

Mutations in PKD1 and PKD2 are the leading cause of autosomal dominant polycystic kidney disease (ADPKD). In this Outlook, Ma and Guan discuss the work in this issue of Genes & Development by Cai et al., which reveals new insight into the molecular basis by which PKD1 deficiency leads to cystic kidney pathogenesis.
Keywords: 3D culture, ADPKD, Hippo signaling, RhoA signaling, YAP/TAZ, c-Myc
Abstract
Mutations in PKD1 and PKD2 are the leading cause of autosomal dominant polycystic kidney disease (ADPKD). In this issue of Genes & Development, a report by Cai and colleagues (pp. 781–793) reveals new insight into the molecular basis by which PKD1 deficiency leads to cystic kidney pathogenesis. By using extensive mouse genetic analyses coupled with in vitro cystic assays, the investigators delineate a RhoA–YAP–c-Myc signaling axis as a key downstream from PKD1 deficiency in ADPKD pathogenesis. Their findings provide evidence that the Hippo pathway could be a potential target for treating ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common human monogenic inherited systemic disorder with an occurrence of one in 400–1000 people worldwide (Chebib et al. 2015). The hallmark of ADPKD is the spontaneous formation and continuous enlargement of fluid-filled kidney cysts, culminating in end-stage renal disease (ESRD) in more than half of patients by age 60 (Grantham 2008). ADPKD is caused by mutations in either the PKD1 or PKD2 gene, encoding polycystin 1 (PC1) and PC2, respectively. PC1 and PC2 are multimembrane-spanning proteins that interact with each other (Qian et al. 1997). Loss-of-function mutations in either gene results in similar clinical symptoms (Grantham 2008). Decades after the discovery of PKD1 and PKD2 mutations, multiple signaling pathways have been linked to ADPKD, including Ca2+ signaling, cAMP, SRC, Wnt, and mTOR (Lemos and Ehrlich 2018). However, limited treatment options are currently available, and there is great interest in identifying novel molecular targets for ADPKD. In this issue of Genes & Development, Cai et al. (2018) discovered a functional connection between PKD1 mutations and the Hippo pathway, which is best known for its function in organ size control, tissue homeostasis/regeneration, and cancer. Furthermore, extensive mouse genetic data and human patient sample analyses support a critical role of the Hippo pathway in ADPKD pathogenesis when PKD1 is mutated. This study provides strong evidence supporting a model that RhoA–YAP–c-Myc axis is a direct downstream target of PKD1 mutations, and targeting the Hippo pathway could be valuable in treating PKD1-deficient ADPKD (Fig. 1).
Figure 1.
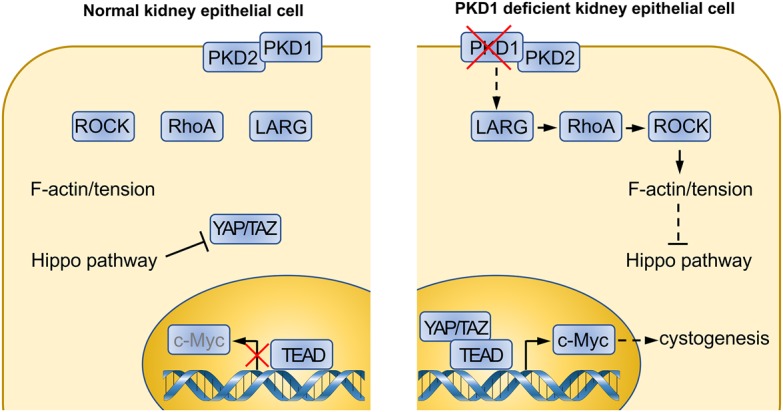
Altered Hippo pathway signaling contributes to the kidney cystogenesis in Pkd1 mutant ADPKD. In kidney tubular epithelial cells, Pkd1 deficiency leads to ADPKD. The procyst phenotype is mediated by hyperactivated YAP/TAZ and up-regulation of its transcriptional target, c-Myc. RhoGEF (guanine nucleotide exchange factor) LARG, RhoA, and ROCK (Rho-associated protein kinase) act downstream from Pkd1 to regulate the Hippo pathway and YAP/TAZ.
