

Abstract
The plant Brassica campestris includes the vegetables turnip and Chinese cabbage, important plants of economic importance. Here, we have analysed the codon usage bias of B. campestris for 116 protein coding genes. Neutrality analysis showed that B. campestris had a wide range of GC3s, and a significant correlation was observed between GC12 and GC3. Nc versus GC3s plot showed a few genes on or proximate to the expected curve, but the majority of points were found to be scattered distantly from the expected curve. Correspondence analysis on codon usage revealed that the position preference of codons on multidimensional space totally depends on the presence of A and T at synonymous third codon position. These results altogether suggest that composition bias along with selection (major) and mutation pressure (minor) affects the codon usage pattern of the protein coding genes in Brassica campestris.
Electronic supplementary material
The online version of this article (10.1007/s10068-017-0285-x) contains supplementary material, which is available to authorized users.
Keywords: Codon usage, Dinucleotide, Selection, Mutation, Brassica campestris
Introduction
Except the two amino acids i.e., methionine and tryptophan (which are encoded by single codons) all other amino acids are encoded by two or more synonymous codons. Variation mainly occurs among synonymous codons ending in cytosine (C) or guanine (G) versus adenine (A) or thymine (T). The choice of synonymous codons is known to be nonrandom and thought to reflect a balance among the forces of selection, mutation and random genetic drift [1, 2]. In contrast, it was reported that translational selection at silent sites played the most important role in shaping codon usage in some plants [3, 4]. This unequal use of synonymous codon within the coding sequences of a genome is defined as codon usage bias (CUB). It has been extensively studied in a wide variety of organisms [5, 6]. However, in recent years, several factors namely gene length [7], gene translation initiation signal [8], expression level [9–12], protein amino acid composition [13], protein structure [14], tRNA abundance [15, 16] and GC composition [17] have been reported to influence the CUB. Genome-wide investigations of codon bias patterns and their causes and consequences are of significant importance for heterologous gene expression [18], gene classification and function prediction [19] as well as for prediction of the pattern of evolution of the organism.
Brassica is a genus of plants in the mustard family (Brassicaceae). Brassica campestris is widely cultivated as a leaf vegetable, a root vegetable, and as an oilseed crop. Almost all parts of the plant are consumed as food, including the root, stems, leaves, flowers and seeds. During the last few years, a large number of plant genes have been cloned and sequenced. This now permits a meaningful analysis and comparison of the codon bias of genes and genomes of higher plants. In contrast to its economic importance, there were a few studies on the codon usage bias of B. campestris [20–22]. In accordance with the previous studies on Brassica genes, here also, we observed the avoidance of CG and TA doublets [23]. Brassica campestris is a self-incompatible crop. Self-pollination reduces recombination rate and GC3s. For the 116 B. campestris genes used in the present study, GC3 showed the highest usage variation followed by GC1 and GC2. Therefore, the increased variation of GC content at synonymous third codon position of the Brassica genes is thought to be due to mutational pressure [24]. Optimization of heterologous protein expression is one of the major research areas of modern biotechnology, for example, efficient enzyme production is a dire need in biotechnology industry [25]. In recent years, the extensive study on codon bias has revealed that codon replacement (usage of some preferred codons) has a significant impact on gene expression levels and protein folding [26]. The present study contributes to better understanding of the evolution of B. campestris genome at the molecular level through codon usage bias and provides basic information for synthetic designing of genes for increased protein production using Brassica genome as a host system. In the present study, we report the detailed codon usage data and analysis of various factors shaping the codon usage patterns in B. campestris genes.
Materials and methods
Sequence data
The coding sequences of B. campestris genes were retrieved from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov). In the present study, a total of 116 coding sequences (cds) were analysed. Only the perfect cds which are exact multiple of three bases were analysed in the present work.
Indices of codon usage
Relative synonymous codon usage (RSCU)
RSCU is the observed frequency of a codon divided by the expected frequency [1]. If all synonymous codons encoding the same amino acid are used equally, RSCU values are close to 1.0, indicating a lack of bias. Moreover, the RSCU value greater than 1.6 is treated as over represented codon and RSCU value lower than 0.6 is considered as under-represented codon
where Xij is the frequency of occurrence of the jth codon for ith amino acid (any Xij with a value of zero is arbitrarily assigned a value of 0.5) and ni is the number of codons for the ith amino acid (ith codon family).
