

Abstract
Chronic exposure to cigarette smoke markedly increases the risk for lung cancer. Regulation of gene expression at the post-transcriptional level by miRNAs influences a variety of cancer-related interactomes. Yet, relatively little is known on the effects of long-term cigarette smoke exposure on miRNA expression and gene regulation. NCI-H292 (H292) is a cell line sensitive to cigarette smoke with mucoepidermoid characteristics in culture. We report, in this study, original observations on long-term (12 months) cigarette smoke effects in the H292 cell line, using microarray-based miRNA expression profiling, and stable isotopic labeling with amino acids in cell culture-based quantitative proteomic analysis. We identified 112 upregulated and 147 downregulated miRNAs (by twofold) in cigarette smoke-treated H292 cells. The liquid chromatography–tandem mass spectrometry analysis identified 3,959 proteins, of which, 303 proteins were overexpressed and 112 proteins downregulated (by twofold). We observed 39 miRNA target pairs (proven targets) that were differentially expressed in response to chronic cigarette smoke exposure. Gene ontology analysis of the target proteins revealed enrichment of proteins in biological processes driving metabolism, cell communication, and nucleic acid metabolism. Pathway analysis revealed the enrichment of phagosome maturation, antigen presentation pathway, nuclear factor erythroid 2-related factor 2-mediated oxidative stress response, and cholesterol biosynthesis pathways in cigarette smoke-exposed cells. In conclusion, this report makes an important contribution to knowledge on molecular changes in a lung cell line in response to long term cigarette smoke exposure. The findings might inform future strategies for drug target, biomarker and diagnostics innovation in lung cancer, and clinical oncology. These observations also call for further research on the extent to which continuing or stopping cigarette smoking in patients diagnosed with lung cancer translates into molecular and clinical outcomes.
Keywords: : lung cancer, biomarkers, diagnostics, miRNA profiling, cigarette smoke
Introduction
Smoking remains the major risk factor for cancer, resulting in more than 30% of global cancer-related deaths and about 90% of lung cancer deaths globally (Landi et al., 2008; Vial, 1986). Approximately, 85% of the lung cancers are non-small-cell lung cancer (NSCLC) (Molina et al., 2008). In the United States, the 5-year relative survival rate is 17% for patients diagnosed with NSCLC and <4% in metastasized cancers (Cetin et al., 2011). It is reported that quitting smoking after the diagnosis of early- and late-stage NSCLC lead to an increase in 5-year survival rates by 37% and 34%, respectively. In other words, nearly twice as many quitters would survive for 5 years compared to continuing smokers (Parsons et al., 2010). Apart from cancer, cigarette smoke is associated with chronic pulmonary diseases, including pulmonary edema, chronic bronchitis, and chronic obstructive pulmonary disease (COPD).
Patients with COPD have increased incidence of lung cancer (Tockman et al., 1987). Even the low-grade COPD without noticeable airflow obstruction is associated with elevated risk of lung cancer (de Torres et al., 2007). Cigarette smoking has also been found to induce a number of genetic and molecular changes in the respiratory tract, including cellular atypia (Franklin et al., 1997) and loss of heterozygosity (Powell et al., 1999; Wistuba et al., 1997), which can occur in cytologically normal airway epithelium (Thiberville et al., 1995; Wistuba et al., 1997). Yet, molecular alterations induced by smoking and the factors that impact the survival rates remain elusive.
Accumulation of both genetic and epigenetic alterations results in sequential morphological changes leading to tumorigenesis (Osada and Takahashi, 2002). Regulation of gene expression at the posttranscriptional level by miRNAs influences a variety of pathways, including developmental and oncogenic pathways (Calin and Croce, 2006; He et al., 2007; Hu et al., 2010; Johnson et al., 2005).
The crucial role of miRNAs in various cellular processes such as differentiation, cell growth, proliferation, and apoptosis has been established by several groups (Kent and Mendell, 2006; Xu et al., 2011). Dysregulation of miRNAs and their targets is associated with hallmarks of cancer, including cytoskeletal remodeling, proliferation, and migration (Huang et al., 2008; Kobayashi et al., 2008; Lee et al., 2005). Distinct miRNA expression profiles have been observed for different cancers (Barker et al., 2009). miRNAs ultimately affect protein expression either by mediating degradation of mRNA or by inhibiting translation (Jacobsen et al., 2013; Yang et al., 2009). Since dysregulation of miRNAs can affect multiple targets leading to diverse functional consequences, it is imperative to study the targets of the dysregulated miRNAs in any disease.
miRNA expression profile of lung cancer has been previously characterized using microarrays (Yanaihara et al., 2006) as well as next-generation sequencing technologies (Ma et al., 2014; Wang et al., 2015). Cigarette smoke has been shown to alter the expression of several genes in various biological models, including mice (Izzotti et al., 2004), rats (Izzotti et al., 2005), and airway epithelial cells from smokers (Spira et al., 2004). In addition, miRNA dysregulation has also been reported in bronchial epithelium of smokers (Schembri et al., 2009) and animal models exposed to cigarette smoke (Izzotti et al., 2009). Recently, lung cancer cells exposed to cigarette smoke revealed the dysregulation of several genes in response to particulate matter and vapor phase components of cigarette smoke (Sekine et al., 2015).
One of the genes that showed enhanced expression in response to cigarette smoke includes HMOX1, an antioxidant gene regulated by the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2). In addition, Nrf2 signaling and DNA damage response pathways were found to be enriched in response to cigarette smoke. Integration of miRNA expression and proteomic data in cigarette smoke-exposed lung cells could lead to the identification of potential signaling mechanisms affected by cigarette smoke. NCI-H292 (H292) is a bronchial epithelial cell line derived from a mucoepidermoid carcinoma established from a nonsmoker. It is one of the widely used cell lines used to study the effect of cigarette smoke as it is sensitive to cigarette smoke and retain their mucoepidermoid characteristics in culture (Newland and Richter, 2008). Several studies have reported the effect of cigarette smoke on NCI-H292 cells to be similar to that of primary human bronchial epithelial cells and the airway epithelium in vivo (Baginski et al., 2006; Luppi et al., 2005; Newland and Richter, 2008; Shao et al., 2004; Thorne et al., 2009).
In vitro cellular models where human lung cells have been exposed to varied doses of cigarette smoke have served as useful tool to interpret disease progression and understand the molecular mechanisms associated with them. Even though these studies have identified few molecular mechanisms by which cigarette smoke may exert its effects, there has been no study on the chronic effect of cigarette smoke in lung cells. It is known that the carcinogenic effect of cigarette smoke in lung cancer is through chronic exposure and not by acute exposure.
In this study, we investigated chronic effects of cigarette smoke on miRNA expression as well as proteomic changes in H292 cells chronically (12 months) exposed to cigarette smoke using microarray and mass spectrometry (MS) approaches.
Materials and Methods
Cell culture and stable isotopic labeling with amino acids in cell culture labeling
H292 cells were obtained from the American Type Culture Collection and grown in Dulbecco's modified Eagle's medium (DMEM) medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), and 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified incubator at 37°C with 5% CO2. These cells were and adapted to stable isotopic labeling with amino acids in cell culture (SILAC) media as previously described (Harsha et al., 2008). Briefly, the cells were maintained in DMEM medium without lysine and arginine supplemented with 10% FBS, 2 mM l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin, 50 mg/l-arginine-13C6 monohydrochloride, and 100 mg/L lysine-13C6 monohydrochloride (heavy) (Cambridge Isotope Laboratories).
To study the chronic effect of cigarette smoke condensate (CSC; Murty Pharmaceuticals, Inc.), H292 cells were subjected to chronic treatment with 0.1% CSC for 12 months (Chang et al., 2011). H292 cells were grown in a smoke-dedicated incubator. Parental cells were grown in regular CO2 incubator. Cells that were grown and passaged in the smoke-dedicated incubator exposed to CSC were labeled as H292-smoke or termed as cigarette smoke-treated cells. Henceforth, the H292 cells exposed to CSC will be referred to as H292-smoke and untreated cells as H292-parental.
