

Cai et al. identified the Hippo signaling effector YAP and its transcriptional target, c-Myc, as promoters of cystic kidney pathogenesis. A comprehensive kinase inhibitor screen identified a signaling pathway involving the RhoGEF LARG, the small GTPase RhoA, and the RhoA effector Rho-associated kinase (ROCK) as a critical signaling module between PKD1 and YAP.
Keywords: 3D culture, ADPKD, Hippo signaling, RhoA signaling, YAP/TAZ, c-Myc
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disorder caused by mutations in PKD1 or PKD2 and affects one in 500–1000 humans. Limited treatment is currently available for ADPKD. Here we identify the Hippo signaling effector YAP and its transcriptional target, c-Myc, as promoters of cystic kidney pathogenesis. While transgenic overexpression of YAP promotes proliferation and tubule dilation in mouse kidneys, loss of YAP/TAZ or c-Myc suppresses cystogenesis in a mouse ADPKD model resulting from Pkd1 deficiency. Through a comprehensive kinase inhibitor screen based on a novel three-dimensional (3D) culture of Pkd1 mutant mouse kidney cells, we identified a signaling pathway involving the RhoGEF (guanine nucleotide exchange factor) LARG, the small GTPase RhoA, and the RhoA effector Rho-associated kinase (ROCK) as a critical signaling module between PKD1 and YAP. Further corroborating its physiological importance, inhibition of RhoA signaling suppresses cystogenesis in 3D culture of Pkd1 mutant kidney cells as well as Pkd1 mutant mouse kidneys in vivo. Taken together, our findings implicate the RhoA–YAP–c-Myc signaling axis as a critical mediator and potential drug target in ADPKD.
In mammals, the Hippo signaling pathway comprises several tumor suppressors (NF2, Sav1, Mst1/2, Lats1/2, etc.) acting through a kinase cascade to affect the phosphorylation and cytoplasmic retention of the oncoproteins YAP/TAZ, transcriptional coactivators that normally partner with the TEF/TEAD family DNA-binding transcription factors to transactivate growth-promoting genes (Pan 2010; Halder et al. 2012; Yu et al. 2015). Much of the initial interest in this pathway stems from its potent role in growth control and tumorigenesis, as YAP/TAZ is activated in a wide variety of human cancers, and YAP activation in mice results in tumorigenesis in multiple mouse tissues (Pan 2010; Halder et al. 2012; Yu et al. 2015). Indeed, genetic mutations in the various components of the Hippo pathways have been linked to a wide spectrum of human diseases such as cancer (NF2) (Yin et al. 2013), immunodeficiency (Mst1) (Abdollahpour et al. 2012; Nehme et al. 2012), and chorioretinal degeneration (TEAD1) (Fossdal et al. 2004).
Autosomal dominant polycystic kidney disease (ADPKD) is a human disease characterized by the development of multiple fluid-filled cysts and massive enlargement of the kidneys, frequently leading to end-stage renal disease (Wilson 2004). One in 500–1000 humans is affected by ADPKD. Eighty-five percent of ADPKD patients carry mutations in the PKD1 gene (encoding polycystin-1 [PC1]), and the rest of individuals harbor mutations in the PKD2 gene (encoding PC2) (Bisceglia et al. 2006; Chapin and Caplan 2010). Limited treatment is currently available for ADPKD, and there is great interest in identifying molecular targets and/or pathways for the development of mechanism-based therapeutics (Patel et al. 2009; Aguiari et al. 2013; LaRiviere et al. 2015). In this study, we identify the Hippo signaling effector YAP and its transcriptional target, c-Myc, as critical mediators of cystic kidney pathogenesis after PKD1 is inactivated. Using a novel three-dimensional (3D) cell culture model for ADPKD, we further identify a LARG–RhoA–ROCK (Rho-associated kinase)–MLC (myosin light chain) pathway as a critical signaling module between PKD1 and YAP. These findings shed light on the mechanisms underlying ADPKD pathogenesis and suggest that targeting this RhoA–YAP–c-Myc signaling axis may serve as a potential therapeutic strategy against ADPKD.
Results
Activation of YAP in human ADPKD and mouse Pkd1 mutant kidney cysts
As an unbiased way to identify effector pathways relevant for the pathogenesis of ADPKD, we previously performed global gene profiling on human PKD1 polycystic kidney cysts compared with minimally cystic tissues using Affymetrix cDNA arrays (Song et al. 2009). Gene set enrichment analysis revealed transcriptional profiles characteristic of loss of kidney epithelial differentiation genes and reactivation of developmental and mitogenic signaling in ADPKD tissues (Song et al. 2009). However, the status of Hippo signaling in ADPKD tissues was not analyzed in this study. To investigate whether YAP/TAZ is activated in ADPKD tissues, we examined an unbiased set of 379 direct YAP/TAZ target genes that were identified previously in MDA-MB-231 human breast cancer cells using ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) and microarray analysis (Zanconato et al. 2015). Using these direct YAP/TAZ targets as a reference, gene set enrichment analysis of ADPKD cysts versus minimally cystic tissues showed that these targets are significantly enriched in ADPKD cysts (Fig. 1A,B). Several known YAP/TAZ targets, including Cyr61, Ctgf, c-Myc, Amotl2, and Axl, are among the top up-regulated gene list (Fig. 1C; Supplemental Tables S1,S2). These findings suggest that YAP/TAZ is activated in ADPKD cysts.
Figure 1.

Activation of YAP in human ADPKD and mouse Pkd1 mutant kidney cysts. (A) Gene set enrichment analysis showing the 10 most significantly enriched signaling pathways by normalized enrichment score in ADPKD cysts compared with minimally cystic tissues. (B) Gene set enrichment analysis of ADPKD cysts versus minimally cystic tissues for the expression of previously reported direct YAP/TAZ target genes. Normalized enrichment score = 2.1389; false discovery rate q = 0.0000. (C) Heat map analysis of 50 direct YAP/TAZ target genes with the most up-regulation in human ADPKD cysts compared with minimally cystic tissues. Tissues from different patients were analyzed. (SC) Small cyst; (MC) medium cyst; (LC) large cyst; (MCT) minimally cystic tissue. The fold change represents the ratio of gene expression in cysts (mean of 13 cysts, including five small cysts, five medium cysts, and three large cycts) over minimally cystic tissues (mean of five minimally cystic tissues). (D–F) YAP (D), TAZ (E), and c-Myc (F) staining in control (Pkd1flox/flox) and Pkd1 mutant (AhCre;Pkd1flox/flox) kidneys at postnatal day 15 (P15). Animals received daily injections of β-naphthoflavone (β-NF) from P8 to P11. Note the cytoplasmic localization of YAP and TAZ in control tubular epithelium and nuclear localization of YAP and TAZ in Pkd1 mutant kidney cysts (compare arrows in the magnified insets). Also note the nuclear staining of c-Myc in Pkd1 mutant kidney cysts, which is absent in the control tubular epithelium (arrows). Bar, 100 µm. (G) Western blot showing increased total YAP and c-Myc protein levels and relatively decreased phosphorylation of YAP (the ratio of p-YAP over YAP) in Pkd1 mutant kidneys. (H) Up-regulation of direct YAP/TAZ target genes in Pkd1 mutant kidneys. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (G,H) Control and Pkd1 mutant mice received daily injections of β-NF from P8 to P11 and were analyzed at P15.