Effective number of codons (Nc)
It is quite often used to quantify the codon bias in one specific gene [27], which is an assessment of non-uniformity of usage within synonymous groups of codons. The values of Nc values can vary from 20 (extreme bias where only one codon is used per amino acid) to 61 (without bias where codons are used in equal probability). If the calculated Nc is greater than 61 (because codon usage is more evenly distributed than expected), it is adjusted to 61. Nc value within the range 20–45 is generally considered as high codon bias
where Fk (k = 2, 3, 4, 6) is the mean of Fk values for the k-fold degenerate amino acids, 2 stands for two amino acids, i.e., met and trp; 9, 1, 5, and 3 stand for the total number of amino acids with degeneracy class of 2, 3, 4, and 6 codons, respectively.
Codon adaptation index (CAI)
Sharp and Li [11] proposed that the codon adaptation index (CAI) is an effective measure of codon bias in prokaryotes [28] and eukaryotes [29]. CAI is a measurement of the relative adaptiveness of the codon usage of a gene towards the codon usage of the highly expressed genes. CAI values range from 0 to 1, with higher value towards 1 means stronger codon usage bias and thus a potentially higher gene expression level. [30]. The CAI is calculated as:
where L is the number of codons in the gene and Wc(k) is the w value for the kth codon in the gene. The CAI defines the frequent codons in highly expressed genes as the translationally optimal codons.
Dinucleotide odds ratio
The odds ratio is generally used to compute the dinucleotides in gene sequences. Odds ratio is the likelihood of observing a dinucleotide in a sequence and is calculated as
where, x and y stand for the nucleotides that form dinucleotide xy; and fx, fy, fxy denote the frequencies of nucleotide x, nucleotide y, and dinucleotide xy, respectively. Karlin et al. [31] showed that dinucleotides with an odds ratio falling outside the range (0.78–1.23) could be considered as being more under-represented-or over-represented dinucleotide than normal.
Correspondence analysis
Correspondence analysis has been successfully used to explore codon usage variation among genes. It is a commonly used multivariate statistical technique, in which all genes were plotted in a 59-dimensional space, according to the usage of the 59 sense codons (excluding codons for Met, Trp and stop codons). The plot was then used to identify the axes which represent the most prominent factors contributing to variation among genes. Major trends within this dataset can be determined using the measures of relative inertia (eigen value) and genes ordered according to their positions along these axes of major inertia.
Statistical analysis
Correlation analysis was carried out using the Spearman’s rank correlation analysis method wrapped in the multianalysis software SPSS version 16.0.
Results and discussion
Nucleotide composition and codon usage in B. campestris
The overall nucleotide composition varies from genome to genome because of intrinsic, organism-specific metabolic process, environmental conditions and the after-effect of neutral process and selection [32]. This difference in nucleotide composition causes the niche intricacy of genomes and results in the observed CUB [33]. We, therefore, analysed the nucleotide compositions of the coding sequences for B. campestris. With reference to previous studies on plant genomes here also the coding sequences were found to compositionally biased towards the usage of AT nucleotide [34, 35], can result to the ascendance of the A/T-ending codons. The GC content of the B. campestris genes varied from 35.92 to 61.35% with a standard deviation (SD) of 4.05. The nucleobase A showed the highest mean ± SD (28.2 ± 3.22) followed by T (24.6 ± 2.44) and G (24.8 ± 2.96). The nucleobase C showed the lowest usage percentage i.e, 22.47 (SD = ± 2.54). To get an unmistakeable picture about the preferences of codon usage, we investigated the coding intensity of the nucleotides at first second and third codon positions. At first codon position, the mean ± SD of G1 was the highest (32 ± 3.93) followed by A and C with T being the lowest. At second codon position, A showed the highest mean ± SD (32.2 ± 3.90) and T at third codon position (29.2 ± 5.06). Therefore, from the composition analysis, it was evident that nucleobase T/G/A might be more favored, suggesting that compositional constraints play the paramount role in the evolution of codons in this species.