Trypsin digestion and basic pH reverse phase liquid chromatography
The CSC-treated and CSC-untreated H292 cells were washed with cold phosphate-buffered saline and lysed in lysis buffer (20 mM HEPES, pH 8.0, 9 M urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerophosphate), sonicated, and centrifuged at 16,000 g at 15°C for 20 min. The protein concentration was determined using the BCA assay (Pierce). Equal amounts of protein from CSC-treated and CSC-untreated H292 cells were reduced with 5 mM dithiothreitol and incubated at 60°C for 20 min. These were further alkylated using 10 mM iodoacetamide for 10 min at room temperature in the dark. The samples were diluted such that urea was <2 M with 20 mM HEPES, pH 8.0.
The samples were subjected to digestion with TPCK-treated trypsin (Worthington Biochemical Corporation) overnight at room temperature. The peptide mixture was acidified using 1% trifluoroacetic acid and desalted using C18 Sep-Pak cartridge (Waters; Cat#WAT051910). The extracted peptides were lyophilized and subjected to basic pH reverse phase chromatography (bRPLC). The samples were reconstituted in bRPLC solvent A [7mM triethyl ammonium bicarbonate (TEABC), pH 9] and separated on a XBridge BEH C18 column (Waters) using a linear increase in gradient from 5% to 100% of 7 mM TEABC with 90% acetonitrile (pH 9) over 30 min. A total of 96 fractions were initially collected and later concatenated to 24 fractions and dried using SpeedVac.
bRPLC fractionation and liquid chromatography–tandem mass spectrometry analysis
The samples were analyzed on an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) interfaced with an Easy-nLC II nanoflow liquid chromatography system (Thermo Scientific). The peptides were reconstituted in 0.1% formic acid and loaded onto a 75 μm × 2 cm Magic C18 AQ (Michrom Bioresources, Inc.), 5 μm, 120 Å trap column. Peptides were resolved on a 75 μm × 20 cm analytical column at a flow rate of 350 nL/min using a linear gradient of 8–30% solvent B (0.1% formic acid in 95% acetonitrile) over 75 min. The total run time, including sample loading and column reconditioning, was 95 min. Data-dependent acquisition was carried out with full scans in 350–1800 m/z range with a mass resolution of 60,000 at 400 m/z. Fifteen most intense precursor ions from a survey scan were selected for MS/MS, fragmented using higher energy collisional dissociation (HCD) with 39% normalized collision energy, and detected at a mass resolution of 15,000 at 400 m/z. Dynamic exclusion was set for 40 sec with a 7 ppm mass window. Internal calibration was carried out using lock mass option (m/z 445.1200025) from ambient air.
Proteomics data analysis
MS-derived data were searched against human RefSeq database (version 65 containing 34,454 entries along with common contaminants) using SequestHT and Mascot (version 2.2) search algorithms through Proteome Discoverer (Thermo Scientific; version 1.4.1.14). The search parameters included carbamidomethylation of cysteine as a fixed modification and N-terminal acetylation, oxidation of methionine, and SILAC labeling (13C6) at lysine and arginine (6.02013 Da) as variable modifications. MS/MS spectra were searched with a precursor mass tolerance of 20 ppm and a fragment mass tolerance of 0.1 Da. Trypsin was specified as the protease and a maximum of two missed cleavages were allowed. The data were searched against decoy database and a 1% false discovery rate was set at the peptide level. Proteins with ratios ≥2-fold were considered to be upregulated where as those ≤0.5-fold were considered to be downregulated.
miRNA microarray analysis
The miRNA microarray profiling was carried out using Affymetrix GeneChip miRNA Arrays (Affymetrix) as per the manufacturer's instructions. Total RNA was extracted from H292 cells and quality assessment of the samples was done using the TaqMan assay. The miRNA targets were biotinylated using an Asuragen developed direct labeling procedure. Once labeled, the miRNA targets were hybridized overnight onto the microarrays following which the arrays were washed and stained using Streptavidin-Phycoerythrin. Arrays were processed using the gene chip operating software (GCOS). Data extraction was from the Affymetrix miRNA QC tool using robust multi-array average (RMA) background correction and median polish summarization. Candidate miRNAs that were increased or decreased in expression with fold changes ≥2-fold following treatment with CSC were considered to be differentially regulated.
Bioinformatics analysis
The identified proteins were sorted with a gene ontology-based functional classification by Human Protein Reference Database (HPRD1) (Keshava Prasad et al., 2009a, 2009b). Molecule class, molecular function, biological process, subcellular localization, tissue expression, and domain information were fetched from HPRD for all proteins. FunRich tool was used for functional enrichment of the 415 dysregulated proteins using the HPRD annotation as backend database (Pathan et al., 2015). The potential targets of differentially expressed miRNAs were predicted using TargetScan algorithm2 (Lewis et al., 2005). These were then compared with the proteomic data to infer the expression of target proteins under chronic smoke exposure. Pathway analysis of the differentially expressed proteins was carried out using QIAGEN's Ingenuity Pathway Analysis (IPA®; QIAGEN Redwood City3).
Identification of dysregulated miRNAs from The Cancer Genome Atlas
miRNA expression of NSCLC (squamous cell carcinoma and adenocarcinoma) and its corresponding clinical information for Illumina GA and HiSeq platform were obtained from The Cancer Genome Atlas (TCGA4). Data used were obtained from the study entitled “miRNA Analysis of TCGA LUSC and LUAD Samples using Illumina.” Patient barcodes were segregated into “smokers” and “nonsmokers” based on the clinical information provided. Cases where no information was available were not used.
Based on the clinical information provided by TCGA, the expression profile of miRNA for each case was obtained using both Illumina platforms. In addition, an in-house PERL script was used to create a matrix of the miRNA expression of each patient across each miRNA for tumor smoker versus nonsmoker from the matrix; a median value of the expression for each miRNA was calculated across patient samples. Differential expression of miRNA is further calculated by taking the ratios of the expression levels of smoker versus nonsmoker.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium5 (Vizcaino et al., 2014) by the PRIDE partner repository with the dataset identifier PXD003086.
Results
Dysregulated miRNAs
We employed a microarray-based approach to carry out miRNA expression analysis of H292 cells chronically treated with cigarette smoke. A schematic representation of the work flow is presented in Figure 1 (left panel). Our analysis led to the identification of 834 miRNAs among which 259 were dysregulated (≥2-fold) in the lung cells treated with cigarette smoke. A total of 112 miRNAs were upregulated and 147 were downregulated in the cigarette smoke-treated cells. A partial list of dysregulated miRNAs is provided in Table 1. A complete list of miRNAs along with corresponding fold changes in smoke-treated cells is provided in Supplementary Table S1.
FIG. 1.

Experimental workflow employed to profile miRNA expression in smoke-exposed H292 cells. RNA was isolated from H292 cells, chronically treated with CSC. Samples were processed further depending on the integrity of the isolated RNA and labeling was carried out. The samples were hybridized on miRNA microarrays. Data were processed further to identify dysregulated miRNAs. Experimental workflow employed to analyze the proteome of smoke-exposed H292 cells. H292 cells were chronically treated with CSC. The cells were adapted to stable isotopic labeling with amino acids in cell culture media. The cells treated with CSC were grown in a medium containing normal arginine and lysine. The untreated cells were grown in a medium containing heavy arginine and lysine. Equal amounts of proteins from both treated and untreated samples were digested. The peptides were fractionated by strong cation exchange chromatography. The fractionated samples were subjected to LC-MS/MS analysis on a LTQ-Orbitrap Velos mass spectrometry. The data were searched and quantitated using the Sequest and Mascot search algorithms. CSC, cigarette smoke condensate; LC-MS/MS, liquid chromatography–tandem mass spectrometry.
Table 1.
