

In this study, Lanz et al. investigated how the Mec1/ATR kinase functions in genome maintenance and replication and, using a novel genetic system to spatially manipulate Mec1 activation and action, show that the ability of Mec1 to suppress genomic instabilities is separate from a novel role in promoting DNA replication. These findings establish that the Mec1/ATR kinase initiates checkpoint signaling, promotes DNA replication, and maintains genetic stability through distinct modes of action.
Keywords: Mec1, ATR, Dna2, Dpb11, DNA replication, gross chromosomal rearrangements
Abstract
The Mec1/ATR kinase coordinates multiple cellular responses to replication stress. In addition to its canonical role in activating the checkpoint kinase Rad53, Mec1 also plays checkpoint-independent roles in genome maintenance that are not well understood. Here we used a combined genetic–phosphoproteomic approach to manipulate Mec1 activation and globally monitor Mec1 signaling, allowing us to delineate distinct checkpoint-independent modes of Mec1 action. Using cells in which endogenous Mec1 activators were genetically ablated, we found that expression of “free” Mec1 activation domains (MADs) can robustly activate Mec1 and rescue the severe DNA replication and growth defects of these cells back to wild-type levels. However, unlike the activation mediated by endogenous activator proteins, “free” MADs are unable to stimulate Mec1-mediated suppression of gross chromosomal rearrangements (GCRs), revealing that Mec1's role in genome maintenance is separable from a previously unappreciated proreplicative function. Both Mec1's functions in promoting replication and suppressing GCRs are independent of the downstream checkpoint kinases. Additionally, Mec1-dependent GCR suppression seems to require localized Mec1 action at DNA lesions, which correlates with the phosphorylation of activator-proximal substrates involved in homologous recombination-mediated DNA repair. These findings establish that Mec1 initiates checkpoint signaling, promotes DNA replication, and maintains genetic stability through distinct modes of action.
DNA replication is an inherently stressful process. Progression of the replication machinery is often impeded by barriers such as DNA adducts, DNA–RNA hybrids, and protein–DNA complexes (Branzei and Foiani 2010; Lambert and Carr 2013). Replication forks may stall or collapse at these obstacles or at DNA lesions, leading to the exposure of stretches of ssDNA, which signal for the activation of the evolutionarily conserved PI3K-like kinase ATR (yeast Mec1) (Zou and Elledge 2003; MacDougall et al. 2007). Mec1/ATR coordinates multiple cellular responses to DNA replication stress through the phosphorylation of a wide range of substrates (Matsuoka et al. 2007; Smolka et al. 2007; Bastos de Oliveira et al. 2015).
In its canonical role, Mec1/ATR initiates the DNA damage checkpoint by phosphorylating and activating the CHK1 kinase (yeast Rad53) (Sanchez et al. 1996; Liu et al. 2000). Once activated, Rad53/CHK1 signaling induces hallmarks of the checkpoint response, including cell cycle arrest, inhibition of origin firing, increased dNTP levels, and the stabilization of stalled replication forks (Santocanale and Diffley 1998; Lopes et al. 2001; Chabes et al. 2003; Cobb et al. 2005; Ge and Blow 2010). The DNA damage checkpoint is necessary for the response to genotoxic stresses, and cells lacking either Mec1/ATR or Rad53/CHK1 are exquisitely sensitive to exogenous genotoxins that damage DNA or stall DNA replication forks (Weinert et al. 1994; Zhou and Elledge 2000).
Mounting evidence reveals that Mec1/ATR also plays crucial roles independently of the DNA damage checkpoint. For example, in budding yeast, the genotoxic sensitivity of rad53Δ, but not mec1Δ, cells can be rescued by the removal of the EXO1 exonuclease (Segurado and Diffley 2008). Mec1 may also directly phosphorylate MCM helicase proteins, priming prereplication complexes for origin firing in a manner redundant to cyclin-dependent kinases (CDKs)/Dbf4-dependent kinase (DDKs) (Randell et al. 2010). Mec1 was also shown to inhibit de novo telomere addition at DNA break sites by phosphorylating Cdc13 (Zhang and Durocher 2010). In addition to these roles, well-documented genetic evidence indicates that Mec1 performs key functions for genome maintenance that are largely checkpoint-independent. Specifically, yeast lacking Mec1 exhibits high genetic instability in the form of gross chromosomal rearrangements (GCRs) during unperturbed cell proliferation, yet yeast lacking downstream checkpoint factors (e.g., rad53Δ) does not (Myung et al. 2001). Despite the importance of this checkpoint-independent role for Mec1, the mechanism by which it suppresses GCRs is unknown.
Activation of Mec1/ATR relies on its recruitment to ssDNA via its binding partner, Ddc2/ATRIP (Rouse and Jackson 2000; Myung et al. 2001). Of importance, Mec1/ATR is mostly inactive upon recruitment and requires interaction with specific stimulatory proteins for activation. In yeast, proteins reported to have the ability to activate Mec1 include Ddc1, Dpb11, and Dna2, all of which are known to associate directly or indirectly with DNA structures formed at replication forks or post-replicative lesions (Mordes et al. 2008b; Navadgi-Patil and Burgers 2008, 2009; Kumar and Burgers 2013). Until recently, only the ortholog of Dpb11, TOPBP1, had been shown to activate ATR in mammals (Kumagai et al. 2006). However, recent reports have revealed that ETAA1, a protein implicated previously in the response to DNA replication stress, can also activate ATR (Bass et al. 2016; Haahr et al. 2016; Lee et al. 2016). Therefore, similar to Mec1 in yeast, ATR can also use distinct proteins for activation. All of these activators possess Mec1 activation domains (MADs)/ATR activation domains (AADs), which are unstructured domains that physically mediate the stimulation of Mec1/ATR kinase activity (Kumagai et al. 2006; Mordes et al. 2008a; Wanrooij et al. 2016). Mec1/ATR activators are critical for the initiation of the DNA damage checkpoint and appear to function in a highly redundant manner, since the complete disruption of Rad53 activation upon exposure to genotoxins requires the concomitant disruption of all endogenous MADs (Kumar and Burgers 2013). We reported recently the unexpected finding that Dna2 and Ddc1–Dpb11 are also required for a replication-correlated mode of Mec1 signaling that is distinguishable from the action of Mec1 in activating Rad53 (Bastos de Oliveira et al. 2015). Replication-correlated signaling is robustly active during normal S phase (independently of genotoxin-induced replication stress) and impinges on ∼80 different substrate proteins (Bastos de Oliveira et al. 2015).
Here, to further investigate Mec1-dependent GCR suppression and potentially other checkpoint-independent functions of Mec1, we developed a combined genetic–phosphoproteomic approach in which we manipulated Mec1 activation and globally monitored the impact on Mec1 signaling output and function. Using this system, we demonstrate that the expression of “free” MADs is sufficient for robust Mec1 kinase activation and supports wild-type-like growth and DNA replication rates. However, “free” MADs are unable to stimulate Mec1-mediated suppression of GCRs, which specifically requires activation via MADs attached to proteins in proximity to DNA lesions (e.g., Ddc1–Dpb11 and Dna2). Strikingly, we found that Mec1's functions in both promoting DNA replication and suppressing GCRs are independent of the downstream checkpoint kinases.
Results
A genetic system for the manipulation of Mec1 activation
Despite the common depiction of Mec1/ATR as a checkpoint kinase, multiple lines of genetic evidence indicate that Mec1 has important checkpoint-independent functions. Yeast cells lacking Mec1 are significantly more genetically unstable than yeast cells lacking downstream checkpoint factors (Fig. 1A; Myung et al. 2001). This is especially apparent in the absence of the related PI3K-like kinase Tel1 (human ATM), which plays partially redundant roles with Mec1 (Morrow et al. 1995). Notably, mec1Δ tel1Δ cells have drastically elevated GCR rates and display a severe growth defect that is not observed in cells lacking Rad53 signaling (Fig. 1A; Supplemental Fig. S1A). How Mec1 operates independently of Rad53 to prevent genomic instability and promote proper cell growth has remained one of the most fundamental knowledge gaps in our understanding of Mec1 function.
Figure 1.