The relative use recurrence of the GC content at three different codon positions i.e. GC1, GC2 and GC3 varies from gene to gene and from genome to genome. The GC1, GC2, and GC3 were 0.51, 0.42, and 0.47, respectively. Differences in GC content among the genes were the highest at third codon position (ranges from 0.30 to 0.79) followed by the second position (ranges from 0.27 to 0.51) and first codon (ranges from 0.43 to 0.62) position. The distinctions in the initial two positions that generally prompt an adjustment in amino-acid composition suggested that a different GC mutation bias leads to different codon choice despite the changes in protein sequences. These results suggest that there might be compositional constraint in the presence of mutation pressure which affects the B. campestris genes. Neutrality plot analysis (Supplementary file Fig. 1) between GC12 and GC3 reveals the relative effect of mutation/selection in shaping the CUB. In contrast to Kawabe and Miyashita 2003 work on dicots [36], a significant correlation was observed between GC12 and GC3 (Pearson r = 0.385, p < 0.01). To test the goodness-of-fit of the regression model GC3 = 0.148GC12 + 0.401 we performed Chi square test (1.58, p < 0.01) at 116-3 = 113 df. The estimated Chi square value was found to be non-significant at p = 0.01, indicating that the regression model has goodness-of-fit. This significantly positive correlation in the neutrality plots indicated that the mutation pressure and selection contribute to the codon bias in B. campestris.
Codon usage and Nc
In order to identify the roles of nucleobase G and C to codon usage bias, the values of Nc was plotted against GC3s of the genes. Nc is a standard parameter used to measure the magnitude of codon usage bias. Nc value of the different genes ranged from 36.5 to 61, denoting there is a strong compositional pressure which results in the observed variation in codon bias among the genes. GC3 showed the highest correlation value with Nc, followed by GC2 and GC1. To assess the relative effect of mutation and selection on genes having variation in codon bias distribution the initial dataset was grouped into two separate groups i.e., the high codon bias gene group (16 cds < Nc = 50) and low codon bias group (100 cds > Nc = 50). Pearson’s correlation between GC3 and Nc was calculated for both the gene groups. The two correlation coefficients differ significantly, high codon bias gene group showed significant correlation value of − 0.44 (p < 0.05) whereas low codon bias gene group showed a correlation value of 0.107 (p < 0.001). These intricate correlations further uncovered the impact of neutral mutation bias which brings about the variation in codon usage among the genes.
From the Fig. 1 it is clearly observed that a relatively higher number of genes scatter distantly from the expected curve, which indicates that GC3s is not the sole determinant of CUB. The discrete distribution of different genes in Nc versus GC3s plot that cannot be explained by mutation alone implies that some other factors, for example, protein structure, gene expression level and so on may have substantial effect in CUB among the genes, independent of the compositional constraints. To get a clearer picture we analysed the frequency distribution of effective number of codon ratio (Supplementary file Fig. 2). Interestingly most of the genes showed the ratio between 0 and 0.1, suggesting that these genes have Nc values lower than expected, though a few genes have Nc greater than the expected value. In our analysis, 8 out of 116 coding sequences showed negative ratio indicating that other factors (e.g., selection) operated at the higher level in a directional way to influence the observed Nc.
Fig. 1.

Distribution of effective number of codons (Nc) and GC3s in B. campestris
Relative synonymous codon usage bias
After investigating the 49,578 codons in 116 coding sequences used in the present study it was observed that amongst the three stop codons TAA (52.58%) was used most frequently followed by TAG and TGA (46.98 and 0.43%, respectively). Based on the occurrence of synonymous codons of all 59 codons measured by their relative synonymous codon usage frequency, 14 and 12 codons were found to be translationally favored and rare codons, respectively. For the six-fold, four-fold and two-fold degenerative amino acids, at least, three, two and one synonymous codons showed RSCU ≥ 1, respectively (Fig. 2). TCT (Ser) and AGA (Arg) had the highest values (1.57 and 2.04, respectively). CTT (Leu) and GTT (Val), GCT (Ala) and GGA (Gly) were used much more frequently than other synonymous codons for the corresponding amino acids (1.44, 1.46, 1.49 and 1.42, respectively). The synonymous third codon positions in the favored codons were found to be occupied by either NNAs or NNTs. Previous studies on AT-rich genomes show a preference for A or T in third codon position [37, 38]. Therefore, the use of A/T nucleobase at third codon position of the favored codons suggests that the compositional constraints are the most paramount factors in shaping the codon usage variation among the genes. The increased usage of NCT and the decreased use of NTA codons are thought to be due to the genomic composition for better expression and stability of the genes, respectively.
Fig. 2.
