

Abstract
Xenopus laevis has long been a popular model for studies of development and, based on the use of cell-free extracts derived from its eggs, as a model for reconstitution of cell cycle regulation and other basic cellular processes. However, work over the last several years has shown that intact Xenopus eggs and embryos are also powerful models for visualization and characterization of cell cycle-regulated cytoskeletal dynamics. These findings were something of a surprise, given that the relatively low opacity of Xenopus eggs and embryos was assumed to make them poor subjects for live-cell imaging. In fact, however, the high tolerance for light exposure, the development of new imaging approaches, new probes for cytoskeletal components and cytoskeletal regulators, and the ease of microinjection make the Xenopus oocytes, eggs, and embryos one of the most useful live-cell imaging models among the vertebrates. In this review, we describe the basics of using X. laevis as a model organism for studying cell division and outline experimental approaches for imaging cytoskeletal components in vivo in X. laevis embryos and eggs.
1 INTRODUCTION
1.1 LIVE IMAGING OF XENOPUS EGGS AND EMBRYOS
As model systems, eggs and embryos of the African clawed frog, Xenopus laevis, are most commonly associated with investigations of development or reconstitution of complex cellular processes in vitro. For good reason: there are literally thousands of studies in which the strengths of the Xenopus model system have been exploited to provide insights into vertebrate developmental biology and basic eukaryotic cell processes. However, the wide-spread appreciation of the strengths of Xenopus eggs and embryos for developmental biology and reconstitution has tended to obscure the fact that live imaging of the intact eggs and embryos is also remarkably useful for studies of cell biology.
It may initially seem that their thickness, yolkiness, and pigment render X. laevis eggs and embryos poor subjects for high-resolution live imaging studies, but the reality is very different. While it is difficult, if not impossible, to image deep into living Xenopus eggs and embryos, many important cell processes are confined to the cell cortex, which is easily accessible to a standard laser scanning confocal microscope (Woolner, Miller, & Bement, 2009). Further, the dynamics of the mitotic spindle can be visualized by the time the embryonic epithelial layer develops in blastulae and gastrulae, as can other important structures such as cell–cell junctions and nascent cilia. And, of course, unlike cultured cell models, the intact embryos come replete with all of the relevant context in the form of extracellular matrix, cell–cell adhesions, forces, and so forth.
Xenopus has several further advantages that allow for imaging studies that would be difficult in other systems. First, the cells are not very sensitive to light, allowing continuous imaging of eggs and embryos for many minutes or even hours with light intensities that would kill most other cells. Second, microinjection is easy because the cells are very robust. Consequently, vetting new fluorescent probes is much easier and more time efficient than in other systems, as are double or even triple injections. Third, their size and hardiness makes them easy to manipulate, a feature that not only permits the kind of physical perturbations that are so useful for studies of morphogenesis (Kim & Davidson, 2013) but also makes mounting for imaging straightforward. Fourth, they develop externally and can be cultured and imaged in simple saline solutions at room temperature, obviating the need for temperature-controlled stages. Fifth, their size and pigmentation can be exploited to permit visualization of important cell cycle and developmental transitions with no more than the aid of a dissecting microscope. Sixth, cell division in the embryonic epithelium is brisk, even after the cell cycle slows down at midblastula, proceeding in a wave-like manner such that any given field of view either has or soon will have many cells undergoing mitosis (Fig. 1).
FIG. 1.
