

In this study, Zhao et al. show that PAPAS, a long noncoding RNA (lncRNA) that is transcribed in an orientation antisense to pre-rRNA, interacts directly with DNA, forming a DNA–RNA triplex structure that tethers PAPAS to a stretch of purines within the enhancer region, thereby guiding associated CHD4/NuRD to the rDNA promoter. Their results provide insight into the function of lncRNAs in guiding chromatin remodeling complexes to specific genomic loci and a phosphorylation-dependent mechanism of CHD4/NuRD-mediated transcriptional regulation.
Keywords: CHD4/NuRD, heat shock, lncRNA, RNA polymerase I transcription, rRNA genes, DNA–RNA triplexes
Abstract
Attenuation of pre-rRNA synthesis in response to elevated temperature is accompanied by increased levels of PAPAS (“promoter and pre-rRNA antisense”), a long noncoding RNA (lncRNA) that is transcribed in an orientation antisense to pre-rRNA. Here we show that PAPAS interacts directly with DNA, forming a DNA–RNA triplex structure that tethers PAPAS to a stretch of purines within the enhancer region, thereby guiding associated CHD4/NuRD (nucleosome remodeling and deacetylation) to the rDNA promoter. Protein–RNA interaction experiments combined with RNA secondary structure mapping revealed that the N-terminal part of CHD4 interacts with an unstructured A-rich region in PAPAS. Deletion or mutation of this sequence abolishes the interaction with CHD4. Stress-dependent up-regulation of PAPAS is accompanied by dephosphorylation of CHD4 at three serine residues, which enhances the interaction of CHD4/NuRD with RNA and reinforces repression of rDNA transcription. The results emphasize the function of lncRNAs in guiding chromatin remodeling complexes to specific genomic loci and uncover a phosphorylation-dependent mechanism of CHD4/NuRD-mediated transcriptional regulation.
There is emerging evidence that the interaction of long noncoding RNAs (lncRNAs) with chromatin-associated proteins impinges on chromatin structure and transcriptional regulation. lncRNAs have been proposed to function in a variety of ways, serving as scaffolds, decoys, guides, or enhancers. They are known to act in cis or in trans; however, the mechanisms underlying lncRNA-mediated changes of chromatin structure in response to environmental signals remain elusive. Many chromatin modifiers are RNA-binding proteins and are targeted to specific chromatin loci by regulatory RNAs (Rinn et al. 2007; Nagano et al. 2008; Zhao et al. 2008; Tsai et al. 2010). Global transcriptome analysis has shown that up to 70% of transcripts have antisense partners that can alter the expression of the respective sense gene. In accord with this notion, strand-specific PCR walking across rDNA revealed low levels of antisense transcripts, termed PAPAS (“promoter and pre-rRNA antisense”). PAPAS is transcribed by RNA polymerase II (Pol II), yielding >10-kb transcripts that cover the gene body and intergenic regions, including the rDNA promoter and upstream enhancer sequences. Notably, the level of PAPAS anti-correlates with rDNA transcription; that is, PAPAS is up-regulated in growth-arrested cells and down-regulated in rapidly proliferating cancer cells. Elevated levels of PAPAS in quiescent cells recruit the histone methyltransferase Suv4-20h2 to the rDNA promoter, leading to trimethylation of histone H4 at Lys20 (H4K20me3), chromatin compaction, and repression of rRNA synthesis (Bierhoff et al. 2014). This study revealed a unique mechanism by which PAPAS regulates the epigenetic signature at the rDNA promoter and ensures shutdown of RNA Pol I transcription when cells become quiescent.
Another PAPAS-based mechanism throttles rRNA synthesis in response to hypo-osmotic stress or heat shock. At elevated temperatures, increased levels of PAPAS recruit CHD4/NuRD (nucleosome remodeling and deacetylation), a protein complex that has important roles in the regulation of gene expression by integrating ATP-dependent chromatin remodeling and histone-modifying activities (Tong et al. 1998; Xue et al. 1998; Zhang et al. 1998). NuRD is composed of several subunits, each with several isoforms, including the ATPase CHD3/4, the protein deacetylases HDAC1/2, metastasis-associated proteins MTA1/2/3, the retinoblastoma-associated histone-binding proteins RBP4/7, p66/68, and MBD2/3, which harbor a methyl-CpG-binding domain. Consistent with a versatile use in transcriptional regulation, NuRD plays a key role in various cellular processes, including cell cycle progression, stem cell biology, DNA damage responses, and maintenance of genome integrity (Denslow and Wade 2007; Lai and Wade 2011; Hu and Wade 2012).
PAPAS interacts with CHD4, the ATPase subunit of NuRD, which triggers epigenetic silencing of rDNA by shifting the promoter-bound nucleosome into a position that is refractory to transcription initiation (Zhao et al. 2016a,b). Here we show that PAPAS directly binds to a promoter-proximal stretch of homopurines in the rDNA enhancer. Tethering PAPAS to DNA facilitates the recruitment of CHD4/NuRD to the rDNA promoter and ensures repression of rRNA synthesis in response to elevated temperature. Of note, increased levels of PAPAS are necessary but not sufficient for CHD4/NuRD-dependent repression of rDNA transcription. Targeting CHD4/NuRD to the rDNA promoter requires heat-induced dephosphorylation of CHD4, which facilitates the interaction with PAPAS. Furthermore, RNA–protein interaction experiments, chemical structure probing, and RNase footprinting show that a single-stranded A-rich motif in PAPAS is recognized by CHD4. Disruption of this sequence leads to loss of CHD4 binding, indicating that the structure of PAPAS covering the rDNA promoter is important for its function. The results decipher the molecular mechanism underlying PAPAS-dependent targeting of CHD4/NuRD to rDNA and reveal that heat stress-induced hypophosphorylation of CHD4 is essential for NuRD recruitment and repression of rDNA transcription.
Results
The interaction of CHD4 with PAPAS is increased upon stress
Repression of pre-rRNA synthesis in response to unfavorable growth conditions is accompanied by up-regulation of PAPAS, a lncRNA that is transcribed in antisense orientation to pre-rRNA. PAPAS represents a heterogeneous 12- to 16-kb RNA population that is synthesized by RNA Pol II from a subpopulation of rDNA repeats covering the coding region, the rDNA promoter, and further upstream enhancer sequences (Bierhoff et al. 2010, 2014). Transcription of PAPAS inversely correlates with the level of 45S pre-rRNA, being low in transformed cells that exhibit high transcriptional activity and high in quiescent and stressed cells in which rDNA transcription is repressed. Stress-induced up-regulation of PAPAS correlates with increased occupancy of the CHD4/NuRD corepressor complex at rDNA, deacetylation of histones, and movement of the promoter-bound nucleosome into a position that is refractory to transcription initiation (Zhao et al. 2016a,b). In accord with these studies, up-regulation of PAPAS in response to heat shock correlates with repression of pre-rRNA synthesis (Fig. 1A; Supplemental Fig. S1A). To examine whether up-regulation of PAPAS is sufficient to recruit CHD4/NuRD to the rDNA promoter and attenuate pre-rRNA synthesis, we took advantage of the CRISPRa approach (Gilbert et al. 2013; Konermann et al. 2015) to target dCas9-VP64 to the 3′ end of the rDNA transcription unit. Although CRISPRa-mediated up-regulation of PAPAS was higher than in heat-shocked cells, elevated levels of PAPAS induced by dCas9-VP64 did not compromise pre-rRNA synthesis (Fig. 1A). Thus, although there is a causal relationship between stress-dependent up-regulation of PAPAS and CHD4-dependent repression of rDNA transcription, elevated levels of PAPAS on their own are not sufficient to down-regulate rDNA transcription.
Figure 1.

