

Abstract
The X-chromosome comprises only about 5 percent of the human genome but accounts for about 15 percent of the genes currently known to be associated with intellectual disability. The early progress in identifying the XLID-associated genes through linkage analysis and candidate gene sequencing has been accelerated with the use of high throughput technologies. In the 10 years since the last update, the number of genes associated with XLID has increased by 96 percent from 72 to 141 and duplications of all 141 XLID genes have been described, primarily through the application of high resolution microarrays and next generation sequencing. The progress in identifying genetic and genomic alterations associated with XLID has not been matched with insights that improve the clinician's ability to form differential diagnoses, that bring into view the possibility of curative therapies for patients, or that inform scientists of the impact of the genetic alterations on cell organization and function.
Keywords: X chromosome, intellectual disability, syndrome, genes, XLID
Introduction
Over the last three decades there has been remarkable progress in the understanding of X-linked intellectual disability (XLID). The X chromosome was targeted for study because of the excess of males among all individuals with intellectually disability (ID) and the availability of numerous families in which ID followed an X-linked pattern of inheritance. Milestones in the early history of XLID were the study of Penrose (1938) on institutionalized individuals, the description of large XLID families by Martin and Bell (1943), Allan et al. (1944) and Renpenning et al. (1962) and subsequent work by Lehrke (1972). Additional strong motivation came from the notion that the X-chromosome might harbor a disproportionate fraction of genes that influence cognitive function (Turner and Partington 1991) and the discovery of the Fragile X syndrome (a posteriori, rediscovery of the Martin-Bell syndrome), the first XLID entity to be regionally mapped to the X-chromosome (Lubs 1969).
Looking back at previous XLID updates published regularly every two years from 1990 to 2000 and again in 2008, it is apparent that great efforts went into classifying conditions whose phenotypes could not be more diverse. Eventually, these efforts failed, with the exception of the distinction of syndromal from nonsyndromal XLID. Even though the line dividing the two classes is blurred, sometimes arbitrary and difficult to defend from a biological standpoint, it has by now acquired quasi-historical value and deserves to be retained. In compiling the current update, no effort was made to categorize conditions and associated genes. In the practical terms of diagnosis and counseling, what is important is to know the causal genes, the mode of transmission and possibly the pathogenic mechanism.
XLID update 2017 includes the enumeration of all currently known syndromes, nonsyndromal conditions (IDX), and genes, with particular attention to the syndromes described and genes assigned since the 2007 XLID update (Chiurazzi et al. 2008). The discovery of XLID genes has, in large measure, moved from the research laboratory to the diagnostic laboratory during the past decade. Linkage analysis and candidate gene testing has been replaced with X-chromosome or exome sequencing. Various microarray technologies have identified duplications of all XLID genes, bringing attention to genomic aberrations that may even outnumber gene sequence variants. A section of this update, not present in previous editions, describes these duplications and comments on their relevance.
Finding the outstanding causal genes, possibly as many as 80, should complete the delineation of XLID at the genetic/genomic level. This may require utilizing newly emerging technologies – higher resolution microarrays, genome and RNA sequencing, metabolomics and epigenomics.
New Entries
Since the last update in 2007, 69 new XLID genes have been reported in the literature (Table I). Most of these have been associated with syndromal entities, both previously reported and novel. The identified 69 XLID genes may actually be less. As pointed out by Piton et al. (2013), for three genes (MAGT1, RAB40AL, NXF5) the databases utilized as “controls” contain too many loss of function mutations. For three other genes (RPL10, ZMYM3, SIZN1), there have been only a single report so that their inclusion as XLID genes needs replication in additional studies, as further discussed below.
Table I. XLID genes identified 2007-2017.
| Year | Gene Symbol |
Gene Name | Locus | Gene OMIM |
How Discovered |
Clinical Description | Function | References |
|---|---|---|---|---|---|---|---|---|
| 2007 | BRWD3 | BROMODOMAIN- AND WD REPEAT-CONTAINING PROTEIN 3 | Xq21.1 | 300553 | X-seq | XLID-Macrocephaly-Large Ears, IDX93 | Transcription factor | Field et al. 2007 |
| 2007 | CUL4B | CULLIN 4B | Xq24 | 300304 | X-seq | XLID-Hypogonadism-Tremor | Cell cycle, ubiquitin cycle, E3 ubiquitin ligase | Tarpey et al. 2007 |
| 2007 | GRIA3 | GLUTAMATE RECEPTOR, IONOTROPIC, AMPA 3 | Xq25 | 305915 | Chr-rea, Exp-Arr, X-seq | Chiyonobu XLID, IDX94 | Signal transduction, ion transport, glutamate signaling pathway | Wu et al. 2007 |