A Partial List of miRNAs Differentially Regulated in H292 Cells Exposed to Cigarette Smoke
| miRNA | Fold change (log2) | Significance |
|---|---|---|
| miR-124-3p | 3.5-Fold upregulated | Tumor-suppressive miRNA in several cancers (Chen et al., 2015; Hunt et al., 2011; Peng et al., 2014) |
| miR-374a-5p | 2.7-Fold downregulated | Promotes cell proliferation, migration, and invasion (Xu et al., 2015) |
| miR-204-5p | 2-Fold downregulated | Inhibits proliferation and invasion (Xia et al., 2014) |
| miR-155-5p | 5-Fold downregulated | Downregulation promotes cell cycle arrest and apoptosis (Zhu et al., 2016) |
| miR-21-5p | 3.5-Fold upregulated | Known to be oncogenic miRNA with important roles in cell proliferation, apoptosis, and invasion (Yamamichi et al., 2009) |
Proteins differentially expressed
To understand how the dysregulated miRNAs affect the expression of their targets, we employed SILAC-based proteomic strategy. Liquid chromatography–tandem mass spectrometry-based quantitative proteomics of H292 parental and H292 smoke cells led to the identification of 3959 proteins. The proteomic workflow used for this study is outlined in Figure 1 (right panel). Among the differentially expressed proteins, 302 proteins were overexpressed (≥2-fold) and 112 proteins were found to be downregulated (≤0.5-fold) in the cigarette smoke-treated H292 cells. A partial list of proteins is provided in Table 2. The complete list of identified proteins and peptides is provided in Supplementary Table S2. MS/MS spectra of a subset of these candidates are provided in Figure 2 and Supplementary Figure S1.
Table 2.
A Partial List of Proteins Differentially Expressed in H292 Cells Exposed to Cigarette Smoke
| Protein name | Gene symbol | Fold change | Function |
|---|---|---|---|
| S100 calcium binding protein A9 | S100A9 | 89.6 | Involved in the regulation of multiple cellular processes, including cell cycle progression and differentiation |
| Cytochrome P450 1A1 | CYP1A1 | 36.6 | Involved in drug metabolism, synthesis of cholesterol, steroids, and other lipids |
| Lipocalin 2 | LCN2 | 29.3 | Plays a role in innate immunity by limiting bacterial growth as a result of sequestering iron-containing siderophores |
| Sterile alpha motif domain containing 9 like | SAMD9L | 22.4 | Acts as a tumor suppressor and plays a key role in cell proliferation and innate immune response to viral infection |
| Ectodysplasin-A | EDA | 17.6 | Acts as a homotrimer and involved in cell–cell signaling during the development of ectodermal organs |
FIG. 2.
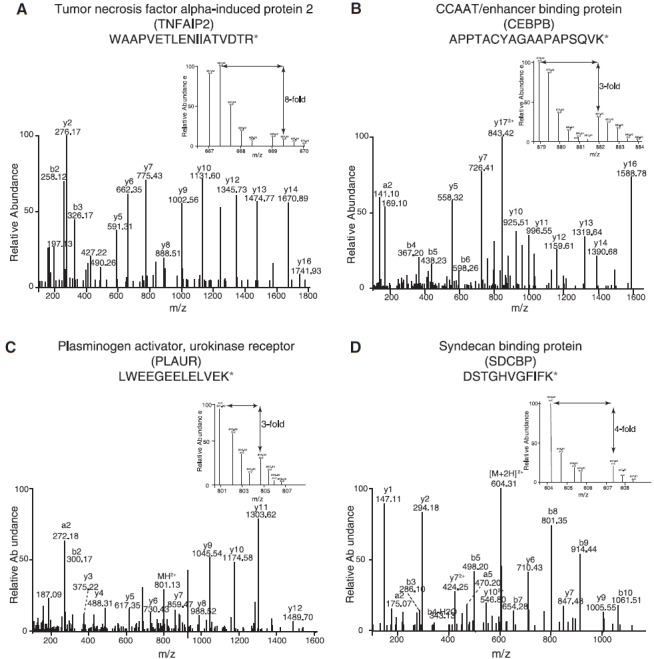
Representative MS/MS spectra depicting the overexpression of (A) TNFAIP2 (eightfold), (B) CEBPB (threefold), (C) PLAUR (threefold) and (D) SDCBP (fourfold). TNFAIP2, tumor necrosis factor alpha-induced protein 2; CEBPB, CCAAT/enhancer binding protein; PLAUR, plasminogen activator, urokinase receptor; SDCBP, syndecan binding protein.
Correlation of miRNA with protein expression
TargetScan (Friedman et al., 2009) and mirTarBase (Chou et al., 2016) were used to determine the targets of dysregulated miRNAs identified for H292 cells treated with cigarette smoke. Only experimentally proven targets were considered for further downstream analysis. As mentioned earlier, 259 miRNAs were dysregulated in the smoke-exposed H292 cells. Among the dysregulated miRNAs, 202/259 miRNAs were dysregulated and identified in our data set only (Fig. 3). Of the 202 miRNAs that were dysregulated and unique to our data set, 132 were present in the TargetScan and mirTarBase databases and were utilized for proven target prediction (Supplementary Table S3). This led to the identification of 6853 targets for H292 data sets. 2431/6853 were identified in our proteomic data.
FIG. 3.

Bioinformatics workflow. Pipeline depicting the analysis of dysregulated miRNA in H292 smoke-exposed cells and non-small-cell lung cancer The Cancer Genome Atlas data sets.
Supplementary Table S3 lists the dysregulated miRNAs in the smoke-exposed H292 cells and their targets. The overall significant directional changes in miRNA and protein expression were found for 39 miRNAs (Fig. 4), which were identified in our data set. A partial list of miRNAs and their target proteins with directional changes is provided in Table 3. A complete list of miRNAs and their targets showing reciprocal expression is provided as Supplementary Table S4A and B.
FIG. 4.

Bubbleplot representing miRNAs and their protein targets, which shows reciprocal expression in H292 smoke-exposed cells. Significant dysregulation in inverse direction among miRNAs and their respective target proteins was observed in 39 miRNAs. Size of the bubble represents fold change.
Table 3.
A Partial List of Dysregulated miRNAs and Their Potential Targets in Cigarette Smoke-Exposed H292 Cells
| miRNA | Fold change (log2) | Target | Ratio |
|---|---|---|---|
| miR-155-5p | 5-Fold downregulated | CEBPB | 2.25 |
| miR-196a-5p | 1.5-Fold downregulated | S100A9 | 89.25 |
| miR-222-3p | 0.8-Fold downregulated | PPP2R2A | 2 |
| HMGA1 | 1.8 | ||
| SOD2 | 9 | ||
| miR-21-5p | 3.5-Fold upregulated | PDCD4 | 0.4 |
Bioinformatics analysis
The identified proteins were classified based on their molecule class, molecular function, biological process, subcellular localization, and tissue expression using HPRD (Keshava Prasad et al., 2009a, 2009b)6. In addition, information pertaining to the presence of signal peptides and domains in identified proteins was also obtained from HPRD. Pathway analysis of proteins that were differentially expressed in response to cigarette smoke exposure was carried out using IPA. Pathway analysis of upregulated proteins led to the identification of several enriched pathways, including phagosome maturation, antigen presentation pathway, NRF2-mediated oxidative stress response, and cholesterol biosynthesis pathways. Similar analysis using downregulated proteins identified the enrichment of retionic acid receptor (RAR) activation pathway.
In addition, we carried out the analysis of differentially expressed proteins using FunRich tool. Subcellular localization-based classification revealed that 47% of the identified proteins localized to cytoplasm, 30% of the identified proteins localized to the lysosome, and 21% of the proteins were present in the endoplasmic reticulum. Further classification based on the biological process showed an enrichment of proteins involved in metabolism (20%), energy pathways (19%), and transport (11%) processes. The gene ontology-based functional annotation of identified proteins is depicted as Figure 5, which shows significant enrichment of lysosomal proteins (30%) in the smoke-exposed lung cells. Other components enriched under gene ontology included endoplasmic reticulum (20%), exosomes (27%), and mitochondria (16%).
FIG. 5.
