

Abstract
Background
The long QT syndrome (LQTS) is an arrhythmogenic disorder of QT interval prolongation that predisposes patients to life-threatening ventricular arrhythmias such as Torsade de pointes (TdP) and sudden cardiac death (SCD). Clinical genetic testing has emerged as the standard of care to identify genetic variants in patients suspected of having LQTS. However, these results are often confounded by the discovery of variants of unknown significance (VUS), for which there is insufficient evidence of pathogenicity.
Objectives
To demonstrate that genome editing of patient-specific induced pluripotent stem cells (iPSCs) can be a valuable approach to delineate the pathogenicity of VUS in cardiac channelopathy.
Methods
Peripheral blood mononuclear cells (PBMCs) were isolated from a carrier with a novel missense variant (T983I) in the KCNH2 (LQT2) gene and an unrelated healthy control subject. iPSCs were generated using an integration-free Sendai virus and differentiated to iPSC-derived cardiomyocytes (iPSC-CMs).
Results
Whole-cell patch clamp recordings revealed significant prolongation of the action-potential duration (APD) and reduced rapidly activating delayed rectifier K+ current (IKr) density in VUS iPSC-CMs compared to healthy control iPSC-CMs. ICA-105574, a potent IKr activator, enhanced IKr magnitude and restored normal APD in VUS iPSC-CMs. Notably, VUS iPSC-CMs exhibited greater propensity to proarrhythmia than healthy control cells in response to high-risk torsadogenic drugs (dofetilide, ibutilide, and azimilide), suggesting a compromised repolarization reserve. Finally, the selective correction of the causal variant in iPSC-CMs using CRISPR/Cas9 gene editing (isogenic control) normalized the aberrant cellular phenotype, whereas the introduction of the homozygous variant in healthy control cells recapitulated hallmark features of the LQTS disorder.
Conclusions
Our results suggest that the KCNH2T983I VUS may be classified as potentially pathogenic.
Keywords: Induced pluripotent stem cells, genome editing, variant of unknown significance, long QT syndrome, arrhythmia
Introduction
Congenital LQTS is a potentially lethal genetic disorder of cardiac repolarization that represents a leading cause of SCD in the young with a prevalence of ~1 in 2,500 among the general population (1). Among 17 known LQTS subtypes, those associated with mutations in ion channel genes KCNQ1 (LQT1), KCNH2 (LQT2) and SCN5A (LQT3) account for 90% of all genotype-positive cases (2). β-adrenergic blockers are the mainstay of treatment for symptomatic LQTS patients, but implantable cardioverter-defibrillator (ICD), left cardiac sympathetic denervation, and sodium channel blocker therapy may also be recommended (3,4).
Despite significant advances in the management of LQTS based on an improved understanding of implicated genes and underlying ion currents, the care of almost 1/3 of LQTS patients remains challenging largely due to low penetrance of clinical symptoms and high variability in phenotypic expression (5). Even multiple family members carrying the same mutation may have different QT intervals and clinical manifestations (6,7). Additionally, the prevalence of asymptomatic individuals with latent LQTS is higher than anticipated (8). This creates a management dilemma of committing patients to lifelong medical therapy that may be risky or unproven, with significant consequences for patient’s quality of life.
An emerging standard of care for LQTS patients uses clinical genetic testing to identify causal variants in the LQTS susceptibility genes. However, such testing can identify >100 novel nonsynonymous coding variants in any given individual. Ascertaining which, if any, of these is a true causal variant is currently one of the major challenges in the treatment of heritable disorders especially when the variant is of unknown significance (VUS) due to inadequate evidence of pathogenicity (9,10). In the era of next generation sequencing, interpretation of growing numbers of discovered unknown variants will represent an even greater challenge to therapy (9).
Currently, there are no reliable platforms to predict a priori whether a given variant predisposes an individual to a disease or whether the coding change is benign. The launch of the Precision Medicine Initiative in 2015 has facilitated rapid advances in technologies such as induced pluripotent stem cells (iPSCs) and clustered regularly interspaced short palindromic repeats (CRISPR) genome editing. The creation of isogenic iPSC lines with single variant changes using CRISPR allows a direct comparison of phenotype at a cellular level. This novel approach of combining these methods can be applied to test the precise phenotypic impact of VUS. Such a strategy may aid in deciphering VUS pathogenicity in LQTS and in tailoring drug treatment and ICD therapy, and could potentially be applied to other inherited cardiac disorders.
By combining patient-specific iPSCs and genome editing, we aimed to develop and validate a human-based platform for elucidating VUS pathogenicity ‘in a dish’ for inherited arrhythmia syndromes and channelopathies. To this end, iPSC lines were derived from a patient carrying the novel variant T983I in the C-terminus of the KCNH2 gene (KCNH2T983I), which encodes a channel that mediates the rapidly activating component of the delayed rectifying potassium current (IKr). Consistent with an LQT2 phenotype, we observed a prolongation of the action potential duration (APD) and reduced IKr density in VUS iPSC-CMs compared to cells derived from a healthy control. VUS iPSC-CMs were more susceptible to proarrhythmic effects of torsadogenic drugs compared to healthy control iPSC-CMs, whereas treatment of cells with an IKr activator restored normal APD. Finally, CRISPR/Cas9 genome editing of VUS iPSCs rescued the observed electrophysiological abnormalities and the introduction of the homozygous variant in a healthy control line recapitulated hallmark LQTS phenotype. Our findings provide important insights into the pathophysiological mechanisms of this previously uncharacterized variant, and suggest that the presence of this mutation could be sufficient to induce LQT2.
Methods
An extended methods section is available in the Online Data Supplement
Generation of iPSC lines
Somatic reprogramming was used to generate iPSC lines from peripheral blood mononuclear cells (PBMCs) of the patient with the KCNH2T983I variant as confirmed by RT-PCR and from a healthy subject using the Sendai virus reprogramming protocol as described previously (11). iPSCs were also derived from an affected patient with a verified LQT2 (KCNH2A561V) mutation (24) as described above. At least 3 colonies were generated from both patients and healthy control subject. All recruitment and consenting procedures conformed to the Stanford Institutional Review Board (IRB) approved protocol.