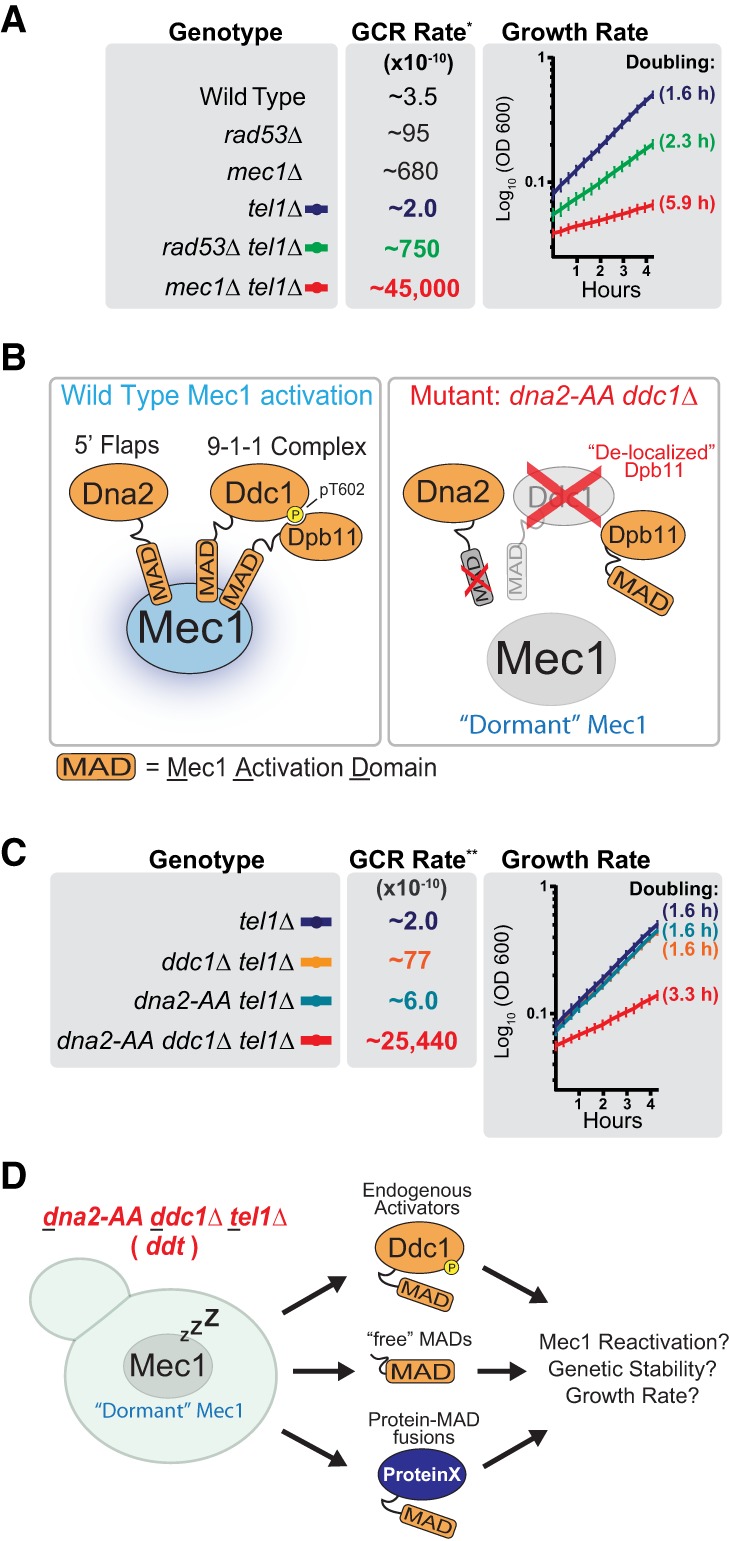
A genetic system for the manipulation of Mec1 activation. (A) GCR rate and log phase growth rate of the indicated strains. The doubling time during log phase growth in YPD medium is indicated in parentheses. All strains are sml1Δ. Error bars represent the standard deviation of at least six replicate cultures. The results depicted are representative of multiple independent experiments. For extended growth curves, see Supplemental Figure S1A. (*) GCR data from Myung et al. (2001). (B) Rationale for the generation of a strain in which removal or disruption of endogenous MADs results in a Mec1 kinase that is largely inactive (“dormant”). The dna2-AA strain contains the W128A and Y130A mutations. (C) GCR rate and log phase growth rate of strains with and without functional endogenous Mec1 activators. The doubling time during log phase growth in YPD medium is indicated in parentheses. All strains are sml1Δ. For extended growth curves, see Supplemental Figure S1A. (**) GCR data from Bastos de Oliveira et al. (2015). (D) Scheme describing the use of the ddt strain for the manipulation of Mec1 activation. All ddt and ddt derivative strains used in this work are sml1Δ unless noted otherwise.
To dissect the checkpoint-independent functions of Mec1, we engineered a genetic system that allows us to manipulate Mec1 activation and, as a result, its action. This system is based on the combined disruption of the Mec1 activators by mutation of the MAD of Dna2 (dna2-AA mutant) and deletion of DDC1, which also leads to delocalized Dpb11 (Navadgi-Patil and Burgers 2009; Balint et al. 2015). In the dna2-AA ddc1Δ strain, Mec1 should be in a “dormant,” largely inactive state (Fig. 1B). Consistent with this prediction and with previous work (Kumar and Burgers 2013; Bastos de Oliveira et al. 2015), deletion of TEL1 in the dna2-AA ddc1Δ strain closely mimics the high GCR rate of mec1Δ tel1Δ cells and also results in a slow growth phenotype (Fig. 1C; Supplemental Fig. S1A). We note that deletion of either DDC1 or DNA2 does not result in high GCR rates or slow growth even when combined with deletion of TEL1 (Fig. 1C). The dna2-AA ddc1Δ tel1Δ strain (referred to here as the ddt strain) forms the basis of our system, as various constructs can be introduced to differentially manipulate the reactivation of “dormant” Mec1 (Fig. 1D). Of note, mec1Δ and rad53Δ are not viable unless dNTP pools are increased via the deletion of SML1 (Zhao et al. 1998). Thus, all ddt and ddt derivative strains were generated in a sml1Δ background (unless indicated otherwise).
Expression of ‘free’ MADs can rescue the growth defect of ddt
Since the endogenous MADs are imbedded within proteins known to localize to DNA lesions and stalled replication forks (Tkach et al. 2012), we first sought to test whether Mec1 requires activation via MADs within their native proteins or whether the expression of “free” untethered MADs is sufficient to promote activation of Mec1 in vivo (Fig. 2A). Despite lacking sequence homology, the MADs of Dpb11, Ddc1, and Dna2 are all predicted to be unstructured in nature and possess two bulky hydrophobic residues that are critical for stimulating Mec1's catalytic activity (Fig. 2A). Remarkably, ectopic expression of any one of the three yeast MADs (from Ddc1, Dpb11, or Dna2) was able to rescue the severe growth defect of ddt cells (Fig. 2B; Supplemental Fig. S1B). In fact, we found that medium-level expression of MADDNA2 (representing amino acids 1–450 of Dna2; CYC1 promoter) rescues the growth of ddt cells nearly as well as the reintroduction of full-length Ddc1 (DDC1 promoter) (Fig. 2B). Importantly, expression of “free” MADs in a strain lacking the Mec1 kinase had no observable effect on growth (Fig. 2C). The extent of the growth rescue correlated with the level of MAD expression (Fig. 2D). However, drastic overexpression of MADDNA2 (ADH1 promoter) began to negatively affect the growth rescue (data not shown); thus, the promoters used in Figure 2B represent the optimal level of growth rescue for each MAD tested. Relative expression levels of MADDNA2 and MADDPB11 (representing amino acids 572–764 of Dpb11) under the control of ADH1 and CPY1 promoters were assessed by Western blot (Supplemental Fig. S1D).
Figure 2.

Mec1 activation via “free” MADs rescues the growth defect of ddt strains independently of downstream checkpoint kinases. (A) A scheme describing the use of the ddt strain to test the effect of expressing “free” MADs in vivo. The cartoon diagram indicates the amino acid residue numbers corresponding to the regions in Dna2, Dpb11, and Ddc1 used as “free” MADs. “W” and “Y” represent the approximate locations of the hydrophobic residues necessary for Mec1 activation. (B) Growth rates of the ddt strain transformed with pRS415 plasmids with the indicated inserts. The doubling time during log phase growth in −Leu medium is indicated in parenthesis. Error bars represent the standard deviation of at least six replicate cultures. The results depicted are representative of multiple independent experiments. For extended growth curves, see Supplemental Figure S1B. (C) The effect of MADDNA2 expression on the growth of ddt and mec1Δ tel1Δ strains. Strains are sml1Δ. (D) Growth of ddt cells transformed with pRS415 plasmids for expression of “free” MADDPB11 using the following promoters of increasing strength: PDPB11, PCYC1, and PADH1. Log phase liquid cultures were concentrated and spotted onto a −Leu plate using a replicator. Strains are sml1Δ. (E) Growth and MMS sensitivity of ddt cells transformed with pRS415 plasmids with the indicated inserts. Of note, since MAD-expressing plasmids are self-selected in the ddt strain, cells were not grown under auxotrophic selection. Similar results were obtained when cells were spotted on −Leu plates (Supplemental Fig. S1C). (F) MMS-induced Rad53 phospho-shift in response to Mec1 activation by “free” MADs or Ddc1. Asynchronous cultures were treated with 0.02% MMS for 3 h. (G) The growth and MMS sensitivity of ddt rad53Δ chk1Δ cells transformed with pRS415 plasmids with the indicated inserts. (H) Mec1 promotes growth independently of the downstream canonical checkpoint.