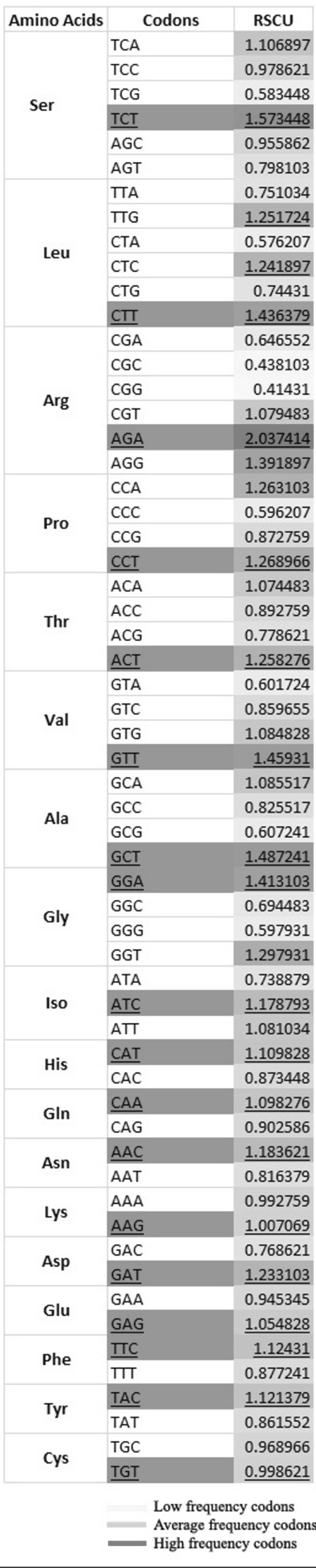
Unequal usage of all synonymous codons (except the three stop codons, ATG and TGG) in B. campestris. The relative usage frequency of codons are listed and colored yellow, orange yellow and red to show low, average and high occurrence frequencies, respectively. The abundant codons from each synonymous group were underlined and highlighted
Dinucleotide odds ratio
As a next step in our analyses, we studied the odds ratio of the 16 dinucleotides. Dinucleotide bias shows significant effect on the usage frequency of codons within the coding region [21]. This index measures the relative abundance of dinucleotides with respect to what would be expected from the random union of mononucleotides [39] and the value 1 is expected when no bias is observed. Dinucleotides with odds ratio ≤ 0.78 are termed as under-represented or suppressed dinucleotides and index value ≥ 1.23 is treated as over- represented dinucleotide. We observed that the dinucleotide TA and CG are significantly underrepresented in the coding sequences (Fig. 3). Porceddu and Camiolo [40] also found similar result in other plant genes. Except the amino acid tyrosine, which is encoded by TAC and TAT codon (both of them contains TA dinucleotide), all other amino acid encoding codons with TA and CG dinucleotide showed under-representation (Fig. 2). The dinucleotides TG, TC, GA, CA and CT are the overrepresented ones. Interestingly, the TG bias in coding sequences mirrors the pattern of CG suppression in the genes of B. campestris. The inadequate representation of CG dinucleotides in plant genome may be attributed to selection pressure against methylation or to the classical methylation deamination mutation mechanism, for enhanced translational speed [21, 41]. The scarcity of TA in plants might be related to mRNA stability and also to evade improper binding of the various factors involved in transcription processes [42].
Fig. 3.

Distribution of 116 genes based on dinucleotide odd ratio values
PR2 bias analysis
PR2 plot is a helpful approach to reveal the presence of any asymmetric mutation and/or selection pressure. To evade a systematic bias from PR2, three stop codons (TAA, TAG, or TGA) and codons ATG (Met), TGG (Trp) and ATA (Ile) were not included in the compositional analyses. Mutation pressure and natural selection are the major factors considered to shape the codon usage pattern. If mutation bias is the cause of codon usage bias, then GC and AT ought to be used proportionally among the degenerate codon groups. In contrast, natural selection for codon choice would not necessarily cause the proportional use of G and C (A and T) [43]. Our result showed that the AT-rich genome of B. campestris uses A and T more frequently than G and C (Fig. 4). The regression coefficient of A3/(A3 + T3) against G3(G3 + C3) is 0.337, indicating a relative neutrality of 0.34 or a relative constraint of 0.66. The observed difference in between C/G and A/T contents within the coding region suggest that both selection and base composition bias mainly contributed to CUB of B. campestris genes.
Fig. 4.