Differentiation of iPSC-CMs
iPSC-CMs were generated using a 2D monolayer differentiation protocol and maintained in a 5% CO2/air environment as previously published (12). Briefly, iPSC colonies were dissociated with 0.5 mM EDTA (Gibco) into single-cell suspension and re-suspended in E8 media containing 10 M Rho-associated protein kinase inhibitor (Sigma). Approximately 100,000 cells were re-plated into Matrigel-coated 6-well plates. iPSCs were next cultured to 85% cell confluence, and then treated for 2 days with 6 μM CHIR99021 (Selleck Chemicals) in RPMI + B27 supplement without insulin to activate WNT signaling and induce mesodermal differentiation. On day 2, cells were incubated in RPMI + B27 without insulin and CHIR99021 (12). On days 3–4, cells were treated with 5 μM IWR-1 (Sigma) to inhibit WNT pathway signaling. On days 5–6, cells were removed from IWR-1 treatment and placed in RPMI + B27 without insulin. From day 7 onwards, cells were placed in RPMI + B27 with insulin until beating was observed. At this point, cells were glucose-starved for 3 days with RPMI + B27 with insulin to purify iPSC-CMs. Following purification, cells were cultured in RPMI + B27 with insulin. When re-plating for future use, iPSC-CMs were dissociated with 0.25% trypsin-EDTA into a single-cell suspension and seeded on Matrigel-coated plates. Purification by starvation was repeated between days 20–23 and differentiated CMs were analyzed on day 30–50.
Genome editing
KCNH2 gRNA (5′-CGCCCGGGATACCTGACAGG-3′) was designed using a CRISPR design tool and sub-cloned into a plasmid vector (pSpCas9(BB)-2A-GFP, Addgene). To introduce the KCNH22948C>T point mutation into a healthy control iPSC line (KCNH2T983I), a single-strand DNA oligonucleotide (ssDNA) was designed to contain a single base-pair substitution (C>T) at the respective position. Similarly, a ssDNA containing a single base-pair substitution (T>C) was designed for the correction of the patient KCNH2T983I mutated iPSC line (KCNH22948C>T). Both ssDNAs also contained point mutations of the protospacer adjacent motif (PAM) sequence to prevent repetitive cutting of genomic DNA by the Cas9 nuclease. The sequence of the ssDNA utilized for gene editing was: 5′ TGCCCTTCTCCAGCCCCAGGCCCCCCGGAGAGCCGCCGGGTGGGGAGCCCCTGATGGAGGACTGCGAGAAGAGCAGCGACATTTGCAATCCTCTGTCAGGTATCCCGGGCGACGGGCGGGCGAGGGA-3′ and the correcting ssDNA oligo was: 5′-TGCCCTTCTCCAGCCCCAGGCCCCCCGGAGAGCCGCCGGGTGGGGAGCCCCTGATGGAGGACTGCGAGAAGAGCAGCGACACTTGCAATCCTCTGTCAGGTATCCCGGGCGACGGGCGGGCGAGGGA-3′.
Statistical analysis
Statistical significance was determined by unpaired Student’s t-test (two-tail), with *denoting p<0.05 and considered to be statistically significant; ** indicates p<0.01, *** indicates p<0.001 when compared to healthy control iPSC-CMs. One-way ANOVA followed by Tukey’s post-hoc tests were used when comparing genome-edited iPSC lines.
Results
Patient characteristics
The patient is a 39-year-old male with a history of palpitations and presyncope. He had multiple ECGs demonstrating QTc from a range of 435 ms to 485 ms, with one very prolonged QTc in recovery phase of exercise testing of 507 ms (Figure 1A–B). Based on the multiple ECGs demonstrating a prolonged QTc in the setting of exertional symptoms and the absence of QT-prolonging medications, the patient was diagnosed with LQTS. The patient’s brother had a history of syncope, but ECG revealed a QTc of 430ms. A maternal cousin suffered a cardiac arrest while playing soccer and had an ICD placed, but was not diagnosed with LQTS. The proband patient’s maternal grandfather had four brothers who all died suddenly under 40 years of age.
Figure 1. Family pedigree and generation of patient-specific iPSC lines.

(A) Pedigree of VUS patient. Proband (arrow) was diagnosed with LQTS after presenting with prolonged QTc (507 ms) on (B) surface electrocardiogram. (C) Schematic representation of KCNH2 channel protein. Red star denotes approximate location of the LQTS variant T983I in the C terminus. cNBHD, cyclic nucleotide binding homology domain (D) Representative immunostaining of pluripotency markers SOX2 (green) and NANOG (red) in iPSC clone derived from the VUS patient. DAPI staining (blue) indicates the nucleus.
Upon genetic testing, patient was found to have a p.Thr983Ile (c.2948 C>T) variant in the KCNH2 gene (Figure 1C) with a population frequency of 0.0001398 in the gnomAD database. Given the population frequency and lack of clear prior phenotypic data, this variant was classified as a variant of unclear significance. Multiple groups have similarly classified this variant of unclear significance based on available data (ClinVar for NM_000238.3(KCNH2):c.2948C>T[p.Thr983Ile]). The unrelated healthy subject was an 18 year old male who had normal ECG and no personal or family history of syncope or sudden cardiac death.
Generation and characterization of iPSC-CM lines
iPSCs were derived from unrelated healthy control and the VUS patient harboring the heterozygous c.2948C>T variant in the KCNH2 gene as described previously (11). Pluripotency of generated iPSC lines was characterized using immunostaining of pluripotency markers such as SOX2 and NANOG (Figure 1D). iPSCs were then differentiated into iPSC-CMs as previously described (12) and characterized using immunostaining for cardiac-specific markers, cardiac Troponin T (cTnT) and sarcomeric α-Actinin (Online Figure 1A). More than 95% of differentiated cells were positively stained for cTnT (Online Figure 1B).
Patient-specific VUS iPSC-CMs displayed an abnormal electrophysiological profile
The action potential (AP) profiles of the healthy control and VUS iPSC-CMs were examined using the standard whole-cell patch clamp technique, and AP parameters such as maximal diastolic potential (MDP), overshoot, action potential amplitude (APA), action potential duration at 50% and 90% repolarization (APD50 and APD90), beating rate, and maximal upstroke velocity (Vmax) were quantified (Online Table 1). In accordance with the previous published reports (13–15), three types of AP morphologies—ventricular-like, atrial-like, and nodal-like— were observed in both healthy control and VUS iPSC-CMs (Figure 2A). The AP properties of healthy control iPSC-CMs were similar to those observed in other human iPSC-CM studies (16–18). For most AP parameters, there was no significant difference between healthy control and VUS iPSC-CMs (p>0.05; Online Table 1). However, both ventricular-like and atrial-like VUS iPSC-CMs displayed significantly prolonged APD50 and APD90, respectively (Figure 2B). The APD90 was 793.7 ± 44.1 and 524.1 ± 47.9 ms in VUS iPSC-CMs compared with 367.2 ± 19.6 ms and 288.7 ± 27.7 ms in healthy control iPSC-CMs for ventricular-like and atrial-like cells, respectively. Additionally, mild spontaneous arrhythmogenic activity characterized by beat irregularity or early after depolarizations (EADs) was observed in VUS iPSC-CMs (Figure 2C). While 12.9% of VUS iPSC-CMs (n = 130) exhibited EADs, no arrhythmogenic activity was observed in healthy control iPSC-CMs (n = 64).