The interaction of CHD4 with PAPAS is enhanced upon heat shock. (A) PAPAS and pre-rRNA levels in NIH3T3 cells cultured in normal conditions (Ctrl), upon heat shock (HS) for 3h at 42°C, or after single guide RNA (sgRNA)-mediated recruitment of dCas9-VP64 (VP64). PAPAS values measured by RT-qPCR were normalized to GAPDH mRNA and are displayed relative to Ctrl. Pre-rRNA was quantified from fluorograms of metabolically labeled cellular RNA shown in Supplemental Figure S1A. n = 3. (B) ChIP-qPCR (chromatin immunoprecipitation [ChIP] combined with quantitative PCR [qPCR]) monitoring CHD4 occupancy at the rDNA promoter upon heat shock (HS) or after dCas9-VP64-mediated up-regulation of PAPAS (VP64). Data were normalized to cells transfected with the empty sgRNA vector. n = 3. (C) CLIP/RT–PCR (cross-linking and immunoprecipitation [CLIP] followed by RT–PCR) monitoring the interaction of CHD4 with PAPAS and the lncRNA Air in normal conditions (Ctrl), upon heat shock (HS), or after dCas9-VP64-mediated up-regulation of PAPAS (VP64). The numbers below represent the amount of CHD4-associated PAPAS normalized to input levels in unperturbed conditions (Ctrl), after heat shock (HS), and after CRISPRa-mediated recruitment of dCas9-VP64 (VP64). (D) Western blot showing binding of Flag-CHD4 (Fl-CHD4) from normal (Ctrl) or heat-shocked (HS) HEK293T cells to empty beads or bead-bound biotinylated PAPAS (−1/−205). (E) Pull-down assay showing binding of radiolabeled PAPAS (−1/−205) to immobilized Fl-CHD4 from untreated HEK293T cells (Ctrl) and cells exposed to heat shock (HS). The Coomassie brilliant blue stain shows equal amounts of bead-bound Fl-CHD4. (F) Western blot showing binding of CHD4 to chromatin from control or heat-shocked HEK293T cells in the absence and presence of RNase A. The bar diagram at the right shows the level of CHD4 binding after normalization to histone H3. n = 3. (G) Pull-down assay monitoring binding of radiolabeled PAPAS (−1/−205) to immobilized Fl-CHD4-N and Fl-CHD4-C. The structure of full-length and truncated CHD4 is depicted below. (H) Northwestern blot showing binding of PAPAS to Fl-CHD4-N. The Ponceau stain at the right shows the amount of filter-bound Fl-CHD4-N and Fl-CHD4-C. (I) CLIP/RT-qPCR comparing binding of PAPAS to full-length Fl-CHD4, Fl-CHD4-N, and Fl-CHD4-C. Data were normalized to input. n = 3. (J) ChIP-qPCR assays monitoring binding of Fl-CHD4, Fl-CHD4-N, and Fl-CHD4-C to the rDNA promoter. n = 4. Error bars represent mean values ± standard deviation (SD).
Chromatin immunoprecipitation (ChIP) experiments confirmed that up-regulation of PAPAS after thermal stress correlates with increased rDNA occupancy of CHD4/NuRD without affecting the overall level of NuRD components (Fig. 1B; Supplemental Fig. S1B). CRISPRa-mediated induction of PAPAS, on the other hand, did not lead to increased binding. These results emphasize that elevated levels of PAPAS are required for NuRD-dependent transcriptional repression upon heat shock but do not affect transcription in unstressed cells.
Previous studies have shown that CHD4 is an RNA-binding protein that exhibits binding preference for the 3′-terminal part of PAPAS, which overlaps the rDNA promoter (Zhao et al. 2016b). To get insight into the molecular mechanism underlying PAPAS-dependent recruitment of CHD4/NuRD in response to heat shock, we monitored CHD4-associated PAPAS by CLIP/RT-PCR (cross-linking and immunoprecipitation [CLIP] followed by RT–PCR). In support of elevated levels of PAPAS targeting CHD4/NuRD to rDNA, the interaction of CHD4 with PAPAS was strongly enhanced at elevated temperatures (Fig. 1C). Binding of other CHD4-associated RNAs that have been identified by genome-wide formaldehyde RNA immunoprecipitation (fRIP) assays (for example, NEAT1 and XIST) (Hendrickson et al. 2016) was also increased, while the interaction with HOTAIR and U2 small nuclear RNA (snRNA) was not affected (Supplemental Fig. S1C). In contrast, CRISPRa did not increase the level of CHD4-associated PAPAS. The finding that the RNA-binding activity of CHD4 is increased upon external stress suggests that stress-induced post-translational modifications are required for PAPAS-dependent targeting of CHD4 to rDNA.
To get insight into the molecular mechanism underlying PAPAS-dependent recruitment of CHD4/NuRD in response to elevated temperatures, we incubated bead-bound PAPAS with lysates of unstressed and heat-shocked HEK293T cells and analyzed PAPAS-associated CHD4 on Western blots. Higher amounts of CHD4 were retained if PAPAS was incubated with lysates from heat-shocked as compared with untreated cells, underscoring that heat stress augments the interaction of PAPAS with CHD4 (Fig. 1D). In a reciprocal approach, we monitored binding of PAPAS to CHD4 upon exposure to elevated temperatures. Again, CHD4 from heat-shocked cells interacted more efficiently with PAPAS than CHD4 from unstressed cells (Fig. 1E). These in vitro assays are in accord with the CLIP data showing increased association of CHD4 with PAPAS upon thermal stress. The weak interaction with “pRNA,” the respective sense counterpart (−205/−1) of PAPAS (Schmitz et al. 2010), was also enhanced, suggesting a functional interplay of PAPAS, pRNA, and NuRD that attenuates rDNA transcription in heat-shock conditions (Supplemental Fig. S1D,E).
The finding that the interaction of CHD4 with PAPAS correlated with rDNA occupancy of CHD4 suggested that RNA might stabilize CHD4/NuRD binding to chromatin. In support of this view, treatment with RNase A significantly decreased the association of CHD4 with chromatin (Fig. 1F). Association of the transcription factor UBF was not affected by RNase A treatment, indicating that binding of NuRD, but not components of the Pol I transcription apparatus, requires RNA.
CHD4 is a large (1912-amino-acid) protein with multiple functional domains and numerous post-translational modifications. To dissect the RNA-binding capacity of CHD4, we performed RNA pull-down assays using the N-terminal part (amino acids 1–700) and the C-terminal part (amino acids 701–1912) of CHD4. The N-terminal part (CHD4-N) efficiently bound to PAPAS, whereas the C-terminal part (CHD4-C) did not display significant RNA-binding activity (Fig. 1G). To substantiate that CHD4 rather than other subunits of the NuRD complex or NuRD-associated proteins mediates RNA binding, we monitored PAPAS–CHD4 interactions on Northwestern blots. After gel electrophoresis and transfer to a nitrocellulose membrane, CHD4 was renatured and incubated with radiolabeled in vitro transcribed PAPAS. Again, CHD4-N, but not CHD4-C, interacted with PAPAS, confirming that CHD4-N harboring the two plant homeodomains (PHDs) and chromodomains mediates the interaction with RNA (Fig. 1H). The interaction of CHD4-N with PAPAS was also augmented at elevated temperatures, underscoring that the N-terminal half of CHD4 behaves similarly to full-length CHD4 (Supplemental Fig. S1F–H).
To assess the RNA-binding specificity of CHD4, we monitored the association of PAPAS with CHD4 after UV cross-linking and immunoprecipitation with anti-Flag antibody. This CLIP experiment substantiated that both full-length CHD4 and CHD4-N, but not CHD4-C, are associated with PAPAS in vivo (Fig. 1I). Parallel ChIP experiments corroborated that both full-length CHD4 and the C-terminally truncated CHD4-N bound to the rDNA promoter, while the C-terminal part harboring the SNF2 and helicase domain did not associate with rDNA (Fig. 1J). Altogether, these results reveal that CHD4 is a strong and specific RNA-binding protein, and the interaction with both RNA and DNA is brought about by the N-terminal part of CHD4, which harbors the PHD and chromodomains.