PR2 bias plot [A3/(A3 + T3) against G3/(G3 + C3)] of B. campestris coding sequences. Average position is x = 0.4328 ± 0.0578, y = 0.5042 ± 0.0970
Gene expression analysis and codon usage bias
Correspondence analysis (COA) of RSCU values of codons in B. campestris genes identified a single major trend in codon usage: the first axis generated by the analysis represented 18.26% of the total variability, whereas the next three axes only account for 11.34, 9.28 and 7.79%, respectively, corroborating that the primary axis was the main factor expounding codon utilization in these genes. The plot of genes on the first two axes [Fig. 5(A)] shows the genes from both the datasets (High and Low CAI) scatters throughout the plot area. The plot of axis 1 against axis 2 represents the distance between each and every genes based on RSCU in multidimensional space. CAI showed low correlation with both the axis coordinates. This equal distribution of genes from both the datasets over the plot area suggests that gene expression level was not responsible for separating genes according to their codon usage along the two axes. The position of each cds along the axis 1 is strongly correlated with its GC, GC3s (r = 0.864 and 0.904 respectively, p < 0.01) and negatively correlated with cds length (r = − 0.135, p < 0.001). Similar correlations were likewise reported in a few plants [44] and these results suggest that natural selection must be the major factor in determining the variation of codon usage bias of these genes [45, 46]. The distribution of codons along the axis 1 and axis 2 showed that the separation of codons on axis 1 totally depends on the presence of A and/or T at the synonymous position [Fig. 5(B)] [47, 48]. Moreover, there was non-significant correlation (r = 0.017) between the gene expression level assessed by CAI and Nc, indicates that CUB is not constrained by translational selection [36]. While significantly negative correlations of CAI with GC3s and GC content was observed (r = − 0.253 and − 0.220, respectively, p < 0.01). The coding intensity of the nucleotides A, T, G and C at synonymous third codon position were also analyzed (r = 0.756, 0.758, 0.780 and 0.687, respectively at p < 0.01). In the AT-rich genome of B. campestris, except the nucleobase C at synonymous third codon position, all the nucleotides showed almost similar correlation value. The significant difference in correlation of C3 with CAI may be due to mutational pressure, results in the observed genomic AT-bias. Furthermore, researchers also proposed that since C3 showed positive correlation with CAI there must be selection pressure in favour of C3 in the AT- rich genomes [49]. Taken together, it can be concluded that the nucleotide composition bias is the essential element impacting codon usage in B. campestris genes.
Fig. 5.

(A) Distribution of B. campestris genes on the plane defined by the first two main axes of the correspondence analysis. Red colored squares and yellow colored diamonds indicate genes with extremely high and low CAI values used as the High and Low datasets, respectively. (B) The distribution of codons on axis 1 versus axis 2. Each codon with A or T at third synonymous codon position showed negative axis 1 value in correspondence analysis
Relationship of codon bias with hydropathicity index and aromaticity score
Different studies have exhibited that the hydropathicity and the aromaticity of encoded proteins determines the pattern of codon usage [30, 50, 51]. In order to investigate whether the same holds great in B. campestris, we conducted a correlation analysis of CUB with gravy and aromaticity score. The correlation coefficients for codon usage with gravy (r = − 0.068, p < 0.001) and aromaticity (r = 0.165, p < 0.05) indicated that CUB was associated with aromaticity of the encoded proteins however not with the hydrophobicity of the amino acids.