Figure 2. Patient-specific VUS iPSC-CMs exhibit characteristic LQTS signatures.
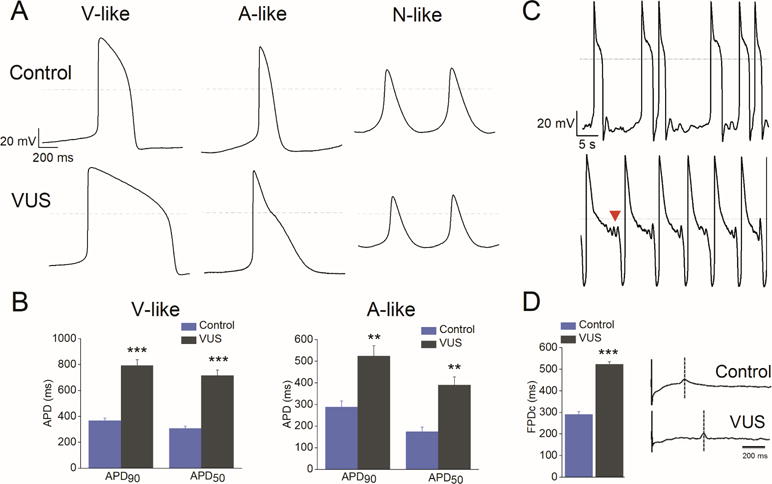
(A) AP recordings from control and VUS iPSC-CMs showing ventricular (V)-like, atrial (A)-like, and nodal (N)-like morphologies. Notice the marked APD prolongation in both V-like and A-like VUS iPSC-CMs. (B) The action potential duration (APD) measured at 50% (APD50) and 90% repolarization (APD90) in VUS (grey, n = 119) and healthy control (blue, n = 49) A-like and V-like iPSC-CMs. ***p<0.001 when compared to control cells. Error bars show S.E.M. (C) Development of spontaneous arrhythmogenicity in VUS iPSC-CMs manifested as beat irregularity (top) or EADs indicated by arrowhead (bottom). (D) Left panel, summary of rate-matched Fridericia’s corrected field potential duration (FPDc) values of VUS iPSC-CMs (grey, n = 7) and healthy control (blue, n = 7). FPDc = FPD/(Beat Period)^0.3333. Error bars show S.E.M. ***p<0.001. Right panel, representative MEA recordings from healthy control (top) and VUS (bottom) iPSC-CM monolayer.
The in vitro cardiac field-potential duration (FPD) on a multielectrode array (MEA) platform has been shown in many studies to correlate with QT interval properties in the ECG (13,14). To evaluate the electrophysiological properties of iPSC-CMs at the multicellular level, we next performed MEA studies and observed that similar to the prolonged APD in patch clamp results, VUS iPSC-CMs had a significantly prolonged cFPD (522.8 ± 11.4 ms; FPD corrected for variations in beating frequency) compared with healthy controls (291.2 ± 12.9 ms) (Figure 2D, p<0.001). Collectively, these data indicate that the patient-specific VUS iPSC-CMs display the characteristic hallmarks of the LQTS phenotype.
VUS iPSC-CMs exhibited decreased IKr density
To investigate the underlying cause of the LQTS phenotype in the patient, ventricular-like iPSC-CMs were subjected to single-cell voltage clamp recordings to measure IKr density. Using the specific inhibitor E-4031, IKr was isolated (Figure 3A) and its magnitude was found to be markedly reduced in VUS iPSC-CMs compared to that in healthy control iPSC-CMs (Figure 3B–C). Tail IKr density was significantly decreased, by 41–44%, at voltages from 0 to +40 mV (p<0.01; Figure 3C).
Figure 3. Single-cell voltage-clamp recordings revealed reduced IKr density in VUS iPSC-CMs.
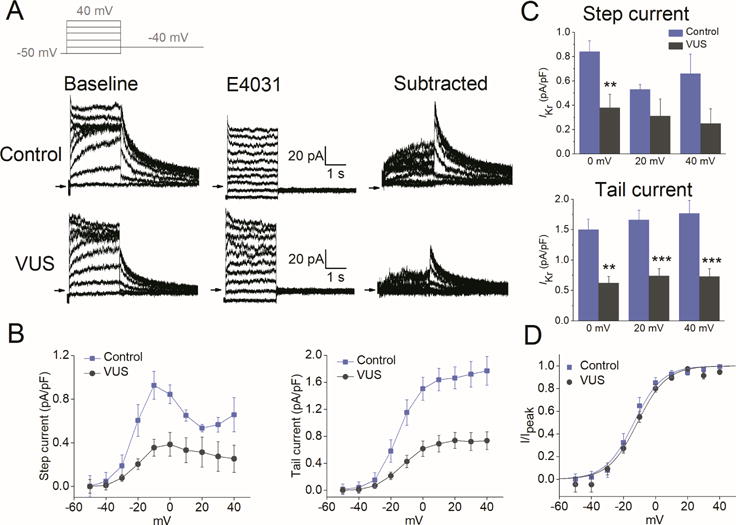
(A) Voltage-clamp recordings of IKr, measured from healthy control (top) and VUS (bottom) iPSC-CMs. Left, baseline recordings. Middle, recording following administration of E-4031 (2 μM). Right, E-4031-sensitive current (IKr) defined by digital subtraction of the two currents. Inset: voltage clamp protocol. (B) Average current–voltage (I–V) relationships for IKr (left, step current measured at the end of the test pulses and right, peak tail current) in control (blue) and VUS (grey) iPSC-CMs. (C) Summary of the step current (top) and peak tail current (bottom) for IKr density in pA/pF from healthy control (blue; n = 4) and VUS iPSC-CMs (grey; n = 8) at membrane potentials of 0, +20, and +40 mV (**p<0.01, ***p<0.001). (D) Average peak tail current normalized to the maximal current following repolarization to −40 mV (I/Ipeak) in healthy control (blue) and VUS (grey) iPSC-CMs. Mean values ± S.E.M. are shown. V1/2 = - 13.55 ± 2.0 mV for VUS vs. −14.27 ± 2.3 mV for healthy control. Comparison of iPSC lines was performed with one-way ANOVA, p<0.05 being considered statistically significant.