To investigate whether PKD1 regulates YAP activity, we characterized a new Pkd1 mutant mouse model using the inducible AhCre driver (AhCre;Pkd1flox/flox) in which Cre expression is regulated by the cytochrome P450 promoter element in response to β-naphthoflavone (β-NF) injection (Ireland et al. 2004). Without β-NF, the AhCre mice showed sporadic Cre activity in the kidney tubular epithelium (Supplemental Fig. S1B). After daily intraperitoneal (i.p.) injections of β-NF from postnatal day 8 (P8) to P11, Cre was active in the majority of the kidney tubular epithelial cells (Supplemental Fig. S1C). Most Pkd1 mutant mice died within 4 wk after β-NF injections. Three weeks after β-NF injections, we observed a dramatic enlargement of Pkd1 mutant kidneys, with massive cyst formation in all segments of the kidney nephrons, including the glomerulus, the proximal tubule, the thin ascending limb, the distal convoluted tubule, and the collecting duct (Supplemental Fig. S1D–F), phenotypically recapitulating a previously reported ADPKD mouse model carrying homozygous Pkd1 mutation (Lu et al. 1997). In contrast to the cytoplasmic localization of YAP and TAZ in normal tubular epithelia of control kidneys, nuclear accumulation of YAP and TAZ was detected in the cystic tubular epithelia of Pkd1 mutant kidneys (Fig. 1D,E), indicating activation of both YAP and TAZ. The increase in YAP/TAZ activity was further evidenced by increased c-Myc staining in Pkd1 mutant cysts (Fig. 1F), increased YAP and c-Myc protein levels (Fig. 1G), and increased mRNA levels of YAP/TAZ target genes such as c-Myc, Cyr61, Ctgf, Col12A1, Amotl2, and Axl in Pkd1 mutant kidneys (Fig. 1H). These results further demonstrated that loss of PKD1 leads to activation of YAP/TAZ in the kidney tubular epithelium.
YAP activation promotes cell proliferation and dilation in the kidney tubular epithelium
Although a previous study also reported that YAP is activated in Pkd1-deficient mouse kidneys and human ADPKD tissues (Happe et al. 2011), both the underlying mechanism and the functional relevance of YAP to cystogenesis remained elusive. To investigate whether YAP plays a causal role in cystogenesis, we first generated a transgenic mouse model that allows doxycycline-inducible YAP overexpression in all segments of the kidney tubular epithelium under the control of a Pax8-rtTA driver (Fig. 2A; Traykova-Brauch et al. 2008). Doxycycline treatment of 4-wk-old YAP transgenic mice resulted in a progressive increase in kidney weight and size (Fig. 2B,C), accompanied by the induction of YAP target genes such as c-Myc, Cyr61, Ctgf, Col12A1, Amotl2, and Axl (Fig. 2D). Histological analysis of YAP-overexpressing kidneys revealed increased YAP protein levels in the tubular epithelium, including both the proximal tubules and collecting ducts, and increased Ki67-positive cells as early as 2 d after doxycycline treatment (Fig. 2E,F; Supplemental Fig. S2A,B). Dilation of the tubular epithelium was observed after 4 d of doxycycline treatment and significantly enlarged by 8 d (Fig. 2G). These dilations developed from the glomerulus, the proximal tubule, the thin ascending limb, and the distal convoluted tubule but not the collecting duct (Fig. 2G; Supplemental Fig. S2C–E). These results indicate that overexpression of YAP induced cell proliferation and dilation in the kidney tubular epithelium, with proliferation preceding tubule dilation.
Figure 2.

YAP activation in renal tubular epithelium leads to kidney tubule dilation. (A) Genetic scheme for analyzing Pax8-rtTA;pTRE2-Yap double-transgenic mice. (B) Progression of kidney enlargement upon YAP induction by doxycycline induction for the indicated time. The ratio of kidney weight (KW) over body weight (BW) was plotted. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (C) Doxycycline induction for 8 d led to increased kidney size in the double-transgenic mice (YapTg) compared with the control. (D) Up-regulation of YAP transcriptional target genes upon YAP overexpression in the kidney. YAP was induced by doxycycline induction for 2d and 8 d, respectively. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (E,F) YAP (E) and Ki67 (F) staining (arrows) showing increased YAP protein levels and cell proliferation in the kidney tubular epithelium after doxycycline treatment of Pax8-rtTA;pTRE2-Yap double-transgenic mice for different times. Note the increased YAP protein levels and Ki67-positive cells as early as 2 d after doxycycline induction. Bar, 100 µm. (G) H&E staining showing the progression of kidney dilations upon YAP induction in the double-transgenic mice. Tubule dilation began at 4 d after doxycycline induction and progressed to more severe phenotypes by 8 d. Arrows mark representative dilations, while the arrowhead indicates a glomerular dilation. Bar, 100 µm.
As a more physiological approach to study the consequences of YAP activation in the kidney, we activated YAP by generating mice deficient for Sav1, a negative regulator of YAP. Without β-NF, AhCre;Sav1flox/flox mice showed no overt kidney phenotypes. Three months after β-NF injections from P8 to P11 as described above, AhCre;Sav1flox/flox mice exhibited kidney tubular epithelial cell hyperproliferation (Supplemental Fig. S3A) and dilation (Supplemental Fig. S3B). Like the YAP transgenic model, the Sav1 mutant tubule dilations were derived from the glomerulus, the proximal tubule, the thin ascending limb, and the distal convoluted tubule but not the collecting duct (Supplemental Fig. S3B–E). We also generated AhCre;Yapflox/flox and AhCre;Sav1flox/flox;Yapflox/flox mice and induced AhCre activity in these mice by β-NF injections. Although the AhCre;Yapflox/flox mice did not show any overt kidney phenotype under the same condition, loss of YAP completely suppressed hyperproliferation and tubule dilation in the Sav1 mutant kidneys (Supplemental Fig. S3A–E), indicating that hyperactivity of YAP is required for cell proliferation and dilation in the kidney tubular epithelium after Hippo signaling is perturbed.
YAP and TAZ are required for cystogenesis after inactivation of PKD1
To analyze the physiological relevance of YAP to cystogenesis in Pkd1 mutant kidneys, we generated AhCre;Pkd1flox/flox;Yapflox/flox mice and compared them with AhCre;Pkd1flox/flox mice. Due to sporadic AhCre activity (Supplemental Fig. S1B), AhCre;Pkd1flox/flox mice developed cysts even in the absence of β-NF injection (Fig. 3A; Supplemental Fig. S4A). Under the same condition, loss of YAP suppressed the increased ratio of kidney over body weight and cyst formation in Pkd1 mutant kidneys (Fig. 3A–C). To examine the contribution of TAZ in cystogenesis in Pkd1 mutant kidneys, we further generated AhCre;Pkd1flox/flox;Yapflox/flox;Tazflox/flox and AhCre;Yapflox/flox;Tazflox/flox mice. A greater suppression of the Pkd1 mutant kidney cyst formation was achieved after loss of both YAP and TAZ in Pkd1 mutant kidneys (Fig. 3A–C), although loss of TAZ alone or TAZ plus YAP did not show any overt kidney phenotype (Fig. 3B; Supplemental Fig. S4B). These results suggest that both YAP and TAZ contribute to cystogenesis in Pkd1-deficient kidneys.
Figure 3.