The growth rescue elicited by ‘free’ MADs in ddt is independent of the downstream checkpoint kinases
We next asked whether “free” MADs were also capable of initiating the checkpoint response upon exposure to genotoxic stress. While expression of full-length Ddc1 in ddt cells conferred resistance to MMS and enabled robust checkpoint activation through the phosphorylation of Rad53, expression of “free” MADs did not (Fig. 2E,F). We next tested whether the growth rescue elicited by “free” MADs was also dependent on the action of the downstream checkpoint kinases Rad53 and Chk1. Surprisingly, deletion of both checkpoint effector kinases did not preclude the growth rescue elicited by “free” MADs or Ddc1 (Fig. 2G). To our knowledge, this is the first definitive demonstration that Mec1 promotes cell growth independently of the canonical checkpoint pathways (Fig. 2H).
Separable checkpoint-independent roles for Mec1 in promoting growth and suppressing GCRs
To test whether, in addition to promoting cell growth, the expression of free MADs also restores genetic stability in ddt cells, we designed a plasmid-compatible GCR assay to assess changes in genetic stability within the ddt strain background (Fig. 3A). Surprisingly, we found that although expression of “free” MADDPB11 or expression of “free” MADDNA2 could rescue the growth defects of ddt cells, they were unable to restore genetic stability to the same extent as Ddc1 expression (Fig. 3B,C), suggesting that Mec1 suppresses GCRs through a mode of action that requires activation via the endogenous activators. The rescue of genetic stability in ddt cells through the expression of Ddc1 was independent of the canonical DNA damage checkpoint, as the downstream kinases Rad53 and Chk1 were dispensable for Ddc1-dependent rescue of GCR suppression (Fig. 3D,E; Supplemental Fig. S2A,B). We were unable to include an empty vector control in the analyses of ddt rad53Δ cells in Figure 3E, as these cells with empty vector form only barely visible microcolonies on +5-FOA/+Can plates (Supplemental Fig. S2C). Taken together, since “free” MADs could almost fully rescue the growth defect of ddt but were unable to restore genetic stability, we conclude that the slow growth phenotype of strains lacking Mec1 and Tel1 signaling is not a consequence of the accumulation of GCRs but results from the disruption of other, currently unknown, functions of Mec1. Collectively, these data demonstrate that Mec1 plays separable checkpoint-independent roles in suppressing GCRs and promoting cell growth (Fig. 3F) and suggest that these roles are distinguished by the context of Mec1 activation.
Figure 3.
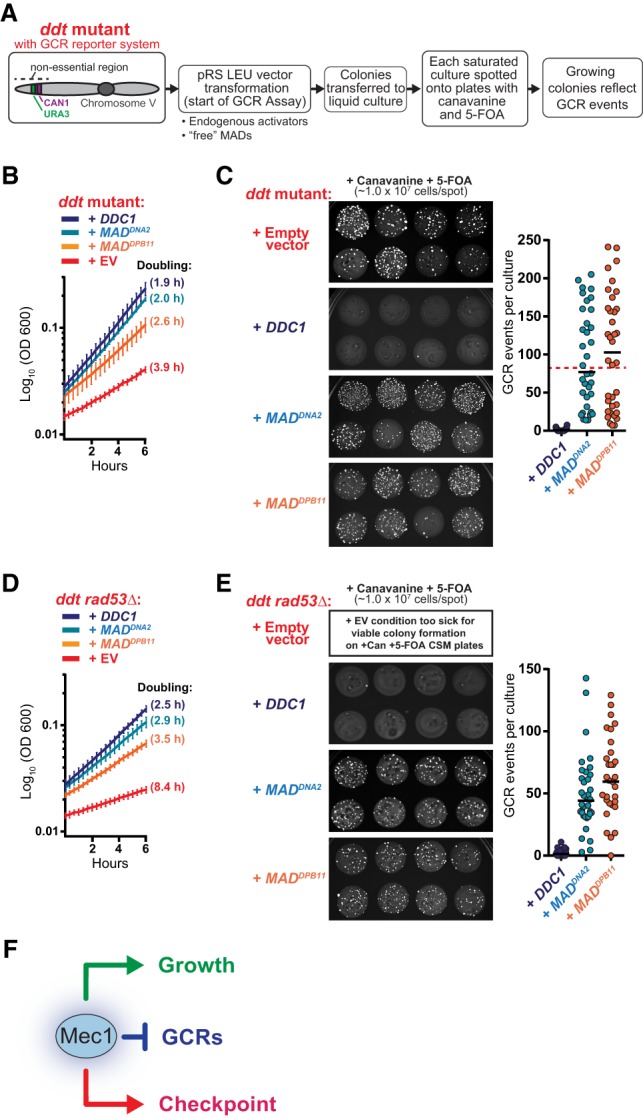
Mec1 activation via “free” MADs rescues the growth defect but not the genetic instability of ddt cells. (A) Experimental design for assessing how different modes of Mec1 reactivation impact the suppression of GCRs in ddt. (B) Growth rate of the ddt strain transformed with pRS415 plasmids with the indicated inserts (as in Fig. 2B; Supplemental Fig. S1B). (C) Measurement of GCR events in the ddt strain transformed with the indicated pRS415 plasmids. Representative spots from independent saturated −Leu cultures carrying the indicated plasmids are shown. The approximate number of GCR events per culture was calculated based on the number of canavanine/5-FOA-resistant colonies per spot. Each data point in the dot plot corresponds to the estimated number of GCR events that occurred in an independent culture. For DDC1, n = 36; for MADDNA2, n = 35; for MADDPB11, n = 36. The red dashed line indicates the approximate median level of GCR events for the ddt strain, as described previously (Bastos de Oliveira et al. 2015). (D,E) Experiments performed as described in C and D, respectively, but using a ddt rad53Δ strain. For extended growth curves, see Supplemental Figure S2A. When carrying an empty vector, the ddt rad53Δ strain grows very poorly in minimal medium and is only capable of forming tiny colonies on +Can +5-FOA plates (Supplemental Fig. S2C). (F) A model depicting separable roles for Mec1 in GCR suppression, checkpoint signaling, and growth.
Recruitment of MADs at DNA lesions is required for Mec1-mediated GCR suppression
To further investigate the context of Mec1 activation required for proper GCR suppression, we assessed how the assembly of the Ddc1–Dpb11 complex impacted the ability of Mec1 to suppress GCRs. Ddc1 is a component of the yeast 9-1-1 DNA clamp that is loaded at the 5′ end of dsDNA:ssDNA junctions (Majka et al. 2006). The recruitment of Dpb11 to the 9-1-1 complex is dependent on the Mec1-mediated phosphorylation of Ddc1 at Thr602 (Fig. 4A; Puddu et al. 2008). Since “free” MADs are unable to restore GCR suppression in ddt cells, we hypothesized that the ability of Mec1 to suppress GCRs relies on its efficient local activation at DNA lesions. To test this hypothesis, we asked whether disrupting the recruitment of Dpb11 to the 9-1-1 complex might impair the ability of Mec1 to suppress GCRs. Consistent with this hypothesis, reintroduction of a ddc1 phospho-mutant (ddc1-T602A; Fig. 4A) rescued the growth defects of ddt cells (Fig. 4B; Supplemental Fig. S1B) but could not restore genetic stability to the extent of wild-type DDC1 (Fig. 4C). Furthermore, we performed a similar experiment using a ddt strain that also lacked the endogenous MAD of Dpb11 (ddt dpb11-601) (Fig. 4A; Navadgi-Patil and Burgers 2008) and found that, in these cells, expression of wild-type DDC1 or ddc1-T602A does not efficiently rescue GCRs (Fig. 4D,E; Supplemental Fig. S3). Together, these findings suggest that the recruitment of MADs at DNA lesions—specifically at the 5′ ends of dsDNA:ssDNA junctions—promotes a localized mode of Mec1 activation that is required for GCR suppression and cannot be recapitulated with “free” MADs.
Figure 4.
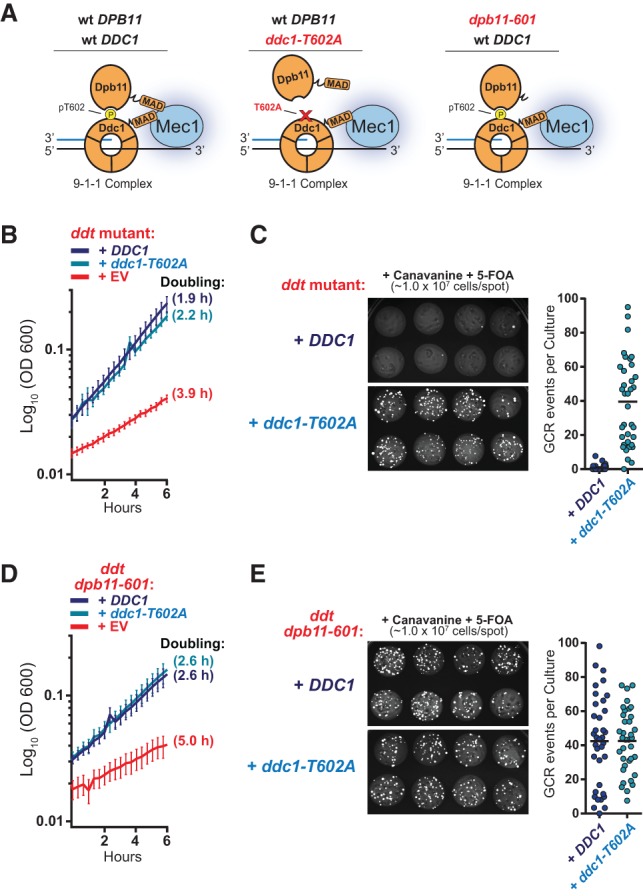
Mec1-mediated GCR suppression requires the recruitment of MADs to the 9-1-1 complex. (A) A schematic representation for how the Ddc1–Dpb11 activator complex assembles with Mec1 at DNA lesions with 5′ junctions and the expected impact of the indicated mutations. For simplicity, RPA and Ddc2 are not depicted. (B) The growth rate of the ddt strain transformed with pRS415 plasmids with the indicated inserts. DDC1 and ddc1-T602A were expressed under the DDC1 promoter. Error bars represent the standard deviation of at least six replicate cultures. The results depicted are representative of multiple independent experiments. For extended growth curves, see Supplemental Figure S1B. (C) Measurement of GCR events in the ddt strain transformed with pRS415 plasmids with the indicated inserts. The experiment was performed as described in Figure 3A. (D,E) Experiments performed as described in B and C, respectively, with a ddt strain in which endogenous Dpb11 is lacking its MAD domain (dpb11-601). For extended growth curves, see Supplemental Figure S3.
