to the phosphodiester backbone and ribose groups of the duplex (Fig. 1B). This suggests that SF3b1/Hsh155 may be functioning to recognize duplex formation or features of the U2/BS duplex itself. This observation is consistent with utilization of nonconsensus BS in yeast and humans, as sequence-specific contacts at this step would constrain the versatility of the spliceosome to utilize different BS. One exception is binding of the BP-A, which is not paired in the U2/BS duplex but instead sequestered within a pocket (Supplemental Fig. 6) situated at the interface of HEAT 15 and 16. The lack of nucleotide-specific interactions within the pocket likely explains in part why other nucleotides can also be used in place of BP-A (Smith et al. 2009).
The data presented here suggest a pathway for BS recognition during splicing (Fig. 6). For both chemical steps in splicing to occur efficiently, it is critical for the U2/BS duplex to contain a bulged nucleotide and for the bulge to be appropriately positioned within the duplex (Query et al. 1994; Smith et al. 2009). One function of SF3b1/Hsh155 might be to ensure that both of these requirements are met upon pairing of the U2 GUAGUA sequence with a cognate RNA. Accommodation of the bulged nucleotide aligns the U2/BS duplex with a set of positively charged amino acids within SF3b1/Hsh155. It is possible that proper accommodation and alignment of the duplex promotes usage of a particular BS (Fig. 6), or that misalignment is not tolerated by Hsh155 and blocks assembly. This agrees well with previous studies of BS recognition using an orthogonal splicing system in yeast that revealed the importance of bulge position within the U2/BS duplex (Smith et al. 2009). In that work, Prp5 mutations could also suppress defects related to mutations in the U2/BS duplex but not those due to changes in bulge position. This suggests that SF3b1/Hsh155 and Prp5 may collaborate to ensure proper structure of the U2/BS duplex, with SF3b1/Hsh155 playing a more significant role in identifying the position of the bulged nucleotide.
FIGURE 6.
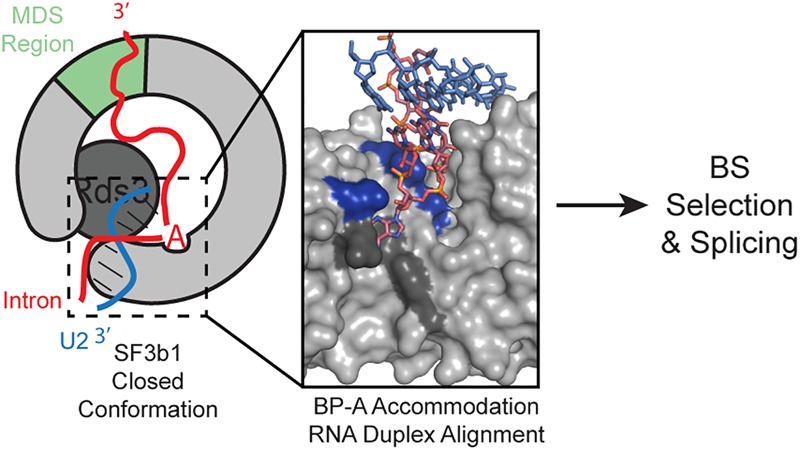
U2/BS duplex recognition by SF3b1 and PHF5A during splicing. A properly closed conformation of SF3b1/Hsh155 is associated with accommodation of the bulged, BP-A of the U2/BS duplex within the hydrophobic pocket of SF3b1/Hsh155 (dark gray) and alignment of the phosphodiester backbone of the intron strand of the U2/BS duplex with a set of positively charged amino acids within the C-terminal HEAT domain. N-terminal HEAT repeats and PHF5A/Rds3 contact the U2/BS duplex. Formation of this complex promotes selection of a particular BS and splicing (pbd: 5GM6) (Yan et al. 2016). Note that Rds3 was omitted from the inset for clarity.