Stress-dependent dephosphorylation increases the RNA-binding efficiency of CHD4
As protein phosphorylation is a central post-translational modification in cellular signaling, we sought to explore whether reversible phosphorylation might regulate the interaction of CHD4 with RNA. To test this, we performed pull-down experiments monitoring the interaction of CHD4-N with PAPAS in the absence and presence of phosphatase inhibitors. Binding of CHD4-N to PAPAS was decreased in the presence of phosphatase inhibitors, indicating that phosphorylation inhibits the interaction of CHD4 with RNA. CHD4-N from heat-stressed cells, on the other hand, retained elevated binding activity regardless of whether it was purified in the presence of phosphatase inhibitors or was treated with calf intestine phosphatase (Fig. 2A; Supplemental Fig. S2A). The finding that binding of CHD4 from control cells was decreased if dephosphorylation was prevented indicates that, in normal conditions, CHD4 is hyperphosphorylated, and its interaction with PAPAS is weak. Under stress conditions, CHD4 is dephosphorylated, which enhances the interaction with RNA. In support of this view, Western blots monitoring phosphorylation of CHD4-N with anti-phospho-serine antibody revealed a marked decrease in serine phosphorylation in heat-shocked cells (Fig. 2B), suggesting that the kinase that targets CHD4 is inactivated upon exposure to elevated temperatures. Alternatively, the counteracting phosphatase could be activated, leading to hypophosphorylation of CHD4, which augments the interaction with PAPAS.
Figure 2.

Heat-induced dephosphorylation enhances RNA binding of CHD4. (A) Pull-down assays showing binding of radiolabeled PAPAS to immobilized Fl-CHD4-N from untreated (Ctrl) or heat-shocked (HS) HEK293T cells. Where indicated, Fl-CHD4-N was purified in the absence or presence of phosphatase inhibitors (phos-stop) or treated with calf intestine phosphatase (CIP). (B) Western blot with anti-phospho-serine antibody comparing phosphorylation of CHD4-N in control and heat-shocked HEK293T cells. Expression of Fl-CHD4 was monitored with anti-Flag antibody. (C) Pull-down assay showing binding of radiolabeled PAPAS to immobilized Fl-CHD4-N from control and heat-shocked HEK293T cells that were pretreated for 12 h with 5 µM CX-4945. (D) Pull-down assay showing binding of radiolabeled PAPAS to immobilized wild-type Fl-CHD4-N and mutant Fl-CHD4-N/S3A from untreated or heat-shocked HEK293T cells. (E) CLIP/RT-qPCR comparing binding of PAPAS to Flag-tagged wild-type CHD4, CHD4/S3A, and CHD4/S3E in HEK293T cells. Data were normalized to input. n = 3. (F) ChIP-qPCR assays monitoring binding of wild-type CHD4 and mutants CHD4/S3A and CHD4/S3E to the rDNA promoter. Data were normalized to wild-type CHD4. n = 3. (G) ChIP-qPCR monitoring the occupancy of HDAC1 and MBD2 at the rDNA promoter. n = 3. (H) ChIP-qPCR monitoring acetylated histone H4 at the rDNA promoter. n = 3. Data were normalized to bound histone H4. Error bars represent mean values ± SD.
CHD4 is phosphorylated at multiple sites (Bouazoune and Brehm 2005; Polo et al. 2010; Urquhart et al. 2011). To identify the sites that are dephosphorylated upon heat shock, we compared the phosphorylation pattern of CHD4 in normal and heat-shocked cells by mass spectrometry. This analysis revealed that three serine residues—Ser308, Ser310, and Ser428—were dephosphorylated upon thermal stress. Two of these sites comprise genuine CK2 target motifs, suggesting that, in normal conditions, CK2 phosphorylates CHD4. Significantly, treatment with the CK2 inhibitor CX-4945 markedly increased the interaction of CHD4 with PAPAS, supporting that CK2-dependent phosphorylation impairs the RNA-binding activity of CHD4 (Fig. 2C; Supplemental Fig. S2B).
To confirm the impact of phosphorylation on CHD4 activity, we generated mutants in which Ser308, Ser310, and Ser428 were replaced by alanine (CHD4-N/S3A) or glutamic acid (CHD4-N/S3E). Immunoblots with phospho-CK2 substrate antibody (pS/pTDXE) revealed reduced phosphorylation in both mutants, indicating that CK2 targets these serine residues (Supplemental Fig. S2C). The interaction of CHD4-N/S3A with PAPAS was comparable with the more efficient binding activity of wild-type CHD4-N after heat shock, underscoring that hypophosphorylation enhances the interaction of CHD4 with RNA (Fig. 2D; Supplemental Fig. S2D–F).
Further support for the impact of phosphorylation on CHD4–PAPAS interaction was provided by CLIP experiments, which revealed stronger interaction of CHD4/S3A with PAPAS than of wild-type CHD4 (Fig. 2E). Unexpectedly, the association of the phospho-mimetic mutant CHD4/S3E with PAPAS was not reduced but remained similar to that of wild-type CHD4 (Supplemental Fig. S2E). Likewise, while rDNA occupancy of CHD4/S3A was more than twofold stronger than CHD4, binding of CHD4/S3E was comparable with parental CHD4 (Fig. 2F; Supplemental Fig. S2F). The finding that CHD4/S3E did not mimic the phenotype of the phosphorylated protein (i.e., did not exhibit reduced binding activity) is probably due to the fact that the majority of cellular CHD4 is hyperphosphorylated. Therefore, replacement of phospho-serines by glutamic acid residues does not decrease the binding activity of CHD4. rDNA occupancy of other CHD4/S3A-associated NuRD components, such as HDAC1 and MBD2, was also increased, demonstrating that PAPAS recruits the NuRD complex rather than CHD4 alone (Fig. 2G). Increased levels of HDAC1 recruited by CHD4/S3A correlated with decreased levels of acetylated histone H4 (ac-H4) (Fig. 2H) and reduced pre-rRNA synthesis (Supplemental Fig. S2G). These results reveal that heat-induced dephosphorylation of CHD4 augments the interaction with PAPAS. Binding to PAPAS, in turn, is a prerequisite for targeting CHD4/NuRD to the rDNA promoter and shifting the promoter-bound nucleosome into a position that is refractory to transcription initiation (Li et al. 2006; Xie et al. 2012; Zhao et al. 2016b).
CHD4 binds to an A-rich loop structure
Previous studies have shown that CHD4 binds to PAPAS comprising nucleotides −1/−205 relative to the transcription start site (TSS) of the rDNA transcription unit (Zhao et al. 2016b). To confine the region of PAPAS that is recognized by CHD4, we assayed truncated versions of PAPAS for their capability to interact with bead-bound CHD4. These pull-down assays revealed that CHD4 interacts with PAPAS comprising nucleotides −1/−105. Deletion of 35 nucleotides (nt) from the 5′ end of PAPAS (−36/−160) did not affect the interaction with CHD4, whereas deletion of 67 nt abolished CHD4 binding, indicating that the sequence between −36 and −67 is critical for the interaction with CHD4/NuRD (Fig. 3A).
Figure 3.

CHD4 binds to A-rich single-stranded regions in PAPAS. (A) Pull-down assays comparing binding of truncated radiolabeled PAPAS to immobilized Fl-CHD4. (B) Secondary structure determination of PAPAS using dimethylsulfate (DMS) and SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) probing. Synthetic PAPAS treated with 1% DMS, 50 mM 2-methylnicotinic acid imidazolide (NAI), or DMSO was reverse-transcribed using a 5′ end-labeled primer (mrDNA −205/−185), and cDNA was analyzed on a 6% sequencing gel. Lanes T, C, G, and A represent sequencing ladders of mouse rDNA using the same 5′-labeled primer. (C) Secondary structure of PAPAS derived from SHAPE/DMS probing. (D) Pull-down experiment comparing binding of radiolabeled truncated or mutant versions of PAPAS with immobilized Fl-CHD4 or Fl-CHD4-N. (E) Pull-down/competition experiment. Increasing amounts (10-fold, 33-fold, and 100-fold molar excess) of synthetic RNA comprising sequences −21/−51 of wild-type PAPAS, mutant PAPAS (A/U), or an unrelated A-rich or G-rich RNA were included in pull-down assays to compete for binding of CHD4 to radiolabeled PAPAS (−1/−205). (F) RNase I footprinting assay showing binding of CHD4 to PAPAS. 5′-labeled PAPAS (−1/−205) was incubated with increasing amounts of Fl-CHD4-N and partially digested with RNase I, and the cleavage products were analyzed on a sequencing gel.