In the next set of experiments, we overexpressed the wild type or the variant KCNH2 channel in Xenopus laevis oocytes and recorded IKr using the two-electrode voltage-clamp (TEVC) technique. Representative recordings from oocytes injected with either wild-type (hERG WT) or VUS KCNH2 (hERG T983I) cRNA are shown in Online Figure 2A. We observed a similar reduction in peak IKr density (~53%) in TEVC recordings (p<0.05, Online Figure 2B). These electrophysiological findings were consistent with the channel surface expression in iPSC-CMs. Using Western blot analysis, the differential expression of the mature complex- glycosylated 155 kDa protein band in the VUS iPSC-CMs was found to be significantly reduced compared to healthy control iPSC-CMs, suggesting that mutant channels were not trafficked normally to the cell membrane (Online Figure 3). In addition, the total KCNH2 protein levels were also found to be reduced in VUS iPSC-CMs compared to control iPSC-CMs.
To further determine if the KCNH2 variant could reduce IKr density in VUS iPSC-CMs by altering channel-gating kinetics, we analyzed voltage dependence of activation parameters (half-maximal voltage (V1/2) and slope factor (k)). Interestingly, these parameters were similar between the two groups (V1/2 = −13.55 ± 2.0 mV for VUS vs. −14.27 ± 2.3 mV for healthy control; Figure 3D), suggesting that the variant had no effect on IKr channel gating. Moreover, biophysical analysis of hERG WT and hERG T983I from Xenopus recordings also confirmed that the mutant channels exhibited largely similar gating kinetics as the WT channels (Online Figure 2C).
ICA-105574 enhanced IKr and rescued abnormal electrophysiological phenotype in VUS iPSC-CMs
An interesting approach to treat LQTS is by directly targeting the deficiency in net repolarizing current (19). Recently, Zhang et al. reported that a type II IKr activator that attenuates channel inactivation shortens APD in iPSC-CMs from LQT1 patients in a dose-dependent manner, whereas a type I compound that slows channel deactivation was ineffective (20). ICA-105574 (ICA) is a type II IKr activator that dramatically increases IKr amplitude in heterologous expression cells, shortens APD in guinea pig ventricular myocytes (21), and shortens QT intervals in isolated guinea-pig hearts as well as in anesthetized dogs (22). However, the effectiveness of ICA in normalizing APD prolongation and suppressing arrhythmia in more physiological human diseased iPSC-CMs has not been previously investigated. Our results indicate that ICA is capable of reversing the disease phenotype in VUS iPSC-CMs by drastically increasing the magnitude of IKr and shortening APD. Notably, the effects of ICA were not limited to VUS iPSC-CMs, but were extended to healthy control iPSC-CMs (Figure 4A). The current voltage relationships of IKr step currents and tail currents are shown in Figure 4B. ICA (1 μM) increased currents recorded at +40 mV in VUS iPSC-CMs from a basal current density of 1.67 ± 0.23 to 7.16 ± 2.2 pA/pF, a 4.3-fold increase (p<0.001; Figure 4C). It is important to note here that as ICA is a selective IKr agonist, we did not use E4031 in these experiments to isolate IKr.
Figure 4. ICA-105574 enhanced IKr density and normalized prolonged APD in VUS iPSC-CMs.
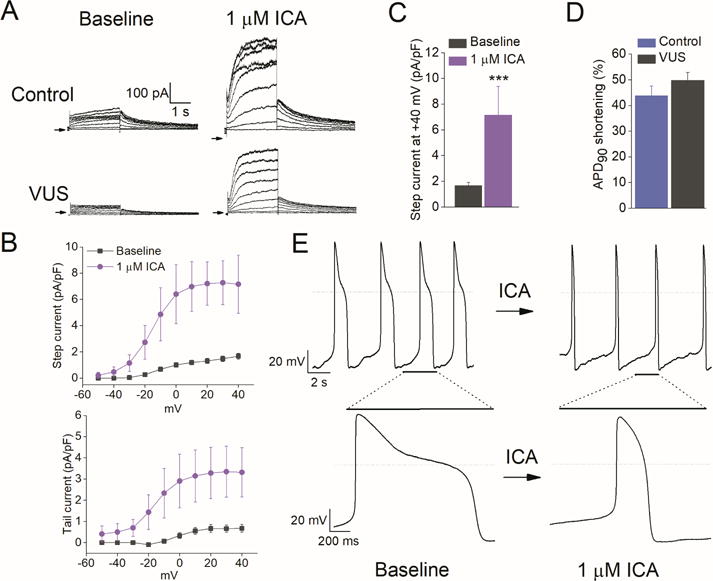
(A) Representative IKr traces in healthy control (top) and VUS (bottom) iPSC-CMs at baseline (left) and after application of 1 μM ICA-105574, a KCNH2 channel agonist. (B) Average I-V curves for IKr step current (top) and peak tail current (bottom) density in pA/pF at baseline (grey) and after application of 1 μM ICA-105574 (pink) in VUS iPSC-CMs. (C) Summary of average step current density at +40 mV for baseline and 1 μM ICA-105574 in VUS iPSC-CMs (n = 7, ***p<0.001). (D) Summary of % APD90 shortening induced by ICA-105574 for healthy control and VUS iPSC-CMs (n = 7, p>0.05). (E) AP recordings at baseline (left) and in the presence of 1 μM ICA-105574 (right). Note the severely prolonged APD in VUS iPSC-CMs before application of ICA-105574. Expanded view highlighting a single AP is shown at the bottom.
In the next set of experiments, we evaluated the effect of 1 μM ICA on ventricular APD from healthy control and VUS iPSC-CMs. As anticipated, the application of ICA produced a rapid shortening of APD in both healthy control and VUS iPSC-CMs, as quantified from the percentage reductions in APD90 (Figure 4D-E). For 1 μM ICA, APD90 was shortened by 43.8 ± 3.8 % in healthy control iPSC-CMs (n = 3) and 49.9 ± 3.0 % in VUS iPSC-CMs (n = 7, p>0.05). Our findings suggest that ICA can normalize APD prolongation and prevent cellular arrhythmias in LQTS iPSC-CMs; however, the potential for excessive QT shortening and pro-arrhythmic risks should be carefully considered before further developing type II IKr agonists.