While the data presented here implicate the RNA duplex binding region as an important determinant of BS usage, other regions of SF3b1/Hsh155 can have similar effects. We and others have observed that mutation of HEAT repeats in Hsh155 far from the U2/BS duplex binding site influences BS selection (Supplemental Fig. 7; Tang et al. 2016; Carrocci et al. 2017). Indeed, many cancer-associated mutations in SF3b1 alter BS usage in both yeast and human cells despite being located far from the duplex binding site (Darman et al. 2015; Alsafadi et al. 2016). We previously proposed a mechanism for SF3b1/Hsh155 in BS selection in which different mutations could alter the equilibrium between open and closed conformations of the protein in a step distinct from the Prp5-dependent proofreading (Xu and Query 2007; Liang and Cheng 2015; Carrocci et al. 2017). Our work here expands on this by noting that proper recognition of the U2/BS duplex and accommodation of the BP-A may be necessary for formation of a stable structure. This stable structure may not require full closure of Hsh155 and contacts to both RNA strands of the U2/BS duplex since the snRNA contacts and N-terminal domains are nonessential in yeast (Figs. 1, 2). Additional stability may arise from interactions with Rds3, which also contacts the U2/BS duplex and interacts extensively with Hsh155 (Supplemental Fig. 6). Conformational dynamics of SF3b1/Hsh155 may be influenced by the structure of the U2/BS duplex and the contacts it can or cannot make with the protein.
Finally, we note that the Hsh155 mutations studied here may alter other steps in splicing in addition to spliceosome assembly. We have previously shown that MDS-linked Hsh155 mutations interact genetically with mutations in the Prp2 ATPase, which is responsible for release of the SF3 proteins from the U2/BS duplex during spliceosome activation (Lardelli et al. 2010; Liu and Cheng 2012; Bao et al. 2017a; Carrocci et al. 2017). We proposed that mutations that stabilize the closed complex of Hsh155 may favor assembly but inhibit activation, while mutations that stabilize the open complex may do the opposite. Perturbation of the Hsh155 U2/BS duplex binding site can also result in genetic interactions with the cold-sensitive Prp2 Q548N mutant (Supplemental Fig. 5). Molecular interpretations of these data are difficult at this stage since little is known about how ATP hydrolysis by Prp2 correlates with specific structural transitions. It is possible that how the U2/BS is accommodated by Hsh155 also correlates with demand of the machinery for Prp2 ATPase activity during activation. The particular impact of Hsh155 mutation on splicing of a given transcript may therefore depend on whether assembly or activation is limiting for the overall process.
Inhibition of yeast splicing by pladienolide B
Given the high conservation of SF3b1, including the inhibitor binding site, it is surprising that PB fails to inhibit yeast splicing in vitro even at µM concentrations—several hundred-fold higher than required to inhibit splicing in human nuclear extract (Effenberger et al. 2014; Finci et al. 2018). It is unlikely that failure of PB to inhibit yeast splicing stems from differences in yeast and humans spliceosome assembly pathways since humanized Hsh155 alone confers PB sensitivity to the yeast splicing machinery. One possibility is that while the proposed PB binding site (and BP-A pocket) is highly similar between yeast and humans (Supplemental Fig. 6), differences in inhibition stem from subtle alterations to amino acids at these locations. Indeed, mutation of V1078 to alanine or isoleucine in human cells confers resistance to inhibition and this position corresponds to N747 in yeast (Teng et al. 2017). Intriguingly, N747A strongly alters the splicing of pre-mRNAs with nonconsensus BS (Fig. 3D). Amino acid variation of Hsh155/SF3b1 may therefore contribute to differences in both BS usage and sensitivity to inhibitors. In the future, systematic mutation of the BP-A pocket and surrounding region may prove useful for deducing the minimal structural requirements for PB binding and inhibition.
The mechanisms by which PB and other SF3b1 ligands inhibit splicing in vivo are not yet well-understood since the splicing of different introns is inhibited to different degrees by the same drug and related drugs perturb the splicing of different sets of introns (Corrionero et al. 2011; Effenberger et al. 2017; Teng et al. 2017; Vigevani et al. 2017; Finci et al. 2018; Seiler et al. 2018). One possibility is that SF3b1 inhibitors can bind to SF3b1 whenever the binding site is not occupied by the U2/BS duplex. In this case, drug inhibition results in competition between introns for a limited pool of U2 snRNPs not bound by these compounds (Effenberger et al. 2017). The introns most sensitive to inhibition would consequently be those that are the least competitive for functional U2 recruitment. Such competition among introns for the splicing machinery has been documented in yeast and more recently in human cells (Munding et al. 2013; Paolella et al. 2017). Alternatively, the inhibitor binding site in SF3b1 may only become available for drug binding transiently during BS recognition, as it has previously been proposed that U2 undergoes multiple conformational changes at this stage of spliceosome assembly (Wiest et al. 1996; Perriman and Ares 2010; Folco et al. 2011; Effenberger et al. 2016). In this scenario, sensitivity of particular introns to drug inhibition may stem from how well the transcription and splicing machinery promotes these rearrangements and the lifetime of the inhibitor-sensitive complex on a particular transcript. Chemical inhibition of splicing in yeast containing humanized Hsh155 may be able to distinguish between these possibilities as well as prove generally useful for many other studies of gene expression.