To conclude, in the present study, synonymous codon usage among B. campestris genes mainly appears to be the result of base composition bias, where selection pressure plays a dominant role over mutation pressure. PR2 plot showed that the gene does not equally use A/T/G/C-ending codons. The over-representation of AT over GC in the degenerate codon positions in our current analysis further reflects the fact that selection pressure has played an important role in driving the CUB of B. campestris. COA on RSCU and codon usage further supports the PR2 analysis. The present study has revealed the codon usage pattern at molecular level for the Brassica genes. Therefore, from this in near future we can design synthetic gene for efficient oil production [52], better biotic [53] and abiotic [54] stress tolerance etc., based on codon usage patterns.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to the University Grants Commission, New Delhi, India, for providing a UGC-BSR Fellowship to carry out this research work. We are also grateful to the Assam University, Silchar, Assam, India for providing the research facility.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Sharp PM, Li W-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. Journal of molecular evolution. 1986;24(1–2):28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 2.Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129(3):897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fennoy SL, Bailey-Serres J. Synonymous codon usage in Zea mays L. nuclear genes is varied by levels of C and G-ending codons. Nucleic acids research. 1993;21(23):5294–5300. doi: 10.1093/nar/21.23.5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiapello, H., et al., Codon usage and gene function are related in sequences of Arabidopsis thaliana. Gene, 1998. 209(1): p. GC1–GC38. [DOI] [PubMed]
- 5.Hershberg R, Petrov DA, Nachman MW. General rules for optimal codon choice. PLoS Genet. 2009;5(7):e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Molecular biology and evolution. 1985;2(1):13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95(16):9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184(20):5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10(22):7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp PM, Li WH. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24(1–2):28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 11.Sharp PM, Li WH. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharp PM, Tuohy TM, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14(13):5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lobry JR, Gautier C. Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res. 1994;22(15):3174–3180. doi: 10.1093/nar/22.15.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Onofrio G, Ghosh TC, Bernardi G. The base composition of the genes is correlated with the secondary structures of the encoded proteins. Gene. 2002;300(1–2):179–187. doi: 10.1016/S0378-1119(02)01045-4. [DOI] [PubMed] [Google Scholar]
- 15.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151(3):389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 16.Ikemura T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J Mol Biol. 1982;158(4):573–597. doi: 10.1016/0022-2836(82)90250-9. [DOI] [PubMed] [Google Scholar]
- 17.Sueoka N, Kawanishi Y. DNA G + C content of the third codon position and codon usage biases of human genes. Gene. 2000;261(1):53–62. doi: 10.1016/S0378-1119(00)00480-7. [DOI] [PubMed] [Google Scholar]
- 18.Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends in biotechnology. 2004;22(7):346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Zhao L, et al. Characterization of codon usage bias in the dUTPase gene of duck enteritis virus. Progress in natural science. 2008;18(9):1069–1076. doi: 10.1016/j.pnsc.2008.03.009. [DOI] [Google Scholar]
- 20.Paul P, Chakraborty S. Codon usage bias analysis for the coding sequences of Camellia sinensis and Brassica campestris. African Journal of Biotechnology. 2016;15(8):236–251. doi: 10.5897/AJB2015.15111. [DOI] [Google Scholar]
- 21.De Amicis F, Marchetti S. Intercodon dinucleotides affect codon choice in plant genes. Nucleic acids research. 2000;28(17):3339–3345. doi: 10.1093/nar/28.17.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camiolo S, Melito S, Porceddu A. New insights into the interplay between codon bias determinants in plants. DNA Research. 2015;22(6):461–470. doi: 10.1093/dnares/dsv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Sharma R. Codon usage in Brassica genes. Journal of Plant Biochemistry and Biotechnology. 1995;4(2):113–115. doi: 10.1007/BF03262965. [DOI] [Google Scholar]
- 24.Elhaik, E. and T. Tatarinova, GC3 biology in eukaryotes and prokaryotes. arXiv preprint arXiv:1203.3929, 2012.
- 25.Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics—developing a new class of drugs. Nature reviews Drug discovery. 2014;13(10):759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 26.Elena, C., et al., Expression of codon optimized genes in microbial systems: current industrial applications and perspectives. Frontiers in microbiology, 2014. 5. [DOI] [PMC free article] [PubMed]