Phosphoproteomics delineate context-dependent Mec1 activation
To further dissect Mec1's checkpoint-independent functions in promoting growth and suppressing GCRs, we used quantitative mass spectrometry (MS) analysis of phosphosubstrates (QMAPS) (Bastos de Oliveira et al. 2015, 2018) to globally monitor the phosphorylation of Mec1 substrates in response to activation via “free” MADs or endogenous activators. Consistent with their ability to efficiently rescue the growth rates of ddt cells, we found that “free” MADs robustly stimulate the kinase activity of Mec1, promoting the phosphorylation of dozens of Mec1 substrates (Fig. 5A). For almost all detected substrates (including Mec1 autophosphorylation at Ser38), the level of phosphorylation promoted by “free” MADs was comparable with, or even higher than, the level achieved when Mec1 was activated via Ddc1–Dpb11 (Fig. 5B, see “growth-correlated” substrates), which strongly suggests that the inability of the “free” MADs to rescue genetic stability in ddt is not due to a lesser amount of overall Mec1 kinase activity. Strikingly, activation via the Ddc1–Dpb11 complex, but not via “free” MADs, promoted robust phosphorylation of the Mec1 substrates Rtt107 and Slx4 (Fig. 5B, see substrates “correlated with GCR suppression”), which are protein scaffolds that have multiple roles in genome maintenance, including homologous recombination (HR)-mediated DNA repair (Ii and Brill 2005; Liberi et al. 2005; Balint et al. 2015; Hang et al. 2015; Dibitetto et al. 2016; Liu et al. 2017). The unique ability of the Ddc1–Dpb11 complex to promote the phosphorylation of Rtt107 and Slx4 by Mec1 was also readily detected in untreated asynchronous cultures (Fig. 5C).
Figure 5.
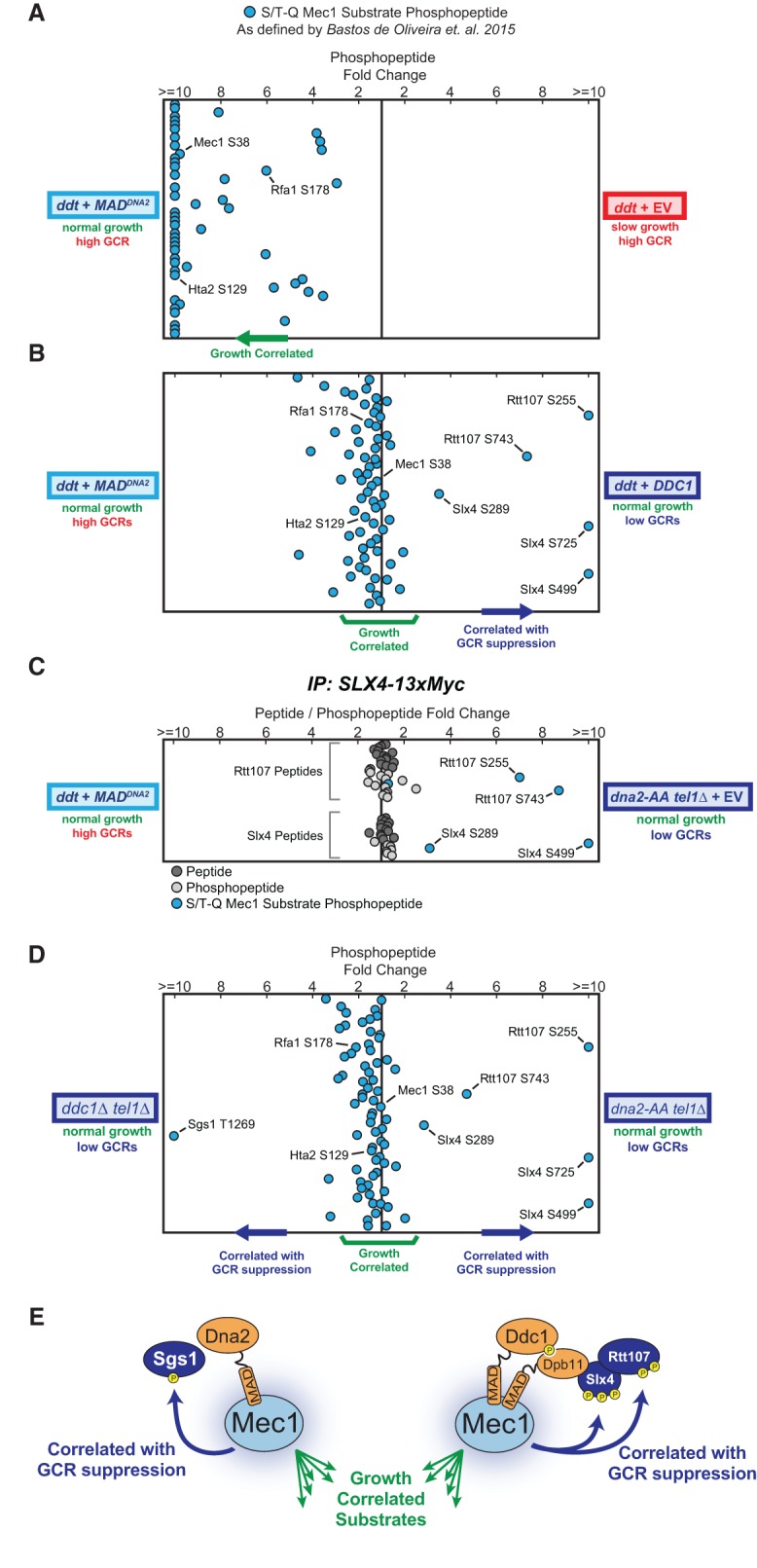
Quantitative phosphoproteomic profiles of Mec1 signaling in response to different modes of activation. QMAPS showing the relative abundance of pS/pT-Q Mec1 substrates. For each plot, each dot corresponds to a phosphopeptide containing a Mec1/Tel1-dependent and Rad53-independent phosphorylation event (as defined in Bastos de Oliveira et al. 2015). The horizontal position of each dot on a plot is determined by the relative fold abundance change of the phosphopeptide in the two conditions being compared (indicated at the left and right of the plots in A–D). The Y-axis is arbitrary. In A, B, and D, cells were synchronized in S phase by treating with 0.03% MMS for 2 h. All presented phosphopeptides were detected and quantified in at least two independent experiments (complete data are shown in Supplemental Table S1). (C) Quantitative analysis of Slx4 and Rtt107 phosphopeptides from asynchronous and untreated cells. Myc-tagged Slx4 was immunoprecipitated from the indicated strains/plasmids and phosphopeptides enriched by immobilized metal affinity chromatography (IMAC). The plot depicts the fold change of all Rtt107 and Slx4 peptides and phosphopeptides detected at least twice. For the QMAPS in D, using the list of Mec1 targets reported previously in Bastos de Oliveira et al. (2015), we did not identify a substrate specifically targeted in ddc1Δ tel1Δ cells (via Dna2-induced Mec1 signaling). We therefore searched for additional pS/pT-Q phosphopeptides in the data set, which revealed the indicated Sgs1 phosphopeptide. (E) Model for activator-directed phosphorylation of Mec1's GCR-correlated substrates.
Since Slx4 and Rtt107 form a protein complex that directly interacts with Dpb11 at DNA lesions (Ohouo et al. 2010, 2013; Gritenaite et al. 2014; Balint et al. 2015; Cussiol et al. 2015), we hypothesized that the endogenous Mec1 activators (in this case, Dpb11) are able to direct Mec1 phosphorylation to specific lesion-proximal substrates. Consistent with this model, we found that while deletion of DDC1 selectively impaired phosphorylation of Slx4–Rtt107, mutation of Dna2 selectively impaired the Mec1 phosphorylation of Sgs1, a RecQ DNA helicase shown to associate with Dna2 (see Fig. 5D, comparison of the phosphoproteome of ddc1Δ tel1Δ cells with the phosphoproteome of dna2-AA tel1Δ cells) (Cejka et al. 2010; Yimit et al. 2015). Furthermore, the results also suggest that Mec1 activation via Ddc1–Dpb11 or Dna2 may suppress GCRs through redundant mechanisms, since the presence of either endogenous activator alone is sufficient for maintaining genetic stability (Fig. 1C).