MATERIALS AND METHODS
Parental yeast strains used were described previously (Carrocci et al. 2017). Supplemental Tables 1 and 2 contain detailed lists of plasmids and strains used in this study. Yeast transformation and growth was carried out using standard techniques and media.
Western blotting
Total protein was isolated by trichloroacetic acid precipitation and western blots were performed as previously described (Carrocci et al. 2017). α-FLAG-HRP, α-actin, and α-mouse-HRP antibodies were purchased from Sigma Aldrich, AMB Millipore, and Bio-rad AbD Serotech, respectively. Blots were developed using Clarity Western ECL substrate (Bio-rad) and imaged using an LAS4000 Imager (GE Healthcare Life Sciences).
Site-directed mutagenesis
Point mutants were generated using inverse polymerase chain reaction (PCR) with Phusion DNA polymerase (New England Biolabs). Chimeric Hsh155 plasmids were made by restriction enzyme cloning using a yeast codon optimized sequence for SF3b1 purchased from IDT, and all plasmids were confirmed by sequencing. The sequence exchanged is shown in Supplemental Table 3.
In vitro splicing assays
Splicing extracts were prepared from yeast strains derived from BJ2168 and prepared as previously described (Ansari and Schwer 1995; Hoskins et al. 2011). Extract was flash frozen in liquid nitrogen and stored at −80°C until use. Capped, [32P]-labeled RP51a pre-mRNA substrate was transcribed using T7 RNA polymerase and was gel purified prior to use (Crawford et al. 2008). Splicing reactions consisted of 100 mM KPi pH 7.3, 3% (w/v) PEG 8000, 2.5 mM MgCl2, 1 mM DTT, 2 mM ATP, 40 U Murine RNase Inhibitor (New England Biolabs), 300 pM [32P]-labeled pre-mRNA, and 40% (v/v) whole cell extract. Splicing reactions were performed at room temperature and stopped after 45 min by addition of 2 mg/mL Proteinase K, 2.5% (w/v) SDS, and 1 mM EDTA. The samples were heated to 60°C for 10 min followed by addition of deionized formamide loading buffer. Samples were resolved on a 12% polyacrylamide (19:1 acrylamide:bisacrylamide), 8 M urea gel with 1× tris/borate/EDTA (TBE) buffer. Gels were dried and imaged using a storage phosphorscreen and Typhoon FLA 9500 phosphorimager (GE Healthcare Life Sciences). Gels were quantified using ImageJ software (Schneider et al. 2012).
Splicing inhibition assays
PB was purchased from Santa Cruz Biotechnology. PB was dissolved in DMSO and used at concentrations ranging from 0.1–20 μM. Extract was incubated with PB for 10 min at room temperature prior to the addition of pre-mRNA to initiate splicing. DMSO alone was added to samples not containing PB. Relative fraction spliced was calculated as [(MPB + LPB)/(MPB + LPB + PPB)]/[(MUntreated + LUntreated)/(MUntreated + LUntreated + PUntreated)] for individual strains to account for differences in extract activity. Relative 1st step activity was calculated as: [(LPB)/(LPB + PPB)]/[(LUntreated)/(LUntreated + PUntreated)]. M is mature, L is lariat intermediate, and P is pre-mRNA and the subscripts “PB” and “Untreated” indicate results obtained in the presence or absence of PB, respectively.
Temperature sensitivity and growth assays
Yeast were grown to mid-log phase, adjusted to OD600 = 0.5 and plated on YPD media in 10-fold serial dilutions. Plates were incubated for 2 d at 30°C, and 37°C, 3 d at 23°C, and 10 d at 16°C prior to imaging. Assays examining the functionality of Hsh155 mutants and truncations compared growth to WT Hsh155.