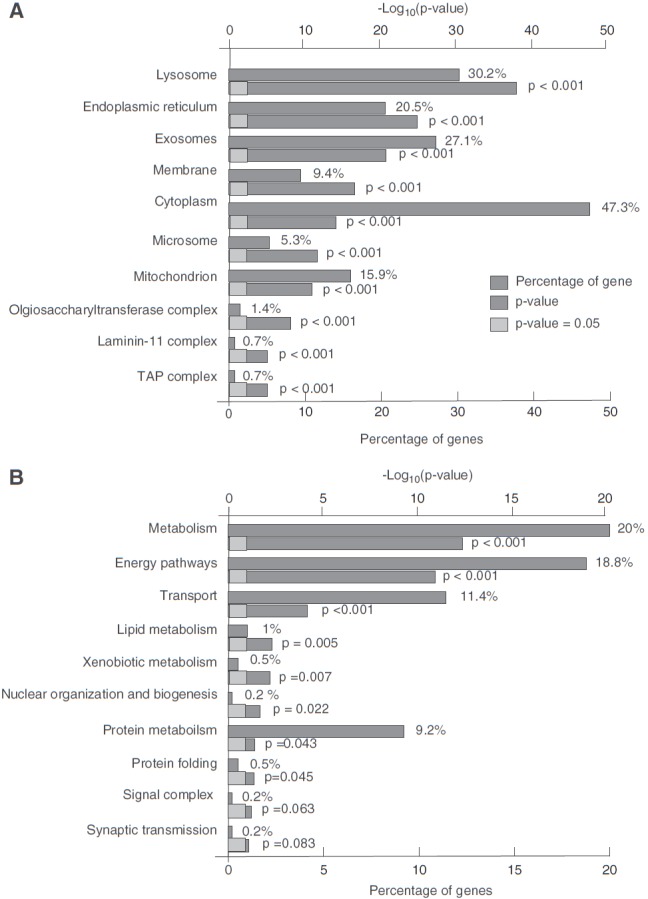
Gene ontology-based functional annotation of proteins identified in H292 cell lines. (A) Classification of proteins based on subcellular localization. (B) Classification of proteins based on biological process.
Comparison to the TCGA datasets of dysregulated miRNAs in NSCLC
Apart from studying the miRNA dysregulation in H292 cells in response to cigarette smoke, we also compared our data set with NSCLC miRNA data set of TCGA. We fetched miRNA-seq raw data from TCGA repository for NSCLC. miRNA expression was calculated in smokers compared to nonsmokers. In total, we identified 933 miRNAs (Fig. 3). We performed an F-test between smokers and nonsmokers using p-value to select all the potential miRNAs expressed. We found that 606 miRNAs had a significant p-value (≤0.05) and 263/606 miRNAs were dysregulated in TCGA data sets (Supplementary Fig. S2A, B). We then compared the miRNAs dysregulated in TCGA dataset with our miRNA data pertaining to the H292 cells treated chronically with cigarette smoke.
Fifty-seven miRNAs were dysregulated and common between H292 (smoke) and TCGA data set (Fig. 3 and Supplementary Fig. S2B); among these, 26 miRNAs showed concordance expression between the two data sets (Supplementary Fig. S3A, B). Supplementary Table S5A and B lists the dysregulated miRNAs in NSCLC (adenocarcinoma and squamous cell carcinoma, respectively) in TCGA data sets with their targets.
Discussion
miRNAs are one of the major regulators of gene expression. They are known to regulate several key cellular processes, including proliferation, differentiation, and apoptosis (Bueno and Malumbres, 2011). Dysregulation of miRNAs have been widely associated with the development of various cancers, including lung cancer (Guz et al., 2014; Hayes et al., 2014). Cigarette smoke has been identified as one of the risk factors associated with the development of lung cancer. Several research groups have shown in both human and rat cell models that exposure of cigarette smoke resulted in the dysregulation of miRNAs (Izzotti et al., 2009, 2010; Schembri et al., 2009; Vucic et al., 2014). However, the exact mechanisms by which cigarette smoke induces the dysregulation of miRNAs are not fully understood.
Against the above overarching scientific context, we carried out microarray-based miRNA expression profiling of cigarette smoke-treated H292 cells. In addition, we also carried out a SILAC-based proteomic analysis to identify differentially expressed proteins in H292 cells after exposure to cigarette smoke. The identification of reciprocal expression of miRNAs and their targets in H292 cells exposed to cigarette smoke provides a unique molecular basis to further explore the biological role of miRNAs in NSCLC. Integrative analyses identified both predicted and experimentally validated miRNA-target pairs, but only experimentally validated targets were considered.
We identified several miRNAs previously reported to be dysregulated in lung cancer corroborating our approach. miR-124 has been reported to be dysregulated in both highly invasive subcell lines and NSCLC (Sun et al., 2015). miR-124 (miR-124-3p) was found to be upregulated in our data set by about sevenfold in response to cigarette smoke. We observed an overexpression of miR-21 (hsa-miR-21-3p) and miR-192 (hsa-miR-192-5p) in the H292 smoke cells. Expression levels of serum miR-21 and miR-192 were previously reported to be significantly higher in stage I NSCLC patients when compared to controls (Qi et al., 2014b).
The reduced expression of miR-125a (hsa-miR-125a-3p; identified as downregulated in our study) has been shown to be strongly associated with NSCLC dedifferentiation in tissue and serum samples (Zhu et al., 2014). It has also been reported to be a key miRNA responsible for the activation of p53 and induction of apoptosis in human lung cancer cells (Jiang et al., 2013). Low expression of miR-374a (hsa-miR-374a-5p; downregulated by 5.4-fold in this study) has been associated with poor patient survival in early-stage NSCLC (Vosa et al., 2011). It has also been known to promote metastasis in breast cancer cells through the Wnt/β-catenin signaling cascade (Cai et al., 2013). Xia et al. (2014) have reported involvement of miR-204 (hsa-miR-204-5p) in NSCLC development as its expression was decreased in tumor samples.
Furthermore, overexpression of miR-204 led to reduced cell proliferation and invasion, increased expression of E-cadherin, and decreased expression of N-cadherin and Vimentin (Xia et al., 2014). In our data set, miR-204 (hsa-miR-204-5p) was found to be downregulated by about 4-fold in response to cigarette smoke. Similarly, miR-101, miR-125a, miR-140, miR-145, and miR-27b, which were found to be downregulated in the H292 cells chronically treated with cigarette smoke, have been reported to be downregulated in lung adenocarcinoma (Yanaihara et al., 2006).
Apart from miRNAs reported to be dysregulated in NSCLC, we also identified several miRNAs reported to be downregulated in other cancers, but identified and reported for first time in NSCLC and cigarette smoke. These include miR-214, miR-215, miR-342, miR-34b, miR-378, and miR-424 in colon cancer (Gaur et al., 2007; Schetter et al., 2008), miR-194 in breast cancer (Iorio et al., 2005), miR-195 in hepatocellular carcinoma (Murakami et al., 2006), miR-152 and miR-155 in pancreatic cancer (Volinia et al., 2006), and miR-32 in uterine leiomyoma (Wang et al., 2007). A total of four differentially expressed miRNAs (miR-146, miR-27, miR-877, and miR-186) have been known to be involved in metastatic melanoma (Qi et al., 2014a). We observed a downregulation of miR-27 and miR-877 in H292 cells treated with cigarette smoke.
Consistent with the cancer biology literature, many miRNAs identified to be dysregulated in our study showed reciprocal relationship with their targets. PPP2R2A has been shown to inhibit Akt phosphorylation in a cohort of NSCLC tumors and is a target of miR-222. HMGA1 is also reported to be regulated by miR-222 (Zhang et al., 2011). We found miR-222 to be downregulated by 0.5-fold and its targets PPP2R2A (twofold) and HMGA1 (1.8-fold) were overexpressed in response to cigarette smoke.