| 2007 | HSD17B10 (HADH2) | 17-BETA-HYDROXYSTEROID DEHYDROGENASE X | Xp11.22 | 300256 | L-can | XLID-Choreoathetosis | Lipid metabolism | Lenski et al. 2007 |
| 2007 | MED12 (HOPA) | MEDIATOR COMPLEX SUBUNIT 12NOTE: MEDIATOR COMPLEX SUBUNIT 12 | Xq13.1 | 300188 | L-can | Opitz FG, Lujan, IDX67 | Transcription regulation, RNA polymerase II transcription mediator activity, ligand-dependent nuclear receptor transcription coactivator activity, vitamin D receptor and thyroid hormone receptor binding | Risheg et al. 2007 |
| 2007 | NDUFA1 | NADH-UBIQUINONE OXIDOREDUCTASE 1 ALPHA SUBCOMPLEX, 1 | Xq24 | 300078 | Mol-Fu | Mitochondrial Complex 1 Deficiency | Energy production, oxidoreductase activity | Fernandez-Moreira et al. 2007 |
| 2007 | NXF5 * | NUCLEAR RNA EXPORT FACTOR 5 | Xq22.1 | 300319 | Chr-rea | XLID-Short Stature-Muscle Wasting | mRNA processing, mRNA export from nucleus | Froyen et al. 2007 |
| 2007 | PORCN | PORCUPINE, DROSOPHILA, HOMOLOG | Xp11.23 | 300651 | Chr-rea (del) | Goltz | Wnt receptor signaling pathway, acyltransferase activity, integral to membrane of endoplasmic reticulum | Wang et al. 2007 |
| 2007 | PRPS1 | PHOSPHORIBOSYLPYROPHOSPHATE SYNTHETASE I | Xq22.3 | 311850 | L-can | Arts, PRPS1 Superactivity | Ribonucleotide monophosphate biosynthesis | de Brouwer et al. 2007 |
| 2007 | RPL10 * | RIBOSOMAL PROTEIN L10 | Xq28 | 312173 | X-seq | Autism | Protein synthesis, ribosomal protein | Klauck et al. 2006 |
| 2007 | UPF3B | UPF3, YEAST, HOMOLOG OF, B | Xq24 | 300298 | X-seq | Lujan/FG Phenotype, IDX62 | mRNA catabolism, nonsense-mediated decay | Tarpey et al. 2007 |
| 2007 | ZDHHC9 | ZINC FINGER DHHC DOMAIN-CONTAINING PROTEIN 9 | Xq26.1 | 300646 | X-seq | XLID-Macrocephaly-Marfanoid Habitus | Raymond et al. 2007 | |
| 2008 | HUWE1 | HECT, UBA, AND WWE DOMAINS-CONTAINING PROTEIN 1 | Xp11.22 | 300697 | CMA | XLID-Macrocephaly, Juberg-Marsidi-Brooks, IDX17, IDX31 | Ubiquitin-protein ligase, mRNA transport | Froyen et al. 2008 |
| 2008 | PCDH19 | PROTOCADHERIN 19 | Xq22.1 | 300460 | L-can | Epilepsy-Intellectual Disability Limited to Females | Dibbens et al. 2008 | |
| 2008 | SLC9A6 | SOLUTE CARRIER FAMILY 9, MEMBER 6 | Xq26.3 | 300231 | L-can | Christianson, X-linked Angelman-like | Sodium-hydrogen antiporter activity, lysosome organization and biogenesis, regulation of endosome volume | Gilfillan et al. 2008 |
| 2008 | SIZN1 *(ZCCHC12) | SMAD-INTERACTING ZINC FINGER PROTEIN 1 | Xq24 | 300701 | L-can | IDX | Modulates BMP signalling influences forebrain cholinergic neurons | Cho et al. 2008 |
| 2009 | MAGT1 * | MAGNESIUM TRANSPORTER 1 | Xq21.1 | 300715 | L-can, X-seq | IDX95 | Magnesium transporter with N-glycosylation sites and putative phosphorylation sites | Molinari et al. 2008 |
| 2009 | MBTPS2 | MEMBRANE-BOUND TRANSCRIPTION FACTOR PROTEASE, SITE 2 | Xp22.12 | 300294 | L-can | Ichthyosis Follicularis, Atrichia, Photophobia (IFAP) | Protease activity, activates signaling proteins | Oeffner et al. 2009 |
| 2009 | NSDHL | NAD(P)H STEROID DEHYDROGENASE-LIKE PROTEIN | Xq28 | 300275 | L-can | CK (microcephaly, pachygyria, facial dysmorphism, seizures), also in CHILD syndrome | Sterol metabolism | du Souich et al. 2009 |
| 2009 | SYP | SYNAPTOPHYSIN | Xp11.23 | 313475 | X-seq | IDX96 | Membrane protein of small synaptic vesicles | Tarpey et al. 2009 |
| 2009 | ZNF711 | ZINC FINGER PROTEIN 711 | Xq21.1 | 314990 | X-seq | IDX97 | Binds to a subset of PHF8 target genes | Tarpey et al. 2009 |
| 2010 | IQSEC2 | IQ MOTIF- AND SEC7 DOMAIN-CONTAINING PROTEIN 2 | Xp11.22 | 300522 | X-seq | IDX1, IDX18, and other Nonsyndromal XLID | Regulation of vesicular transport and organelle structure | Shoubridge et al. 2010 |
| 2010 | PTCHD1 | PATCHED DOMAIN-CONTAINING PROTEIN 1 | Xp22.11 | 300828 | CMA | Autism-XLID | Transmembrane protein related to hedgehog receptors | Noor et al. 2010 |
| 2010 | RAB39B | RAS-ASSOCIATED PROTEIN | Xq28 | 300774 | L-can | XLID-Macrocephaly-Seizures -Autism, Waisman-Laxova, IDX72 | Formation and maintenance of synapse | Giannandrea et al. 2010 |
| 2011 | CLCN4 | CHLORIDE CHANNEL 4 | Xp22.2 | 302910 | X-seq | IDX15, IDX49 | Chloride transport | Veeramah et al. 2013 |
| 2011 | CNKSR2 | CONNECTOR ENHANCER OF KSR 2 | Xp22.12 | 300724 | CMA | XLID-Microcephaly-Seizures | Stimulates MAPK signalling | Houge et al. 2012 |
| 2011 | EIF2S3 | EUKARYOTIC TRANSLATION INITIATION FACTOR 2, SUBUNIT 3 | Xp22.11 | 300161 | X-seq | MEHMO | Initiates translation | Borck et al. 2012 |
| 2011 | HCFC1 | HOST CELL FACTOR C1 | Xq28 | 300019 | X-seq | IDX3 | Cell proliferation | Huang et al. 2012 |
| 2011 | HDAC6 | HISTONE DEACETYLASE 6 | Xp11.23 | 300272 | L-can | Chassaing-Lacombe Chondrodysplasia | Tubulin deacetylase | Simon et al. 2010 |
| 2011 | HDAC8 | HISTONE DEACETYLASE 8 | Xq13.1 | 300269 | Mol-Fu, X-seq | Cornelia de Lange, X-linked | Chromatin cohesion | Deardorff et al. 2012; Harakalova et al. 2012 |
| 2011 | LAS1L | LAS1-LIKE RIBOSOME BIOGENESIS FACTOR | Xq12 | 300964 | X-seq | Wilson-Turner | Nucleolar protein, cell proliferation and ribosome biogenesis | Hu et al. 2016 |