ACT1-CUP1 copper assays
ACT1-CUP1 reporters and growth assays have been described previously (Lesser and Guthrie 1993; Carrocci et al. 2017). Briefly, yeast strains expressing WT or mutant proteins and ACT1-CUP1 reporters were grown to mid-log phase in the appropriate media to maintain selection for the plasmids, adjusted to OD600 = 0.5 and equal volumes were spotted onto plates containing 0, 0.025, 0.05, 0.075, 0.1, 0.15, 0.2, 0.25, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.25, or 2.5 mM CuSO4. Plates were scored after 3 d growth at 30°C.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eric Montemayor (UW-Madison) for help in creating Supplemental Figure 6. We thank Manny Ares (UC Santa Cruz), Silvia Buonamici (H3 Biomedicine), Melissa Jurica (UC Santa Cruz), Corina Maeder (Trinity University), and Charles Query (Albert Einstein College of Medicine) for helpful discussions. We thank Allison Didychuk and Clarisse van der Feltz for their comments on the manuscript. We thank Brandon Nikolai for technical help with Supplemental Figure 5. This work was supported by the National Institute of General Medical Sciences, National Institutes of Health (R00 GM086471, R01 GM112735 to A.A.H.; T32-GM08349, R01- HL133406 to T.J.C.); a Shaw Scientist Award, Greater Milwaukee Foundation; a Beckman Young Investigator Award (Arnold and Mabel Beckman Foundation); startup funding from the University of Wisconsin-Madison, Wisconsin Alumni Research Foundation (WARF), and the Department of Biochemistry; and a William H. Peterson Fellowship (T.J.C.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.065664.118.
REFERENCES
- Agrawal AA, Yu L, Smith PG, Buonamici S. 2017. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev 48: 67–74. [DOI] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, Schwer B. 1995. SLU7 and a novel activity, SSF1, act during the PRP16-dependent step of yeast pre-mRNA splicing. EMBO J 14: 4001–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao P, Höbartner C, Hartmuth K, Lührmann R. 2017a. Yeast Prp2 liberates the 5′ splice site and the branch site adenosine for catalysis of pre-mRNA splicing. RNA 23: 1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao P, Will CL, Urlaub H, Boon KL, Lührmann R. 2017b. The RES complex is required for efficient transformation of the precatalytic B spliceosome into an activated B complex. Genes Dev 31: 2416–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Dybkov O, Haselbach D, Leelaram MN, Will CL, Urlaub H, Kastner B, Lührmann R, Stark H. 2017. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 170: 701–713.e11. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Vigevani L, Valcárcel J. 2012. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov 11: 847–859. [DOI] [PubMed] [Google Scholar]
- Boudrez A, Beullens M, Waelkens E, Stalmans W, Bollen M. 2002. Phosphorylation-dependent interaction between the splicing factors SAP155 and NIPP1. J Biol Chem 277: 31834–31841. [DOI] [PubMed] [Google Scholar]
- Brow DA. 2002. Allosteric cascade of spliceosome activation. Annu Rev Genet 36: 333–360. [DOI] [PubMed] [Google Scholar]
- Carrocci TJ, Zoerner DM, Paulson JC, Hoskins AA. 2017. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res 45: 4837–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Moore MJ. 2014. The spliceosome: disorder and dynamics defined. Curr Opin Struct Biol 24: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Miñana B, Valcárcel J. 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ. 2008. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA 14: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu C, Schmitzová J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Lührmann R, Pena V. 2016. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell 64: 307–319. [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. [DOI] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. 2016. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16: 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Anderson DD, Bray WM, Prichard BE, Ma N, Adams MS, Ghosh AK, Jurica MS. 2014. Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J Biol Chem 289: 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS. 2016. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 22: 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Urabe VK, Jurica MS. 2017. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA 8: e1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finci LI, Huang X, Zhou Q, Tsai J, Teng T, Agrawal A, Chan B, Irwin S, Karr C, Cook A, et al. 2018. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev 32: 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Coil KE, Reed R. 2011. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev 25: 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould GM, Paggi JM, Guo Y, Phizicky DV, Zinshteyn B, Wang ET, Gilbert WV, Gifford DK, Burge CB. 2016. Identification of new branch points and unconventional introns in Saccharomyces cerevisiae. RNA 22: 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Potashkin J, Reed R. 1998. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 18: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habara Y, Urushiyama S, Tani T, Ohshima Y. 1998. The fission yeast prp10+ gene involved in pre-mRNA splicing encodes a homologue of highly conserved splicing factor, SAP155. Nucleic Acids Res 26: 5662–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NL, Senapathy P. 1990. Distribution and consensus of branch point signals in eukaryotic genes: a computerized statistical analysis. Nucleic Acids Res 18: 3015–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, Neugebauer KM. 2017. Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol 18: 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Moore MJ. 2012. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci 37: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ. 2011. Ordered and dynamic assembly of single spliceosomes. Science 331: 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JL, Kielkopf CL. 2017. Splicing factor mutations in myelodysplasias: insights from spliceosome structures. Trends Genet 33: 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf CL. 2017. Insights from structures of cancer-relevant pre-mRNA splicing factors. Curr Opin Genet Dev 48: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli RM, Thompson JX, Yates JR III, Stevens SW. 2010. Release of SF3 from the intron branch point activates the first step of pre-mRNA splicing. RNA 16: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C. 1993. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WW, Cheng SC. 2015. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev 29: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HL, Cheng SC. 2012. The interaction of Prp2 with a defined region of the intron is required for the first splicing reaction. Mol Cell Biol 32: 5056–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CW, Kaida D, Nishimura S, Matsuyama A, Yashiroda Y, Taoka H, Ishigami K, Watanabe H, Nakajima H, Tani T, et al. 2007. Inhibition of splicing and nuclear retention of pre-mRNA by spliceostatin A in fission yeast. Biochem Biophys Res Commun 364: 573–577. [DOI] [PubMed] [Google Scholar]
- McPheeters DS, Muhlenkamp P. 2003. Spatial organization of protein-RNA interactions in the branch site-3′ splice site region during pre-mRNA splicing in yeast. Mol Cell Biol 23: 4174–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Andersen SB, Brunck ME, Haerty W, Crawford J, Taft RJ, Nielsen LK, Dinger ME, Mattick JS. 2015. Genome-wide discovery of human splicing branchpoints. Genome Res 25: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munding EM, Shiue L, Katzman S, Donohue JP, Ares M Jr. 2013. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol Cell 51: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, et al. 2017. Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife 6: e23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Ares M. 2010. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell 38: 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaschka C, Lin PC, Nagai K. 2017. Structure of a pre-catalytic spliceosome. Nature 546: 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D, Huang L, Wlodaver A, Andrade J, Staley JP. 2016. Sequencing of lariat termini in S. cerevisiae reveals 5′ splice sites, branch points, and novel splicing events. RNA 22: 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query CC, Moore MJ, Sharp PA. 1994. Branch nucleophile selection in pre-mRNA splicing: evidence for the bulged duplex model. Genes Dev 8: 587–597. [DOI] [PubMed] [Google Scholar]
- Rauhut R, Fabrizio P, Dybkov O, Hartmuth K, Pena V, Chari A, Kumar V, Lee CT, Urlaub H, Kastner B, et al. 2016. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science 353: 1399–1405. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Agafonov DE, Schmitzová J, Hartmuth K, Fabrizio P, Lührmann R. 2015. Dynamic contacts of U2, RES, Cwc25, Prp8 and Prp45 proteins with the pre-mRNA branch-site and 3′ splice site during catalytic activation and step 1 catalysis in yeast spliceosomes. PLoS Genet 11: e1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. 2018. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Konarska MM, Query CC. 2009. Insights into branch nucleophile positioning and activation from an orthogonal pre-mRNA splicing system in yeast. Mol Cell 34: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Rodriguez-Santiago S, Wang J, Pu J, Yuste A, Gupta V, Moldón A, Xu YZ, Query CC. 2016. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev 30: 2710–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. 2017. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun 8: 15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thickman KR, Swenson MC, Kabogo JM, Gryczynski Z, Kielkopf CL. 2006. Multiple U2AF65 binding sites within SF3b155: thermodynamic and spectroscopic characterization of protein–protein interactions among pre-mRNA splicing factors. J Mol Biol 356: 664–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigevani L, Gohr A, Webb T, Irimia M, Valcárcel J. 2017. Molecular basis of differential 3′ splice site sensitivity to anti-tumor drugs targeting U2 snRNP. Nat Commun 8: 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wang C, Chua K, Seghezzi W, Lees E, Gozani O, Reed R. 1998. Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes Dev 12: 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest DK, O'Day CL, Abelson J. 1996. In vitro studies of the Prp9.Prp11.Prp21 complex indicate a pathway for U2 small nuclear ribonucleoprotein activation. J Biol Chem 271: 33268–33276. [DOI] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Persp Biol 3: a005207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Query CC. 2007. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28: 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2016. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 353: 904–911. [DOI] [PubMed] [Google Scholar]
- Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane K, Hamaguchi M, Iwata M, et al. 2011. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J 278: 4870–4880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.