VUS iPSC-CMs displayed enhanced susceptibility to dofetilide-induced proarrhythmia
We next investigated the proarrhythmic liability of torsadogenic drugs chosen from the Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative (23) on healthy control and VUS iPSC lines. We tested three high-risk drugs with known QT prolongation effects: dofetilide, ibutilide, and azimilide. These class III antiarrhythmic drugs selectively block KCNH2 (human ether-a-go-go related gene or hERG) channels. Dofetilide induced arrhythmogenicity in both healthy control and VUS iPSC-CMs; however, VUS iPSC-CMs were significantly more susceptible to the proarrhythmic effects of the blocker (Figure 5A–C). We observed a cut-off concentration (10 nM) at which dofetilide was found to induce EADs in VUS iPSC-CMs, but not in healthy control iPSC-CMs (Figure 5A–B). We also investigated the effects of dofetilide on KCNH2 channels harboring a verified (classical) LQT2 mutation (LQT2-A561V) (24) by generating iPSC-CMs from an affected patient. iPSC-CMs harboring this mutation were found to have diminished IKr, prolonged repolarization durations, and elevated baseline arrhythmogenesis (Online Figure 4). Similar to our findings with VUS iPSC-CMs, patient-specific LQT2-A561V iPSC-CMs also displayed arrhythmias starting at 10 nM (Figure 5A–B), a concentration that was not proarrhythmic for healthy control iPSC-CMs. The enhanced sensitivity of LQT2-A561V and VUS iPSC-CMs to dofetilide-induced toxicity were likely related to drug-induced exacerbation of the baseline arrhythmias in diseased lines as consistent with previous studies (16). Healthy control iPSC-CMs that did not exhibit baseline arrhythmia or APD prolongation required higher doses of dofetilide before exhibiting cardiotoxicity at the single cell level.
Figure 5. Patient-specific VUS iPSC-CMs and classical pathogenic LQT2-A561V iPSC-CMs displayed enhanced susceptibility to dofetilide-induced proarrhythmia.

(A) Representative recordings showing dose-dependent effect of dofetilide (Dof) on AP from healthy control (left), VUS (middle), and classical pathogenic LQT2-A561V (right) iPSC-CMs (black = baseline; red = 3 nmol/L; blue = 10 nmol/L; green = 30 nmol/L; orange = 100 nmol/L). Arrowheads indicate dofetilide-induced EADs. VUS and LQT2-A561V iPSC-CMs were observed to exhibit dofetilide-induced EADs at lower concentrations (10 nmol/L) as compared with control iPSC-CMs (30 nmol/L). (B) Aligned APs showing the effects of dofetilide on control, VUS, and LQT2-A561V iPSC-CMs, respectively (n = 4). (C) Percentage increase in steady-state prolongation of APD90 by dofetilide at different concentrations for control, VUS, and LQT2-A561V iPSC-CMs. *p<0.05.
Similar to dofetilide, the application of ibutilide (5 nM), a drug used to treat atrial flutter and atrial fibrillation, elicited arrhythmogenicity that manifested as EADs only in VUS iPSC-CMs (Online Figure 5A and 5C). Enhanced arrhythmogenicity with azimilide (1 μM), a drug that is currently in clinical trials, also manifested as EADs only in VUS iPSC-CMs (Online Figure 5B–C). Taken together, these findings indicate that VUS iPSC-CMs, which were characterized by an elevated baseline of arrhythmia, exhibited a particular propensity for drug-induced EADs by IKr blockade. This high frequency of baseline arrhythmias and electrophysiological abnormalities observed in LQT2-A561V and VUS iPSC-CMs may be explained by imbalances in ion channel homeostasis caused by mutations in the KCNH2 gene.
Genome-edited homozygous iPSC-CMs (VUShom) displayed exacerbated LQTS phenotype
To confirm the role of the KCNH2T983I variant in the generation of an LQTS phenotype, and to assess whether introducing a homozygous variant results in further exacerbation of the arrhythmic phenotype compared to the heterozygous patient iPSC-CMs, we next introduced the homozygous variant in healthy control cells using the CRISPR/Cas9 genome-editing technology (Online Figures 6 and 7). Whole-cell current clamp recordings were performed from single beating iPSC-CMs to obtain spontaneous APs (Figure 6A). The key AP parameters were collected and assessed (Online Table 1). The ventricular-like and atrial-like iPSC-CMs derived from genome edited homozygous line (VUShom) exhibited significantly longer APD90 compared to healthy control line (605.3 ± 72.2 ms vs 288.7 ± 27.7 ms for atrial-like and 1207.9 ± 198.3 ms vs 367.2 ± 19.6 ms for ventricular-like cells, respectively; p<0.01, Figure 6B). Importantly, as anticipated the VUShom iPSC-CMs displayed a severe arrhythmogenic phenotype (Figure 6C) more akin to that observed in the verified LQT2-A561V iPSC-CMs such as the development of EADs or triggered beat (45.2% in VUShom vs. 12.9% in VUS vs. 46% in LQT2-A561V iPSC-CMs). In conclusion, consistent with our hypothesis regarding the role of this novel variant as an underlying cause of LQTS, VUShom iPSC-CMs faithfully recapitulated a more severe arrhythmic disease phenotype.
Figure 6. Genome-edited homozygous iPSC-CMs (VUShom) displayed potentiated LQTS phenotype.

(A) Representative AP recordings from VUShom iPSC-CMs showing V-like, A-like, and A-like-EAD waveforms. Notice the marked APD prolongation in both V-like and A-like iPSC-CMs. (B) The APD90, and APD50 values in control (blue, n = 49), VUS (grey, n = 119), and VUShom (orange, n = 29) atrial-like and ventricular-like iPSC-CMs. **p<0.01, ***p<0.001 when compared to healthy control iPSC-CMs. Error bars show S.E.M. (C) Development of spontaneous arrhythmogenicity in VUShom iPSC-CMs manifested as EADs (left) or triggered beat (right).