Collectively, these QMAPS data indicate that “free” MADs can activate Mec1 to an extent similar to, or even greater than, the endogenous activators (Fig. 5B, growth-correlated substrates). Moreover, the results reveal that in addition to facilitating the phosphorylation of all growth-correlated substrates, the endogenous activators are able to direct Mec1 phosphorylation to particular proteins that associate specifically with each activator (Fig. 5E) and that this “activator-directed” phosphorylation strongly correlates with GCR suppression. Overall, these findings provide a biological rationale for the evolutionary integration of MADs within select endogenous proteins.
Rationally designed protein-MAD fusions can direct Mec1 phosphorylation and restore genetic stability in ddt cells
To further interrogate the model that “activator-directed” Mec1 phosphorylation is important for promoting GCR suppression, we fused MADs directly to the substrates whose phosphorylation correlates with GCR suppression (Rtt107 and Sgs1), with the rationale that these chimeras would bypass the need for the endogenous Mec1 activators (Fig. 6A). Consistent with this rationale, introduction of MAD-Rtt107 into ddt cells promoted the preferential phosphorylation of both Rtt107 and Slx4 (Fig. 6B), mimicking the introduction of DDC1 (Fig. 5B). Similarly, a Sgs1-MAD fusion promoted its own phosphorylation (Fig. 6C). These changes in phosphopeptide abundance are not due to dramatic changes in protein expression, as the abundance of several non-S/T-Q phosphopeptides in Rtt107 and Sgs1 is not strongly affected (Supplemental Fig. S4A). Remarkably, these unnatural MAD fusion proteins were able to suppress genetic instability in ddt cells (Fig. 6D). As a control, the mutation of key hydrophobic residues within the MAD domains of Sgs1-MAD or MAD-Rtt107 impaired their ability to suppress GCRs (Supplemental Fig. S4B). Taken together, these results are consistent with the interpretation that these fusions, as opposed to “free” MADs, are capable of directing Mec1 phosphorylation in a manner similar to the endogenous activators. We note that the observed rescue seems highly specific to MAD-Rtt107 and Sgs1-MAD, as fusion of MADs with other proteins associated with DNA replication and chromatin did not restore genetic stability to the extent of MAD-Rtt107 or Sgs1-MAD (Fig. 6E). Notably, all MAD fusions tested could robustly rescue growth and activate Mec1 (Fig. 6E, top panel; Supplemental Fig. S5). Interestingly, both MAD-Msh6 and MAD-Ctf4 fusions were able to specifically promote robust phosphorylation of S/T-Q sites in Msh6 and Ctf4, respectively (Supplemental Fig. S5), although these phosphorylation events do not seem to be involved in GCR suppression. Moreover, of all MAD fusions tested, the Sgs1-MAD fusion promoted the weakest rescue of the growth defect of ddt cells (Fig. 6E, top panel) despite supporting the most efficient suppression of GCRs. In agreement with this observation, QMAPS comparing Mec1 activation via Sgs1-MAD or “free” MAD shows that the latter promotes more efficient phosphorylation of several growth-correlated substrates (Fig. 6C).
Figure 6.
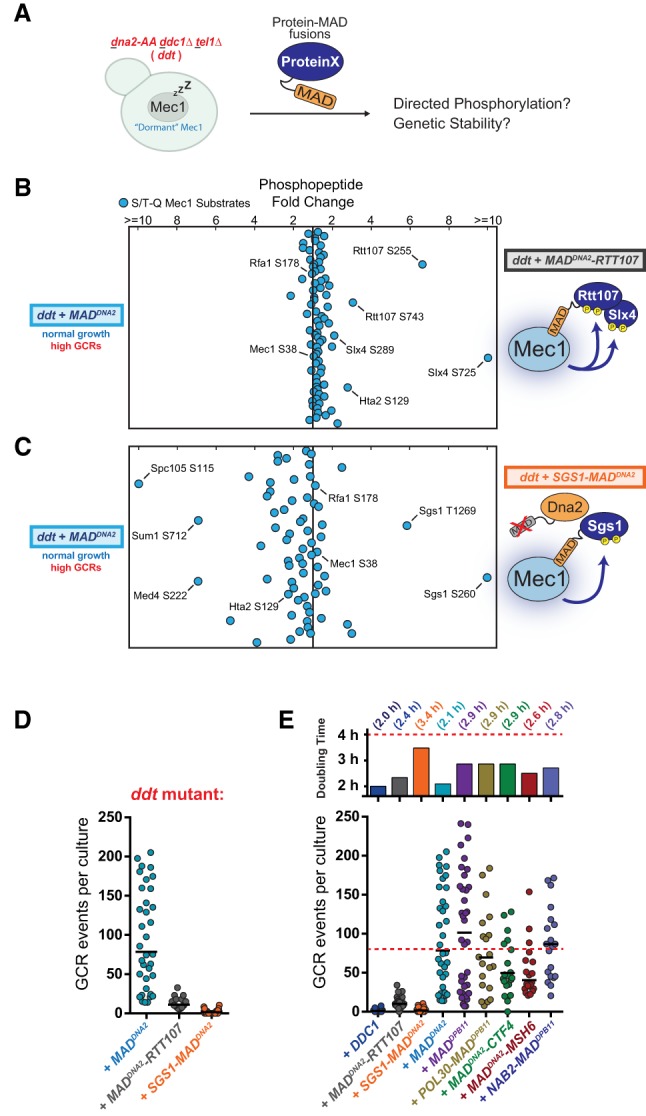
Fusing MADs to Rtt107 or Sgs1 stimulates their phosphorylation and can restore genetic stability in ddt cells. (A) Rationale for testing whether engineered protein-MAD fusions can direct Mec1 signaling and suppress GCRs in ddt cells. (B) QMAPS comparing Mec1 signaling activated by “free” MADDNA2 versus a MADDNA2-RTT107 fusion. Cells were synchronized in S phase using 0.03% MMS. (C) QMAPS comparing Mec1 signaling activated by “free” MADDNA2 versus a SGS1-MADDNA2 fusion. Cells were synchronized in S phase using 0.03% MMS. (D) Measurement of GCR events in the ddt strain transformed with the indicated pRS415 plasmids. MADDNA2-RTT107 and SGS1-MADDNA2 were expressed from the ADH1 promoter. The experiment was performed as described in Figure 3A. (E) Measurement of GCR events and doubling time of the ddt strain transformed with pRS415 plasmids with the indicated inserts. The red dashed line indicates the approximate median level of GCR events for the ddt strain. For extended growth curves and QMAPS demonstrating the ability of these protein-MAD fusions to activate Mec1, see Supplemental Figures S4C and S5.
A noncanonical role for Mec1 in promoting DNA replication
Reactivation of Mec1 in ddt cells promoted a drastic increase in growth rate, but the mechanism underlying this proproliferative effect is unclear. Since Mec1 plays an established role in the regulation of dNTP levels via the downstream checkpoint kinases Rad53 and Dun1 (Zhao and Rothstein 2002; Chabes et al. 2003; Giannattasio and Branzei 2017), we tested whether the growth rescue conferred by Mec1 reactivation in ddt cells requires Rad53 or Dun1. We generated a ddt strain that has additional deletions of both RAD53 and DUN1 and found that the removal of both downstream kinases did not preclude the growth rescue elicited by the introduction of “free” MADs or Ddc1 (Fig. 7A,B; Supplemental Fig. S6A). Consistent with this finding, the presence of the ribonucleotide reductase inhibitor protein Sml1 did not prevent MAD-dependent growth rescue in ddt cells (Supplemental Fig. S6B). To rule out the possibility that Mec1 is increasing dNTP levels in a manner independent of the checkpoint kinases and beyond the increase resulting from the deletion of SML1, we directly measured dNTP concentrations in ddt rad53Δ and ddt rad53Δ dun1Δ cells expressing “free” MADs, Ddc1, or an empty vector. We found that neither “free” MADs nor Ddc1 increased the concentrations of dNTPs in these cells (Fig. 7C). We also assessed the involvement of telomere regulation and found that restoring telomere length in ddt cells does not affect MAD-dependent growth rescue (Supplemental Fig. S7A,B). Collectively, these data demonstrate that Mec1 promotes growth independently of the downstream checkpoint kinases and through a means other than increasing global dNTP pools. The growth rescue is associated with a faster progression of cells through S phase, as revealed by FACS analysis of asynchronous cultures showing that Mec1 activation induced by “free” MADs reduces the accumulation of S-phase cells (Fig. 7D).
Figure 7.