| 2011 | NAA10 | N-ALPHA-ACETYLTRANSFERASE 10, NatA CATALYTIC SUBUNIT | Xq28 | 300013 | X-seq | N-Alpha-Acetyltransferase Deficiency | N-terminal acetylation | Rope et al. 2011 |
| 2011 | RAB40AL | RAS-ASSOCIATED PROTEIN RAB40A-LIKE | Xq22.1 | 300405 | X-seq | Martin-Probst (?) Questioned: Hum Mutat 35:1171, 2014 | Ras-like GTPase protein | Bedoyan et al. 2012 |
| 2011 | RBM10 | RNA-BINDING MOTIF PROTEIN 10 | Xp11.3 | 300080 | X-seq | TARP | RNA-binding | Johnston et al. 2014 |
| 2011 | THOC2 | THO COMPLEX, SUBUNIT 2 | Xq25 | 300395 | X-seq | IDX12, IDX35 | mRNA transcription or export | Kumar et al. 2015 |
| 2012 | AIFM1 | APOPTOSIS-INDUCING FACTOR, MITOCHONDRIA-ASSOCIATED, 1 | Xq26.1 | 300169 | X-seq | Charcot-Marie-Tooth disease, Cowchock variant and XLID-spondyloepimetaphyseal dysplasia | Induces apoptosis | Rinaldi et al. 2012 |
| 2012 | ALG13 | ALG13, S. CEREVISIAE, HOMOLOG | Xq23 | 300776 | WES | CDG1s | Glycosylation | Timal et al. 2012; de Ligt et al. 2012 |
| 2012 | CCDC22 | COILED-COIL DOMAIN-CONTAINING PROTEIN 22 | Xp11.23 | 300859 | X-seq | Cardiofacioskeletal (like 3C/Ritscher-Schinzel) | Unknown | Voineagu et al. 2012 |
| 2012 | CLIC2 * | CHLORIDE INTRACELLULAR CHANNEL 2 | Xq28 | 300138 | X-seq | XLID-Cardiomegaly-Seizures | Regulating ryanodine receptor channel activity | Takano et al. 2012 |
| 2012 | EBP | EMOPAMIL-BINDING PROTEIN | Xp11.23 | 300205 | Met-Fu | Variable manifestations but XLID-Aggression in one family | Enzyme in cholesterol metabolism | Furtado et al. 2010 |
| 2012 | KDM6A | LYSINE-SPECIFIC DEMETHYLASE 6A | Xp11.3 | 300128 | Chr-rea | Kabuki syndrome 2 | Histone demethylase and methyltransferase | Lederer et al. 2012 |
| 2012 | PIGA | PHOSPHATIDYLINOSITOL GLYCAN ANCHOR BIOSYNTHESIS CLASS A PROTEIN | Xp22.2 | 311770 | X-seq | XLID-Brain iron accumulation | Enzyme, signal transduction pathway, adhesion molecules | Johnston et al. 2012 |
| 2012 | TMLHE | EPSILON-TRIMETHYLLYSINE HYDROXYLASE | Xq28 | 300777 | X-seq | Autism-ID | Enzyme in carnitine synthesis | Celestino-Soper et al. 2011 |
| 2013 | BCAP31 | B-CELL RECEPTOR-ASSOCIATED PROTEIN 31 | Xq28 | 300398 | X-seq | XLID-microcephaly-dystonia | ER & golgi structure and metabolism | Cacciagli et al. 2013 |
| 2013 | SSR4 | SIGNAL SEQUENCE RECEPTOR, DELTA | Xq28 | 300090 | WES | XLID-glycosylation defect | Glycosylation | Losfeld et al. 2014 |
| 2013 | WDR45 | WD REPEAT-CONTAINING PROTEIN 45 | Xp11.23 | 300526 | WES | Neurodegeneration with brain iron accumulation-XLID | Autophagy and other cellular functions | Haack et al. 2012; Saitsu et al. 2013 |
| 2013 | ZC4H2 | ZINC FINGER C4H2 DOMAIN-CONTAINING PROTEIN | Xq11.2 | 300897 | WES, Chr-rea, CMA | Wieacker-Wolff, Miles-Carpenter | Axon guidance | Hirata et al. 2013 |
| 2014 | HMGB3 | HIGH MOBILITY GROUP BOX 3 | Xq28 | 300193 | WES | Microphthalmia 13 | DNA replication, transcription, nucleosome assembly | Scott et al. 2014 |
| 2014 | KIF4A | KINESIN FAMILY MEMBER 4A | Xq13.1 | 300521 | WES | IDX100 | Moves proteins along microtubule | Willemsen et al. 2014 |
| 2014 | MID2 | MIDLINE 2 | Xq22.3 | 300204 | L-can, WES | IDX101 | Enzyme, ubiquity ligase E3 microtubule stabilization | Geetha et al. 2014 |
| 2014 | USP9X | UBIQUITIN-SPECIFIC PROTEASE 9, X-LINKED | Xp11.4 | 300072 | X-seq | IDX99 | Neuronal migration growth | Homan et al. 2014 |
| 2014 | ZMYM3 * | ZINC FINGER, MYM-TYPE 3 | Xq13.1 | 300061 | X-seq | XLID-aortic stenosis-hypospadias | Component of histone deacetylase-containing multiple protein complexes | Philips et al. 2014 |
| 2015 | KLHL15 | KELCH-LIKE 15 | Xp22.11 | 300980 | X-seq | Mild to moderate cognitive impairment, facial dysmorphism, IDX103 | Uncertain | Mignon-Ravix et al. 2014 |
| 2015 | DDX3X | DEAD/H BOX 3, X-LINKED | Xp11.4 | 300160 | WES | Variable cognitive, neurologic and nonneurologic manifestations, IDX102 | RNA helicase | Snijders Blok et al. 2015 |
| 2015 | FRMPD4 * | FERM AND PDZ DOMAINS-CONTAINING PROTEIN 4 | Xp22.2 | 300838 | X-seq | XLID-Aphasia-seizures, IDX104 | Regulates spine morphogenesis | Hu et al. 2016 |
| 2015 | MSL3 | MALE-SPECIFIC LETHAL 3, DROSOPHILA, HOMOLOG | Xp22.2 | 300609 | WES | MSL3-Related XLID | Transcription regulation, chromatin modifier | Thevenon et al. 2015 |
| 2015 | NONO | NON-POU DOMAIN-CONTAINING OCTAMER-BINDING PROTEIN | Xq13.1 | 300084 | WES | Mircsof-Langouët | Regulation of transcription (activation, repression, splicing, pre-mRNA processing, RNA transport) | Mircsof et al. 2015 |
| 2015 | RBMX | RNA-BINDING MOTIF PROTEIN, X CHROMOSOME | Xq26.3 | 300199 | WES | Shashi | RNA binding | Shashi et al. 2015 |
| 2015 | RLIM (RNF12) | RING FINGER PROTEIN, LIM DOMAIN-INTERACTING | Xq13.2 | 300379 | X-seq | IDX with variable microcephaly, behaviour abnormalities and other manifestation, IDX61 | Enzyme, ubiquity ligase E3 | Tonne et al. 2015 |
| 2015 | RNF113A | RING FINGER PROTEIN 113A | Xq24 | 300951 | WES | Trichothiodystrophy 5 | Gene regulation, DNA repair | Corbett et al. 2015 |
| 2015 | TAF1 | TAF1 RNA POLYMERASE II, TATA BOX-BINDING PROTEIN-ASSOCIATED FACTOR, 250-KD | Xq13.1 | 313650 | WGS | XLID-Craniofacial-Caudal | Key role in initiating transcription | O'Rawe et al. 2015 |