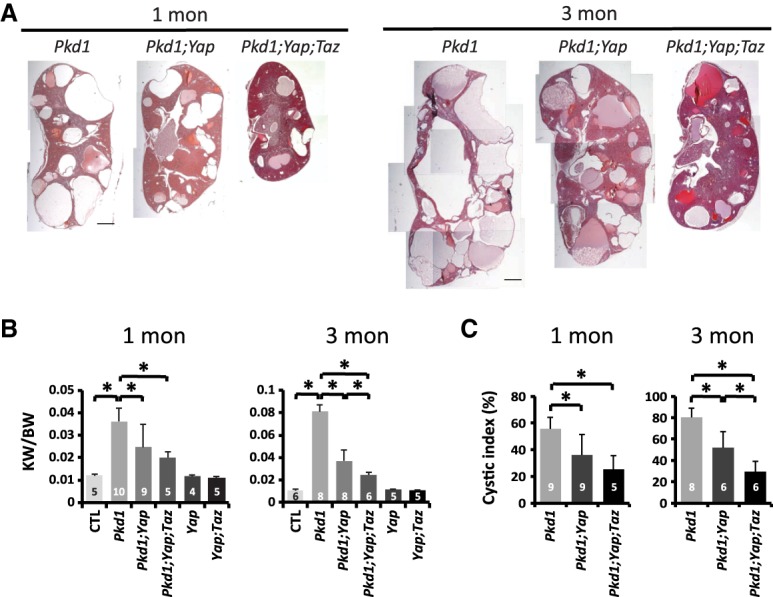
Suppression of Pkd1 mutant kidney cystogenesis by inactivation of YAP and TAZ. (A) H&E staining of the kidneys in 1- and 3-mo-old Pkd1 mutant (AhCre;Pkd1flox/flox), Pkd1;Yap double-mutant (AhCre;Pkd1flox/flox;Yapflox/flox), and Pkd1;Yap;Taz triple-mutant (AhCre;Pkd1flox/flox;Yapflox/flox;Tazflox/flox) mice. Bar, 1 mm. (B) The ratio of kidney weight (KW) over body weight (BW) in control (Pkd1flox/flox; 1 mo, n = 5; 3 mo, n = 6), Pkd1 mutant (1 mo, n = 10; 3 mo, n = 8), Pkd1;Yap double-mutant (1 mo, n = 9; 3 mo, n = 8), Pkd1;Yap;Taz triple-mutant (1 mo, n = 5; 3 mo, n = 6), Yap mutant (AhCre;Yapflox/flox; 1 mo, n = 4; 3 mo, n = 5), and Yap;Taz double-mutant (AhCre;Yapflox/flox;Tazflox/flox; 1 mo, n = 5; 3 mo, n = 5) mice. Data are mean ± SD. (*) P < 0.05, t-test. (C) The cystic index, calculated as the percentage of cystic area over total area on kidney sections, in Pkd1 mutant (1 mo, n = 9; 3 mo, n = 8), Pkd1;Yap double-mutant (1 mo, n = 9; 3 mo, n = 6), and Pkd1;Yap;Taz triple-mutant (1 mo, n = 5; 3 mo, n = 6) mice. Data are mean ± SD. (*) P < 0.05, t-test.
Characterization of c-Myc as a direct transcriptional target of YAP that is functionally required for cystogenesis in both Sav1 and Pkd1 mutant kidneys
Among the YAP target genes induced in the Pkd1 mutant kidneys and the YAP transgenic kidneys, c-Myc is particularly noteworthy, since its overexpression in mouse kidneys is known to induce tubular epithelial cell proliferation and cystogenesis (Trudel et al. 1991). Interestingly, our microarray analysis revealed a more than eightfold induction of c-Myc in the human PKD1 polycystic kidney cysts compared with minimally cystic tissues (Fig. 1C), and c-Myc targets were among the top enriched pathways in ADPKD cysts (Fig. 1A). Similarly, c-Myc was induced in the PKD1 mutant (Fig. 1H), the YAP transgenic (Fig. 2D), and the Sav1 mutant (Fig. 4A) mouse kidneys. c-Myc protein levels were also increased in Pkd1 mutant (Fig. 1F,G), YAP transgenic (Supplemental Fig. S5A), and Sav1 mutant (Supplemental Fig. S5B) mouse kidneys, and the elevated c-Myc expression in the Sav1 or Pkd1 mutant kidneys was suppressed by inactivation of YAP or coinactivation of YAP/TAZ (Supplemental Fig. S5B,C). These findings implicate c-Myc as a potential transcriptional target of YAP in the kidneys. Consistent with this possibility, ChIP in TCMK-1 mouse kidney cells revealed specific binding of YAP to one of the TEAD consensus sites in the c-Myc promoter and another site in the first intron of the c-Myc locus (Fig. 4B,C). Mutagenesis of these YAP/TEAD-binding sites abolished their enhancer activity in a luciferase reporter assay in response to YAP and TEAD2 coexpression (Fig. 4D). These results suggest that the YAP/TEAD complex directly regulates c-Myc transcription by associating with c-Myc’s promoter and first intron. Importantly, loss of c-Myc completely suppressed tubule dilation and hyperproliferation in the Sav1 mutant kidneys even though inactivation of c-Myc itself did not show any overt kidney phenotypes (Fig. 4E–G; Supplemental Fig. S6A–C). Besides the kidney, β-NF also induced AhCre activity in the liver, which allowed us to characterize multiple tissues in the same animal. Similar to the kidney, loss of c-Myc also greatly suppressed the Sav1-deficient liver phenotypes, including hepatomegaly, hyperproliferation, and tumor formation (Supplemental Fig. S7). Thus, despite the myriad of YAP target genes that has been reported, our findings implicate c-Myc as the only transcriptional target of YAP whose functional contribution to YAP activity has been demonstrated clearly.
Figure 4.

c-Myc is an essential transcriptional target of YAP and a critical effector of cystogenesis in Pkd1 mutant kidneys. (A) Up-regulation of c-Myc in Sav1 mutant (AhCre;Sav1flox/flox) kidneys 3 mo after P8–P11 β-NF injections. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (B) Schematic representation of the mouse c-Myc locus showing ∼5 kb of promoter, exons (black boxes), and introns. Red bars indicate potential YAP/TEAD-binding sites. (C) ChIP assay in TCMK-1 cells showing YAP binding to site 2 in the c-Myc promoter and site 6 in the first intron of c-Myc. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (D) Luciferase reporter assay in HEK293 cells for enhancers containing either wild-type or mutated TEAD-binding sites in the c-Myc locus. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (E,F) H&E (E) and Ki67 (F) staining in control (Sav1flox/flox), Sav1 mutant (AhCre; Sav1flox/flox), Sav1;c-Myc double-mutant (AhCre;Sav1flox/flox;c-Mycflox/flox), and c-Myc mutant (AhCre;c- Mycflox/flox) kidneys 3 mo after P8–P11 β-NF injections. Arrows mark representative kidney dilations (E) and proliferating cells (F), while the arrowhead indicates a glomerular dilation (E). Bar, 100 µm. (G) Quantification of Ki67-positive cells in control, Sav1 mutant, Sav1;Yap double-mutant (AhCre;Sav1flox/flox;Yapflox/flox), Yap mutant (AhCre; Yapflox/flox), Sav1;c-Myc double-mutant, and c-Myc mutant kidneys (as in Fig. 4F; Supplemental Fig. S3A). Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (H) H&E staining of the kidneys in 1- and 3-mo-old Pkd1 mutant and Pkd1;c-Myc double-mutant (AhCre;Pkd1flox/flox;c-Mycflox/flox) mice. Bar, 1 mm. (I) The ratio of kidney weight (KW) over body weight (BW) in control (1 mo, n = 5; 3 mo, n = 4), Pkd1 mutant (1 mo, n = 9; 3 mo, n = 5), Pkd1;c-Myc double-mutant (1 mo, n = 7; 3 mo, n = 10), and c-Myc mutant (AhCre; c-Mycflox/flox) (1 mo, n = 5; 3 mo, n = 5) mice. Data are mean ± SD. (*) P < 0.05, t-test. (J) The cystic index in Pkd1 mutant (1 mo, n = 9; 3 mo, n = 5) and Pkd1;c-Myc double-mutant (1 mo, n = 7; 3 mo, n = 9) kidneys. Data are mean ± SD. (*) P < 0.05, t-test.
The essential role of c-Myc for cystogenesis in Sav1 mutant kidneys prompted us to investigate the functional requirement of c-Myc in cystogenesis in our AhCre;Pkd1flox/flox model (Fig. 3). Strikingly, while inactivation of c-Myc itself did not produce any overt kidney phenotypes, loss of c-Myc significantly suppressed the increased ratio of kidney weight over body weight and cyst formation in Pkd1 mutant kidneys (Fig. 4H–J). Taken together, these results implicate YAP and its downstream target, c-Myc, as important promoters of cystogenesis induced by Pkd1 inactivation.