Genome-edited corrected iPSC-CMs (VUScorr) elicited phenotypic rescue of repolarization abnormality
We next utilized CRISPR/Cas9-mediated genome editing to correct the KCNH2T983I variant implicated in the presented VUS to validate the pathogenicity of the suspected causative variant (Online Figures 6 and 7). The resultant VUScorr iPSC-CMs had a correction of the causative KCNH2 variant (rs397514446) and served as an isogenic control line. As shown in Figure 7, the VUScorr iPSC-CMs displayed a marked reduction in APD prolongation compared to the VUS iPSC-CMs, without any incidence of EADs or triggered activity, such that the AP recordings resembled those from healthy control lines (APD90 of 452.62 ± 3.7 and 332.2 ± 61.1 ms in VUScorr vs 367.17 ± 19.6 and 288.73 ± 27.7 ms in healthy control for ventricular- and atrial-like cells, respectively; Figure 7A–B). Similar findings were obtained in extracellular MEA recordings (cFPD of 297.4 ± 4.0 ms vs 291.2 ± 12.9 ms) that revealed a reversal of the LQTS phenotype (Figure 7C). Moreover, voltage clamp recordings of IKr from VUScorr demonstrated a significant increase in tail current density compared to the VUS line (1.46 ± 0.24 vs 0.73 ± 0.13 pA/pF, p<0.01) (Figure 7D–F). Furthermore, no change was observed in voltage dependence of IKr activation parameters (V1/2 and k) when compared to healthy control or VUS iPSC-CMs (Figure 7G). We also recorded intracellular calcium concentration ([Ca2+]i) transients (elicited by electrical stimulation at 0.5 Hz) from the VUScorr iPSC-CMs and compared it to the VUS iPSC-CMs. Notably, in comparison to VUS, VUScorr iPSC-CMs displayed normal electrical (non-arrhythmic) activity (Online Figure 8A), lower diastolic [Ca2+]i, and acceleration of cytosolic [Ca2+]i decay as indicated by a faster rate constant and shorter decay tau (p<0.001, Online Figure 8B–E). Taken together, these findings suggest that genome-edited correction of the KCNH2T983I variant influences the IKr expression and reverses the abnormal phenotype in VUS iPSC-CMs.
Figure 7. Genome-edited corrected iPSC-CMs (VUScorr) showed rescue of abnormal phenotype.
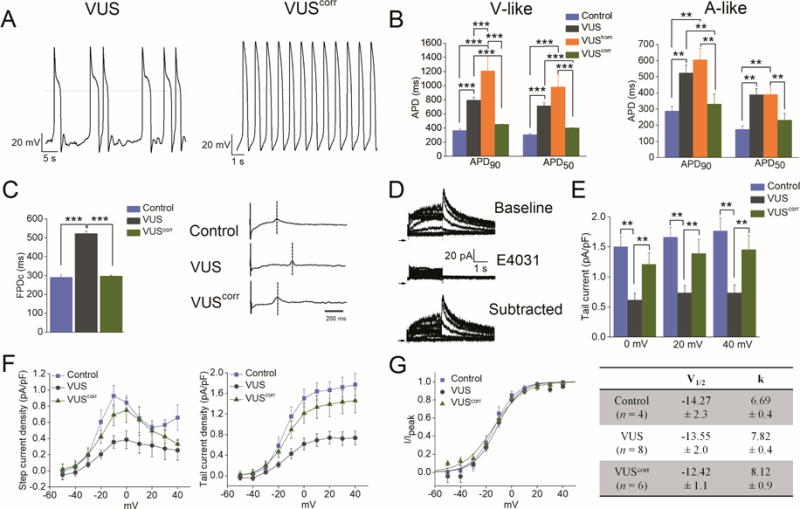
(A) Representative AP recordings from (left) VUS and (right) VUScorr iPSC-CMs. (B) Summary of APD90 and APD50 from control (blue), VUS (grey), VUShom (orange), and VUScorr(green) iPSC-CMs (**p<0.01, ***p<0.001). (C) Left panel, summary of rate-matched Fridericia’s corrected field potential duration (FPDc) values from control (blue), VUS (grey), and VUScorr (green) iPSC-CMs (n = 7). FPDc = FPD/(Beat Period)^0.3333. Error bars show S.E.M. ***p<0.001. Right panel, representative MEA recordings from control (top), VUS (middle), and VUScorr (bottom) iPSC-CM monolayer. (D) Single cell voltage-clamp recordings of IKr, measured from VUScorr iPSC-CMs. Top, baseline recordings. Middle, recording following administration of E-4031 (2 μM). Bottom, E-4031-sensitive current (IKr) defined by digital subtraction of the two currents. (E) Summary of peak tail IKr density in pA/pF from control (blue; n = 4), VUS (grey; n = 8), and VUScorr (green; n = 6) iPSC-CMs at membrane potentials; 0, +20, and +40 mV (**p<0.01). (F) Average I-V curves for IKr step current (left) and tail current (right) density in pA/pF for control (blue), VUS (grey), and VUScorr (green) iPSC-CMs. (G) Left panel, average peak tail current normalized to the maximal current following repolarization to −40 mV (I/Ipeak) in control (blue), VUS (grey), and VUScorr (green) iPSC-CMs. Right panel, summary of V1/2 and k parameters of IKr compared for control, VUS, and VUScorr iPSC-CMs. Mean values ± S.E.M. are shown. Comparison of iPSC lines was performed with one-way ANOVA, p<0.05 being considered statistically significant.
Discussion
In recent years, genetic testing has emerged as a routine clinical tool (25) to perform cascade screening of LQTS patients. While these tests have significantly improved the rapid identification of disease-causing variants in patients, the results are often inconclusive due to incomplete penetrance and variable expressivity (26). This issue is further compounded by the identification of hundreds of variants of unknown significance (VUS) in LQTS-susceptibility genes, which is currently based on combining data from population frequency and in silico modeling, along with published data on co-segregation and functional studies in heterologous expression systems (9,10).
Identification of VUS designation in LQTS is clinically important, as these variants may be used to guide genotype-specific management or facilitate rapid screening of potentially at-risk relatives. Unfortunately, this can put candidate patients, families, and ordering physicians in a frustrating state of clinical limbo (27,28). Under such circumstances, patient-specific electrophysiological characterization of these variants to distinguish pathogenic mutations from benign rare variants is crucial not only for accurate diagnosis but also for appropriate clinical management. This new approach represents a significant advancement for precision medicine in the management of LQTS disorders.
Recent advances in CRISPR genome editing and iPSC-CM platforms can provide a valuable clinical application to decipher the pathogenicity of VUS and better predict arrhythmia risk, but this approach remains unproven to date. Since the original discovery of iPSC technology in 2007, iPSCs have proven to be a robust model for studying various cardiac disease mechanisms (29–31), for drug screening (16,32,33) and for validating a KCNJ2 variant (10), despite their relative immaturity with respect to human adult cardiomyocytes. Although iPSC-based models for verified LQT2 mutations such as severely symptomatic A614V (13), A561T (15) and asymptomatic R176W (14) mutations in KCNH2 have been previously reported, VUS that lack any functional data or family history were not studied for LQT2 using the iPSC platform.