| 2015 | USP27X | UBIQUITIN-SPECIFIC PROTEASE 27, X-LINKED | Xp11.23 | 3000975 | X-seq | Variable cognitive and speech impairment and behavioural abnormalities, IDX105 | Peptidase | Hu et al. 2016 |
| 2016 | EFHC2 | EF-HAND DOMAIN (C-TERMINAL)-CONTAINING PROTEIN 2 | Xp11.3 | 300817 | L-can | IDX74 | Calcium binding and other unknown functions | Startin et al. 2015 |
| 2016 | GRASP1 | GRIP1-ASSOCIATED PROTEIN 1 | Xp11.23 | 300408 | X-seq | XLID-Short Stature-Spasticity | Synaptic function and neuronal connectivity | Chiu et al. 2017 |
| 2016 | HNRNPH2 | HETEROGENEOUS NUCLEAR RIBONUCLEOPROTEIN H2 | Xp22.1 | 300610 | WES | Bain XLID, Female Limited | Nuclear localization | Bain et al. 2016 |
| 2016 | SLC25A5 | SOLUTE CARRIER FAMILY 25 (MITOCHONDRIAL CARRIER, ADENINE NUCLEOTIDE TRANSLOCATOR), MEMBER A5 | Xq24 | 300150 | CMA | IDX70 | Mitochondrial exchange of ADP/ATP | Vandewalle et al. 2013 |
| 2016 | STAG2 | STROMAL ANTIGEN 2 | Xq25 | 300826 | CMA | STAG2-Related XLID | Chromatin cohesion | Leroy et al. 2016 |
| 2017 | GPKOW | G-PATCH DOMAIN AND KOW MOTIFS | Xp11.23 | 301003 | X-seq | XLID-Microcephaly-Early lethality | Spliceosome component | Carroll et al. 2017 |
| 2017 | OGT | O-LINKED N-ACETYLGLUCOSAMINE TRANSFERASE | Xq13.2 | 300255 | WES | XLID-Faciogenital | Post translational modification of nucleocytoplasmic proteins | Vaidyanathan et al. 2018 |
Association with XLID has been challenged (Piton et al. 2013) and not subsequently resolved (n=7)
Chr-rea = chromosome rearrangement
WES = whole exome sequencing
WGS = whole genome sequencing
Exp-Arr = expression array
L-can = linkage and candidate gene testing
CMA = array-comparative genomic hybridization
Met-Fu = exploitation of metabolic alteration
Mol-Fu = exploitation of molecular finding
X-seq = sequencing of X-chromosome exons
Among the remaining 63 XLID validated genes identified since 2007, there are 10 (16%) which are associated with the broad process of transcription and an almost equal number, 8 (13%), are involved with either ubiquitination or metabolism. The ubiquitination category was hardly represented among genes reported prior to 2007. Additionally, there is a large group of genes, 13, whose protein function does not fit clearly into an ontology group either because the function is not yet known (e.g. ZC4H2), or its function is not classified (e.g. HMGB3), or because the function could place it into two separate groups (e.g. STAG2).
With the increased annotation of the genome using both genomic and RNA sequencing, at least 6 XLID genes have been found to have anti-sense transcripts (SYP, PTCHD1, THLHE, USP27X, SLC25A5 and HCFC1). It is quite possible that alterations in the noncoding portions of these genes might adversely affect the function, if there is one, of their anti-sense RNA. This may be true for genes cloned before 2007, including FMR1.
Listing of All Known Genes
All XLID genes known to date (141) are listed by year of discovery in Supplementary Table I (S1). The position on the X chromosome of genes associated with XLID syndromes is depicted in Fig. 1. Perusal of the list calls for some comments and considerations. Three syndromes initially reported as X-linked have been found to be autosomal and have been retired. These are Zollino syndrome, initially believed to be X-linked based on one pedigree and subsequently found to be due to an unbalanced 1;12 translocation (Zollino et al. 1992, 2003), Roifman syndrome, initially reported to be X-linked and subsequently found to be caused by a deletion on 2q (Roifman 1999, Merico et al. 2015), and Wittwer syndrome, initially mapped to Xp22.3 and later found to be caused by an unbalanced 4;17 translocation (Wittwer et al. 1996, Wieland et al. 2014).
Figure 1.
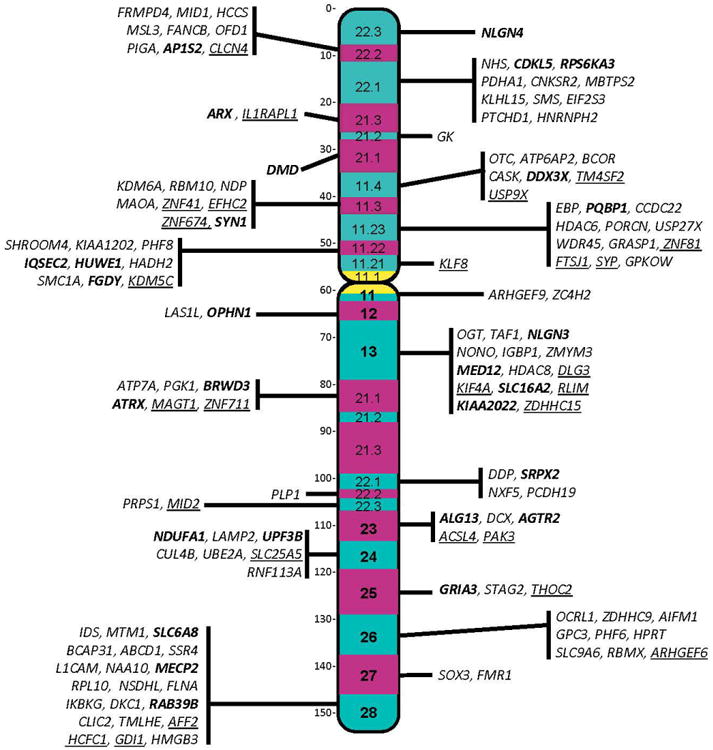
Regional locations of genes associated with XLID syndromes. Bolded genes have also been associated with nonsyndromal XLID. Genes underlined have been challenged by Piton et al. 2013. Color figure can be viewed in the online issue.
Several syndromes previously listed in XLID updates have been removed because the limited evidence that they have intellectual disability as a primary manifestation or that they are X-linked. These include XLID-retinoschisis (312700), X-linked hypoparathyroidism (307700), Giuffrè-Tsukahara (603438), Hyde-Forster (300064), Homfray seizures-contractures, XLID-precocious puberty, and XLID-thyroid aplasia-cutis verticis gyrata. Two additional syndromes have been removed because they appear to be contiguous gene syndromes (XLID-choroideremia-ectodermal dysplasia and XLID-retinitis pigmentosa).
As alluded to above, in 2013, Piton et al. challenged the validity of 25 of the 106 genes then reported to be associated with XLID. Five of the challenged genes were considered highly questionable, five questionable and fifteen in need of replication. None of the 5 highly questionable gene assignments has been confirmed. Of the 5 questionable genes, only ATP6AP2 has been confirmed. Of the 15 assignments in need of replication, additional cases with MAOA, HCFC1, CCDC22, CNKSR2, KIAA2022, NAA10 and SHROOM4 sequence variants have now been published, leaving 17 of the 25 challenged genes unresolved. These genes are identified by an asterisk (*) in Table S1.