Initially, the investigators performed gene set enrichment analysis (GSEA) on a previous human ADPKD polycystic kidney expression-profiling data set and discovered a strong enrichment of YAP/TAZ target genes. YAP/TAZ are transcriptional coactivators that mediate the majority of the functional output (if not the entire functional output) of the Hippo pathway. YAP/TAZ are inhibited by Hippo pathway-dependent phosphorylation and cytoplasmic localization. The investigators validated their findings in a transgenic ADPKD mouse model by spatiotemporally targeting Pkd1 in the kidney tubular epithelium. Pkd1 deletion caused strong YAP/TAZ dephosphorylation and nuclear localization, with a corresponding increase of YAP/TAZ target gene expression. Thus, Pkd1 deficiency causes constitutive activation of YAP/TAZ during kidney cystogenesis.
The investigators then explored the functional relevance of YAP/TAZ activation in ADPKD. First, they asked whether activating YAP alone is sufficient to induce cystogenesis in the kidney. By using a drug-inducible YAP overexpression model in kidney tubular epithelium, dilation of the tubular epithelium was observed, which was also accompanied by increased cell proliferation. Second, the investigators deleted Sav1, a core component of the Hippo pathway and a negative regulator of YAP/TAZ, and observed phenotypes similar to YAP overexpression. Furthermore, the hyperproliferation and kidney tubule dilation phenotypes induced by Sav1 mutants are completely blocked by loss of YAP. Moreover, loss of both YAP and TAZ greatly suppressed cyst formation by Pkd1 deficiency. Together, these observations strongly support a key role of the Hippo pathway and a role for YAP/TAZ in cystogenesis associated with Pkd1 mutant kidneys.
The avidity of YAP/TAZ activation for the Pkd1 mutant ADPKD prompted the investigators to investigate its transcriptional downstream effector. Among the genes induced by Pkd1 deficiency, c-Myc is noteworthy because its activation in the kidney was reported to induce cystogenesis and tubular epithelial cell proliferation (Trudel et al. 1991). Cai et al. (2018) showed that c-Myc is a direct transcription target of YAP/TAZ. Strikingly, loss of c-Myc significantly suppressed the cyst formation phenotype in Sav1 mutants as well as in Pkd1 mutant kidneys, thereby defining a Hippo–YAP–c-Myc axis involved in kidney cystogenesis caused by Pkd1 deficiency.
Although the core components of the Hippo pathway and YAP/TAZ have been studied extensively, the upstream pathway is more complex and not as well defined (Yu et al. 2015). To elucidate the mechanism by which PKD1 deficiency activates YAP/TAZ, the investigators established an in vitro three-dimensional (3D) culture system that mimics PKD1-deficient cyst formation. Through an unbiased screening for kinase inhibitors, they discovered five chemicals that could inhibit cyst formation. Interestingly, all five inhibitors share a common target: the Rho-associated protein kinase (ROCK), which is a downstream effector of Rho and mediates the formation of actin stress fibers. Indeed, RhoA, MLC (myosin light chain), and F-actin are all activated and show apical enrichment in Pkd1-deficient mouse kidney cysts as well as in human ADPKD cyst samples. In addition, further studies revealed that RhoGEF (guanine nucleotide exchange factor) LARG links Pkd1 deficiency to high RhoA activation. Reducing LARG protein levels inhibited RhoA activity as well as cyst formation induced by Pkd1 mutation in vitro. These data fit nicely with the known function of RhoA in Hippo regulation. RhoA has been shown to activate YAP/TAZ by inhibiting the Hippo pathway, although the precise molecular mechanism is unclear.
Finally, to test the importance of RhoA–ROCK signaling in vivo, Cai et al. (2018) determined the effect of the ROCK inhibitor Y-27632 in the Pkd1-deficient ADPKD mouse model. Remarkably, Y-27632 treatment decreased YAP activity and c-Myc expression as well as ameliorated the Pkd1 mutant phenotypes, including both epithelial cell proliferation and cyst formation. These genetic and pharmacological data support a key role of RhoA and ROCK in ADPKD associated with PKD1 mutation.