To fine-map the sequence and/or structure that dictates the interaction of CHD4 with PAPAS, we determined the secondary structure of PAPAS by chemical structure probing. For this, we treated in vitro transcribed PAPAS (−1/−205) with either dimethylsulfate (DMS; which selectively methylates unpaired adenosines and cytosines) or 2-methylnicotinic acid imidazolide (NAI; which modifies all nucleotides in unstructured RNA regions). The adducts of the RNA backbone terminate reverse transcription, thus enabling detection of modified nucleotides by primer extension with radiolabeled DNA primers and electrophoresis on sequencing gels along with DNA sequencing ladders. The experimentally derived DMS and NAI reactivity, together with measurements of RNase T1 sensitivity, showed that the analyzed part of PAPAS is highly structured (Fig. 3B; Supplemental Fig. S3A). Significantly, the region that is required for CHD4 binding exhibits high reactivity in both DMS and NAI probing, suggesting that it is unstructured and contained in a single-stranded loop (Fig. 3C).
To examine the impact of PAPAS structure on the interaction with CHD4, we generated mutants in which the stem structures were disrupted. Unexpectedly, neither mutations that destroy base pairing of stem regions of PAPAS nor deletion of the apical stem–loop structure (PAPASΔ−87/−105) impaired binding to CHD4 (Fig. 3D; Supplemental Fig. S3B), indicating that sequences downstream from nucleotide −87 do not have a major impact on binding specificity. We therefore reasoned that the A-rich sequence (24 adenines within 42 nt) contained in the large 5′-terminal loop is recognized by CHD4. To test this, we either deleted a part of the A-rich sequence (PAPASΔ−21/−51) or replaced several adenine residues with uridines (PAPAS-A/U). Although the overall secondary structure of PAPAS-A/U remained unaffected by these mutations, the interaction with CHD4 was markedly decreased (Fig. 3D; Supplemental Fig. S3C). Deletion of nucleotides −21/−51 abolished the interaction, emphasizing the importance of the A-rich sequence for CHD4 binding.
Furthermore, we used synthetic RNAs as competitors in CHD4/PAPAS interaction assays. We incubated bead-bound CHD4 with radiolabeled PAPAS in the absence or presence of short RNAs comprising nucleotides −21/−51 of wild-type PAPAS or PAPAS-A/U. As controls, we used another A-rich and G-rich RNA in the binding competition assays. Both the short wild-type RNA and the A-rich foreign RNA efficiently competed for CHD4 binding to radiolabeled PAPAS, while PAPAS-A/U mutations and the G-rich RNA did not affect the interaction with CHD4 (Fig. 3E).
To reinforce that CHD4 recognizes the A-rich sequence of PAPAS, we performed RNase footprinting experiments, a technique used for probing RNA–protein interactions at single-nucleotide resolution. After incubation of 5′ end-labeled PAPAS with immunopurified CHD4-N and partial digestion with RNase I, the digestion products were analyzed on sequencing gels. Increasing amounts of CHD4-N shielded PAPAS from cleavage by RNase I, the three protected regions exactly overlapping A-rich single-stranded sequences, supporting that CHD4 binds to the unstructured part of PAPAS comprising short stretches of adenosines (Fig. 3F; Supplemental Fig. S3D).
PAPAS binds to the rDNA enhancer through a stretch of homopurines
Next we examined the molecular mechanism underlying PAPAS-dependent targeting of CHD4/NuRD to the rDNA promoter. One attractive possibility would be that PAPAS exerts its cis-regulatory function while being tethered to the same allele from which it is transcribed. Sequence-specific tethering of RNA to complementary DNA sequences can be achieved by forming DNA–RNA hybrids (R loops) or by DNA–RNA triplex structures, thus facilitating guidance of RNA-associated proteins to specific genomic sites.
In mice, PAPAS is transcribed in an orientation antisense to rDNA, covering the gene body and sequences upstream of the TSS, including several repeated elements that enhance Pol I transcription initiation (Fig. 4A; Kuhn et al. 1990). The enhancer repeats are separated by stretches of homothymidine residues. The complementary purine-rich strand should have a high potential to form triple-helical DNA–RNA structures with antisense RNA, suggesting that tethering PAPAS to such putative triplex-forming regions (TFR) would represent an intriguing mechanism to target CHD4/NuRD to rDNA.
Figure 4.

PAPAS is tethered to rDNA by DNA–RNA triplex formation. (A) Schematic representation of a murine rDNA transcription unit. Enhancer repeats are depicted as purple boxes, and terminator elements are shown as orange boxes. The 3′-terminal part of PAPAS used to study CHD4 interactions (−1/−205) is indicated by a blue line. The En3-TFR and the corresponding PAPAS sequences are shown below. (B) RT-qPCR comparing the levels of chromatin-bound PAPAS (−36/−135), 7SK, and GAPDH pre-mRNA from control (Ctrl) and heat-shocked (HS) NIH3T3 cells. Values were normalized to 18S rRNA. n = 3. (C) DNA capture assay showing binding of genomic DNA digested with AccI and SalI to biotinylated PAPAS fragments #1 and #2. PAPAS-associated DNA was monitored by qPCR amplifying rDNA sequences −365/−221 and −135/−36. Data represent DNA recovery normalized to input. n = 3. The locations of the two RNAs used for the capture experiment are depicted by a purple and a gray line. (D) Binding of biotinylated PAPAS (−275/−336) to PCR fragments #1 and #2 comprising En3-TFR (−365/−221) and intergenic spacer (IGS16) sequences. PAPAS-associated DNA was monitored by qPCR. Data represent DNA recovery normalized to samples without RNA. n = 3. (E) Synthetic RNAs comprising PAPAS sequences −234/−333 or −182/−333 and a control RNA (IGS16 RNA) were incubated with 0.25 pmol of a double-stranded 32P-labeled exonuclease I-treated oligonucleotide comprising En3-TFR (Supplemental Table S1), and formation of DNA–RNA triplexes was monitored by electrophoretic mobility shift assay (EMSA). (F) Competition experiment showing that transfection of synthetic RNA comprising the En3-TFR sequence (−275/−336) reduces binding of heat-induced endogenous PAPAS (−234/−333) to chromatin. Values are displayed relative to mock transfected cells. n = 3. (G) ChIP-qPCR monitoring CHD4 occupancy in heat-shocked NIH3T3 cells transfected with synthetic PAPAS or IGS16 RNA as in F. Values were normalized to input. n = 3. Data are presented as mean ± SEM.
As PAPAS is associated with chromatin (Bierhoff et al. 2014), we first examined whether heat shock affects the association of PAPAS with chromatin. To this end, we prepared chromatin from untreated and heat-shocked cells and monitored the level of bound PAPAS by RT-qPCR. In support of PAPAS exerting its function on chromatin, these experiments revealed that the amount of chromatin-bound PAPAS was markedly increased after heat shock (Fig. 4B). Normalization of chromatin-associated PAPAS to cellular PAPAS in normal and heat-stressed cells revealed similar amounts of chromatin-bound PAPAS (Supplemental Fig. S4A), underscoring that cellular PAPAS is associated with chromatin.
To examine whether PAPAS is capable of binding directly to rDNA, we incubated fragmented genomic DNA with biotinylated PAPAS derivatives comprising either the TFR (RNA#1) or sequences around the TSS (RNA#2), and monitored PAPAS-associated rDNA after binding to streptavidin beads. To increase the resolution of the capture assay, we separated the rDNA promoter from the enhancer by digestion of genomic DNA with AccI and SalI, which cleave within the upstream terminator T0 between the rDNA promoter and the enhancer elements (Grummt et al. 1986). This experimental approach revealed that PAPAS comprising nucleotides −275/−336 captured the corresponding region in the rDNA enhancer that contains the TSS-proximal T stretch, termed En3-TFR (Maldonado et al. 2018). RNA comprising PAPAS sequences from +5 to −55 did not capture significant amounts of rDNA (Fig. 4C). These results indicate that the 3′-terminal part of PAPAS harboring nucleotides −275/−336 interacts with the rDNA enhancer in a sequence-specific manner.