Furthermore, a comment on penetrance is in order. Although 183 conditions are classified as XLID syndromes, it is appropriate to note that not all individuals with one of these diagnoses will have ID. Exemplifying this is the inconsistent or even infrequent occurrence of intellectual disability among individuals with Incontinentia Pigmenti, Dyskeratosis Congenita, Oralfacialdigital I, Duchenne Muscular Dystrophy, Simpson-Golabi-Behmel, Aarskog and Nance-Horan syndromes among others. In similar fashion, the severity of ID may vary considerably within certain syndromes e.g., Pelizaeus-Merzbacher, Hunter and Christian syndromes.
In Fig. 2, a histogram illustrates the growing knowledge of XLID syndromes and respective causal genes from 1988 to date. The number of known syndromes has increased from 83 to 183, and the number of cloned genes associated with XLID syndromes from 5 to 113. If we inspect the last column (but this is also true for the others), the apparent discrepancy between 136 syndromes with gene assigned and only 113 genes cloned, requires an explanation. This is due to the fact that a number of genes were found to be responsible for more than one syndrome. Genes that have been linked to three or more XLID syndromes include PQBP1, ATRX, ARX, FLNA, AP1S2 and MED12. The allelic PQBP1-associated disorders include Renpenning, Sutherland-Hann, Hamel Cerebro-Palato-Cardiac, Golabi-Ito-Hall, and Porteous syndromes, all of which have a degree of phenotypic concordance. Strong phenotypic overlap may also be noted among the entities associated with ATRX variants (Alpha-Thalassemia Intellectual Disability, Chudley-Lowry, Holmes-Gang, Carpenter-Waziri, and Arch Fingerprints-Hypotonia syndromes) and the entities associated with AP1S2 variants (Fried syndrome, XLID-Hydrocephaly-Basal Ganglia Calcifications, and Turner XLID syndrome). This is not the case for ARX variants, which have been found in the disparate phenotypes of Partington syndrome, West syndrome, X-linked lissencephaly with abnormal genitalia, hydranencephaly with abnormal genitalia, Proud syndrome, Ohtahara syndrome as well as nonsyndromal XLID (IDX 29, 32, 33, 36, 38, 43, 54, 76, 87). Similar phenotypic discordance has been found among the FLNA-associated disorders (otopalatodigital I and II syndromes, periventricular heterotopias, spondylometaphyseal dysplasia and Melnick-Needles syndrome) and the MED12-associated disorders (Opitz FG, Lujan, and Ohdo syndromes). As to the mapped and unmapped syndromes, totaling 47, some were described in families now lost to follow-up and do not imply that another 47 genes await discovery. It is quite possible that a number of these syndromes could be explained by genes discovered after their initial description.
Figure 2.
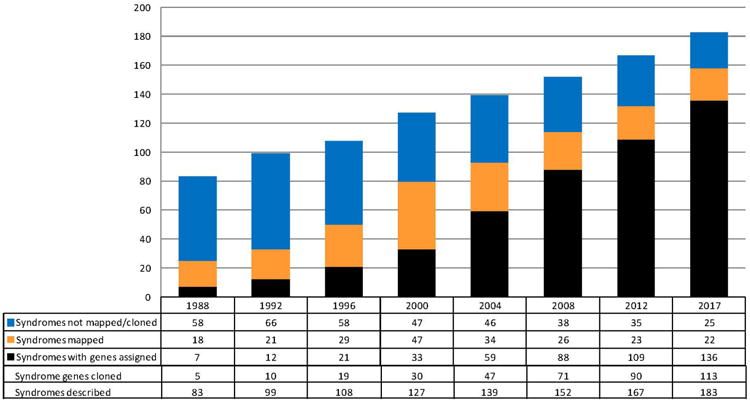
Progress in identifying XLID syndromes and associated genes, 1988-2017. Color figure can be viewed in the online issue.
As to the families with nonsyndromal XLID which have received IDX numbers, their total is currently 105 (Figure 3). For 67 of these families, the genes have been cloned, 33 have been mapped but the genes have not been identified and 5 have reserved IDX numbers (IDX8, 50, 69, 83 and 86) but have not been published. Twenty-eight of the “IDX genes” in Figure 3 have also been associated with XLID syndromes. Sequence variants in 11 genes (KLF8, AFF2, SLC6A8, NDUFA1, ALG13, SRPX2, ATRX, NLGN3, FGDY, CDKL5, and NLGN4) have been found in families with IDX but in whom IDX numbers were never assigned.
Figure 3.
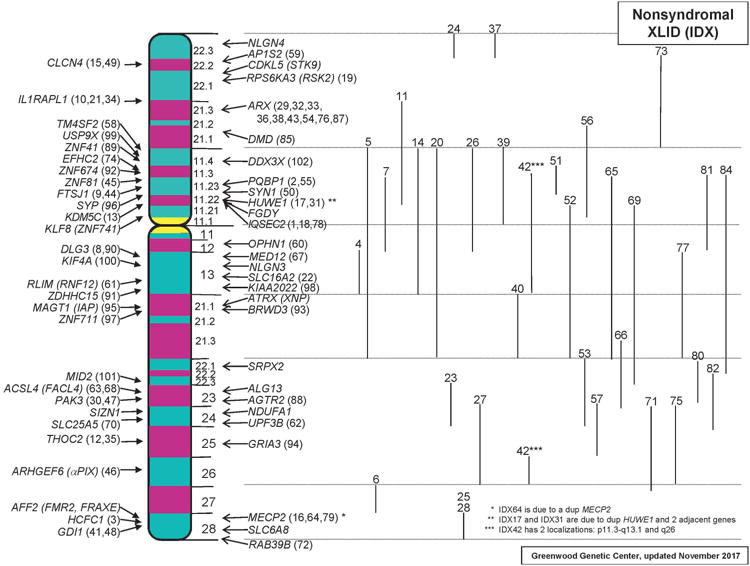
Families with nonsyndromal XLID which have received IDX numbers. Their total is currently 105. Linkage limits for IDX families for which the genes have not been identified are shown to the right. Color figure can be viewed in the online issue.
Duplication of Xlid Of Genes
Segmental duplications involving all protein-coding regions of XLID-associated genes on the p and q arms of the X chromosome have been reported. They vary in size from a few kilobases to many megabases and do not cross the centromere. Relative to phenotypic consequences of duplications, their informativeness is inversely proportional to the length and the number of genes encompassed within the duplicated segment. Among those duplications which appear to be clinically important, marked skewing of X-inactivation in females is typical.