A kinase inhibitor screen identifies the ROCK as a promoter of cystogenesis in 3D-cultured Pkd1 mutant mouse kidney inner medullary collecting duct (mIMCD3) cells
After demonstrating a critical role for YAP–c-Myc signaling in kidney cystogenesis, we wished to examine the molecular mechanism by which PKD1 is linked to YAP regulation. To this end, we established a novel 3D cell culture system that models PKD1-dependent cystogenesis. When cultured in a mixture of collagen matrix and Matrigel, mIMCD3 cells initially formed extended cell cords and aggregates followed by the hollowing of the cell cords and aggregates to form lumen-containing tubules and cysts, respectively (Supplemental Fig. S8A,B). A mixture of cell cords, tubules, and cysts was apparent after a 6-d culture of mIMCD3 cells, with cell cords and tubules collectively representing the predominant structures (Fig. 5A,B). Strikingly, when independent clones of Pkd1 knockout mIMCD3 cells (generated by CRISPR/Cas9) (see Supplemental Fig. S9A) were cultured under the same conditions, the mutant cells predominantly formed cysts instead of cords/tubules (Fig. 5B). Taking advantage of the 3D culture of Pkd1 mutant mIMCD3 cells, we conducted an unbiased screen for kinase inhibitors that promoted tubulogenesis in this in vitro model. Among a library of 155 kinase inhibitors, many had no effect on the 3D structures. Others inhibited or promoted the growth of 3D structures without changing the relative abundance of cords/tubules versus cysts (Fig. 5C). Only five chemicals, including Y-27632, HA-1077, H-89, (S)-H-1152, and (S)-Glycyl-H-1152, significantly increased cord/tubule formation while decreasing cyst formation in 3D culture of the PKD1 mutant mIMCD3 cells, apparently in a dosage-dependent manner (Supplemental Fig. S8C). Interestingly, the common target of these five inhibitors is ROCK (Supplemental Fig. S8D), implicating ROCK as a potential mediator of PKD1-dependent cystogenesis.
Figure 5.

Identification of RhoA signaling as a promoter of cystogenesis in 3D-cultured kidney cells. (A) Formation of cell cords (left), lumen-containing tubules (middle), and cysts (right) after culturing mIMCD3 cells in a mixture of collagen and Matrigel for 6 d. (Red) Phalloidin staining; (blue) DAPI staining. Bars, 100 µm. (B) Quantification of the percentage of cords/tubules versus cysts in control and Pkd1 mutant mIMCD3 cells in 3D culture for 6 d. Note the increased cyst formation in two independent clones of Pkd1 mutant mIMCD3 cells. (Co) Cord; (Tu) tubule. Data are mean ± SD. (*) P < 0.01, t-test. (C) A table summarizing the effect of 155 kinase inhibitors on Pkd1 mutant mIMCD3 cells in 3D culture for 6 d. (D) Western blot showing increased GTP-bound RhoA and phosphorylation of MLC in Pkd1 mutant mIMCD3 cells. (E) Tubulogenesis induced by Y-27632 and blebbistatin (both at 10 µM) in Pkd1 mutant mIMCD3 cells in 3D culture for 6 d. (Red) Phalloidin staining; (blue) DAPI staining. Bars, 100 µm. (F) Quantification of the percentage of cords/tubules versus cysts in control and two independent clones of Pkd1 mutant mIMCD3 cells in 3D culture for 6 d in the presence of Y-27632 and blebbistatin (both at 10 µM). (Co) Cord; (Tu) tubule. Data are mean ± SD. (*) P < 0.01, t-test. (G) Staining of RhoA, YAP, MLC, and F-actin in control (Pkd1flox/flox) and Pkd1 mutant (AhCre;Pkd1flox/flox) kidneys as described in Figure 1D. Note the distinct subcellular localization of RhoA, MLC, and F-actin in the control tubular epithelium (diffused in the cytoplasm; arrow) and Pkd1 mutant kidney cysts (enriched on the apical membrane; arrow) as well as the nuclear accumulation of YAP in Pkd1 mutant kidney cysts (arrow). Bars, 50 µm. (H) Staining of RhoA, YAP, MLC, and F-actin in normal human kidneys and kidneys from ADPKD patients. Note the distinct subcellular localization of RhoA, MLC, and F-actin in the normal human kidney epithelium (diffused in the cytoplasm; arrow) and ADPKD patient kidney cysts (enriched on the apical membrane; arrow) as well as the nuclear accumulation of YAP in ADPKD kidney cysts (arrow). Bars, 50 µm.
The RhoA signaling pathway mediates the regulation of YAP by PKD1
The identification of ROCK inhibitors as suppressors of cystogenesis in our 3D culture model is significant, since previous studies in both Drosophila and mammals have implicated ROCK as an upstream activator of YAP. In this context, ROCK functions as a direct effector of the small GTPase RhoA and controls actomyosin contractility by phosphorylating nonmuscle MLC (Riento and Ridley 2003). This RhoA–ROCK–MLC module has been implicated in activating YAP activity in response to diverse biological inputs such as cell adhesion, mechanical force, and GPCR ligands (Dupont et al. 2011; Wada et al. 2011; Yu et al. 2012), the same processes that are also reported to be regulated by PKD1 (Chapin and Caplan 2010; Drummond 2011). We confirmed that in mIMCD3 cells, ROCK inhibition decreased MLC phosphorylation, promoted phosphorylation of Lats1/2 and YAP (Supplemental Fig. S10A), and decreased mRNA levels of YAP target genes (Supplemental Fig. S10B), in agreement with the functional link between RhoA signaling and YAP that was established previously in other cell types (Wada et al. 2011; Yu et al. 2012).
The suppression of cystogenesis in Pkd1 mutant mIMCD3 cells by ROCK inhibitors suggests that RhoA may be activated in these cells. Indeed, when compared with control mIMCD3 cells, Pkd1 mutant mIMCD3 cells showed increased GTP-bound RhoA as well as phosphorylation of MLC (Fig. 5D). Furthermore, similar to ROCK inhibitors, the myosin inhibitor blebbistatin induced tubulogenesis in 3D culture of Pkd1 mutant mIMCD3 cells (Fig. 5E,F). Taken together, these results are consistent with activation of the RhoA–ROCK–MLC signaling pathway upon loss of PKD1.
To further corroborate a link between PKD1 loss and the RhoA–ROCK–MLC pathway, we examined Pkd1 mutant mouse kidneys. It is known that upon activation by guanine nucleotide exchange factors (GEFs), RhoA translocates to the cell membrane and interacts with effector proteins (Bustelo et al. 2007). Concurrent with nuclear accumulation of YAP in Pkd1 mutant kidney cysts, we observed a profound activation of RhoA, as indicated by accumulation of RhoA on the apical membrane of kidney tubular epithelial cells lining the cysts, in contrast to its diffuse cytoplasmic localization in the control kidney tubular epithelial cells (Fig. 5G). MLC and F-actin, the downstream effectors of RhoA, also showed apical enrichment in Pkd1 mutant cysts (Fig. 5G), consistent with activated RhoA signaling. To corroborate the relevance of these findings in human ADPKD, we examined the status of RhoA signaling and YAP in a collection of ADPKD kidneys in comparison with normal human kidneys. Similar to the Pkd1 mutant mice, human ADPKD cysts showed apical accumulation of RhoA, MLC, and F-actin and nuclear accumulation of YAP (Fig. 5H), suggesting that activation of RhoA and YAP is a general feature in ADPKD.
Inhibition of RhoA signaling suppresses cystogenesis in 3D-cultured mIMCD3 cells and Pkd1 mutant mouse kidneys
The activity of small GTPases such as RhoA is regulated by GEFs, which, upon activation, translocate from the cytoplasm to the plasma membrane to catalyze the release of GDP from small GTPases (Meyer et al. 2008; van Unen et al. 2015). To probe the molecular mechanism underlying RhoA activation in Pkd1 mutant kidney cells, we examined the subcellular localization of several RhoGEFs in mouse kidneys, including LARG, p115RhoGEF, AKAP13, p63RhoGEF, GEF-H1, and TRIO (Cook et al. 2014). Interestingly, while the other GEFs were invariably localized in the cytoplasm in control kidney tubular epithelial cells and Pkd1 mutant kidney cysts (Supplemental Fig. S11), LARG showed distinctive membrane enrichment specifically in Pkd1 mutant kidney cysts (Fig. 6A), indicating activation of LARG upon loss of PKD1.
Figure 6.