- 27.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 28.Eyre-Walker A, Bulmer M. Reduced synonymous substitution rate at the start of enterobacterial genes. Nucleic acids research. 1993;21(19):4599–4603. doi: 10.1093/nar/21.19.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Touchon M, Rocha EP. From GC skews to wavelets: a gentle guide to the analysis of compositional asymmetries in genomic data. Biochimie. 2008;90(4):648–659. doi: 10.1016/j.biochi.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 30.Yang X, Luo X, Cai X. Analysis of codon usage pattern in Taenia saginata based on a transcriptome dataset. Parasites & vectors. 2014;7(1):1–11. doi: 10.1186/1756-3305-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlin S, Mrazek J, Campbell AM. Codon usages in different gene classes of the Escherichia coli genome. Mol Microbiol. 1998;29(6):1341–1355. doi: 10.1046/j.1365-2958.1998.01008.x. [DOI] [PubMed] [Google Scholar]
- 32.Foerstner KU, et al. Environments shape the nucleotide composition of genomes. EMBO reports. 2005;6(12):1208–1213. doi: 10.1038/sj.embor.7400538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus research. 2003;92(1):1–7. doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- 34.Morton BR, Levin JA. The atypical codon usage of the plant psbA gene may be the remnant of an ancestral bias. Proceedings of the National Academy of Sciences. 1997;94(21):11434–11438. doi: 10.1073/pnas.94.21.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nie X, et al. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant molecular biology reporter. 2014;32(4):828–840. doi: 10.1007/s11105-013-0691-z. [DOI] [Google Scholar]
- 36.Kawabe A, Miyashita NT. Patterns of codon usage bias in three dicot and four monocot plant species. Genes & genetic systems. 2003;78(5):343–352. doi: 10.1266/ggs.78.343. [DOI] [PubMed] [Google Scholar]
- 37.Saul A, Battistutta D. Codon usage in Plasmodium falciparum. Molecular and biochemical parasitology. 1988;27(1):35–42. doi: 10.1016/0166-6851(88)90022-9. [DOI] [PubMed] [Google Scholar]
- 38.Muto A, Yamao F, Osawa S. The genome of Mycoplasma capricolum. Progress in nucleic acid research and molecular biology. 1986;34:29–58. doi: 10.1016/S0079-6603(08)60492-4. [DOI] [PubMed] [Google Scholar]
- 39.Burge C, Campbell AM, Karlin S. Over-and under-representation of short oligonucleotides in DNA sequences. Proceedings of the National Academy of Sciences. 1992;89(4):1358–1362. doi: 10.1073/pnas.89.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porceddu A, Camiolo S. Spatial analyses of mono, di and trinucleotide trends in plant genes. PloS one. 2011;6(8):e22855. doi: 10.1371/journal.pone.0022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morton BR, et al. Variation in mutation dynamics across the maize genome as a function of regional and flanking base composition. Genetics. 2006;172(1):569–577. doi: 10.1534/genetics.105.049916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kariin S, Burge C. Dinucleotide relative abundance extremes: a genomic signature. Trends in genetics. 1995;11(7):283–290. doi: 10.1016/S0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- 43.Sueoka N, Kawanishi Y. DNA G + C content of the third codon position and codon usage biases of human genes. Gene. 2000;261(1):53–62. doi: 10.1016/S0378-1119(00)00480-7. [DOI] [PubMed] [Google Scholar]
- 44.Liu Q, et al. Synonymous codon usage bias in Oryza sativa. Plant Science. 2004;167(1):101–105. doi: 10.1016/j.plantsci.2004.03.003. [DOI] [Google Scholar]
- 45.Romero H, Zavala A, Musto H. Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic acids research. 2000;28(10):2084–2090. doi: 10.1093/nar/28.10.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Q, Feng Y, Xue Q. Analysis of factors shaping codon usage in the mitochondrion genome of Oryza sativa. Mitochondrion. 2004;4(4):313–320. doi: 10.1016/j.mito.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Chen, H., et al., Mutation and Selection Cause Codon Usage and Bias in Mitochondrial Genomes of Ribbon Worms (Nemertea). PloS one, 2014. 9(1). [DOI] [PMC free article] [PubMed]
- 48.Le TH, McManus DP, Blair D. Codon usage and bias in mitochondrial genomes of parasitic platyhelminthes. The Korean journal of parasitology. 2004;42(4):159–167. doi: 10.3347/kjp.2004.42.4.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Das S, et al. Compositional variation in bacterial genes and proteins with potential expression level. FEBS letters. 2005;579(23):5205–5210. doi: 10.1016/j.febslet.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 50.Yang X, et al. Codon Usage Bias and Determining Forces in Taenia solium Genome. Korean J Parasitol. 2015;53(6):689–697. doi: 10.3347/kjp.2015.53.6.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L, et al. Synonymous codon usage patterns in different parasitic platyhelminth mitochondrial genomes. Genetics and molecular research: GMR. 2013;12(1):587. doi: 10.4238/2013.February.27.8. [DOI] [PubMed] [Google Scholar]
- 52.Tan H, et al. Enhanced seed oil production in canola by conditional expression of Brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in developing seeds. Plant physiology. 2011;156(3):1577–1588. doi: 10.1104/pp.111.175000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed NU, et al. Identification and characterization of stress resistance related genes of Brassicarapa. Biotechnology letters. 2012;34(5):979–987. doi: 10.1007/s10529-012-0860-4. [DOI] [PubMed] [Google Scholar]
- 54.Shanmugam A, et al. Characterization and abiotic stress-responsive expression analysis of SGT1 genes in Brassica oleracea. Genome. 2016;59(4):243–251. doi: 10.1139/gen-2015-0128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.