To examine whether tethering of PAPAS to rDNA is brought about by DNA–RNA triplex formation, we incubated a PCR fragment overlapping the En3-TFR sequence with biotinylated PAPAS and captured PAPAS-associated DNA by binding to streptavidin beads (Fig. 4D). Consistent with the genomic capture assays, PAPAS comprising nucleotides −275/−336 bound to the En3-TFR fragment but not to a fragment from the intergenic spacer (IGS16). The preferential association of PAPAS with DNA harboring the En3-TFR sequence suggests that PAPAS associates with the rDNA enhancer via formation of a triple-helical DNA–RNA structure. This notion is supported by electrophoretic mobility shift assays (EMSAs), which monitor the direct association of PAPAS with rDNA. To this end, a 5′-labeled DNA oligonucleotide comprising the En3-TFR was annealed with a complementary unlabeled DNA oligo to form a DNA duplex. After treatment with exonuclease I to digest the excess of single-stranded oligos, the duplex was incubated with synthetic RNAs comprising PAPAS sequences −234/−333 or −182/−333, and triplex formation was analyzed by gel electrophoresis. Significantly, RNA harboring TFR-containing PAPAS sequences retarded the mobility of the radiolabeled double-stranded oligonucleotide overlapping the En3-TFR, whereas no DNA–RNA complex was formed with a control RNA that does not contain a potential triplex-forming sequence (Fig. 4E).
To validate that tethering of PAPAS to DNA is required for targeting CHD4/NuRD to the rDNA promoter in vivo, we transfected cells with a synthetic PAPAS derivative comprising the En3-TFR sequence (−275/−336) and monitored the association of endogenous PAPAS with chromatin upon heat shock. This competition experiment showed that binding of PAPAS to chromatin was compromised by ectopic PAPAS containing the En3-TFR (Fig. 4F). Transfection of IGS16 RNA, a stress-induced lncRNA from the rDNA IGS (Audas et al. 2012), did not affect the association of PAPAS with chromatin. The release of PAPAS from chromatin by En3-containing RNA correlated with decreased CHD4 occupancy at the rDNA promoter, corroborating the specificity of the competition experiment (Fig. 4G). Altogether, these results substantiate that PAPAS acts as a molecular trafficking system that guides CHD4/NuRD to the rDNA promoter to repress transcription at elevated temperatures.
Discussion
Several studies have highlighted the function of lncRNAs in chromatin remodeling. For example, the lncRNA MANTIS interacts with BRG1, the catalytic subunit of the SWI/SNF chromatin remodeling complex, and this interaction is required for nucleosome remodeling by keeping the ATPase function of BRG1 active (Leisegang et al. 2017). Conversely, binding of the lncRNA SChLAP1 to SNF5 and Myheart to BRG1 antagonizes the genome-wide localization and regulatory functions of the SWI/SNF complex (Prensner et al. 2013; Han et al. 2014). ncRNAs also impact on nucleolar functions in response to environmental stress. Under several stress conditions, transcription of RNAs from the IGS is up-regulated. These IGS transcripts target proteins containing a certain peptide code for sequestration in the nucleolus (Audas et al. 2012; Jacob et al. 2013).
A key player in down-regulation of rDNA transcription in response to external stress is PAPAS, a low abundant lncRNA that is transcribed in an orientation antisense to pre-rRNA. In quiescent cells, elevated levels of PAPAS recruit the histone methyltransferase Suv4-20h2, which mediates H4K20me3 and establishes transcription refractory chromatin structure in growth-arrested cells (Bierhoff et al. 2014). A different PAPAS-dependent mechanism throttles rDNA transcription in response to hypo-osmotic stress or heat shock. Upon exposure to hypotonic stress or elevated temperature, elevated levels of PAPAS recruit CHD4/NuRD, which leads to deacetylation of histones and movement of the promoter-bound nucleosome into the “off” position that precludes transcription initiation (Zhao et al. 2016a,b).
The interaction of CHD4 with RNA has also been demonstrated by a recent high-throughput approach, termed RBR-ID (identification of RNA-binding regions), which identified RNA-binding regions of 803 nuclear RNA-binding proteins by 4SU photocross-linking and mass spectrometry (He et al. 2016). Among the identified peptides was the PHD of CHD4, which supports our finding that the N-terminal part of CHD4 mediates the interaction with PAPAS. Taking advantage of the CRISPRa approach to guide dCas9-VP64 to the rDNA promoter (Gilbert et al. 2013), we found that a moderate increase of PAPAS on its own is not sufficient to recruit CHD4/NuRD to the rDNA promoter. However, the RNA-binding activity of CHD4 was markedly increased upon heat shock, indicating that stress-induced post-translational modifications of CHD4 regulate the interaction with RNA. In support of this view, mass spectrometric analysis showed heat-induced dephosphorylation of CHD4 at three serine residues (Ser308, Ser310, and Ser428), two of which (Ser310 and Ser428) are contained in a CK2 consensus sequence. These results are consistent with studies in Drosophila melanogaster showing that the N-terminal part of dMi-2/CHD4 is phosphorylated by CK2, hypophosphorylation increasing nucleosome-stimulated ATPase and ATP-dependent nucleosome remodeling activities (Bouazoune and Brehm 2005). Although CK2 is a ubiquitously expressed constitutively active protein kinase, brief exposure to elevated temperature has been shown to affect both the location and the activity of CK2 (Gerber et al. 2000; Davis et al. 2002). After heat shock, the CK2α subunit is concentrated in the granular compartment of nucleoli, whereas the CK2β subunit is localized at the periphery of the condensed chromatin. This indicates that detention of the CK2 subunits in defined nuclear compartments physically separates CK2 from its target proteins, thus preventing phosphorylation of certain proteins and augmenting phosphorylation of others. Accordingly, heat-dependent sequestration of CK2 in the granular compartment of nucleoli prevents phosphorylation of CHD4. As a consequence, the PAPAS-dependent recruitment of CHD4/NuRD to the rDNA promoter is stimulated, and transcription initiation is compromised.
In addition to NuRD-dependent changes in chromatin structure, the basal transcription factor TIF-IA is also inactivated at elevated temperatures. Phosphorylation and dephosphorylation of TIF-IA at two serine residues (Ser170/172) occurs during each round of transcription, phosphorylation of Ser170/172 by protein kinase CK2 being required for the dissociation of TIF-IA from Pol I after transcription initiation and promoter escape (Bierhoff et al. 2008). Inhibition of CK2 upon exposure to elevated temperatures impairs this step, thereby preventing transcription initiation. These results reveal that cells have evolved at least two mechanisms to throttle ribosome biogenesis and guarantee cell survival in response to heat stress, involving PAPAS-dependent recruitment of NuRD (which shifts the promoter-bound nucleosome into a transcription-refractive position) and inactivation of the essential Pol I transcription factor TIF-IA (Zhao et al. 2016b).
Analogous to the domain structure of proteins, lncRNAs have modular structures. Deletion or mutation of a critical module often results in loss of RNA function. Genome-wide studies suggested that lncRNAs are more structured than mRNAs but less structured than rRNAs (Wan et al. 2011; Ding et al. 2014; Quinn and Chang 2016). With the exception of a few well-studied candidates, very little is known about the structure of lncRNAs and their mode of action. To unravel the mechanism underlying PAPAS-mediated guidance of CHD4/NuRD to rDNA, we used DMS and SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) methods to probe the structure of PAPAS that is recognized by CHD4. As the low abundance of PAPAS makes it difficult to probe its structure in vivo, we used in vitro synthesized PAPAS for chemical probing and RNase footprinting experiments. Although in vivo experiments will be required to complement the present study, the results demonstrate that CHD4 binds to a stretch of A residues within a large single-stranded loop. Similar A-rich sequences are present in other CHD4-associated RNAs identified by genome-wide fRIP-seq (fRIP combined with sequencing) (Hendrickson et al. 2016), suggesting that sequence-dependent targeting is a general mechanism that cells use to recruit CHD4/NuRD to distinct genomic loci and alter the chromatin structure in response to external signals.