Inhibition of RhoA signaling suppresses cystogenesis in 3D-cultured mIMCD3 cells and Pkd1 mutant mouse kidneys. (A) LARG immunostaining in control and Pkd1 mutant kidneys as described in Figure 1D. Note the diffuse cytoplasmic localization of LARG in control tubular epithelia and the enrichment of LARG on plasma membranes in Pkd1 mutant kidney cysts (arrows). Bars, 50 µm. (B) Western blot showing decreased GTP-bound RhoA by siRNA knockdown of Larg in Pkd1 mutant mIMCD3 cells. Cells were analyzed 72 h after siRNA transfection. (C) Tubulogenesis induced by knockout of RhoA or Larg in Pkd1 mutant mIMCD3 cells in 3D culture for 6 d. (Red) Phalloidin staining; (blue) DAPI staining. Bars, 100 µm. (D) Quantification of the percentage of cords/tubules versus cysts in control and Pkd1 mutant mIMCD3 cells in 3D culture for 6 d with or without knockout of RhoA or Larg. Two independent clones of RhoA or Larg knockout were analyzed. (Co) Cord; (Tu) tubule. Data are mean ± SD. (*) P < 0.01, t-test. (E) A diagram showing the signaling pathway identified in this study as a critical effector signaling axis for cystogenesis after PKD1 inactivation, which includes LARG, RhoA, ROCK, MLC, YAP/TAZ, and c-Myc. The specific mechanisms by which PKD1 suppresses LARG or MLC activates YAP/TAZ remain to be determined (dashed lines). (F) Experimental scheme for inhibiting RhoA signaling in Pkd1 mutant mice. Control and AhCre;Pkd1flox/flox mice were injected with β-NF daily from P8 to P11, accompanied by daily injections of 1× PBS (vehicle) or the ROCK inhibitor Y-27632 from P8 to P14. Kidneys were harvested at P15 and are analyzed in G–M. (G) Western blot showing decreased total YAP and c-Myc protein levels and relatively increased phosphorylation of YAP (the ratio of p-YAP over YAP) after 20 mg/kg Y-27632 treatment in Pkd1 mutant kidneys. (H) Y-27632 suppressed the expression of YAP transcriptional target genes in Pkd1 mutant kidneys. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. Various dosages of Y-27632 were used. (I) Ki67 staining. Y-27632 decreased the number of proliferating cells in cystic tubular epithelia in Pkd1 mutant kidneys. Bar, 100 µm. (J) Quantification of Ki67-positive cells in cystic tubular epithelia in Pkd1 mutant kidneys treated with different dosages of Y-27632. Data are mean ± SD. n = 3. (*) P < 0.01, t-test. (K) H&E staining of Pkd1 mutant kidneys treated with different dosages of Y-27632. Bar, 1 mm. (L) Quantification of kidney weight (KW) over body weight (BW) ratio in control (n = 6) and untreated Pkd1 mutant (n = 7) mice and Pkd1 mutant mice treated with Y-27632 at 1 mg/kg (n = 4), 10 mg/kg (n = 6), 20 mg/kg (n = 9), or 50 mg/kg (n = 3). Data are mean ± SD. (*) P < 0.05, t-test. Note that administration of 50 mg/kg Y-27632 caused lethality in most of the treated mice, and therefore only three survivors were used for statistics. (M) Quantification of the kidney cystic index in the mice described in L. Data are mean ± SD. (*) P < 0.01, t-test.
Next, we investigated whether activation of LARG is responsible for RhoA activation after loss of PKD1. Consistent with this hypothesis, siRNA knockdown of Larg largely rescued the increased level of GTP-bound RhoA in Pkd1 mutant mIMCD3 cells (Fig. 6B). Furthermore, introduction of RhoA or Larg mutation (by CRISPR/Cas9) (see Supplemental Fig. S9B,C) into the Pkd1 mutant mIMCD3 cells dramatically suppressed cystogenesis and induced tubulogenesis in the 3D culture model (Fig. 6C,D). Taken together, these results uncover a signaling module involving the RhoGEF LARG, RhoA, ROCK, and MLC in mediating YAP activation and cystogenesis upon PKD1 inactivation (Fig. 6E).
To test the importance of RhoA signaling in ADPKD-related cystogenesis in vivo, we intraperitoneally injected the ROCK inhibitor Y-27632 into the β-NF-inducible AhCre;Pkd1flox/flox mouse model (Fig. 6F). Inhibition of RhoA signaling by Y-27632 in the Pkd1 mutant kidneys resulted in decreased YAP and c-Myc protein levels, increased YAP phosphorylation (Fig. 6G), and decreased mRNA levels of YAP target genes, including c-Myc, Cyr61, Ctgf, Col12A1, Amotl2, and Axl (Fig. 6H), suggesting that Y-27632 suppressed YAP activity in Pkd1 mutant kidneys. Remarkably, Y-27632 treatment also ameliorated the Pkd1 mutant phenotype, as indicated by the reduced number of Ki67-positive cystic epithelial cells (Fig. 6I,J), decreased cyst formation, and decreased ratio of kidney weight over body weight (Fig. 6K–M). These results further support the functional importance of activated RhoA–YAP signaling to cystogenesis in Pkd1-deficient kidneys.
Discussion
Although kidney cysts are observed more frequently in the collecting duct in human ADPKD patients, cysts can develop in all segments of the kidney nephron, including the glomerulus, the proximal tubule, the thick ascending limb, the distal convoluted tubule, and the collecting duct (Verani and Silva 1988; Devuyst et al. 1996; Terryn et al. 2011). Our AhCre;Pkd1flox/flox mouse model has confirmed that loss of PKD1 can induce cystogenesis in all segments of the kidney nephron. In contrast, tubular dilations induced by transgenic YAP overexpression or loss of Sav1 were observed mainly in the glomerulus, the proximal tubule, the thick ascending limb, and the distal convoluted tubule, suggesting that YAP activation alone induces a milder phenotype than loss of PKD1. Nevertheless, inactivation of YAP or coinactivation of YAP/TAZ suppressed the cystic phenotype in PKD1 mutant kidneys, implicating the Hippo signaling pathway as an important effector of kidney homeostasis and ADPKD pathogenesis.
YAP and its paralog, TAZ, usually function redundantly in development and tissue homeostasis (Nishioka et al. 2009; Xin et al. 2013; Yi et al. 2016). However, YAP and TAZ were reported to have distinct functions during embryonic kidney development: While loss of YAP impairs nephrogenesis, deletion of TAZ results in cystic tubules (Hossain et al. 2007; Makita et al. 2008; Reginensi et al. 2013), therefore raising the question of whether YAP and TAZ have similar or distinct roles during ADPKD pathogenesis. In our study, we found that, like YAP, TAZ was also activated in Pkd1 mutant kidney cysts. Although inactivation of TAZ in the AhCre;Tazflox/flox mice did not exhibit any overt kidney phenotype, loss of TAZ enhanced the effect of YAP deficiency in suppressing cyst formation in Pkd1 mutant kidneys. Thus, as in many other tissues, YAP and TAZ function redundantly to promote cystogenesis in Pkd1-deficient kidneys in adult mice. Whether the distinct functions of TAZ in kidney cystogenesis (suppressing cystogenesis during embryonic development while enhancing cystogenesis in Pkd1 mutant models) are due to different timing (embryonic versus postnatal) or contexts (normal development versus ADPKD) requires more studies in the future.
We characterized c-Myc as a direct transcriptional target of YAP that critically contributes to kidney cystogenesis. YAP and c-Myc have similar functions in many biological processes, such as cell proliferation, differentiation, apoptosis, and metabolism. Interestingly, it has long been appreciated that overexpression of c-Myc in mouse kidneys can induce tubular cysts (Trudel et al. 1991). Although activation of c-Myc has been observed in several PKD animal models (Trudel 2015), it was unclear whether c-Myc contributes to ADPKD pathogenesis. Our genetic analysis demonstrates that as a direct target gene of YAP, c-Myc not only is essential for Sav1 mutant kidney and liver phenotypes but also functions as a critical mediator of cystogenesis in Pkd1 mutant kidneys. Given that the suppression of Pkd1 mutant phenotypes by loss of c-Myc was stronger overall than the suppression by loss of YAP/TAZ, c-Myc may promote cystogenesis by integrating other pathways besides Hippo, such as Wnt and ERK, both of which have been implicated in the pathogenesis of ADPKD and are also known to regulate c-Myc transcription in other tissues.