Mec1 promotes and is activated by DNA synthesis. (A,B) Growth rate of the ddt rad53Δ dun1Δ strain transformed with the indicated pRS415 plasmids. For B, the doubling time during log phase growth in −Leu medium is indicated in parenthesis. Error bars represent the standard deviation from at least six replicate cultures. The results depicted are representative of multiple independent experiments. For extended growth curves, see Supplemental Figure S6A. (C) Measurement of dNTP pools in ddt rad53Δ dun1Δ cells transformed with the indicated pRS415 plasmids. (D) FACS analysis of the ddt rad53Δ dun1Δ strain carrying the same plasmids as in A. Asynchronous cultures were captured in log phase growth. G1 arrest and release of ddt rad53Δ dun1Δ strain proved technically challenging because of its severe sickness. (E) The cellular processes associated with Mec1's growth-correlated substrates (as defined in Fig. 5A,B). (F) QMAPS comparing α-factor-arrested tel1Δ cultures mock-treated or treated with 0.4 µg/mL 4-NQO for 50 min. Representative FACS plots for each condition are displayed inside a square below the plot. A Rad53 phospho-shift for experimental conditions assessed in the QMAPS is displayed below the plot. (G) QMAPS comparing tel1Δ cells released from G1 into S phase for 45 min with G1-arrested cultures that were treated with 0.4 µg/mL 4-NQO for 50 min. Representative FACS plots for each condition are displayed inside a square below the plot. A Rad53 phospho-shift for experimental conditions assessed in the QMAPS is displayed below the plot. (H) Model for the separable modes of Mec1 action.
Mec1's proreplicative function is uncoupled from downstream checkpoint signaling during active DNA synthesis
In its canonical checkpoint function, Mec1 phosphorylates Rad53, which in turn inhibits the firing of origins of replication (Santocanale and Diffley 1998). It is important to mention that, while Rad53 also up-regulates dNTPs as part of an emergency response to facilitate DNA repair (Chabes et al. 2003), the overall consequence of triggering Mec1–Rad53 checkpoint signaling is an anti-replicative effect, as judged by a reduction in bulk DNA synthesis within an S phase (Zegerman and Diffley 2010). In this context, the discovery of a proreplicative function for Mec1 reveals an interesting paradox: Mec1 is seemingly able to both promote and inhibit DNA replication through the phosphorylation of either its growth-correlated substrates or Rad53, respectively. This observation led us to more carefully examine the differences between how these two seemingly antagonistic modes of Mec1 signaling are induced. The phosphorylation of Rad53 by Mec1 is generally thought to require exposure of ssDNA, which serves as a recruitment platform. One simple scenario that demonstrates contextual requirements for Mec1's phosphorylation of Rad53 can be seen with 4-NQO treatment in G1-arrested cells. Nucleotide excision repair-dependent processing of 4-NQO-induced DNA lesions generates short ssDNA gaps in G1, which are sufficient for checkpoint activation (Giannattasio et al. 2004). We therefore wondered whether 4-NQO-derived ssDNA exposure in G1 could also stimulate the same set of substrates found to be phosphorylated in growth-correlated Mec1 signaling. In addition to promoting Rad53 hyperphosphorylation, 4-NQO treatment of G1-arrested cells also resulted in robust phosphorylation of the same set of Mec1's growth-correlated substrates (Fig. 7F), suggesting that 4-NQO-derived ssDNA exposure in G1 is sufficient for the activation of both the checkpoint and checkpoint-independent modes of Mec1 signaling. “Free” MADs could also activate Mec1 in response to 4-NQO treatment in G1 (Supplemental Fig. S8), although not as well as Ddc1–Dpb11 (Supplemental Fig. S8), suggesting that “free” MADs may require more extensive ssDNA exposure for full Mec1 activation (as seen in S-phase) (Fig. 5A,B). Importantly, in the absence of 4-NQO treatment, most Mec1 substrate phosphopeptides are below the level of detection in G1-arrested cells (Supplemental Table S5). Therefore, our findings are consistent with a model in which ssDNA exposure is required for activating the different modes of Mec1 signaling.
Based on the model that ssDNA is required for activation of Mec1 signaling, we postulated that proreplicative Mec1 signaling could be activated by dynamic ssDNA exposure during active DNA synthesis. Consistent with this model, our phosphoproteomic analysis revealed that cells undergoing normal S phase display a level of phosphorylation of growth-correlated substrates similar to the level induced by 4-NQO treatment in G1 (Fig. 7G). These findings are congruent with our recent work showing that Mec1 is highly active during normal S phase and that this activity is correlated with the extent of DNA synthesis and independent of S-phase CDK activity (Bastos de Oliveira et al. 2015). Interestingly, Rad53 phosphorylation and activation were robustly stimulated in G1 in the presence of 4-NQO but not during normal S phase (Fig. 7G), revealing that growth-correlated Mec1 signaling is uncoupled from downstream checkpoint signaling during normal DNA replication. Taken together, these results are consistent with a model in which Mec1's proreplicative function is induced by active DNA replication forks in a manner that does not trigger the anti-replicative checkpoint cascade. Surprisingly, as conveyed in Figures 2 and 3, this proreplicative function is also separable from Mec1's role in GCR suppression (Fig. 7H).
Discussion
Almost two decades ago, mec1Δ tel1Δ was identified as one of the most genetically unstable yeast strains. These cells have dramatically elevated rates of aneuploidy (McCulley and Petes 2010) and undergo chromosome rearrangements at an extremely high frequency (Myung et al. 2001), often as a consequence of HR between repetitive DNA elements (Vernon et al. 2008). Despite extensive investigation, the mechanism by which the Mec1/ATR and Tel1/ATM kinases suppress GCRs has remained elusive, representing one of the most fundamental knowledge gaps in our mechanistic understanding of genome maintenance. Moreover, mec1Δ tel1Δ cells exhibit a severe growth defect, which has commonly been attributed to their extreme genetic instability and an inability to activate Rad53. Here we report the unexpected finding that the roles of Mec1 in suppressing GCRs and promoting cell proliferation are entirely separable and, importantly, independent of canonical checkpoint signaling (Fig. 7H).
Activator-directed phosphorylation and the suppression of genetic instability
Our approach to manipulate Mec1 reactivation in ddt cells revealed that activator-directed phosphorylation, whether mediated by the endogenous activators (Dna2, Dpb11, and Ddc1) or chimeric protein-MAD fusions, is a critical requirement for the suppression of GCRs. Since Dna2 associates with 5′ DNA flaps (Wanrooij and Burgers 2015) and since Ddc1–Dpb11 associate with 5′ DNA junctions (Majka et al. 2006), our results are consistent with the model that the recruitment of MAD-containing proteins to these structures is required to promote a localized mode of Mec1 signaling that can suppress GCRs (Supplemental Fig. S9). Notably, we found that Mec1 activation by Dna2 or Ddc1–Dpb11 preferentially directs Mec1 phosphorylation to Sgs1 and Slx4–Rtt107, respectively, which are known to associate with the activator that promotes their phosphorylation. Thus, we propose that MAD-containing proteins direct Mec1 to phosphorylate nearby substrates, which, in our phosphoproteomic analyses, are correlated with, and presumably involved in, the suppression of GCRs. Because either Dna2 or Ddc1–Dpb11 alone is sufficient for genetic stability in a tel1Δ background, the revelation that they promote the phosphorylation of distinct Mec1 substrates suggests that they may act in parallel to suppress GCRs. Moreover, since both Slx4–Rtt107 and Sgs1 play roles in DNA end resection and processing of HR intermediates (Shor et al. 2002; Mimitou and Symington 2008; Zhu et al. 2008; Dibitetto et al. 2016), we speculate that they may represent substrates in functionally redundant pathways for Mec1-dependent control of HR-mediated DNA repair. To test this model and define the set of substrates involved in GCR suppression, it will be important to recapitulate the high GCR rates seen in mec1Δ tel1Δ cells by mutating the precise combination of Mec1-dependent phosphorylation sites. However, we expect that the generation of such a mutant strain will be quite challenging. In addition to the potential issue of functional redundancy in GCR-correlated phosphorylation sites, we note that technical limitations are likely preventing the identification of all relevant GCR-correlated phosphorylation events, as MS is able to detect phosphorylation only within MS-compatible peptides. Moreover, since Tel1 is capable of partially suppressing the genetic instability that results from the absence of Mec1, it will be important to investigate whether Tel1 can redundantly phosphorylate the same substrates that are targeted by Mec1 to suppress GCRs or acts as part of an independent backup pathway.
Our findings may also have interesting evolutionary implications. The fact that “free” MADs are unable to stimulate Mec1-dependent GCR suppression provides a biological rationale for the evolutionary integration of MADs within select endogenous proteins. Also, the observation that specific MAD fusions (MAD-Rtt107 and Sgs1-MAD) can rescue the genetic stability of ddt cells indicates that the ability of MAD-containing proteins to direct Mec1 phosphorylation is the utmost feature dictating their functionality as a Mec1/ATR activator. This insight may help explain why not all Mec1 activators in yeast show direct conservation in higher eukaryotes. In fact, the only conserved activator of ATR is the homolog of Dpb11: TOPBP1. A second ATR activator, ETAA1, was identified recently, though it has no clear ortholog in simpler eukaryotes. Interestingly, Bass et al. (2016) demonstrated recently that ETAA1 displays a genetically epistatic relationship to BLM helicase, the mammalian ortholog of Sgs1. Thus, despite being activated by evolutionarily unrelated proteins in different eukaryotic organisms, ATR may still suppress genetic instability via mechanisms that are highly conserved.