Duplication of 137 of the 141 XLID genes have been identified in males and duplications of 4 XLID genes (KDM6A, ZNF674, RBM10, and KLF8) have been found only in females. Typically, in these cases, the entire XLID gene is duplicated, often with complete or partial duplication of adjacent genes. Duplication of KLF8, the XLID gene on the p arm closest to the centromere, has been found only in large duplications that involve the entire p arm (Tuck-Muller et al. 1993).
The phenotypic consequences of duplication of XLID genes are protean. In the first instance, the duplication may be associated with a phenotype identical or similar to that associated with a loss of function mutation or deletion of the gene. Such is the case for duplication of the PLP1 gene which results in Pelizaeus-Merzbacher syndrome. In the second instance, duplication of an XLID gene may result in a distinct phenotype but one quite different from loss of function mutations in the same gene. Duplication of MECP2 appears to be the most common duplication of this type but others include duplication of STAG2, HUWE1 and OCRL (van Esch et al. 2005; Friez et al. 2006, 2016; Froyen et al. 2007, 2008; Leroy et al. 2016). Intermediate between these phenotypic consequences are duplications of the ATRX gene which are associated with some manifestations of the Alpha-Thalassemia Intellectual Disability syndrome (short stature, genital anomalies, intellectual disability, hypotonia) but lack the typical facial features seen with loss of function variants in ATRX (Lugtenberg et al. 2009).
Duplications of certain XLID-associated genes (IKBKG, ARX) and certain X chromosome regions (Xp21.33, Xq21.33) do not appear associated with neurodevelopmental abnormalities although they may be associated with other somatic manifestations (van Asbeck et al. 2014; Popovici et al. 2014; Maurin et al. 2017).
Duplication of PLP1
Sequence variants in the coding or splice site regions of PLP1 are found in less than half of families with Pelizaeus-Merzbacher syndrome. More commonly, the condition is caused by duplications of the entire gene (Mimault et al. 1999; Regis et al. 2008). The severity of the signs and symptoms – nystagmus, initial hypotonia progressing to spastic paraplegia, ataxia, tremors, dystonia and other abnormal movements of the limbs, white matter dysmyelination, and basal ganglia deposits – do not appear to be related to the size of the duplications.
Duplication of HUWE1
Sequence variants in HUWE1 have been associated with Juberg-Marsidi syndrome, Brooks syndrome, Turner XLID-macrocephaly syndrome, and a family in which males had moderate ID and normal facial appearance (Friez et al 2016; Turner et al. 1994; Froyen et al. 2008). Duplication of HUWE1, usually associated with a HSD17B10 duplication, has been associated with ID of moderate severity, limited speech or dysarthria, facial dysmorphism (hypertelorism, upslanted palpebral fissures, synophrys, open mouth) and usually normal prenatal and postnatal growth measurements (Froyen et al 2008; Whibley et al. 2010; Orivoli et al. 2016). Two families (IDX17 and IDX31) had no distinctive dysmorphism and had normal growth. Seizures occurred in several patients, one individual had submucous cleft palate and two boys had first-degree hypospadias.
Duplication of STAG2
A number of duplications of Xq25, a gene poor region have been reported in males and females. STAG2, which encodes a subunit of the cohesin complex, is completely duplicated in these cases and adjacent genes (XIAP, THOC2, GRIA3, SH2D1A, and TENM1) are variably duplicated completely or partially (Kumar et al. 2015). The phenotype is dominated by ID of variable severity. Other common features include normal stature and head circumference, malar flatness, thick lip vermilion, prognathism, facial hypotonia and behavioral problems. Seizures and autism occur in a third or less. In contrast, individuals with deletions or sequencing variants of STAG2 have more severe developmental failure, growth impairment, microcephaly and midline malformations including holoprosencephaly or other CNS anomalies, facial clefting, and ocular colobomas.
Duplication of MECP2
Rett syndrome, due to deletions or sequence variants in MECP2 and characterized by a period of normal development in infancy followed by microcephaly and episodic but unrelenting course of neurological deterioration, loss of purposeful hand use, seizures, and spasticity, differs quite substantially from the manifestations of patients with MECP2 duplications. From birth, patients with duplications have hypotonia which later in childhood progresses to spasticity, absent or limited speech and ambulation, and recurrent respiratory infections which often requires tracheostomy and ventilator care. They have severe ID often complicated by seizures, and most die prior to age 25 years (Meins et al. 2005; van Esch et al. 2005; Friez et al. 2006, Vignoli et al. 2012; Lim et al. 2017). Although initially described as an X-linked recessive syndrome in males, more recent reports have confirmed occurrence in females, generally expressed as infantile hypotonia progressing to spasticity, severe ID and neurodevelopmental manifestations including autism spectrum disorder (Scott Schwoerer et al. 2014; Fieremans et al. 2014).
Duplication of OCRL
Lowe syndrome, caused by sequence variants or deletions of OCRL, is characterized by early onset cataracts, depressed muscle tone and reflexes, aminoaciduria, and ID. Duplication of OCRL has been described in 3 families, all in the company of duplications of adjacent genes (Møller et al. 2014; Schroer et al. 2012). They have had neurodevelopmental abnormalities, ID, autism, and seizures but in no other respect has the phenotype of Lowe syndrome been seen among these boys.
Concluding Remarks
In spite of the progress made over the past 30 years, much remains to be done before the XLID field may be considered fully defined. Perhaps as many as 80 additional genes could still be implicated in XLID. Discovery of pathogenic mechanisms probably represents the next biggest challenge, requiring intense efforts by several laboratories and proportionately large financial support. Is this justifiable? The question is legitimate, in view of the rarity of each individual condition, with the exception of the fragile X syndrome and a small number of other XLID syndromes, and of the limited resources available. On the other hand, if the effort conducted so far were meant to be, as in fact it is, preparatory to the ultimate goal, namely the cure, or at least the improvement, of XLID, it would not make sense to halt studies now. The current state of XLID knowledge has little to offer to further such an ultimate goal. As for most genetic diseases, few are the therapeutic successes. Enzyme infusions, stem cell and bone marrow transplantation, drugs and dietary management have been partially effective in a small number of conditions, e.g. ornithine transcarbamoylase deficiency, Hunter syndrome, adrenoleukodystrophy and phosphoglycerate kinase deficiency. A more widely applicable intervention, gene therapy, is on the horizon, with the recent development of delivery vectors that appear to reach target tissues and avoid safety concerns of the past (Hocquemuller et al. 2016), but at the present time it continues to be a hope for the future.