Liu et al. (2009) showed that ectopic transfection of miR-222 reduced the expression of MMP1 and SOD2 in oral tongue squamous cell carcinoma cell lines. Our data indicate a nine and sevenfold increased expression of MMP1 and SOD2, respectively, in response to cigarette smoke. miR-204 (hsa-miR-204-5p; downregulated by fourfold), which acts as a tumor suppressor, has been reported to be downregulated in renal clear cell carcinoma. It has been reported to be regulated by von Hippel-Lindau tumor-suppressor gene through its target, MAP1LC3B (Mikhaylova et al., 2012).
miR-145 is reported to inhibit cell proliferation of human lung adenocarcinoma by negative regulation of EGFR and NUDT1 (Cho et al., 2011). We found miR-145 (hsa-miR-145-3p) downregulated by 4.6-fold and also identified its target EGFR downregulated by 0.4-fold in response to cigarette smoke. In many cancers, including glioblastoma (Chen et al., 2008a), oral squamous cell carcinoma (Reis et al., 2010), and cholangiocarcinoma (Liu et al., 2012), programmed cell death 4 (PDCD4), a tumor-suppressor gene, was shown as a target of miR-21, and its expression is downregulated by miR-21. PDCD4 was downregulated in our study, while miR-21 (hsa-miR-21-5p) was upregulated. miR-183 (hsa-miR-183-5p; upregulated by 4.6-fold) has been shown to inhibit apoptosis and promote proliferation and invasion of gastric cancer cells by targeting PDCD4 (Gu et al., 2014).
Similarly, miR-34a (hsa-miR-34a-5p), which has been previously shown to suppress cell proliferation and metastasis and targets CD44 in human renal carcinoma cells (Yu et al., 2014), was found to be upregulated in our data set. Interestingly, contrary to the existing literature, we also identified several miRNAs that were downregulated in our data set and showed reciprocal correlation with their targets. These include miR-155 (hsa-miR-155-5p), which was downregulated by about 10-fold, and its target CEBPB is upregulated by 2.25-fold in our data. Salemi et al. (2015) reported miR-155 to be upregulated and its target CEBPB as downregulated in acute myeloid leukemia.
Similarly, miR-196a was shown to be upregulated and its target S100A9 was reported as downregulated in Barrett's metaplasia-dysplasia-invasive adenocarcinoma (Maru et al., 2009), while our data revealed downregulation of miR-196a (hsa-miR-196a-5p) by about 1.5-fold and its target S100A9 upregulated by 89.25-fold. Such observations indicate that the expression of miRNAs may be either tissue specific or cigarette smoke has its own unique mechanism of regulation of miRNAs and their corresponding targets, which needs further investigation and is beyond the scope of this article.
Apart from dysregulation of miRNAs, we also focused on the regulation of proteins, which are the key targets of miRNAs. Pathway analysis using IPA indicated an enrichment of pathways, including NRF2-mediated oxidative stress response, cholesterol biosynthesis, antigen presentation, and phagosome maturation pathways. In addition, our gene ontology data indicated a significant enrichment of lysosomal proteins in the smoke-treated cells. Nrf2, a transcription factor, has been known to regulate redox homeostasis to protect the cell from oxidative damage (Fujihara et al., 2008). Chronic cigarette smoke exposure has been previously observed to induce oxidative damage in murine retinal pigmented epithelial cells (Cano et al., 2010).
In addition, Nrf2 has been known to increase sulfiredoxin-1 (Srx1) expression during cigarette smoke exposure-induced oxidative stress in murine lung cells (Singh et al., 2009), which in turn has been reported to protect cells from oxidative injury in the lungs. Nrf2-deficient mice have been found to be highly susceptible to cigarette smoke-induced emphysema (Iizuka et al., 2005). Furthermore, cigarette smoking among heavy smokers has been observed to repress the Nrf2 pathway expression in peripheral mononuclear cells through the recruitment of NFκB (Garbin et al., 2009).
Our analysis found the enrichment of cholesterol biosynthesis pathway in response to cigarette smoke exposure. Cigarette smoke exposure has been reported to impair plasma lipid profile as well as reverse cholesterol transport in murine models (Zong et al., 2015). It was found that cigarette smoke significantly decreased plasma high-density lipoprotein (HDL) cholesterol levels, while increasing plasma low-density lipoprotein (LDL) cholesterol levels in smokers (Meenakshisundaram et al., 2010). A study investigating the lipid profiles among 274 smokers found a significant rise in LDL and a fall in HDL levels. Interestingly, another study has indicated that the production of progesterone, a cholesterol derivative, was inhibited by cigarette smoke alkaloids (Gocze and Freeman, 2000). Smoking cessation has been found to improve total HDL levels (Gepner et al., 2011).
We observed enrichment of the antigen presentation pathway in response to cigarette smoke. Literature evidence indicates that cigarette smoke decreases immunoproteasome function of immune cells isolated from COPD patients and impairs immunoproteasome-specific MHC I antigen presentation (Kammerl et al., 2016). In addition, long-term cigarette smoke exposure has been shown to suppress the development of functional dendritic cells, which are antigen-presenting cells, suggesting that cigarette smoke exposure may lead to impaired immune response to infection (Givi et al., 2015).
Apart from the pathways enriched, we also observed an enrichment of lysosomal proteins in the smoke-exposed lung cells. Cancer cells are known to demonstrate alterations of the lysosomal compartment that have pro-oncogenic effects, including tumor growth, migration, invasion, and angiogenesis (Mohamed and Sloane, 2006). Furthermore, literature evidence indicates increased numbers of autophagosomes in tissues from patients with COPD and in lung epithelial cells exposed to cigarette smoke extract (Chen et al., 2008b; Kim et al., 2008). Although there is an increase in the number of autophagosomes in alveolar macrophages of smokers, there is defect or decrease in the autophagy function. This defect in autophagy function in smokers leads to mitochondrial dysfunction, increased accumulation of cigarette smoke particulates, and decreased clearance of pathogens (Monick et al., 2010).
Among the lysosomal proteins identified in our study are acid ceramidase (ASAH1) and LAMP1 and 2. ASAH1 has been reported to be associated with the cigarette smoke-mediated increase in sphingosine (Sph) synthesis (Titz et al., 2016). Furthermore, acid ceramidase (ASAH1) has been proposed as a chemotherapeutic target in NSCLC to overcome resistance to the antitumoral effect of choline kinase α inhibition as it synergistically sensitizes lung cancer cells (Ramirez de Molina et al., 2012). In addition to acid ceramidase, we also found an increased expression and enrichment of lysosome-associated membrane glycoproteins (LAMP1 and LAMP2). LAMP1 plays a decisive role in lung cancer metastasis by promoting interactions with organ extracellular matrix and basement membrane (Agarwal et al., 2015).
Furthermore, LAMP1 has been reported to be overexpressed in A549 lung cancer cells (Su et al., 2016) and known as a target of miR-373, which is downregulated in lung cancer tissues and cell lines (Seol et al., 2014). Similarly, higher expression of LAMP2 has been reported in bronchoalveolar lavage of lung cancer cells (Li et al., 2013) and its higher expression was related to high histology grade in lung cancer patients (Giatromanolaki et al., 2015). miR-487b-5p is known to regulate temozolomide resistance in lung cancer cells through LAMP2-medicated autophagy (Bao et al., 2016).
Other lysosomal proteins identified in our data include gamma-glutamyl hydrolase, cathepsin, and calnexin. Gamma-glutamyl hydrolase expression is correlated with poor prognosis in lung and breast cancers (He et al., 2004; Shubbar et al., 2013). Similarly, cathepsin B has been proposed as a potential prognostic and therapeutic marker for lung squamous cell carcinoma (Gong et al., 2013) and its expression in pulmonary adenocarcinomas and squamous cell carcinomas predicts poor prognosis (Cordes et al., 2009). Calnexin has been proposed as a diagnostic marker in lung cancer (Kobayashi et al., 2015). The recapitulation of existing literature on lung cancer by our cellular model indicates that our cellular model system is robust to study the molecular changes induced by cigarette smoke and our data indicate that cigarette smoke leads of impaired functioning of lysosomal proteins.