Growth-correlated Mec1 signaling and the promotion of DNA replication
Perhaps the most unexpected result from our present work is that the slow growth phenotype of cells lacking Mec1 and Tel1 signaling is separable from their genetic instability. This finding has far-reaching implications, especially for investigating how Mec1/ATR signaling functionally interfaces with the DNA replication machinery. Of importance, our genetic system for manipulation of Mec1 activation now provides the means to study the proreplicative functions of Mec1 signaling independently of its role in GCR suppression. We expect that future investigations using this genetic system will allow the careful dissection of the mechanism by which Mec1 promotes DNA replication and cell growth.
The finding of a proreplicative role for Mec1 is consistent with our previous report of a replication-correlated mode of Mec1 signaling that is robustly active during the progression of normal S phase (Bastos de Oliveira et al. 2015). This work also showed that Mec1 activation in S phase is associated with active DNA synthesis and not with the buildup of S-phase CDK activity, supporting the model that replication-correlated signaling is triggered by ssDNA exposure. It is possible that dynamic ssDNA exposure during lagging strand synthesis is conducive for the recruitment of Mec1 and its activators and the consequent activation of Mec1. Importantly, since these replication intermediates are highly dynamic, the checkpoint adaptor proteins (Mrc1 and Rad9) that transduce Mec1 phosphorylation to Rad53 may be unable to assemble, which would explain the uncoupling between Mec1 signaling and Rad53 activation (Fig. 7G; Bastos de Oliveira et al. 2015). Such uncoupling helps explain how Mec1 could play a proreplicative role during unchallenged S phase despite also being able to negatively regulate DNA replication upon replication stress (Santocanale and Diffley 1998).
It is intriguing that Tel1 seems capable of playing a proreplicative role in a manner redundant to Mec1 (note that ddc1Δ dna2-AA strain is significantly healthier than ddc1Δ dna2-AA tel1Δ) (Kumar and Burgers 2013). This is an unexpected finding, since Tel1 is not commonly viewed as a replication fork-associated kinase. Nonetheless, many of the growth-correlated targets identified here can also be targeted by Tel1 (Bastos de Oliveira et al. 2015). We speculate that Tel1 might also be activated by dynamic replication intermediates and redundantly phosphorylate a subset of growth-correlated substrates to promote DNA replication. One alternate explanation is that cells lacking Mec1 naturally accumulate DSBs, which can lead to a constitutive level of Tel1 signaling that results in the phosphorylation of substrates that mediate the progrowth function.
The mechanism by which Mec1 signaling promotes DNA replication is currently unclear. One attractive possibility is that Mec1 can also increase dNTP levels independently of downstream checkpoint signaling, as the up-regulation of dNTP levels has been shown to be sufficient for faster fork speed in S phase (Poli et al. 2012). However, our results in Figure 7C indicate that the proreplicative function of Mec1 highlighted here does not involve the up-regulation of dNTP pools. Alternatively, the Bell laboratory (Randell et al. 2010) has proposed that Mec1 can directly phosphorylate MCM proteins and prime them for firing, a role that may also be consistent with the proreplicative functions that we uncovered here. However, the Bell laboratory (Randell et al. 2010) demonstrated that Mec1's participation in origin priming was entirely redundant to DDK, the canonical priming kinase. Since DDK is functional in our system, the proreplicative Mec1 function delineated by our work is likely independent of origin priming via MCM phosphorylation, although Mec1 could still potentially regulate origins in some other, as yet unknown manner. Based on the identity of the growth-correlated targets that we identified (Fig. 7E), it is logical to hypothesize that Mec1 may promote DNA replication by regulating transcription and chromatin. One possibility is that Mec1 regulates the passage of replication forks through obstacles formed by engaged transcriptional complexes or chromatin. A recent study investigating Mec1's role in limiting transcription–replication conflicts supports this possible model (Poli et al. 2016). In the future, it will be interesting to monitor the dynamics of fork movement across the entire genome in ddt cells expressing “free” MADs or carrying an empty vector. While it is possible that Mec1 promotes DNA replication via regulation of a specific substrate within our list of growth-correlated substrates, we favor the model that Mec1 exerts this proreplicative function through the control of multiple substrates.
We expect that, similar to its checkpoint function, Mec1's proreplicative function is also conserved in mammalian ATR. It may be a challenge to generate a system capable of separating ATR's genome maintenance and checkpoint functions from a potential proreplicative function. Haahr et al. (2016) demonstrated recently that overexpression of just the AAD of ETAA1 can hyperactivate ATR in human cells, and overexpression of the AAD of TOPBP1 has been shown to result in cellular senescence (Toledo et al. 2008). However, experiments analogous to what we presented here need to be performed in cells with a “dormant” ATR (lacking functional AADs of TOPBP1 and ETAA1) so that a growth-rescuing effect can be appreciated. We also note that the dependence on ssDNA exposure for ATR activation by “free” AADs has yet to be formally tested. Finally, since ATR inhibitors are in preclinical trials for cancer treatment, our findings may also have implications for clinical studies, especially if ATR inhibitors exert their anti-cancer effects in part by inhibiting a proreplicative function of ATR in cancer cells.
Materials and methods
Yeast strains
A complete list of yeast strains used in this study is in Supplemental Table S8. The strain background for all yeast used in this study is S288C. Gene deletions were carried out using standard PCR-based strategies to amplify resistance cassettes with appropriate flanking sequences homologous to the target gene. All genetic manipulations were verified by PCR. Spores generated from diploids were genotyped via PCR and/or replica plating. Primer sequences for gene deletions are available on request.
Antibodies
Rad53 was detected via Western blot using the EL7.E1 antibody. The expression of free MADs containing a 3xHA epitope tag was detected via Western blot using the 12CA5 anti-HA antibody.
Plasmid construction
A complete list of the plasmids used in this study is in Supplemental Table S7. MADs from all three Mec1 activators in yeast were cloned into pRS415 vectors containing various promoters for expression in yeast. MADs used in this study were as follows: MADDPB11, MADDNA2, and MADDDC1 (representing amino acids 572–764 of Dpb11, amino acids 1–450 of Dna2, and amino acids 341–562 of Ddc1).
MAD-protein fusions (used in Fig. 6) were generated by Gibson assembly. See Supplemental Figure S10 for a schematic that details the MAD-containing plasmids used in this study and the cloning strategy used to generate the MAD-protein fusions. Additional information regarding the vectors is available on request.
Measurement of GCR events
GCR-compatible strain ddt was obtained as a spore from a heterozygous ddt diploid and frozen immediately. All GCR assays began with a pRS415 plasmid transformation of yeast freshly streaked from the frozen stock. The transformed yeast cells were spread on −Leu plates. Transformants were grown into visible colonies, and, 64–72 h after plasmid transformation, individual colonies were transferred to 2 mL of −Leu liquid culture. After ∼48 h, 0.5 mL of saturated culture (∼10 million yeast cells) was spun down, washed with 300 µL of autoclaved ddH2O, resuspended in 150 µL of autoclaved ddH2O, and spotted onto plates containing 5-FOA and canavanine (plates described by Putnam and Kolodner 2010). For sicker ddt derivative strains (e.g., ddt rad53Δ), 1 mL of saturated culture was used per spot. In parallel with each GCR experiment, multiple saturated cultures were serially diluted 200,000× and plated onto YPD plates to determine the average population viability. After several days, the appearance of 5-FOA- and canavanine-resistant colonies in a spot was used to calculate the number of GCR events that occurred in a single culture. The number of GCR events in a culture was calculated using the equation m[1.24 + ln(m)] − r = 0, where r is the number of 5-FOAr Canr yeast in a culture, and m is the estimated number of GCR events (Putnam and Kolodner 2010).
The number of GCR events per culture was normalized to the average population viability for each experiment.
Growth curves
Yeast was grown to saturation in YPD medium or transformed with pRS415 (LEU) plasmid and grown to saturation in −Leu minimal medium. Saturated YPD cultures and minimal medium cultures were diluted 200× and 100×, respectively, and loaded into a Bioscreen C honeycomb plate. Cultures were grown at 30°C with constant agitation using a Bioscreen C plate reader. Optical density measurements were made every 20 min at 600 nm over a period of 48 h. Each curve represents a single experiment and was constructed from between six and nine replicate cultures. Error bars represent the standard deviation of the replicate cultures. All curves are representative results from at least two independent experiments.
MS-based phosphoproteomics
Methodologies detailing the identification of Mec1/Tel1 substrate phosphopeptides and the construction of QMAPS can be found in our previous work (Bastos de Oliveira et al. 2015). For stable isotope labeling of amino acids in cell culture (SILAC), yeast strains auxotrophic for lysine and arginine were grown in −Arg −Lys synthetic dropout medium supplemented with either normal L-arginine and L-lysine (light culture) or L-lysine 13C6, 15N2 and L-arginine 13C6, 15N4 (heavy culture) as described previously (Ohouo et al. 2013). Unless indicated otherwise, QMAPS were constructed from at least two independent phosphoproteomic experiments. Unless indicated otherwise, only phosphopeptides detected and quantified in at least two independent experiments were included in the QMAPS plots. For statistical analysis, P-values were calculated for all Mec1 substrates using the Mann-Whitney statistical test (see Supplemental Tables S1, S2, S5). In this case, the P-value represents the probability that a given phosphopeptide fold change is significant relative to fold changes seen in the total population of detected phosphopeptides. More detailed information on the QMAPS is in the Supplemental Tables S1–S6.