In the present study, healthy control and VUS iPSCs were differentiated into functional cardiomyocytes, and detailed electrophysiological analysis was performed. Consistent with the patient’s clinical phenotype, VUS iPSC-CMs recapitulated the hallmark features of LQTS disorder, including a delayed repolarization and mild arrhythmogenicity compared to the healthy control. Although APD prolongation was observed in 54% of total cells recorded, only 12.9% of cells exhibited arrhythmogenic activity that manifested as EADs. These findings suggest that similar to studies on asymptomatic R176W by Lahti et al. (14) and in contrast to more severe phenotype reported by Itzhaki et al. (13) and Matsa et al. (15), the T983I variant exhibits moderate arrhythmogenicity. Moreover, MEA analysis performed on a monolayer of cells revealed a significant prolongation of the FPDc in VUS iPSC-CMs compared to healthy control cells, corroborating the single cell patch clamp results. Finally, calcium imaging studies also confirmed an increased incidence of arrhythmia and higher diastolic calcium in VUS iPSC-CMs compared to isogenic control iPSC-CMs.
To further investigate the underlying mechanism of the variant pathogenicity, single cell voltage-clamp recordings of IKr were performed. We observed a 41–44% decrease in IKr tail current density in VUS vs. healthy control iPSC-CMs. The IKr density values obtained in our control lines (1.46 – 1.77 pA/pF) are in agreement with IKr previously reported in iPSC-CMs (0.55–1.9 pA/pF) (13,14). Importantly, biophysical analysis of hERG WT (control) and hERG T983I (VUS) further revealed that the mutant channels exhibited largely similar voltage dependence of gating as the WT channels based on the activation parameters. Furthermore, Western blot data confirmed that the underlying mechanism of the variant pathogenicity is reduced total hERG protein and deficiency of mature channel trafficking to the cell surface.
In previous reports, LQTS iPSC-CMs have been shown to exhibit a higher sensitivity to proarrhythmia due to their already compromised repolarization reserve when compared to healthy control cells (14,16). Consistent with the repolarization reserve hypothesis (34), VUS iPSC-CMs displayed enhanced proarrhythmicity to CiPA high-risk torsadogenic drugs such as dofetilide, ibutilide, and azimilide. In contrast, treatment with a recently discovered hERG channel activator (ICA-105574) enhanced IKr density and normalized APD in VUS iPSC-CMs, suggesting a potential pharmacotherapy for LQT2-specific patients. Collectively, these findings support the notion that IKr is implicated in the disease mechanism encoded by the KCNH2T983I variant.
A unique advantage of genome editing technology in disease modeling is the ability to study variants in an isogenic background to rule out phenotypic variability from epigenetic differences or unknown genetic modifiers. In addition, in a reciprocal approach, genome editing can be used to correct known variants in iPSC lines to generate isogenic controls for comparison, rather than using unrelated samples or unaffected family members (35). Using the CRISPR/Cas9 genome editing approach, we first introduced the homozygous variant T983I in a healthy control line and then corrected the variant in the patient line followed by examination of the electrophysiological properties of both genome-edited lines. Similar to our findings in the parent lines, the genome edited homozygous line (VUShom) exhibited characteristic LQTS phenotype with significantly prolonged APDs and generation of baseline arrhythmias such as EADs and triggered activity. In line with our hypothesis, and similar to the verified LQT2 mutation A561V, the VUShom iPSC-CMs had a higher percentage of baseline arrhythmias compared to heterozygous patient VUS line. In contrast, the corrected line (VUScorr) exhibited properties similar to the healthy control line, confirming the causative role of the underlying single KCNH2 mutation in the patient.
In summary, we generated iPSC-CMs from a VUS carrier of LQT2 and demonstrated their abnormal electrophysiological phenotype. We delineated VUS pathogenicity by using genome editing to selectively correct the variant in KCNH2 to normalize the aberrant cellular phenotype, and finally introduced the homozygous KCNH2 variant in an otherwise healthy individual to exacerbate the disease phenotype (Central Illustration). These findings provide the first report of utilizing iPSC-CMs and CRISPR genome editing technology as tools to examine the pathogenicity of VUS in LQTS. This stands to add to the yield of clinical genetic testing in this population and advance the care of patients with LQTS to improve their quality of life and appropriately manage their risk of SCD. In the future, this approach can be expanded to other channelopathies to accelerate progress toward realizing the promise of precision medicine in inherited arrhythmia.
Central Illustration. Patient-in-a-dish platform for elucidating VUS pathogenicity.
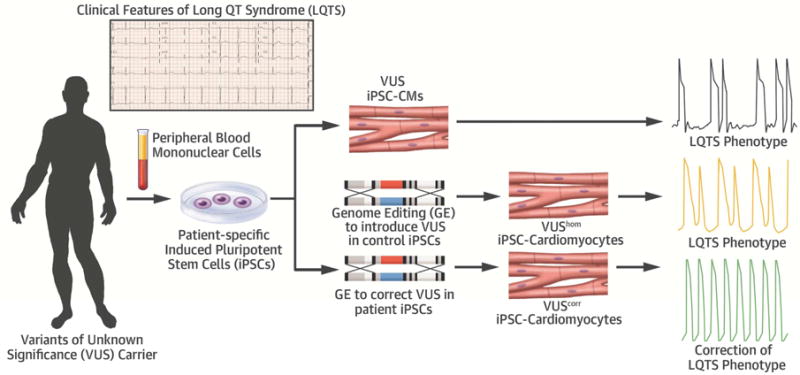
Patient-specific iPSC-CMs generated from a VUS (KCNH2T983I) carrier exhibited prolonged APD due to reduced IKr density compared to healthy control (black trace). Introduction of the homozygous variant in the healthy control iPSCs recapitulated a severe LQTS phenotype (orange trace) whereas correction of the VUS in patient iPSCs rescued the observed electrophysiological abnormalities (green trace). Thus genome editing of iPSC-CMs can potentially offer a unique precision medicine approach to decipher VUS pathogenicity in a dish. This robust approach may bring a major advancement in the care of LQTS patients to improve their quality of life and appropriately manage their risk of sudden death.
Supplementary Material
CLINICAL PERSPECTIVES.
Competency in Medical Knowledge
Selective genome editing of patient-specific cardiomyocytes derived from induced pluripotent stem cells (iPSCs) can identify pathogenic variants in an animal model of arrhythmogenic cardiac channelopathy and correct the aberrant cellular phenotype.
Translational Outlook
Future studies based on human iPSCs and genome editing could provide a personalized approach to drug therapy for patients with congenital long QT syndrome and other inherited conditions associated with cardiac arrhythmias.