The Hippo signaling pathway can be regulated by diverse biological inputs such as cell polarity, adhesion, mechanical force, and GPCR ligands. Many of these signals have been suggested to impact YAP activity by regulating cytoskeletal tension through a pathway involving the small GTPase RhoA, nonmuscle myosin II, and F-actin in cultured cells (Dupont et al. 2011; Wada et al. 2011; Yu et al. 2012), although the exact mechanism by which YAP is regulated by cytoskeletal tension remains elusive. Our kinase inhibitor screen in 3D-cultured Pkd1 mutant mIMCD3 cells identified a RhoA signaling pathway involving the RhoGEF LARG, RhoA, ROCK, and MLC as a molecular link between PKD1 and YAP (Fig. 6E). We further confirmed that this RhoA–YAP signaling pathway is activated in both Pkd1 mutant mouse kidneys and human ADPKD tissues. Since cystogenesis in ADPKD is characterized by abnormal accumulation of luminal fluid, which increases hydrostatic pressure within the growing cysts and mechanical stress of the cyst-lining epithelial cells (Takiar and Caplan 2011), this may provide a feed-forward mechanism that further enhances RhoA–YAP signaling and drives cystogenesis. An unanswered question from our present study concerns the specific mechanism by which PKD1 regulates LARG function. Along this line, we note that previous studies have linked PKD1 to trimeric G proteins, integrins, and cilia function (Chapin and Caplan 2010; Drummond 2011). Whether any of these entities is required for PKD1-mediated regulation of LARG activity requires future studies.
Finally, we found that chemical inhibition of RhoA–YAP signaling suppressed cyst formation after PKD1 inactivation both in vitro and in vivo. In general, the suppression of cystogenesis by chemical inhibitors is milder than genetic intervention of the signaling components (Y-27632-treated Pkd1 mutant mIMCD3 cells vs. Pkd1;Larg and Pkd1;RhoA double-mutant mIMCD3 cells in 3D culture and Y-27632-treated PKD1 mutant kidneys vs. Pkd1;Yap;Taz triple-mutant and Pkd1;c-Myc double-mutant kidneys). Consistent with these results, nuclear YAP, TAZ, and c-Myc were still detectable in some of the remaining cysts in Pkd1 mutant kidneys after Y-27632 treatment. This likely is due to the incomplete nature of chemical suppression as compared with genetic knockout. While more potent inhibitors targeting RhoA signaling may provide improved suppression, the 3D culture of Pkd1 mutant mIMCD3 that we established also offers a powerful system for unbiased high-throughput chemical screens against ADPKD in the future.
In summary, our studies identify the Hippo signaling effector YAP/TAZ and its transcriptional target, c-Myc, as critical mediators of cystic kidney pathogenesis. Not only is YAP/TAZ activated in human and mouse ADPKD tissues, loss of YAP/TAZ or c-Myc also suppresses cystogenesis in a mouse ADPKD model resulting from Pkd1 deficiency. Using a novel 3D cell culture model for ADPKD, we further identified a LARG–RhoA–ROCK–MLC pathway as a critical signaling module between PKD1 and YAP. Supporting the pathological importance of the signaling module, RhoA and its effectors are activated in human and mouse ADPKD tissues, and chemical or genetic inhibition of RhoA signaling suppresses cystogenesis in both the cell-based cystogenesis model in vitro and Pkd1 mutant mouse kidneys in vivo. Thus, the RhoA–YAP–c-Myc signaling axis represents a critical mediator in ADPKD and a potential target for therapeutic intervention.
Materials and methods
Mice
Pax8-rtTA (Traykova-Brauch et al. 2008) and Rosa26-LacZ (Soriano 1999) mice were from the Jackson Laboratory. AhCre mice (Ireland et al. 2004) were kindly provided by Dr. Douglas J. Winton. pTRE2-Yap (Chen et al. 2014), Pkd1flox (Piontek et al. 2004), Sav1flox (Cai et al. 2010), Yapflox (Zhang et al. 2010), Tazflox (Xin et al. 2013), and c-Mycflox (de Alboran et al. 2001) mice were described previously. These mice were maintained on a mixed genetic background of C57BL/6J and 129/Sv.
The Pax8-rtTA;pTRE2-Yap double-transgenic mice were generated by breeding Pax8-rtTA with pTRE2-Yap single-transgenic mice. To induce YAP expression in the kidney, mice at 4 wk of age were fed 0.02 mg/mL doxycycline (LKT Laboratories) in drinking water supplemented with 2.5% sucrose. Wild-type, Pax8-rtTA, or pTRE2-Yap single-transgenic mice fed doxycycline were used as a control. Mice with Pkd1, Sav1, Yap, Taz, or c-Myc specifically deleted in the kidney were generated by breeding Pkd1flox, Sav1flox, Yapflox, Tazflox, or c-Mycflox mice with AhCre mice. Four daily i.p. injections of 80 mg/kg β-NF (Sigma) dissolved in corn oil were performed in animals from P8 to induce Cre expression. To inhibit RhoA signaling, seven daily i.p. injections of Y-27632 (Enzo Life Sciences) dissolved in sterile 1× PBS were performed in animals from P8. Both male and female mice were used. The animals used in phenotypic analyses are listed in Supplemental Table S3. The animals used in Figure 5 were generated at the University of Texas Southwestern Medical Center, and the rest were generated at Johns Hopkins University. Animal protocols were approved by the Institutional Animal Care and Use Committee of Johns Hopkins University and the University of Texas Southwestern Medical Center.
Cell culture analysis
ON-TARGETplus mouse Larg siRNA was from Dharmacon. Transfection reagents Lipofectamine RNAi-MAX and Lipofectamine 3000 were from Life Technologies. The kinase screening library (no. 10505) was from Cayman Chemical, and chemicals were used at a final concentration of 10 µM in DMEM/F12 medium. Y-27632 dihydrochloride was from Enzo Life Sciences. HA-1077 dihydrochloride and H-89 dihydrochloride were from Sigma. H-1152 dihydrochloride was from Fisher Scientific. Glycyl-H-1152 dihydrochloride was from R&D Systems. Blebbistatin was from Sigma. These chemicals were used in Figure 5, E and F, and Supplemental Figure S8C.
TCMK-1 and HEK293 cells (both from American Type Culture Collection [ATCC]) were cultured at 37°C in DMEM with 10% fetal bovine serum and penicillin/streptomycin (Invitrogen). ChIP assays were performed according to a previously described protocol (Wang et al. 2009). Briefly, ∼5 × 106 TCMK-1 cells for each assay were cross-linked with 1% formaldehyde and sonicated to an average fragment size between 200 and 500 base pairs. Two micrograms of antibody (rabbit IgG or YAP antibody) and 50 µL of protein G agarose were used in each ChIP assay. The immunoprecipitated DNA was quantified using real-time PCR. All values were normalized to the input. YAP antibody (NB110-58358) was purchased from Novus Biologicals. Consensus YAP–TEAD-binding sites in c-Myc’s promoter and first intron were predicted by MatInspector. The primers for analyzing the ChIP DNA are listed in Supplemental Table S4.
For luciferase assay, HEK293 cells were transfected with plasmids, and analysis was performed 24 h later. Transfection reagent Effectene from Qiagen was used. We performed the experiments at least three times and show representative results. The YAP/TEAD-binding site-containing c-Myc promoter and intron fragments were amplified with specific primers (Supplemental Table S4) and mouse liver genomic DNAs as the template and subcloned into pBV-Luc reporter plasmids (Addgene, 16539) (He et al. 1998). Mutagenesis of the YAP/TEAD-binding sites (Supplemental Table S4) was performed using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's protocol.
mIMCD3 cells (ATCC) were cultured at 37°C in DMEM/F12 with 10% fetal bovine serum and penicillin/streptomycin (Invitrogen). Pkd1, RhoA, and Larg knockout mIMCD3 cells were generated as described (Ran et al. 2013). Briefly, the guide oligos 5′-CACCGGCTACCCTGGTGGGGCCCCA-3′ and 5′-AAACTGGGGCCCCACCAGGGTAGCC-3′ for Pkd1, 5′-CACCGTCGTGTGCTCGTCATTCCGA-3′ and 5′-AAACTCGGAATGACGAGCACACGAC-3′ for RhoA, and 5′-CACCGGCAATCTTTGGGTAGCCGTT-3′, and 5′-AAACAACGGCTACCCAAAGATTGCC-3′ for Larg were annealed and cloned into pSpCas9(BB)-2A-Puro vector before being transfected into mIMCD3 cells. Puromycin (1 µg/mL) was used for selection after transfection. Surviving cells were diluted in DMEM/F12 at a concentration of 0.5 cells per 100 µL and plated in 96-well plates. Colonies derived from single cells were expanded, and mutant colonies were selected by genome sequencing and/or Western blot.