A key issue in epigenetic research regards the mechanism by which lncRNA acts as a scaffold to guide associated proteins to specific genomic sites. Up-regulation of PAPAS in response to elevated temperatures correlated with enhanced occupancy of CHD4/NuRD at the rDNA promoter and association of PAPAS with chromatin, suggesting that PAPAS is tethered to chromatin either via binding to proteins or by physical association with rDNA. Our results show that PAPAS interacts directly with the rDNA enhancer. The murine rDNA enhancer repeats are separated by T stretches of different lengths (Kuhn et al. 1990; Maldonado et al. 2018). Intriguingly, at active rRNA genes, these repeats wrap around UBF, which allows PAPAS to bind to the T stretches that separate individual enhancer repeats. We used several approaches to prove that PAPAS physically associates with the rDNA enhancer in vitro and in vivo. The results show that a polyadenine sequence in the 3′-terminal part of PAPAS binds to the T stretch closest to the Pol I TSS, dubbed En3-TFR. Significantly, ectopic RNA harboring the En3-TFR sequence efficiently competed for PAPAS binding to rDNA, emphasizing the in vivo relevance of triplex-mediated anchoring of PAPAS to rDNA. The physical association of PAPAS with dsDNA is in accord with recent genome-wide computational analysis and several functional studies showing that lncRNAs can localize to specific DNA sequences by forming DNA–RNA triplex structures (Schmitz et al. 2010; Grote et al. 2013; Mondal et al. 2015; O'Leary et al. 2015; Postepska-Igielska et al. 2015; Kalwa et al. 2016; Singh et al. 2017). RNA binding has obvious benefits for chromatin-modifying proteins with a broad genome-wide binding profile, allowing lncRNAs to generate locus-specific chromatin profiles that enable fine-tuning of target gene expression. Altogether, our results reveal a previously unsuspected mechanism of rDNA transcription in response to stress, showing that heat-induced PAPAS is tethered to rDNA by direct binding to rDNA, which leads to specific recruitment of CHD4/NuRD (Fig. 5).
Figure 5.
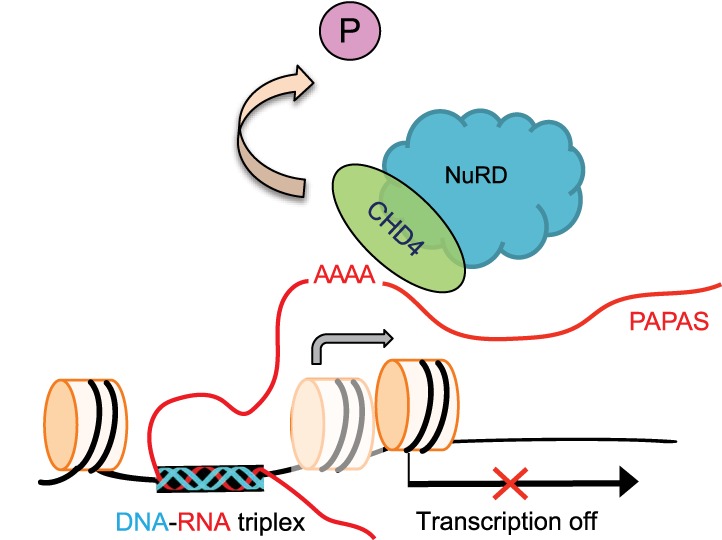
Cartoon illustrating PAPAS-dependent repression of rDNA transcription in response to heat shock. Thermal stress induces transcription of PAPAS and dephosphorylation of CHD4. Hypophosphorylation facilitates binding of CHD4/NuRD to an unstructured A-rich region of PAPAS that covers the rDNA promoter. PAPAS is tethered to the adjacent rDNA enhancer via DNA–RNA triplex formation, thereby targeting CHD4/NuRD to rDNA, where it shifts the promoter-bound nucleosome into a position that does not allow transcription initiation.
Materials and methods
Plasmids, siRNAs, and antibodies
Expression plasmids for Flag-CHD4 have been described (Xie et al. 2012). CHD4-N (amino acids 1–700) and CHD4-C (amino acids 701–1912) were generated by PCR and inserted into pFlag-CMV-6c. Plasmids for dCas9/VP64-mediated transcription activation (single guide RNA [sgRNA] cloning backbone [no. 61424], MS2-P65-HSF1_GFP [no. 61423], and dCAS9-VP64_GFP [no. 61422]) were from Addgene. To activate PAPAS transcription by dCas9/VP64, sgRNAs targeting the minus strand of mouse rDNA were used (GenBank: BK000964.3, sgRNA#1: 13838–13857, sgRNA#2: 13599–13618, and sgRNA#3: 13631–13650) (Supplemental Table S1). Antibodies against CHD4 (ab70469) and HDAC1 (ab7028) were from Abcam, anti-phosphoserine antibodies were from Santa Cruz Biotechnology (sc-81514), the phospho-CK2 substrate motif [(pS/pT)DXE] antibody was from Cell Signaling (8738), α-Flag antibodies (M2) were from Sigma-Aldrich, the anti-acetyl-Histone H4 antibody (06-866) was from Upstate Biotechnology, and anti-MBD2 (NB100-81657) was from Novus Biologicals.
Cell culture and transfection
NIH3T3 and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). For heat shock, cells were transferred to prewarmed DMEM (42°C) and incubated for 3 h at 42°C. Lipofectamine 3000 was used for DNA transfection. Cells were harvested 36–48 h after transfection. In competition experiments, cells were transfected with synthetic RNAs using TransIT-TKO (Mirus), incubated overnight, and harvested after 3 h of heat shock at 42°C. For metabolic labeling of pre-rRNA, 0.2 × 106 NIH3T3 cells were cultured for 3 h at 37°C or 42°C in medium containing 5 μCi/mL 3H-uridine. Two micrograms of cellular RNA was separated on a 1% agarose gel in 20 mM MOPS (pH 7.0), 5 mM sodium acetate, and 1 mM EDTA and transferred to a Hybond-N+ nylon membrane (GE Healthcare), and labeled RNA was visualized by fluorography using EN3HANCE (PerkinElmer).
RNA analysis and in vitro transcription
Cellular RNA was isolated with TRI reagent (Sigma), treated with DNase I (Sigma), and analyzed by reverse transcription using M-MLV reverse transcriptase (Life Technologies). For CLIP experiments, PAPAS cDNA was synthesized using a primer encompassing mouse rDNA sequences from −205 to −185 fused to the T7 promoter. PAPAS cDNA was amplified by PCR using a T7 forward primer and a rDNA-specific reverse primer.
In vitro transcripts were generated using T7-fused rDNA oligos hybridized to complementary T7-containing DNA oligos or PCR products as templates (Supplemental Tables S1, S2) using MEGAscript T7 transcription kit (Ambion). To radiolabel or biotinylate PAPAS, transcription assays were supplemented with 20 µCi of [32P]UTP or 2.5 nM biotin-16-dUTP (Roche). Transcripts were purified with the RNeasy minikit (Qiagen) and analyzed by electrophoresis on polyacrylamide–urea gels.
In vivo RNA–protein interaction assays
Cells overexpressing Flag-tagged CHD4 (Fl-CHD4) were UV cross-linked (150 mJ/cm2 at 254 nm) using a Stratalinker (Stratagene). Nuclei were isolated and lysed in RIPA buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% NP-40, 0.5% sodium deoxycholate, 0.05% SDS, 1 mM DTT, and protease and phosphatase inhibitors. After sonication and preclearing, protein–RNA complexes were immunoprecipitated overnight at 4°C in RIPA buffer using anti-Flag M2 beads (Sigma-Aldrich). Lysates from cells transfected with the empty vector served as a negative control. The immunoprecipitates were washed three times with RIPA buffer containing 600 mM NaCl and once with 1 M urea. Protein-bound RNA was eluted by digestion with proteinase K (Roche), purified with TRI reagent, and analyzed by RT-qPCR. The fraction of coprecipitated RNA is presented as the percentage of input (upon subtraction of the IgG background).