Acknowledgments
We thank Katy C Claiborn for critical reading of the manuscript. We gratefully acknowledge funding support from National Institutes of Health (NIH) T32 EB009035 (PG), NIH R01 HL113006, NIH R01 HL128170, and NIH R24 HL117756 (JCW).
ABBREVIATIONS
- AP
Action potential
- APD
Action potential duration
- Cas9
CRISPR associated protein 9
- CiPA
Comprehensive in Vitro Proarrhythmia Assay
- CRISPR
Clustered regularly interspaced short palindromic repeats
- EAD
Early afterdepolarization
- hERG
human ether-a-go-go related gene
- ICD
Implantable cardioverter defibrillator
- IKr
Rapidly activating delayed rectifier K+ current
- iPSC
Induced pluripotent stem cells
- iPSC-CMs
Induced pluripotent stem cell-derived cardiomyocytes
- LQTS
Long QT syndrome
- MEA
Multielectrode array
- PBMC
Peripheral blood mononuclear cell
- SCD
Sudden cardiac death
- VUS
Variant of unknown significance
- VUScorr
Genome edited corrected VUS
- VUShom
Genome edited homozygous VUS
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors have no conflicts of interest to disclose.
References
- 1.Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–76. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 4.Anderson ME. “We are not alone”: ion channel mutations in a long QT syndrome cohort. Heart Rhythm. 2005;2:1106–7. doi: 10.1016/j.hrthm.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 5.Giudicessi JR, Ackerman MJ. Genetic testing in heritable cardiac arrhythmia syndromes: differentiating pathogenic mutations from background genetic noise. Curr Opin Cardiol. 2013;28:63–71. doi: 10.1097/HCO.0b013e32835b0a41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–33. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 7.Kaufman ES, Priori SG, Napolitano C, et al. Electrocardiographic prediction of abnormal genotype in congenital long QT syndrome: experience in 101 related family members. J Cardiovasc Electrophysiol. 2001;12:455–61. doi: 10.1046/j.1540-8167.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- 8.Marjamaa A, Salomaa V, Newton-Cheh C, et al. High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann Med. 2009;41:234–40. doi: 10.1080/07853890802668530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, Mendenhall JL, Kroncke BM, et al. Predicting the functional impact of KCNQ1 variants of unknown significance. Circulation: Cardiovascular Genetics. 2017:10. doi: 10.1161/CIRCGENETICS.117.001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gélinas R, El Khoury N, Chaix M-A, et al. Characterization of a human induced pluripotent stem cell–derived cardiomyocyte model for the study of variant pathogenicity. Validation of a KCNJ2 mutation. 2017:10. doi: 10.1161/CIRCGENETICS.117.001755. [DOI] [PubMed] [Google Scholar]
- 11.Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–62. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lian X, Hsiao C, Wilson G, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848–57. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 14.Lahti AL, Kujala VJ, Chapman H, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5:220–30. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsa E, Dixon JE, Medway C, et al. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J. 2014;35:1078–87. doi: 10.1093/eurheartj/eht067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang P, Lan F, Lee AS, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Liang P, Lan F, et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64:451–9. doi: 10.1016/j.jacc.2014.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fatima A, Kaifeng S, Dittmann S, et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS One. 2013;8:e83005. doi: 10.1371/journal.pone.0083005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanguinetti MC. HERG1 channel agonists and cardiac arrhythmia. Curr Opin Pharmacol. 2014;15:22–7. doi: 10.1016/j.coph.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Zou B, Yu H, et al. Modulation of hERG potassium channel gating normalizes action potential duration prolonged by dysfunctional KCNQ1 potassium channel. Proc Natl Acad Sci U S A. 2012;109:11866–71. doi: 10.1073/pnas.1205266109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerlach AC, Stoehr SJ, Castle NA. Pharmacological removal of human ether-a-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574) Mol Pharmacol. 2010;77:58–68. doi: 10.1124/mol.109.059543. [DOI] [PubMed] [Google Scholar]
- 22.Asayama M, Kurokawa J, Shirakawa K, et al. Effects of an hERG activator, ICA-105574, on electrophysiological properties of canine hearts. J Pharmacol Sci. 2013;121:1–8. doi: 10.1254/jphs.12220fp. [DOI] [PubMed] [Google Scholar]
- 23.Cavero I, Holzgrefe H. Comprehensive in vitro Proarrhythmia Assay, a novel in vitro/in silico paradigm to detect ventricular proarrhythmic liability: a visionary 21st century initiative. Expert Opin Drug Saf. 2014;13:745–58. doi: 10.1517/14740338.2014.915311. [DOI] [PubMed] [Google Scholar]
- 24.Mehta A, Sequiera GL, Ramachandra CJ, et al. Re-trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovasc Res. 2014;102:497–506. doi: 10.1093/cvr/cvu060. [DOI] [PubMed] [Google Scholar]
- 25.Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amin AS, Pinto YM, Wilde AA. Long QT syndrome: beyond the causal mutation. J Physiol. 2013;591:4125–39. doi: 10.1113/jphysiol.2013.254920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ackerman MJ. Genetic purgatory and the cardiac channelopathies: Exposing the variants of uncertain/unknown significance issue. Heart Rhythm. 2015;12:2325–31. doi: 10.1016/j.hrthm.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Giudicessi JR. Machine Learning and Rare Variant Adjudication in Type 1 Long QT Syndrome. Circulation: Cardiovascular Genetics. 2017:10. doi: 10.1161/CIRCGENETICS.117.001944. [DOI] [PubMed] [Google Scholar]
- 29.Lan F, Lee AS, Liang P, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun N, Yazawa M, Liu J, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang P, Sallam K, Wu H, et al. Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of Brugada Syndrome. J Am Coll Cardiol. 2016;68:2086–2096. doi: 10.1016/j.jacc.2016.07.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris K, Aylott M, Cui Y, Louttit JB, McMahon NC, Sridhar A. Comparison of electrophysiological data from human-induced pluripotent stem cell-derived cardiomyocytes to functional preclinical safety assays. Toxicol Sci. 2013;134:412–26. doi: 10.1093/toxsci/kft113. [DOI] [PubMed] [Google Scholar]
- 33.Mehta A, Ramachandra CJA, Singh P, et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur Heart J. 2017 doi: 10.1093/eurheartj/ehx394. [DOI] [PubMed] [Google Scholar]
- 34.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 35.Bassett AR. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. 2017 doi: 10.1007/s00335-017-9684-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.