3D culture for mIMCD3 cells and immunofluorescence
mIMCD3 cells were cultured in 3D gel mixture. Each 100 µL of the gel mixture contains 1× DMEM/F12, 35 µL of collagen I (ThermoFisher Scientific, A1048301), 15 µL of Matrigel (Fisher Scientific, 354230), 25 mM NaHCO3, 20 mM HEPES, and 5 mM NaOH. Trypsinized mIMCD3 cells were mixed well with the gel mixture prepared on ice. One-hundred microliters of gel mixture containing 1000 cells was transferred into each well of 96-well plates and incubated for 30 min at 37°C to solidify. One-hundred microliters of fresh DMEM/F12 with 10% fetal bovine serum was added on top of the gel mixture and replaced every other day. Chemicals were diluted in DMEM/F12 medium at the indicated concentration. The 3D structures were observed under an inverted microscope. A total of 150–400 3D structures in each well was examined for each experiment, and the experiment was repeated three times. A total of 450–1200 3D structures from three independent experiments was analyzed for each data point.
For immunofluorescence of mIMCD3 3D culture, cells in the gel mixture were cultured on eight-well chamber slides (ThermoFisher Scientific, 154534). For each well, 200 µL of the gel mixture and 250 µL of culture medium were used. Immunofluorescence was performed as described (Elia and Lippincott-Schwartz 2009) and imaged using a Zeiss LSM880 inverted confocal microscope system.
RhoA activation assay
GTP-bound RhoA was detected using a RhoA activation assay kit (Cytoskeleton, BK036) according to the manufacturer's protocol. Briefly, mIMCD3 cells were lysed and centrifuged. A fraction of the supernatant was saved for analysis of total RhoA, and the rest was incubated with rhotekin's Rho-binding domain-conjugated beads for 1 h at 4°C on a rocker. The beads were washed, precipitated, and subjected to Western blot analysis.
Western blotting
Cultured cells were lysed and analyzed. Primary antibodies used for Western blot were rabbit anti-MLC2 (1:1000; Cell Signaling, 8505), anti-p-MLC2 (1:1000; Cell Signaling, 3674), anti-Lats1 (1:1000; Cell Signaling, 3477), anti-p-Lats (Yu et al. 2010), anti-YAP (1:1000; Cell Signaling, 4912), anti-p-S397-YAP (1:1000; Cell Signaling, 13619), and anti-c-Myc (1:1000; Abcam, ab32072) and mouse anti-RhoA (1:500; Cytoskeleton, Inc., ARH03) and anti-Actin (1:20,000; Millipore, MAB1501R).
Human renal tissue analysis
Formalin-fixed paraffin-embedded tissue blocks were obtained from archival human normal and ADPKD kidneys at the PKD Research Biomaterials and Cellular Models Core in the Kidney Institute at the Kansas University Medical Center and were sectioned at 5 µm. Primary antibodies used for staining were rabbit anti-YAP (1:100; Epitomics, 2060-1) and anti-MLC2 (1:10; Cell Signaling, 8505) and mouse anti-RhoA (1:50; Cytoskeleton, Inc., ARH03) and anti-Actin (1:200; EMD Millipore, MAB1501R). Cy3-conjugated goat anti-rabbit and Alexa488-conjugated goat anti-mouse secondary antibodies (Molecular Probes) were used for immunofluorescence. In total, five normal samples and five ADPKD kidneys were analyzed. The use of surgically discarded human kidney tissues was approved by the Institutional Review Board at the Kansas University Medical Center. Informed consent was obtained from all subjects.
Mouse histological analysis and immunohistochemistry
Mouse kidneys were collected, fixed overnight in 4% paraformaldehyde in 1× PBS, embedded in paraffin, and sectioned at 5 µm. Sections were stained with hematoxylin–eosin for histological analysis. Immunohistochemistry was performed according to the manufacturer's protocols. Primary antibodies used for immunohistochemistry were rabbit anti-Ki67 (1:1000; Novocastra, NCL-Ki67p), anti-THP (1:500) (Yu et al. 2007), anti-YAP (1:100; Cell Signaling, 4912), anti-TAZ (1:100; Cell Signaling, 4883), anti-c-Myc (1:100; Abcam, ab32072), anti-MLC2 (1:10; Cell Signaling, 8505), anti-LARG (1:100; Abcam, ab136072), anti-GEF-H1 (1:100; Cell Signaling, 4076), anti-p115RhoGEF (1:100; Cell Signaling, 3669), anti-AKAP13 (1:100; Sigma, SAB140866), anti-p63RhoGEF (1:100; Sigma, SAB2701512), and anti-TRIO (1:100; Sigma, HPA064664) and mouse anti-RhoA (1:50; Cytoskeleton, Inc., ARH03) and anti-Actin (1:200; EMD Millipore, MAB1501R). Biotinylated LTL (1:400; B-1325) and DBA (1:400; B-1035) and fluorescin-conjugated LTL (1:400; FL-1321) and DBA (1:400; FL-1031) were purchased from Vector Laboratories. For Ki67, LTL, THP, and DBA staining, the signals were developed using the ABC kit purchased from Vector Laboratories according to manufacturer's suggestions. Cy3-conjugated goat anti-rabbit and Alexa488-conjugated goat anti-mouse secondary antibodies (Molecular Probes) were used for immunofluorescence.
For LacZ reporter analysis, mouse kidneys were embedded in OCT and frozen-sectioned at 10 µm. After being warmed to room temperature, the sections were fixed by 4% paraformaldehyde and stained in X-gal solution containing 1 mg/mL X-gal, 2 mM MgCl2, 5 mM K3[Fe(CN)6], and 5 mM K4[Fe(CN)6] in 1× PBS. Finally, the sections were counterstained with eosin, dehydrated, and mounted for photographing.
For quantification of cystogenesis, mouse kidneys were longitudinally sectioned. The cystic index was calculated as the percentage of cystic area over total area on kidney sections.
Microarray analysis
Microarray analysis of renal cysts in comparison with minimally cystic tissues and normal kidneys was described previously (Song et al. 2009). Briefly, mRNA were extracted from small cysts (n = 5) <1 mL, medium cysts (n = 5) between 10 and 25 mL, and large cysts (n = 3) >50 mL that were dissected from five human PKD1 kidneys within 30 min of nephrectomy; snap-frozen in liquid N2; and stored at −80°C. Minimally cystic tissue (n = 5), which might have contained a few microscopic cysts from the renal cortex, was obtained as PKD control tissue from the same kidneys and similarly processed. Additionally, noncancerous renal cortical tissue from nephrectomized kidneys with isolated renal cell carcinoma (KIDNEY; n = 3) was used as normal control tissue. Global gene expression profiling was performed using the Human Genome U133 Plus 2.0 array (Affymetrix), and the data were analyzed using published protocols (Song et al. 2009). Significance Analysis of Microarrays (SAM) was used to identify differentially expressed genes with a false discovery rate (q-value) of ≤1%. The expression of 379 known YAP/TAZ target genes (Zanconato et al. 2015) in cysts (mean of 13 cysts, including five small cysts, five medium cysts, and three large cysts), minimally cystic tissues (mean of five minimally cystic tissues), and KIDNEYs (mean of three KIDNEYs) is summarized in Supplemental Table S1. Up-regulated (red) and down-regulated (green) YAP/TAZ target genes in cysts versus minimally cystic tissues are listed in Supplemental Table S2. Gene set enrichment analysis of ADPKD cysts versus minimally cystic tissues was applied to determine the relative representation of targets of various signaling pathways that were significantly up-regulated in ADPKD cysts (Subramanian et al. 2005).