Altogether, Pan and colleagues (Cai et al. 2018) have identified a RhoA–ROCK–Hippo–YAP/TAZ–c-Myc signaling axis that is both required and sufficient to promote kidney cystogenesis in a Pkd1 mutant ADPKD mouse model (Fig. 1). Mechanistically, Pkd1 deficiency promotes cytoskeletal tension through a LARG–RhoA–ROCK pathway, which activates YAP/TAZ by inhibiting the Hippo pathway. The dephosphorylated YAP/TAZ are then translocated into the nucleus and bind with their transcription factor, TEAD, to induce expression of target genes, including c-Myc, which contributes to cystic kidney pathogenesis. These findings may aid future therapy, such as targeting ROCK or inhibiting YAP/TAZ for potential ADPKD treatment.
Despite the significant advances made in this study, several questions remain. For instance, Cai et al. (2018) used a homozygous Pkd1 deletion mouse model, which showed a rapidly appearing and severe kidney cystic phenotype. However, patients with ADPKD are typically heterozygous, with one mutant allele, and disease progression is much slower. Similar studies using a Pkd1 heterozygous mouse model might be warranted, as the heterozygous Pkd1 mutation may better recapitulate genetic alterations in human ADPKD. Moreover, depletion of both YAP and TAZ does not completely block kidney cystogenesis and pathology in the Pkd1 mutant mice, indicating that, in addition to YAP/TAZ hyperactivation, other alterations also play an important role in ADPKD caused by PKD1 deficiency.
In addition, what is the molecular link between Pkd1 deficiency and LARG–RhoA–YAP activation? An inhibitor targeting Src retards the renal cystic phenotype in a Pkd1 heterozygous mouse model and has entered a phase 2 clinical trial to test its efficiency in ADPKD patients (Elliott et al. 2011). Previous reports have shown that Src can activate YAP (Enomoto and Igaki 2013; Kim and Gumbiner 2015; Taniguchi et al. 2015). As multiple mechanisms have been proposed for YAP regulation by Src, it is tempting to speculate about how Src is involved in YAP activation in ADPKD. Furthermore, multiple signaling pathways have been proposed to be deregulated and play a functional role in ADPKD, including Ca2+, cAMP, MAPK, JAK, SRC, STAT, Wnt, and mTOR (Lemos and Ehrlich 2018). Understanding how different signaling pathways, including the RhoA–YAP–c-Myc axis, are interconnected in PKD1 deficiency will be critical to fully comprehend ADPKD.
Another interesting question raised by the present study is whether targeting the RhoA–YAP–c-Myc axis can block both the initiation and progression of ADPKD pathogenesis. Cai et al. (2018) targeted the RhoA–YAP–c-Myc axis from the start, when Pkd1 is deleted. Clinically, therapeutic intervention will not be applied until ADPKD symptoms are apparent in the patient. Therefore, it is important to test whether targeting this axis at a later stage could also ameliorate the renal manifestations. Finally, since Pkd2 mutations produced a similar pathology, it is important to test whether Pkd2 mutations similarly affect the Hippo pathway.
Competing interest statement
K.-L.G. is a cofounder of and has equity interest in Vivace Therapeutics, Inc. The terms of this arrangement have been reviewed and approved by the University of California at San Diego in accordance with its conflict of interest policies.
Acknowledgments
K.-L.G. is supported by National Institutes of Health grants GM51586 and CA196878.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.316570.118.
References
- Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, Pan D. 2018. A RhoA–YAP–c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev (this issue) 10.1101/gad.315127.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebib FT, Sussman CR, Wang X, Harris PC, Torres VE. 2015. Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat Rev Nephrol 11: 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott J, Zheleznova NN, Wilson PD. 2011. c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol Cell Physiol 301: C522–C529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto M, Igaki T. 2013. Src controls tumorigenesis via JNK-dependent regulation of the Hippo pathway in Drosophila. EMBO Rep 14: 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham JJ. 2008. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 359: 1477–1485. [DOI] [PubMed] [Google Scholar]
- Kim NG, Gumbiner BM. 2015. Adhesion to fibronectin regulates Hippo signaling via the FAK–Src–PI3K pathway. J Cell Biol 210: 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos FO, Ehrlich BE. 2018. Polycystin and calcium signaling in cell death and survival. Cell Calcium 69: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. 1997. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16: 179–183. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. 2015. A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 519: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel M, D'Agati V, Costantini F. 1991. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int 39: 665–671. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. 2015. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]