Conclusions and Outlook
This study used an integrated approach to analyze miRNA and protein expression profiles of non-small-cell lung cancer cells chronically exposed to cigarette smoke. Previous studies have shown that continued cigarette smoking by patients with lung cancer receiving chemoradiotherapy is associated with decreased survival (Videtic et al., 2003). We identified an altered expression of several miRNAs as well as proteins in response to cigarette smoke. Pathway analysis using our proteomics data set revealed enrichment of several pathways in response to cigarette smoke, including NRF2-mediated oxidative stress response, cholesterol biosynthesis pathways.
Cigarette smoke has been associated with defects in the autophagy/lysosomal pathway, which in turn lead to accumulation of damaged proteins and particulate matter, mitochondrial damage, and impaired innate immune responses. These results indicate that chronic exposure to cigarette smoke alters miRNA-dependent pathways in NSCLC, potentially aggravating the disease.
In summary, this report makes an important contribution to knowledge on molecular changes in a lung cell line in response to long-term cigarette smoke exposure. The findings might also inform the future strategies for molecular drug target, biomarker and diagnostics innovation in lung cancer, and clinical oncology.
Supplementary Material
Abbreviations Used
- bRPLC
basic pH reverse phase chromatography
- CEBPB
CCAAT/enhancer binding protein
- COPD
chronic obstructive pulmonary disease
- CSC
cigarette smoke condensate
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- FBS
fetal bovine serum
- GCOS
gene chip operating software
- HBEC
human bronchial epithelial cells
- HCD
higher energy collisional dissociation
- HDL
high-density lipoprotein
- HPRD
Human Protein Reference Database
- IPA
ingenuity pathway analysis
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LDL
low-density lipoprotein
- NRF2
nuclear factor erythroid-2 related factor 2
- NSCLC
non-small-cell lung carcinoma
- PDCD4
programmed cell death 4
- PLAUR
plasminogen activator, urokinase receptor
- SDCBP
syndecan binding protein
- SILAC
stable isotopic labeling with amino acids in cell culture
- Srx1
sulfiredoxin-1
- TCGA
The Cancer Genome Atlas
- TEABC
triethyl ammonium bicarbonate
- TNFAIP2
tumor necrosis factor alpha-induced protein 2
- TPCK
tosylphenylalenyl chloromethyl ketone
Acknowledgments
We thank the Department of Biotechnology (DBT), Government of India, for research support to the Institute of Bioinformatics (IOB), Bangalore. IOB is supported by DBT Program Support on Neuroproteomics and infrastructure for proteomic data analysis (BT/01/COE/08/05). This work was supported by the Science and Engineering Research Board, Department of Science and Technology, Government of India, grant “Role of miRNAs in promoting lung cancer in response to cigarette smoke (SR/FT/LS-113/2011),” the NCI's Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036), and FAMRI-funded 072017_YCSA.
J.A. and A.R. are recipients of Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), New Delhi, India. A.A.K. is a recipient of a Senior Research Fellowship from Indian Council of Medical Research (ICMR). miRNA expression of NSCLC and its corresponding clinical information for Illumina GA and HiSeq platform were obtained from The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm) as noted under the “Materials and Methods” section.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
- Agarwal AK, Srinivasan N, Godbole R, et al. (2015). Role of tumor cell surface lysosome-associated membrane protein-1 (LAMP1) and its associated carbohydrates in lung metastasis. J Cancer Res Clin Oncol 141, 1563–1574 [DOI] [PubMed] [Google Scholar]
- Baginski TK, Dabbagh K, Satjawatcharaphong C, and Swinney DC. (2006). Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. Am J Respir Cell Mol Biol 35, 165–174 [DOI] [PubMed] [Google Scholar]
- Bao L, Lv L, Feng J, et al. (2016). miR-487b-5p regulates temozolomide resistance of lung cancer cells through LAMP2-medicated autophagy. DNA Cell Biol 35, 385–392 [DOI] [PubMed] [Google Scholar]
- Barker EV, Cervigne NK, Reis PP, et al. (2009). microRNA evaluation of unknown primary lesions in the head and neck. Mol Cancer 8, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno MJ, and Malumbres M. (2011). MicroRNAs and the cell cycle. Biochim Biophys Acta 1812, 592–601 [DOI] [PubMed] [Google Scholar]
- Cai J, Guan H, Fang L, et al. (2013). MicroRNA-374a activates Wnt/beta-catenin signaling to promote breast cancer metastasis. J Clin Invest 123, 566–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, and Croce CM. (2006). MicroRNA signatures in human cancers. Nat Rev Cancer 6, 857–866 [DOI] [PubMed] [Google Scholar]
- Cano M, Thimmalappula R, Fujihara M, et al. (2010). Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res 50, 652–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin K, Ettinger DS, Hei YJ, and O'Malley CD. (2011). Survival by histologic subtype in stage IV nonsmall cell lung cancer based on data from the Surveillance, Epidemiology and End Results Program. Clin Epidemiol 3, 139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X, Ravi R, Pham V, Bedi A, Chatterjee A, and Sidransky D. (2011). Adenylate kinase 3 sensitizes cells to cigarette smoke condensate vapor induced cisplatin resistance. PLoS One 6, e20806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Wang XY, Li C, and Xu SJ. (2015). Downregulation of microRNA-124 predicts poor prognosis in glioma patients. Neurol Sci 36, 131–135 [DOI] [PubMed] [Google Scholar]
- Chen Y, Liu W, Chao T, et al. (2008a). MicroRNA-21 down-regulates the expression of tumor suppressor PDCD4 in human glioblastoma cell T98G. Cancer Lett 272, 197–205 [DOI] [PubMed] [Google Scholar]
- Chen ZH, Kim HP, Sciurba FC, et al. (2008b). Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 3, e3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WC, Chow AS, and Au JS. (2011). MiR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR and NUDT1. RNA Biol 8, 125–131 [DOI] [PubMed] [Google Scholar]
- Chou CH, Chang NW, Shrestha S, et al. (2016). miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44, D239–D247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes C, Bartling B, Simm A, et al. (2009). Simultaneous expression of Cathepsins B and K in pulmonary adenocarcinomas and squamous cell carcinomas predicts poor recurrence-free and overall survival. Lung Cancer 64, 79–85 [DOI] [PubMed] [Google Scholar]
- de Torres JP, Bastarrika G, Wisnivesky JP, et al. (2007). Assessing the relationship between lung cancer risk and emphysema detected on low-dose CT of the chest. Chest 132, 1932–1938 [DOI] [PubMed] [Google Scholar]
- Franklin WA, Gazdar AF, Haney J, et al. (1997). Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest 100, 2133–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, and Bartel DP. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujihara M, Nagai N, Sussan TE, Biswal S, and Handa JT. (2008). Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS One 3, e3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbin U, Fratta Pasini A, Stranieri C, et al. (2009). Cigarette smoking blocks the protective expression of Nrf2/ARE pathway in peripheral mononuclear cells of young heavy smokers favouring inflammation. PLoS One 4, e8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur A, Jewell DA, Liang Y, et al. (2007). Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res 67, 2456–2468 [DOI] [PubMed] [Google Scholar]
- Gepner AD, Piper ME, Johnson HM, Fiore MC, Baker TB, and Stein JH. (2011). Effects of smoking and smoking cessation on lipids and lipoproteins: Outcomes from a randomized clinical trial. Am Heart J 161, 145–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giatromanolaki A, Kalamida D, Sivridis E, et al. (2015). Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 90, 98–105 [DOI] [PubMed] [Google Scholar]
- Givi ME, Folkerts G, Wagenaar GT, Redegeld FA, and Mortaz E. (2015). Cigarette smoke differentially modulates dendritic cell maturation and function in time. Respir Res 16, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocze PM, and Freeman DA. (2000). Cytotoxic effects of cigarette smoke alkaloids inhibit the progesterone production and cell growth of cultured MA-10 Leydig tumor cells. Eur J Obstet Gynecol Reprod Biol 93, 77–83 [DOI] [PubMed] [Google Scholar]