In the meanwhile, a number of outstanding issues continue to challenge clinicians, scientists and families alike. Clinicians must consider in the differential diagnosis a large number of syndromes when evaluating a male with ID and somatic, neurologic, or behavior manifestations and an almost equally large number of possibilities when evaluating a male with ID and no other manifestations. Family history does not help in isolated cases, but can be very informative in kindreds with multiple affected individuals. Physical examination continues to be the basic tool of clinical diagnosis, now aided by a number of genetic databases, including OMIM, POSSUM and Face2Gene. Recognition and use of the human phenotype ontology will bring consistency in describing the clinical manifestations of the various XLID disorders (Robinson et al. 2008).
For scientists, the unresolved issues are no less daunting. While acknowledging the disproportionate progress of identifying ID-associated alterations on the X-chromosome in comparison to ID-associated alterations on the autosomes, knowledge about the impact of these alterations on cellular organization and functions remains limited (Vissers et al. 2010, Dekker and Mirny 2016, Kochinke et al. 2016, Schwarzer et al. 2017). Although sequence variants of uncertain pathogenicity continue to distress laboratory and clinical geneticists, a more systematic approach to their resolution with functional studies including protein modeling, enzyme analysis, metabolomics, RNAseq, immunofluorescence studies of the protein in cell cultures and animal models is being formulated in many laboratories. Recently emerged technologies promise to identify alterations of the X-chromosome, especially involving the noncoding regions of the genome, for those XLID entities which have not been resolved with currently used technologies. These include whole genome sequencing, higher resolution microarray analysis, and structural rearrangement detection (Bionano Genomics).
As to the families, the main services which can be offered continue to be estimation of recurrence risks, preimplantation and prenatal diagnosis, ultrasonographic monitoring of at risk pregnancies, best possible neonatal treatment in cases requiring intensive care, habilitation therapies and advocacy for their needs. The development of treatments which significantly ameliorate the signs and symptoms of the XLID disorders has been disappointingly slow and current clinical trials have limited outcome goals. Abandonment of the notion that the brain is not accessible and development of methods to produce adequate amounts of native genes or CRISPR-cas engineered genes are only two of the major hurdles that gene therapy must overcome before reaching the bedside of patients with XLID disorders (Hocquemiller et al. 2016).
Most X-linked disorders are rare and thus access to multiple cases will be limited. Utilization of GeneMatcher to locate other cases with variants in XLID genes helps to connect researchers and clinicians which stimulate the pursuit of functional studies. In the end, it should not be forgotten that the alliance between physicians, scientists and families has spawned some of the greatest successes in discovering the causes of genetic diseases and delineating their phenotypes.
Supplementary Material
Footnotes
Conflicts of Interest: All authors declare that there are no conflicts of interest.
References
- Allan W, Herndon CN, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. American Journal of Mental Deficiency. 1944;48:325–334. [Google Scholar]
- Chiurazzi P, Schwartz CE, Gecz J, Neri G. XLMR genes: update 2007. European Journal of Human Genetics. 2008;16:422–434. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed] [Google Scholar]
- Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell. 2016;164:1110–1121. doi: 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieremans N, Bauters M, Belet S, Verbeeck J, Jansen AC, Seneca S, et al. Froyen G. De novo MECP2 duplications in two females with intellectual disability and unfavorable complete skewed X-inactivation. Human Genetics. 2014;133:1359–1367. doi: 10.1007/s00439-014-1469-6. [DOI] [PubMed] [Google Scholar]
- Friez MJ, Brooks SS, Stevenson RE, Field M, Basehore MJ, Ades LC, et al. Schwartz CE. HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: the results of an X-chromosome exome sequencing study. BMJ Open. 2016;6:e009537. doi: 10.1136/bmjopen-2015-009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, et al. Stevenson RE. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- Froyen G, Corbett M, Vandewalle J, Jarvela I, Lawrence O, Meldrum C, et al. Gecz J. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. American Journal of Human Genetics. 2008;82:432–443. doi: 10.1016/j.ajhg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froyen G, Van Esch H, Bauters M, Hollanders K, Frints SG, Vermeesch JR, et al. Marynen P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Human Mutation. 2007;28:1034–1042. doi: 10.1002/humu.20564. [DOI] [PubMed] [Google Scholar]
- Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Human Gene Therapy. 2016;27:478–496. doi: 10.1089/hum.2016.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, et al. Schenck A. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. American Journal of Human Genetics. 2016;98:149–164. doi: 10.1016/j.ajhg.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Corbett MA, Van Bon BW, Gardner A, Woenig JA, Jolly LA, et al. Gecz J. Increased STAG2 dosage defines a novel cohesinopathy with intellectual disability and behavioral problems. Human Molecular Genetics. 2015;24:7171–7181. doi: 10.1093/hmg/ddv414. [DOI] [PubMed] [Google Scholar]
- Lehrke R. A theory of X-linkage of major intellectual traits. Response to Dr. Anastasi and to the Drs. Nance and Engel. American Journal of Mental Deficiency. 1972;76:626–631. [PubMed] [Google Scholar]
- Leroy C, Jacquemont ML, Doray B, Lamblin D, Cormier-Daire V, Philippe A, et al. Malan V. Xq25 duplication: the crucial role of the STAG2 gene in this novel human cohesinopathy. Clinical Genetics. 2016;89:68–73. doi: 10.1111/cge.12567. [DOI] [PubMed] [Google Scholar]
- Lim Z, Downs J, Wong K, Ellaway C, Leonard H. Expanding the clinical picture of the MECP2 Duplication syndrome. Clinical Genetics. 2017;91:557–563. doi: 10.1111/cge.12814. [DOI] [PubMed] [Google Scholar]
- Lubs HA. A marker X chromosome. American Journal of Human Genetics. 1969;21:231–244. [PMC free article] [PubMed] [Google Scholar]
- Lugtenberg D, de Brouwer AP, Oudakker AR, Pfundt R, Hamel BC, van Bokhoven H, Bongers EM. Xq13.2q21.1 duplication encompassing the ATRX gene in a man with mental retardation, minor facial and genital anomalies, short stature and broad thorax. American Journal of Medical Genetics Part A. 2009;149A:760–766. doi: 10.1002/ajmg.a.32742. [DOI] [PubMed] [Google Scholar]