Phosphorylation analysis of Slx4 immunoprecipitates
Slx4-13xMyc was immunoprecipitated from ddt cells expressing MADDNA2 or dna2-AA tel1Δ cells carrying an empty vector. Cells were harvested in the absence of any exogenous agents while growing asynchronously. Phosphopeptides were enriched from the immunoprecipitated protein. Immunoprecipitation–IMAC (immobilized metal affinity chromatography)–MS steps were performed as described by Bastos de Oliveira et al. (2015).
FACS analysis of cell cycle progression
Cells were collected while growing asynchronously in log phase, fixed in 70% ethanol, and stored at −20°C. Yeast cells were harvested by centrifugation, ethanol was removed, and the cell pellet was resuspended in 50 mM sodium citrate followed by treatment with 200 µg/mL RNase A (Qiagen) for 1 h at 37°C and 500 µg/mL proteinase K (Invitrogen) for 1 h at 42°C. Finally, cells were incubated in the presence of Sytox Green (Molecular Probes) for 2 h. FACS profiles were analyzed using a BD Accuri C6 flow cytometer and CFlow software.
Measurement of dNTP levels
Logarithmically growing yeast cells with OD 600 between 0.3 and 0.4 were harvested on nitrocellulose membrane and suspended immediately in an ice-cold mixture of 12% TCA and 15 mM MgCl2. Cells were vortex-mixed for 15 min at 4°C and then centrifuged at 14,000 rpm for 1 min at 4°C. The supernatant was neutralized with a freon–trioctylamine mix and processed as described previously (Jia et al. 2015).
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Proteomics Identifications) (Vizcaíno et al. 2016) partner repository with the data set identifier PXD009734.
Supplementary Material
Acknowledgments
We thank Beatriz S. Almeida for technical support, Tobias Doerr for guidance with our growth curve analyses, Jim Haber and Thomas Petes for plasmids and reagents, and Grant Brown for insightful feedback on the manuscript. This work is supported by grants from the National Institute of Health (R01-GM097272) to M.B.S., and Swedish Cancer Society and Swedish Research Council grants to A.C.
Author contributions: M.C.L., S.O., and E.J.S. performed experiments. M.C.L. and M.B.S. designed the study, performed data analysis, and wrote the paper. S.S. and A.C. designed and performed the experiments for measuring dNTP pools and helped revise the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.308148.117.
References
- Balint A, Kim T, Gallo D, Cussiol JR, Bastos de Oliveira FM, Yimit A, Ou J, Nakato R, Gurevich A, Shirahige K, et al. 2015. Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J 34: 2182–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D. 2016. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, Doerfler L, Schmidt KH, Yu H, Smolka MB. 2015. Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell 57: 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Lanz M, Smolka MB. 2018. Quantitative analysis of DNA damage signaling responses to chemical and genetic perturbations. Methods Mol Biol 1672: 645–660. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. 2010. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208–219. [DOI] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC. 2010. DNA end resection by Dna2–Sgs1–RPA and its stimulation by Top3–Rmi1 and Mre11–Rad50–Xrs2. Nature 467: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. 2003. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112: 391–401. [DOI] [PubMed] [Google Scholar]
- Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM. 2005. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19: 3055–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB. 2015. Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34: 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibitetto D, Ferrari M, Rawal CC, Balint A, Kim T, Zhang Z, Smolka MB, Brown GW, Marini F, Pellicioli A. 2016. Slx4 and Rtt107 control checkpoint signalling and DNA resection at double-strand breaks. Nucleic Acids Res 44: 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, Blow JJ. 2010. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 191: 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Branzei D. 2017. S-phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell Mol Life Sci 74: 2361–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M. 2004. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J 23: 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, et al. 2014. A cell cycle-regulated Slx4–Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev 28: 1604–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M, Mailand N. 2016. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18: 1196–1207. [DOI] [PubMed] [Google Scholar]
- Hang LE, Peng J, Tan W, Szakal B, Menolfi D, Sheng Z, Lobachev K, Branzei D, Feng W, Zhao X. 2015. Rtt107 is a multi-functional scaffold supporting replication progression with partner SUMO and ubiquitin ligases. Mol Cell 60: 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ii M, Brill SJ. 2005. Roles of SGS1, MUS81, and RAD51 in the repair of lagging-strand replication defects in Saccharomyces cerevisiae. Curr Genet 48: 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Marjavaara L, Buckland R, Sharma S, Chabes A. 2015. Determination of deoxyribonucleoside triphosphate concentrations in yeast cells by strong anion-exchange high-performance liquid chromatography coupled with ultraviolet detection. Methods Mol Biol 1300: 113–121. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. 2006. TopBP1 activates the ATR–ATRIP complex. Cell 124: 943–955. [DOI] [PubMed] [Google Scholar]
- Kumar S, Burgers PM. 2013. Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev 27: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Carr AM. 2013. Replication stress and genome rearrangements: lessons from yeast models. Curr Opin Genet Dev 23: 132–139. [DOI] [PubMed] [Google Scholar]
- Lee YC, Zhou Q, Chen J, Yuan J. 2016. RPA-binding protein ETAA1 is an ATR activator involved in DNA replication stress response. Curr Biol 26: 3257–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. 2005. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev 14: 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cussiol JR, Dibitetto D, Sims JR, Twayana S, Weiss RS, Freire R, Marini F, Pellicioli A, Smolka MB. 2017. TOPBP1Dpb11 plays a conserved role in homologous recombination DNA repair through the coordinated recruitment of 53BP1Rad9. J Cell Biol 216: 623–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561. [DOI] [PubMed] [Google Scholar]
- MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. 2007. The structural determinants of checkpoint activation. Genes Dev 21: 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majka J, Binz SK, Wold MS, Burgers PM. 2006. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J Biol Chem 281: 27855–27861. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166. [DOI] [PubMed] [Google Scholar]
- McCulley JL, Petes TD. 2010. Chromosome rearrangements and aneuploidy in yeast strains lacking both Tel1p and Mec1p reflect deficiencies in two different mechanisms. Proc Natl Acad Sci 107: 11465–11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, Cortez D. 2008a. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 22: 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Nam EA, Cortez D. 2008b. Dpb11 activates the Mec1–Ddc2 complex. Proc Natl Acad Sci 105: 18730–18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. 1995. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 82: 831–840. [DOI] [PubMed] [Google Scholar]
- Myung K, Datta A, Kolodner RD. 2001. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104: 397–408. [DOI] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. 2008. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J Biol Chem 283: 35853–35859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. 2009. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell 36: 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB. 2010. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39: 300–306. [DOI] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB. 2013. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493: 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbe L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Gerhold CB, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner KP, et al. 2016. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev 30: 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, Lazzaro F, Giannattasio M, Plevani P, Muzi-Falconi M. 2008. Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol Cell Biol 28: 4782–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Kolodner RD. 2010. Determination of gross chromosomal rearrangement rates. Cold Spring Harb Protoc 2010: pdb.prot5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP. 2010. Mec1 is one of multiple kinases that prime the Mcm2-7 helicase for phosphorylation by Cdc7. Mol Cell 40: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP. 2000. LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. EMBO J 19: 5801–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. 1996. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360. [DOI] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615–618. [DOI] [PubMed] [Google Scholar]
- Segurado M, Diffley JF. 2008. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev 22: 1816–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R. 2002. Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162: 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Zhou H. 2007. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci 104: 10364–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, et al. 2012. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol 14: 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. 2008. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev 22: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon M, Lobachev K, Petes TD. 2008. High rates of ‘unselected’ aneuploidy and chromosome rearrangements in tel1 mec1 haploid yeast strains. Genetics 179: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016. 2016 update of the PRIDE database and related tools. Nucleic Acids Res 44: D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij PH, Burgers PM. 2015. Yet another job for Dna2: checkpoint activation. DNA Repair (Amst) 32: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij PH, Tannous E, Kumar S, Navadgi-Patil VM, Burgers PM. 2016. Probing the Mec1ATR checkpoint activation mechanism with small peptides. J Biol Chem 291: 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. 1994. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8: 652–665. [DOI] [PubMed] [Google Scholar]
- Yimit A, Riffle M, Brown GW. 2015. Genetic regulation of Dna2 localization during the DNA damage response. G3 5: 1937–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. 2010. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Durocher D. 2010. De novo telomere formation is suppressed by the Mec1-dependent inhibition of Cdc13 accumulation at DNA breaks. Genes Dev 24: 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rothstein R. 2002. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc Natl Acad Sci 99: 3746–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ. 2000. The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Proteomics Identifications) (Vizcaíno et al. 2016) partner repository with the data set identifier PXD009734.