In vitro protein–RNA interaction assays
To monitor the interaction of CHD4 with PAPAS, in vitro labeled RNA was denatured for 2 min at 95°C and renatured in RNA folding buffer (10 mM Tris-HCl at pH 7.0, 10 mM MgCl2, 100 mM KCl, 2 U/µL RNasin) for 30 min at 37°C. Fl-CHD4 was immobilized on M2 beads and incubated with radiolabeled PAPAS (Supplemental Table S2) for 1 h at room temperature in RIPA buffer. After washing, bound RNA was eluted by digestion with proteinase K, precipitated, and analyzed by phosphorimaging after electrophoresis on 6% polyacrylamide–urea gels. Alternatively, biotinylated PAPAS was immobilized on streptavidin Dynabeads (Life Technologies) and incubated for 4 h at 4°C with 1 mg of nuclear extract from HEK293T cells expressing Fl-CHD4. Beads were washed with RIPA buffer containing 600 mM NaCl, and RNA-bound proteins were eluted with SDS and analyzed on immunoblots.
For Northwestern blot analyses, purified proteins were separated by SDS–polyacrylamide electrophoresis and transferred to a nitrocellulose membrane (GE Healthcare). After renaturation in 10 mM Tris-HCl (pH 6.8), 25 mM NaCl, 1 mM EDTA, 0.04% BSA, and 0.04% NP40 overnight at 4°C, the filter was incubated for 2 h at 37°C in the same buffer containing 6 ng of 32P-labeled synthetic PAPAS (−1/−205) and washed three times with buffer containing 300 mM NaCl, 20 mM Tris-HCl (pH 8.0), and 1% NP40, and bound RNA was visualized by phosphorimaging.
To monitor CHD4–PAPAS interaction by filter-binding assays, gel-purified PAPAS was renatured in RNA folding buffer (10 mM Tris-HCl at pH 7.0, 10 mM MgCl2, 100 mM KCl, 2 U/µL RNasin) and incubated with CHD4-N in TBS/10% glycerol for 1 h at room temperature. The formation of protein–RNA complexes was assayed by filtering the incubation mixture through a nitrocellulose membrane (Protran, Schleicher and Schuell) using a manifold I system (Whatman). Membrane-bound protein–RNA complexes were monitored by phosphorimaging.
ChIP assays
Cells were cross-linked with 1% formaldehyde for 10 min and quenched with 125 mM glycine, and isolated chromatin was sonicated in a Bioruptor Pico (Diagenode) in buffer containing 1% SDS, 50 mM Tris-HCl (pH 8.0), and 10 mM EDTA to obtain an average fragment length of 200–500 base pairs (bp). After dilution with 5 vol of ChIP buffer (15 mM Tris-HCl at pH 8.0, 180 mM NaCl, 1.2 mM EDTA, 1.2% Triton X-100, 0.01% SDS) and preclearing, 20 µg of chromatin was incubated with 1–5 µg of antibodies immobilized on protein A/G Sepharose overnight at 4°C. Protein–DNA complexes were washed in buffer A (20 mM Tris-HCl at pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100) followed by two washes with buffer B containing 500 mM NaCl, one wash with buffer C (10 mM Tris-HCl at pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40), and two washes with TE buffer. Protein–DNA complexes were eluted with either 100 mM NaHCO3,/1% SDS or 0.2 µg/µL Flag peptide. After reversal of the cross-link for 6 h at 65°C and digestion with proteinase K, DNA was purified and quantified by qPCR using primers that amplify the murine rDNA promoter (Supplemental Table S1).
Secondary structure probing by DMS/SHAPE
SHAPE was performed as described (Steen et al. 2011; Spitale et al. 2013). Briefly, 200–500 ng of in vitro synthesized PAPAS was dissolved in RNase-free water, denatured for 2 min at 95°C, and refolded in 10 mM Tris-HCl (pH 7.0), 10 mM MgCl2, 100 mM KCl, and 2 U/µL RNasin. After treatment for 3 min at room temperature with 1% DMS (Sigma-Aldrich), the reaction was quenched by addition of 0.5 M DTT. Parallel samples were treated with DMSO as a control. For NAI treatment, RNA was incubated with 50 mM NAI for 3 and 6 min at 37°C and immediately purified. The modified RNA was reverse-transcribed using a 5′ end-labeled forward primer (mrDNA−205/−183) (Supplemental Table S1), and cDNA was analyzed by electrophoresis on 6% sequencing gels. DNA sequence ladders were generated with the USB Thermo Sequenase cycle sequencing kit (Thermo Fisher Scientific). The secondary structure was generated with the Mfold Web server (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form) and ViennaRNA Web services (http://rna.tbi.univie.ac.at/forna).
RNase footprinting
5′ end-labeled PAPAS (−1/−205) was purified on 6% polyacrylamide–urea gels, denatured for 2 min at 95°C, and renatured for 30 min at 37°C in RNA folding buffer (10 mM Tris-HCl at pH 7.0, 10 mM MgCl2, 100 mM KCl, 2 U/µL RNasin). One-hundred femtomoles of PAPAS was incubated for 1 h at room temperature with increasing amounts of CHD4-N in buffer containing 40 mM Tris-HCl (pH 7.5), 2 mM MgCl2, 40 mM KCl, 75 mM NaCl, 0.01% NP40, and 5% glycerol. After digestion with 0.01 U of RNase I (Thermo Scientific) for 5 min at room temperature, the reaction was stopped with SDS, and RNA was purified and analyzed by electrophoresis on 6% polyacrylamide–urea gels (Nilsen 2014).
Isolation of chromatin-associated RNA
Chromatin was extracted from 107 NIH3T3 nuclei by incubation for 5 min in 500 µL of glycerol/urea buffer (25% glycerol, 20 mM Tris-HCl at pH 7.4, 187.5 mM KCl, 0.5 M urea, 0.5% NP-40, 5 mM MgCl2, 1 mM DTT). After centrifugation at 1200g for 3 min, chromatin was resuspended in 1 mL of glycerol/sucrose buffer (10% glycerol, 340 mM sucrose, 10 mM Tris-HCl at pH 7.4, 10 mM KCl, 5 mM MgCl2, 1 mM DTT) and centrifuged again (Werner and Ruthenburg 2015). RNA from chromatin was recovered by TRI reagent (Sigma) according to the manufacturer's instructions. PAPAS cDNA was synthesized using a forward primer comprising nucleotides −355/−337 with respect to the TSS of the pre-rRNA-coding region.
Capture of PAPAS-associated DNA
Genomic DNA was fragmented by sonication and digestion with AccI and SalI to an average size of 200–300 bp. DNA (7.5 µg) was incubated with 20 pmol of in vitro transcribed PAPAS (Supplemental Table S1, S2) for 40 min at 37°C in triplex buffer (10 mM Tris-HCl at pH 7.4, 50 mM KCl, 5 mM MgCl2) supplemented with 0.8 U/µL RNasin (Promega). DNA–RNA complexes were bound to MyOne Streptavidin C1 Dynabeads (Invitrogen). After three washes with triplex buffer containing 0.05% Tween20 and 0.5% NP-40 and one wash with triplex buffer containing 0.05% Tween20, RNA-associated DNA was eluted with 25 ng/µL RNase and 5 mU/µL RNase 1 (Thermo Scientific) for 30 min at 37°C. DNA was recovered by phenol/chloroform extraction and analyzed by qPCR.
To monitor binding of PAPAS to defined DNA fragments, PCR fragments were generated using GoTaq DNA polymerase (Promega). Four-hundred femtomoles of exonuclease I-treated PCR fragments was incubated with 20 pmol of biotinylated PAPAS for 1 h at 37°C in triplex buffer containing 10 mM Tris-HCl (pH 7.4), 50 mM KCl, 5 mM MgCl2, 0.8 U/µL RNasin (Promega), and 10 µg of salmon sperm DNA (Invitrogen). After binding to streptavidin beads, washing, and elution, PAPAS-associated DNA was recovered by phenol/chloroform extraction and analyzed by qPCR.
EMSA
Five picomoles of a DNA oligonucleotide comprising the En3-TFR sequence of PAPAS was 5′-radiolabeled, annealed with the complementary oligonucleotide, and then treated with 0.4 U/µL exonuclease I for 30 min at 37°C to digest the excess of single-stranded oligonucleotides. Labeling of the DNA oligo corresponding to the PAPAS sequence did not allow visualization of DNA–RNA heteroduplexes. After incubation of 0.25 pmol of duplex oligos with 25 pmol of synthetic RNA for 1 h at 37°C in triplex buffer supplemented with 5 µg of salmon sperm DNA and 2.5 U/µL RNasin, triplex formation was monitored by electrophoresis on 8% polyacrylamide gels containing 8 mM MgCl2. Sequences of DNA oligos and RNAs are shown in Supplemental Table S1.