- Gong F, Peng X, Luo C, et al. (2013). Cathepsin B as a potential prognostic and therapeutic marker for human lung squamous cell carcinoma. Mol Cancer 12, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Gao T, Shen J, et al. (2014). MicroRNA-183 inhibits apoptosis and promotes proliferation and invasion of gastric cancer cells by targeting PDCD4. Int J Clin Exp Med 7, 2519–2529 [PMC free article] [PubMed] [Google Scholar]
- Guz M, Rivero-Muller A, Okon E, et al. (2014). MicroRNAs-role in lung cancer. Dis Markers 2014, 218169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsha HC, Molina H, and Pandey A. (2008). Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat Protoc 3, 505–516 [DOI] [PubMed] [Google Scholar]
- Hayes J, Peruzzi PP. and Lawler S. (2014). MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol Med 20, 460–469 [DOI] [PubMed] [Google Scholar]
- He L, He X, Lim LP, et al. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Varticovski L, Bowman ED, et al. (2004). Identification of carboxypeptidase E and gamma-glutamyl hydrolase as biomarkers for pulmonary neuroendocrine tumors by cDNA microarray. Hum Pathol 35, 1196–1209 [DOI] [PubMed] [Google Scholar]
- Hu Z, Chen X, Zhao Y, et al. (2010). Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J Clin Oncol 28, 1721–1726 [DOI] [PubMed] [Google Scholar]
- Huang Q, Gumireddy K, Schrier M, et al. (2008). The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 10, 202–210 [DOI] [PubMed] [Google Scholar]
- Hunt S, Jones AV, Hinsley EE, Whawell SA, and Lambert DW. (2011). MicroRNA-124 suppresses oral squamous cell carcinoma motility by targeting ITGB1. FEBS Lett 585, 187–192 [DOI] [PubMed] [Google Scholar]
- Iizuka T, Ishii Y, Itoh K, et al. (2005). Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10, 1113–1125 [DOI] [PubMed] [Google Scholar]
- Iorio MV, Ferracin M, Liu CG, et al. (2005). MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65, 7065–7070 [DOI] [PubMed] [Google Scholar]
- Izzotti A, Bagnasco M, Cartiglia C, et al. (2005). Chemoprevention of genome, transcriptome, and proteome alterations induced by cigarette smoke in rat lung. Eur J Cancer 41, 1864–1874 [DOI] [PubMed] [Google Scholar]
- Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM, and De Flora S. (2009). Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J 23, 806–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzotti A, Calin GA, Steele VE, et al. (2010). Chemoprevention of cigarette smoke-induced alterations of MicroRNA expression in rat lungs. Cancer Prev Res (Phila) 3, 62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzotti A, Cartiglia C, Longobardi M, et al. (2004). Alterations of gene expression in skin and lung of mice exposed to light and cigarette smoke. FASEB J 18, 1559–1561 [DOI] [PubMed] [Google Scholar]
- Jacobsen A, Silber J, Harinath G, Huse JT, Schultz N, and Sander C. (2013). Analysis of microRNA-target interactions across diverse cancer types. Nat Struct Mol Biol 20, 1325–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Chang J, Zhang Q, Sun L, and Qiu X. (2013). MicroRNA hsa-miR-125a-3p activates p53 and induces apoptosis in lung cancer cells. Cancer Invest 31, 538–544 [DOI] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, et al. (2005). RAS is regulated by the let-7 microRNA family. Cell 120, 635–647 [DOI] [PubMed] [Google Scholar]
- Kammerl IE, Dann A, Mossina A, et al. (2016). Impairment of immunoproteasome function by cigarette smoke and in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 193, 1230–1241 [DOI] [PubMed] [Google Scholar]
- Kent OA, and Mendell JT. (2006). A small piece in the cancer puzzle: MicroRNAs as tumor suppressors and oncogenes. Oncogene 25, 6188–6196 [DOI] [PubMed] [Google Scholar]
- Keshava Prasad TS, Goel R, Kandasamy K, et al. (2009a). Human protein reference database—2009 update. Nucleic Acids Res 37, D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad TS, Kandasamy K, and Pandey A. (2009b). Human protein reference database and human proteinpedia as discovery tools for systems biology. Methods Mol Biol 577, 67–79 [DOI] [PubMed] [Google Scholar]
- Kim HP, Wang X, Chen ZH, et al. (2008). Autophagic proteins regulate cigarette smoke-induced apoptosis: Protective role of heme oxygenase-1. Autophagy 4, 887–895 [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Nagashio R, Jiang SX, et al. (2015). Calnexin is a novel sero-diagnostic marker for lung cancer. Lung Cancer 90, 342–345 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Lu J, Cobb BS, et al. (2008). Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc Natl Acad Sci U S A 105, 1949–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi MT, Dracheva T, Rotunno M, et al. (2008). Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One 3, e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim HK, Chung S, Kim KS, and Dutta A. (2005). Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem 280, 16635–16641 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, and Bartel DP. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 [DOI] [PubMed] [Google Scholar]
- Li QK, Shah P, Li Y, et al. (2013). Glycoproteomic analysis of bronchoalveolar lavage (BAL) fluid identifies tumor-associated glycoproteins from lung adenocarcinoma. J Proteome Res 12, 3689–3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CZ, Liu W, Zheng Y, et al. (2012). PTEN and PDCD4 are bona fide targets of microRNA-21 in human cholangiocarcinoma. Chin Med Sci J 27, 65–72 [PubMed] [Google Scholar]
- Liu X, Yu J, Jiang L, et al. (2009). MicroRNA-222 regulates cell invasion by targeting matrix metalloproteinase 1 (MMP1) and manganese superoxide dismutase 2 (SOD2) in tongue squamous cell carcinoma cell lines. Cancer Genomics Proteomics 6, 131–139 [PMC free article] [PubMed] [Google Scholar]
- Luppi F, Aarbiou J, van Wetering S, et al. (2005). Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: Role of glutathione. Respir Res 6, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Mannoor K, Gao L, et al. (2014). Characterization of microRNA transcriptome in lung cancer by next-generation deep sequencing. Mol Oncol 8, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maru DM, Singh RR, Hannah C, et al. (2009). MicroRNA-196a is a potential marker of progression during Barrett's metaplasia-dysplasia-invasive adenocarcinoma sequence in esophagus. Am J Pathol 174, 1940–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meenakshisundaram R, Rajendiran C, and Thirumalaikolundusubramanian P. (2010). Lipid and lipoprotein profiles among middle aged male smokers: A study from southern India. Tob Induc Dis 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova O, Stratton Y, Hall D, et al. (2012). VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 21, 532–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed MM, and Sloane BF. (2006). Cysteine cathepsins: Multifunctional enzymes in cancer. Nat Rev Cancer 6, 764–775 [DOI] [PubMed] [Google Scholar]
- Molina JR, Yang P, Cassivi SD, Schild SE, and Adjei AA. (2008). Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 83, 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monick MM, Powers LS, Walters K, et al. (2010). Identification of an autophagy defect in smokers' alveolar macrophages. J Immunol 185, 5425–5435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Yasuda T, Saigo K, et al. (2006). Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 25, 2537–2545 [DOI] [PubMed] [Google Scholar]
- Newland N, and Richter A. (2008). Agents associated with lung inflammation induce similar responses in NCI-H292 lung epithelial cells. Toxicol In Vitro 22, 1782–1788 [DOI] [PubMed] [Google Scholar]
- Osada H, and Takahashi T. (2002). Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene 21, 7421–7434 [DOI] [PubMed] [Google Scholar]
- Parsons A, Daley A, Begh R, and Aveyard P. (2010). Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: Systematic review of observational studies with meta-analysis. BMJ 340, b5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan M, Keerthikumar S, Ang CS, et al. (2015). FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 15, 2597–2601 [DOI] [PubMed] [Google Scholar]