Quantitative real-time PCR
RNAs from kidneys were extracted using the TRIzol reagent (Invitrogen). RNA was reverse-transcribed using the iScrip cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was performed using the iQ SYBR Green Supermix (Bio-Rad) on a CFX96 real-time system (Bio-Rad). Primers for real-time PCR are listed in Supplemental Table S4.
Statistical analyses
All samples or animals were included in the statistical analyses. The sample size (n ≥ 3) was not predetermined by any statistical method. No randomization was used. For histological and luciferase analyses, we performed the experiments at least three times and show representative results. None of the analyses were blinded to the group allocation. Comparisons were made using two-tailed t-tests. The variance was similar between groups that were statistically compared. Significance was set at P < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Darren Wallace of the PKD Research Biomaterials and Cellular Models Core in the Kidney Institute at the Kansas University Medical Center for providing the human renal tissues. This study was supported in part by grants from the Department of Defense (PR130920) and the National Institute of Diabetes and Digestive and Kidney Diseases (DK098424, DK095036, and DK111611). This work was supported by a pilot grant from the Baltimore Polycystic Kidney Disease Research and Clinical Core Center (P30 DK090868). D.P. is an investigator of the Howard Hughes Medical Institute.
Author contributions: J.C., F.Q., and D.P. designed and interpreted the study. J.C. performed experiments with X.S. for microarray analysis and with W.W. for ChIP assays. J.C. and D.P. analyzed the results. J.C., T.W., Y.P., F.Q., and D.P. wrote the manuscript. All authors commented on the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.315127.118.
References
- Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, Gertz EM, Schambach A, Kreipe HH, Pfeifer D, et al. 2012. The phenotype of human STK4 deficiency. Blood 119: 3450–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiari G, Catizone L, Del Senno L. 2013. Multidrug therapy for polycystic kidney disease: a review and perspective. Am J Nephrol 37: 175–182. [DOI] [PubMed] [Google Scholar]
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. 2006. Renal cystic diseases: a review. Adv Anat Pathol 13: 26–56. [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Sauzeau V, Berenjeno IM. 2007. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29: 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. 2010. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 24: 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin HC, Caplan MJ. 2010. The cell biology of polycystic kidney disease. J Cell Biol 191: 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, Pan D. 2014. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev 28: 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DR, Rossman KL, Der CJ. 2014. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33: 4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW. 2001. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 14: 45–55. [DOI] [PubMed] [Google Scholar]
- Devuyst O, Burrow CR, Smith BL, Agre P, Knepper MA, Wilson PD. 1996. Expression of aquaporins-1 and -2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am J Physiol 271: F169–F183. [DOI] [PubMed] [Google Scholar]
- Drummond IA. 2011. Polycystins, focal adhesions and extracellular matrix interactions. Biochim Biophys Acta 1812: 1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183. [DOI] [PubMed] [Google Scholar]
- Elia N, Lippincott-Schwartz J. 2009. Culturing MDCK cells in three dimensions for analyzing intracellular dynamics. Curr Protoc Cell Biol 43: 4.22.1–4.22.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossdal R, Jonasson F, Kristjansdottir GT, Kong A, Stefansson H, Gosh S, Gulcher JR, Stefansson K. 2004. A novel TEAD1 mutation is the causative allele in Sveinsson's chorioretinal atrophy (helicoid peripapillary chorioretinal degeneration). Hum Mol Genet 13: 975–981. [DOI] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S. 2012. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol 13: 591–600. [DOI] [PubMed] [Google Scholar]
- Happe H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, Peters DJ. 2011. Altered Hippo signalling in polycystic kidney disease. J Pathol 224: 133–142. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. 1998. Identification of c-MYC as a target of the APC pathway. Science 281: 1509–1512. [DOI] [PubMed] [Google Scholar]
- Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W. 2007. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci 104: 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. 2004. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of β-catenin. Gastroenterology 126: 1236–1246. [DOI] [PubMed] [Google Scholar]
- LaRiviere WB, Irazabal MV, Torres VE. 2015. Novel therapeutic approaches to autosomal dominant polycystic kidney disease. Transl Res 165: 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, Larson C, Brent G, Zhou J. 1997. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 17: 179–181. [DOI] [PubMed] [Google Scholar]
- Makita R, Uchijima Y, Nishiyama K, Amano T, Chen Q, Takeuchi T, Mitani A, Nagase T, Yatomi Y, Aburatani H, et al. 2008. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol 294: F542–F553. [DOI] [PubMed] [Google Scholar]
- Meyer BH, Freuler F, Guerini D, Siehler S. 2008. Reversible translocation of p115–RhoGEF by G(12/13)-coupled receptors. J Cell Biochem 104: 1660–1670. [DOI] [PubMed] [Google Scholar]
- Nehme NT, Pachlopnik Schmid J, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P, Rieux-Laucat F, Lutz P, Picard C, Mahlaoui N, et al. 2012. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119: 3458–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka N, Inoue K, Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N, et al. 2009. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell 16: 398–410. [DOI] [PubMed] [Google Scholar]
- Pan D. 2010. The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Chowdhury R, Igarashi P. 2009. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens 18: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, et al. 2004. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol 15: 3035–3043. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim DS, Pawson T, Wrana J, McNeill H. 2013. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet 9: e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. 2003. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456. [DOI] [PubMed] [Google Scholar]
- Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y. 2009. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18: 2328–2343. [DOI] [PubMed] [Google Scholar]
- Soriano P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70–71. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiar V, Caplan MJ. 2011. Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta 1812: 1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terryn S, Ho A, Beauwens R, Devuyst O. 2011. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 1812: 1314–1321. [DOI] [PubMed] [Google Scholar]
- Traykova-Brauch M, Schonig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, et al. 2008. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel M. 2015. c-Myc signalling in the genetic mechanism of polycystic kidney disease. In Polycystic kidney disease (ed. Li X), pp. 231–257. Codon Publications, Brisbane, Australia. [PubMed] [Google Scholar]
- Trudel M, D'Agati V, Costantini F. 1991. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int 39: 665–671. [DOI] [PubMed] [Google Scholar]
- van Unen J, Reinhard NR, Yin T, Wu YI, Postma M, Gadella TW, Goedhart J. 2015. Plasma membrane restricted RhoGEF activity is sufficient for RhoA-mediated actin polymerization. Sci Rep 5: 14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verani RR, Silva FG. 1988. Histogenesis of the renal cysts in adult (autosomal dominant) polycystic kidney disease: a histochemical study. Mod Pathol 1: 457–463. [PubMed] [Google Scholar]
- Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. 2011. Hippo pathway regulation by cell morphology and stress fibers. Development 138: 3907–3914. [DOI] [PubMed] [Google Scholar]
- Wang W, Huang L, Huang Y, Yin JW, Berk AJ, Friedman JM, Wang G. 2009. Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Dev Cell 16: 764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PD. 2004. Polycystic kidney disease: new understanding in the pathogenesis. Int J Biochem Cell Biol 36: 1868–1873. [DOI] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. 2013. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci 110: 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Lu L, Yanger K, Wang W, Sohn BH, Stanger BZ, Zhang M, Martin JF, Ajani JA, Chen J, et al. 2016. Large tumor suppressor homologs 1 and 2 regulate mouse liver progenitor cell proliferation and maturation through antagonism of the coactivators YAP and TAZ. Hepatology 64: 1757–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. 2013. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 154: 1342–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, Menezes LF, Xu H, Germino GG, Zuo J, et al. 2007. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci 104: 18688–18693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. 2010. Kibra functions as a tumor suppressor protein that regulates hippo signaling in conjunction with Merlin and Expanded. Dev Cell 18: 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al. 2012. Regulation of the Hippo–YAP pathway by G-protein-coupled receptor signaling. Cell 150: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. 2015. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. 2015. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. 2010. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 19: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.