Statistical analysis
Data are reported as mean values from at least three biological replicates, with error bars denoting standard deviation (SD) or mean ± SEM where indicated. Comparisons between two groups were performed using a paired two-tailed Student's t-test (P-values P < 0.05 [*] and P < 0.01 [**]).
Supplementary Material
Acknowledgments
This work has been funded by the Deutsche Forschungsgemeinschaft (GR475/22-2 and SFB1036), CellNetworks (EcTop Survey 2014), and the Baden-Württemberg Stiftung.
Author contributions: Z.Z. and N.S. conducted the experiments and analyzed the data. C.S. generated the CHD4-N/S3A mutant and performed ChIP experiments. I.G. conceived and supervised the project. I.G. and N.S. wrote the manuscript. All authors contributed to the manuscript review.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.311688.118.
References
- Audas TE, Jacob MD, Lee S. 2012. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell 45: 147–157. [DOI] [PubMed] [Google Scholar]
- Bierhoff H, Dundr M, Michels AA, Grummt I. 2008. Phosphorylation by casein kinase 2 facilitates rRNA gene transcription by promoting dissociation of TIF-IA from elongating RNA polymerase I. Mol Cell Biol 28: 4988–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhoff H, Schmitz K, Maass F, Ye J, Grummt I. 2010. Noncoding transcripts in sense and antisense orientation regulate the epigenetic state of ribosomal RNA genes. Cold Spring Harb Symp Quant Biol 75: 357–364. [DOI] [PubMed] [Google Scholar]
- Bierhoff H, Dammert MA, Brocks D, Dambacher S, Schotta G, Grummt I. 2014. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol Cell 54: 675–682. [DOI] [PubMed] [Google Scholar]
- Bouazoune K, Brehm A. 2005. dMi-2 chromatin binding and remodeling activities are regulated by dCK2 phosphorylation. J Biol Chem 280: 41912–41920. [DOI] [PubMed] [Google Scholar]
- Davis AT, Wang H, Zhang P, Ahmed K. 2002. Heat shock mediated modulation of protein kinase CK2 in the nuclear matrix. J Cell Biochem 85: 583–591. [DOI] [PubMed] [Google Scholar]
- Denslow S, Wade P. 2007. The human Mi-2/NuRD complex and gene regulation. Oncogene 26: 5433–5438. [DOI] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. 2014. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696–700. [DOI] [PubMed] [Google Scholar]
- Gerber DA, Souquere-Besse S, Puvion F, Dubois MF, Bensaude O, Cochet C. 2000. Heat-induced relocalization of protein kinase CK2. Implication of CK2 in the context of cellular stress. J Biol Chem 275: 23919–23926. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M. 2013. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 24: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummt I, Kuhn A, Bartsch I, Rosenbauer H. 1986. A transcription terminator located upstream of the mouse rDNA initiation site affects rRNA synthesis. Cell 47: 901–911. [DOI] [PubMed] [Google Scholar]
- Han P, Li W, Lin CH, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin CY, Lin CJ, et al. 2014. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Sidoli S, Warneford-Thomson R, Tatomer DC, Wilusz JE, Garcia BA, Bonasio R. 2016. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol Cell 64: 416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson D, Kelley DR, Tenen D, Bernstein B, Rinn JL. 2016. Widespread RNA binding by chromatin-associated proteins. Genome Biol 17: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Wade PA. 2012. NuRD and pluripotency: a complex balancing act. Cell Stem Cell 10: 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob MD, Audas TE, Uniacke J, Trinkle-Mulcahy L, Lee S. 2013. Environmental cues induce a long noncoding RNA-dependent remodeling of the nucleolus. Mol Biol Cell 24: 2943–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalwa M, Hänzelmann S, Otto S, Kuo C-C, Franzen J, Joussen S, Fernandez-Rebollo E, Rath B, Koch C, Hofmann A. 2016. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res 44: 10631–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517: 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Deppert U, Grummt I. 1990. A 140-base-pair repetitive sequence element in the mouse rRNA gene spacer enhances transcription by RNA polymerase I in a cell-free system. Proc Natl Acad Sci 87: 7527–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AY, Wade PA. 2011. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11: 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisegang MS, Fork C, Josipovic I, Richter FM, Preussner J, Hu J, Miller MJ, Epah J, Hofmann P, Gunther S, Moll F, et al. 2017. Long noncoding RNA MANTIS facilitates endothelial angiogenic function. Circulation 136: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Längst G, Grummt I. 2006. NoRC-dependent nucleosome positioning silences rRNA genes. EMBO J 25: 5735–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R, Filarsky M, Grummt I, Längst G. 2018. Purine- and pyrimidine-triple-helix-forming oligonucleotides recognize qualitatively different target sites at the ribosomal DNA locus. RNA 24: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B, Mitra S, Mohammed A, James AR, Hoberg E. 2015. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–-DNA triplex structures. Nat Commun 6: 7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. 2008. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322: 1717–1720. [DOI] [PubMed] [Google Scholar]
- Nilsen TW. 2014. RNase footprinting to map sites of RNA–protein interactions. Cold Spring Harb Protoc 2014: 677–682. [DOI] [PubMed] [Google Scholar]
- O'Leary VB, Ovsepian SV, Carrascosa LG, Buske FA, Radulovic V, Niyazi M, Moertl S, Trau M, Atkinson MJ, Anastasov N. 2015. Particle, a triplex-forming long ncRNA, regulates locus-specific methylation in response to low-dose irradiation. Cell Rep 11: 474–485. [DOI] [PubMed] [Google Scholar]
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. 2010. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 29: 3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postepska-Igielska A, Giwojna A, Gasri-Plotnitsky L, Schmitt N, Dold A, Ginsberg D, Grummt I. 2015. lncRNA Khps1 regulates expression of the proto-oncogene SPHK1 via triplex-mediated changes in chromatin structure. Mol Cell 60: 626–636. [DOI] [PubMed] [Google Scholar]
- Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, Patel L, Vergara IA, Davicioni E, Erho N, Ghadessi M, et al. 2013. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet 45: 1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Chang HY. 2016. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17: 47–62. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KM, Mayer C, Postepska A, Grummt I. 2010. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24: 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Jalali S, Maiti S, Scaria V. 2017. Genome-wide computational analysis of potential long noncoding RNA mediated DNA:DNA:RNA triplexes in the human genome. J Transl Med 10.1186/s12967-017-1282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY. 2013. RNA SHAPE analysis in living cells. Nat Chem Biol 9: 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KA, Siegfried NA, Weeks KM. 2011. Selective 2′-hydroxyl acylation analyzed by protection from exoribonuclease (RNase-detected SHAPE) for direct analysis of covalent adducts and of nucleotide flexibility in RNA. Nat Protoc 6: 1683–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. 1998. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395: 917–921. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquhart AJ, Gatei M, Richard DJ, Khanna KK. 2011. ATM mediated phosphorylation of CHD4 contributes to genome maintenance. Genome Integr 10.1186/2041-9414-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. 2011. Understanding the transcriptome through RNA structure. Nat Rev Genet 12: 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner MS, Ruthenburg AJ. 2015. Nuclear fractionation reveals thousands of chromatin-tethered noncoding RNAs adjacent to active genes. Cell Rep 12: 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Ling T, Zhou Y, Feng W, Zhu Q, Stunnenberg HG, Grummt I, Tao W. 2012. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc Natl Acad Sci 109: 8161–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. 1998. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 2: 851–861. [DOI] [PubMed] [Google Scholar]
- Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. 1998. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 95: 279–289. [DOI] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. 2008. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Dammert MA, Grummt I, Bierhoff H. 2016a. lncRNA-induced nucleosome repositioning reinforces transcriptional repression of rRNA genes upon hypotonic stress. Cell Rep 14: 1876–1882. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Dammert MA, Hoppe S, Bierhoff H, Grummt I. 2016b. Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucleic Acids Res 44: 8144–8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.