- Peng XH, Huang HR, Lu J, et al. (2014). MiR-124 suppresses tumor growth and metastasis by targeting Foxq1 in nasopharyngeal carcinoma. Mol Cancer 13, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell CA, Klares S, O'Connor G, and Brody JS. (1999). Loss of heterozygosity in epithelial cells obtained by bronchial brushing: Clinical utility in lung cancer. Clin Cancer Res 5, 2025–2034 [PubMed] [Google Scholar]
- Qi M, Huang X, Zhou L, and Zhang J. (2014a). Identification of differentially expressed microRNAs in metastatic melanoma using next-generation sequencing technology. Int J Mol Med 33, 1117–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Yang DY, and Cao J. (2014b). Increased micro-RNA 17, 21, and 192 gene expressions improve early diagnosis in non-small cell lung cancer. Med Oncol 31, 195. [DOI] [PubMed] [Google Scholar]
- Ramirez de Molina A, de la Cueva A, Machado-Pinilla R, et al. (2012). Acid ceramidase as a chemotherapeutic target to overcome resistance to the antitumoral effect of choline kinase alpha inhibition. Curr Cancer Drug Targets 12, 617–624 [DOI] [PubMed] [Google Scholar]
- Reis PP, Tomenson M, Cervigne NK, et al. (2010). Programmed cell death 4 loss increases tumor cell invasion and is regulated by miR-21 in oral squamous cell carcinoma. Mol Cancer 9, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi D, Cammarata G, Agueli C, et al. (2015). miR-155 regulative network in FLT3 mutated acute myeloid leukemia. Leuk Res 39, 883–896 [DOI] [PubMed] [Google Scholar]
- Schembri F, Sridhar S, Perdomo C, et al. (2009). MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci U S A 106, 2319–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetter AJ, Leung SY, Sohn JJ, et al. (2008). MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 299, 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T, Sakaguchi C, and Fukano Y. (2015). Investigation by microarray analysis of effects of cigarette design characteristics on gene expression in human lung mucoepidermoid cancer cells NCI-H292 exposed to cigarette smoke. Exp Toxicol Pathol 67, 143–151 [DOI] [PubMed] [Google Scholar]
- Seol HS, Akiyama Y, Shimada S, et al. (2014). Epigenetic silencing of microRNA-373 to epithelial-mesenchymal transition in non-small cell lung cancer through IRAK2 and LAMP1 axes. Cancer Lett 353, 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao MX, Nakanaga T, and Nadel JA. (2004). Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am J Physiol Lung Cell Mol Physiol 287, L420–L427 [DOI] [PubMed] [Google Scholar]
- Shubbar E, Helou K, Kovacs A, et al. (2013). High levels of gamma-glutamyl hydrolase (GGH) are associated with poor prognosis and unfavorable clinical outcomes in invasive breast cancer. BMC Cancer 13, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Ling G, Suhasini AN, et al. (2009). Nrf2-dependent sulfiredoxin-1 expression protects against cigarette smoke-induced oxidative stress in lungs. Free Radic Biol Med 46, 376–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira A, Beane J, Shah V, et al. (2004). Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A 101, 10143–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Wang K, Li R, et al. (2016). Overexpression of RBM5 induces autophagy in human lung adenocarcinoma cells. World J Surg Oncol 14, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ai X, Shen S, and Lu S. (2015). NF-kappaB-mediated miR-124 suppresses metastasis of non-small-cell lung cancer by targeting MYO10. Oncotarget 6, 8244–8254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiberville L, Payne P, Vielkinds J, et al. (1995). Evidence of cumulative gene losses with progression of premalignant epithelial lesions to carcinoma of the bronchus. Cancer Res 55, 5133–5139 [PubMed] [Google Scholar]
- Thorne D, Wilson J, Kumaravel TS, Massey ED, and McEwan M. (2009). Measurement of oxidative DNA damage induced by mainstream cigarette smoke in cultured NCI-H292 human pulmonary carcinoma cells. Mutat Res 673, 3–8 [DOI] [PubMed] [Google Scholar]
- Titz B, Boue S, Phillips B, et al. (2016). Effects of cigarette smoke, cessation, and switching to two heat-not-burn tobacco products on lung lipid metabolism in C57BL/6 and Apoe−/− mice-an integrative systems toxicology analysis. Toxicol Sci 149, 441–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tockman MS, Anthonisen NR, Wright EC, and Donithan MG. (1987). Airways obstruction and the risk for lung cancer. Ann Intern Med 106, 512–518 [DOI] [PubMed] [Google Scholar]
- Vial WC. (1986). Cigarette smoking and lung disease. Am J Med Sci 291, 130–142 [DOI] [PubMed] [Google Scholar]
- Videtic GM, Stitt LW, Dar AR, et al. (2003). Continued cigarette smoking by patients receiving concurrent chemoradiotherapy for limited-stage small-cell lung cancer is associated with decreased survival. J Clin Oncol 21, 1544–1549 [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, et al. (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103, 2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosa U, Vooder T, Kolde R, et al. (2011). Identification of miR-374a as a prognostic marker for survival in patients with early-stage nonsmall cell lung cancer. Genes Chromosomes Cancer 50, 812–822 [DOI] [PubMed] [Google Scholar]
- Vucic EA, Thu KL, Pikor LA, et al. (2014). Smoking status impacts microRNA mediated prognosis and lung adenocarcinoma biology. BMC Cancer 14, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li Z, Ge Q, et al. (2015). Characterization of microRNA transcriptome in tumor, adjacent, and normal tissues of lung squamous cell carcinoma. J Thorac Cardiovasc Surg 149, 1404–1414.e4 [DOI] [PubMed] [Google Scholar]
- Wang T, Zhang X, Obijuru L, et al. (2007). A micro-RNA signature associated with race, tumor size, and target gene activity in human uterine leiomyomas. Genes Chromosomes Cancer 46, 336–347 [DOI] [PubMed] [Google Scholar]
- Wistuba II, Lam S, Behrens C, et al. (1997). Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst 89, 1366–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Zhu Y, Ma T, et al. (2014). miR-204 functions as a tumor suppressor by regulating SIX1 in NSCLC. FEBS Lett 588, 3703–3712 [DOI] [PubMed] [Google Scholar]
- Xu D, Takeshita F, Hino Y, et al. (2011). miR-22 represses cancer progression by inducing cellular senescence. J Cell Biol 193, 409–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang W, Su N, et al. (2015). miR-374a promotes cell proliferation, migration and invasion by targeting SRCIN1 in gastric cancer. FEBS Lett 589, 407–413 [DOI] [PubMed] [Google Scholar]
- Yamamichi N, Shimomura R, Inada K, et al. (2009). Locked nucleic acid in situ hybridization analysis of miR-21 expression during colorectal cancer development. Clin Cancer Res 15, 4009–4016 [DOI] [PubMed] [Google Scholar]
- Yanaihara N, Caplen N, Bowman E, et al. (2006). Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189–198 [DOI] [PubMed] [Google Scholar]
- Yang Y, Chaerkady R, Beer MA, Mendell JT, and Pandey A. (2009). Identification of miR-21 targets in breast cancer cells using a quantitative proteomic approach. Proteomics 9, 1374–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Li H, Wang J, et al. (2014). miRNA-34a suppresses cell proliferation and metastasis by targeting CD44 in human renal carcinoma cells. J Urol 192, 1229–1237 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma T, Yang S, et al. (2011). High-mobility group A1 proteins enhance the expression of the oncogenic miR-222 in lung cancer cells. Mol Cell Biochem 357, 363–371 [DOI] [PubMed] [Google Scholar]
- Zhu FQ, Zeng L, Tang N, et al. (2016). MicroRNA-155 downregulation promotes cell cycle arrest and apoptosis in diffuse large B-cell lymphoma. Oncol Res 24, 415–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WY, Luo B, An JY, et al. (2014). Differential expression of miR-125a-5p and let-7e predicts the progression and prognosis of non-small cell lung cancer. Cancer Invest 32, 394–401 [DOI] [PubMed] [Google Scholar]
- Zong C, Song G, Yao S, et al. (2015). Cigarette smoke exposure impairs reverse cholesterol transport which can be minimized by treatment of hydrogen-saturated saline. Lipids Health Dis 14, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium5 (Vizcaino et al., 2014) by the PRIDE partner repository with the dataset identifier PXD003086.