- Martin JP, Bell J. A Pedigree of Mental Defect Showing Sex-Linkage. Journal of neurology and psychiatry. 1943;6(3-4):154–157. doi: 10.1136/jnnp.6.3-4.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin ML, Arfeuille C, Sonigo P, Rondeau S, Vekemans M, Turleau C, et al. Malan V. Large Duplications Can Be Benign Copy Number Variants: A Case of a 3.6-Mb Xq21.33 Duplication. Cytogenetic and Genome Research. 2017;151:115–118. doi: 10.1159/000460278. [DOI] [PubMed] [Google Scholar]
- Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. Journal of Medical Genetics. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Roifman M, Braunschweig U, Yuen RK, Alexandrova R, Bates A, et al. Scherer SW. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nature Communications. 2015;6:8718. doi: 10.1038/ncomms9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimault C, Giraud G, Courtois V, Cailloux F, Boire JY, Dastugue B, Boespflug-Tanguy O. Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher Disease: duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The Clinical European Network on Brain Dysmyelinating Disease. American Journal of Human Genetics. 1999;65:360–369. doi: 10.1086/302483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller RS, Jensen LR, Maas SM, Filmus J, Capurro M, Hansen C, et al. Kleefstra T. X-linked congenital ptosis and associated intellectual disability, short stature, microcephaly, cleft palate, digital and genital abnormalities define novel Xq25q26 duplication syndrome. Human Genetics. 2014;133:625–638. doi: 10.1007/s00439-013-1403-3. [DOI] [PubMed] [Google Scholar]
- Orivoli S, Pavlidis E, Cantalupo G, Pezzella M, Zara F, Garavelli L, et al. Piccolo B. Xp11.22 Microduplications Including HUWE1: Case Report and Literature Review. Neuropediatrics. 2016;47:51–56. doi: 10.1055/s-0035-1566233. [DOI] [PubMed] [Google Scholar]
- Penrose LS. Genetic Linkage in Graded Human Characters. Annals of Eugenics. 1938;8:233–237. [Google Scholar]
- Piton A, Redin C, Mandel JL. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. American Journal of Human Genetics. 2013;93:368–383. doi: 10.1016/j.ajhg.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici C, Busa T, Boute O, Thuresson AC, Perret O, Sigaudy S, et al. Philip N. Whole ARX gene duplication is compatible with normal intellectual development. American Journal of Medical Genetics Part A. 2014;164A:2324–2327. doi: 10.1002/ajmg.a.36564. [DOI] [PubMed] [Google Scholar]
- Regis S, Biancheri R, Bertini E, Burlina A, Lualdi S, Bianco MG, et al. Filocamo M. Genotype-phenotype correlation in five Pelizaeus-Merzbacher disease patients with PLP1 gene duplications. Clinical Genetics. 2008;73:279–287. doi: 10.1111/j.1399-0004.2007.00961.x. [DOI] [PubMed] [Google Scholar]
- Renpenning H, Gerrard JW, Zaleski WA, Tabata T. Familial sex-linked mental retardation. Canadian Medical Association Journal. 1962;87:954–956. [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Kohler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. American Journal of Human Genetics. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roifman CM. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: a novel syndrome. Clinical Genetics. 1999;55:103–109. doi: 10.1034/j.1399-0004.1999.550206.x. [DOI] [PubMed] [Google Scholar]
- Schroer RJ, Beaudet AL, Shinawi M, Sahoo T, Patel A, Sun Q, et al. Stevenson RE. Duplication of OCRL and adjacent genes associated with autism but not Lowe syndrome. American Journal of Medical Genetics Part A. 2012;158A:2602–2605. doi: 10.1002/ajmg.a.35566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, et al. Spitz F. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551:51–56. doi: 10.1038/nature24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott Schwoerer J, Laffin J, Haun J, Raca G, Friez MJ, Giampietro PF. MECP2 duplication: possible cause of severe phenotype in females. American Journal of Medical Genetics Part A. 2014;164A:1029–1034. doi: 10.1002/ajmg.a.36380. [DOI] [PubMed] [Google Scholar]
- Tuck-Muller CM, Martinez JE, Batista DA, Kearns WG, Wertelecki W. Duplication of the short arm of the X chromosome in mother and daughter. Human Genetics. 1993;91:395–400. doi: 10.1007/BF00217366. [DOI] [PubMed] [Google Scholar]
- Turner G, Gedeon A, Mulley J. X-linked mental retardation with heterozygous expression and macrocephaly: pericentromeric gene localization. American Journal of Medical Genetics. 1994;51:575–580. doi: 10.1002/ajmg.1320510456. [DOI] [PubMed] [Google Scholar]
- Turner G, Partington MW. Genes for intelligence on the X chromosome. Journal of Medical Genetics. 1991;28:429. doi: 10.1136/jmg.28.6.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asbeck E, Ramalingam A, Dvorak C, Chen TJ, Morava E. Duplication at Xq28 involving IKBKG is associated with progressive macrocephaly, recurrent infections, ectodermal dysplasia, benign tumors, and neuropathy. Clinical Dysmorphology. 2014;23:77–82. doi: 10.1097/MCD.0000000000000038. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, et al. Froyen G. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. American Journal of Human Genetics. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignoli A, Borgatti R, Peron A, Zucca C, Ballarati L, Bonaglia C, Canevini MP. Electroclinical pattern in MECP2 duplication syndrome: eight new reported cases and review of literature. Epilepsia. 2012;53:1146–1155. doi: 10.1111/j.1528-1167.2012.03501.x. [DOI] [PubMed] [Google Scholar]
- Vissers LE, de Vries BB, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. Journal of Medical Genetics. 2010;47:289–297. doi: 10.1136/jmg.2009.072942. [DOI] [PubMed] [Google Scholar]
- Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK, et al. Raymond FL. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. American Journal of Human Genetics. 2010;87:173–188. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland I, Schanze D, Schanze I, Volleth M, Muschke P, Zenker M. A cryptic unbalanced translocation der(4)t(4;17)(p16.1;q25.3) identifies Wittwer syndrome as a variant of Wolf-Hirschhorn syndrome. American Journal of Medical Genetics Part A. 2014;164A:3213–3214. doi: 10.1002/ajmg.a.36765. [DOI] [PubMed] [Google Scholar]
- Wittwer B, Kircheisen R, Leutelt J, Orth U, Gal A. New X-linked mental retardation syndrome with the gene mapped tentatively in Xp22.3. American Journal of Medical Genetics. 1996;64:42–49. doi: 10.1002/(SICI)1096-8628(19960712)64:1<42::AID-AJMG6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Zollino M, Colosimo C, Zuffardi O, Rossi E, Tosolini A, Walsh CA, Neri G. Cryptic t(1;12)(q44;p13.3) translocation in a previously described syndrome with polymicrogyria, segregating as an apparently X-linked trait. American Journal of Medical Genetics Part A. 2003;117A:65–71. doi: 10.1002/ajmg.a.10068. [DOI] [PubMed] [Google Scholar]
- Zollino M, Mastroiacovo P, Zampino G, Mariotti P, Neri G. New XLMR syndrome with characteristic face, hypogenitalism, congenital hypotonia and pachygyria. American Journal of Medical Genetics. 1992;43:452–457. doi: 10.1002/ajmg.1320430168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.