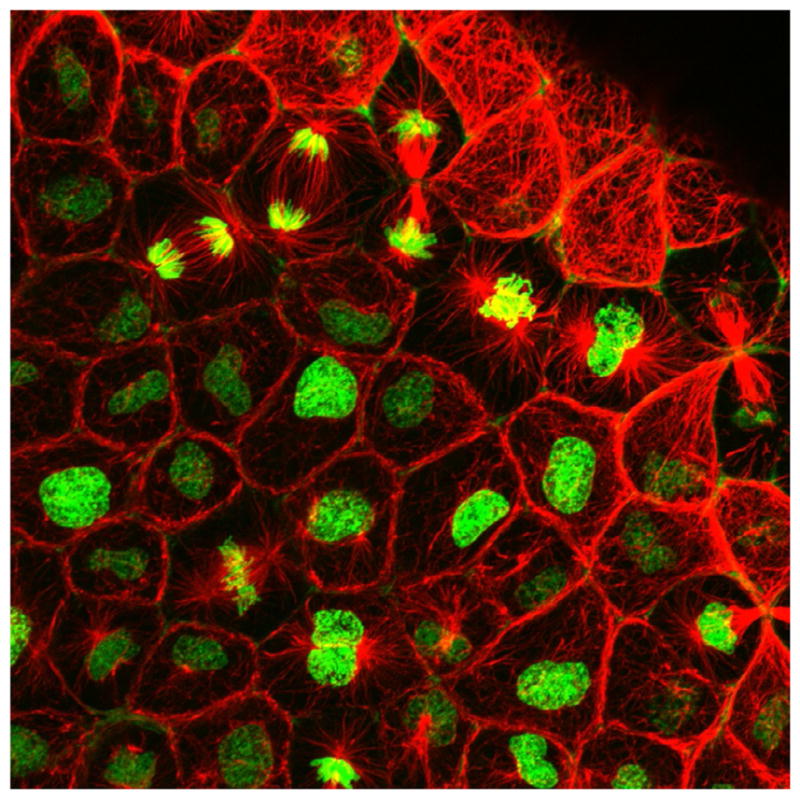
A confocal micrograph of an embryonic Xenopus epithelium expressing RNA encoding fluorescent histone H2B (green), and microtubules labeled with immunofluorescence (mouse antitubulin, red). Note the abundance of cells in different stages of mitosis.
Below, we will first provide examples where live-cell imaging of Xenopus eggs and early embryos has been used to reveal important or unexpected features of meiosis and mitosis, and then describe the basic methods of imaging these processes. However, it should be kept in mind that live imaging in this system also has an excellent track record of discovery in other areas of cell biology. For example, exocytosis-triggered actin coating of secretory granules was first discovered in Xenopus eggs (Sokac, Co, Taunton, & Bement, 2003; Yu & Bement, 2007) but is now known to accompany the exocytosis of secretory granules in many systems (Miklavc et al., 2012; Milberg et al., 2017; Rousso, Schejter, & Shilo, 2016; Sokac & Bement, 2006). Similarly, local activation of Rho GTPases and cytoskeletal mobilization during cell repair were first described in Xenopus oocytes and embryos (Benink & Bement, 2005; Clark et al., 2009; Mandato & Bement, 2001, 2003) and subsequently demonstrated for repair of many different cell types (Abreu-Blanco, Verboon, & Parkhurst, 2011, 2014; Kono, Saeki, Yoshida, Tanaka, & Pellman, 2012; Lin et al., 2012; McDade, Archambeau, & Michele, 2014; Nakamura, Verboon, & Parkhurst, 2017). More recently, live imaging of early Xenopus embryos has been used to investigate the dynamics of epithelial cell–cell junctions (Higashi, Arnold, Stephenson, Dinshaw, & Miller, 2016; Reyes et al., 2014), formation of cilia (Chung et al., 2014; Kim et al., 2010), and dynamic remodeling of the epithelial layer (Sedzinski, Hannezo, Tu, Biro, & Wallingford, 2016). In short, if a given cellular process occurs within several micrometers of the embryo surface, this model can be used to study it.
1.2 MEIOSIS
During meiotic maturation in Xenopus eggs, pigment granules are cleared from the approximate middle of the animal pole and the meiotic spindles assemble in this region (known as the white spot). White spot formation has proven highly useful to those interested in cell cycle dynamics. For example, in the 1970s and 1980s, researchers who were interested in the regulation of meiotic maturation made extensive use of white spot formation as a readout for cell cycle reentry (Maller, 1990). More recently, Ferrell and colleagues exploited this feature of Xenopus meiosis to demonstrate that the meiotic reentry decision is controlled by a bistable switch (Ferrell and Machleder, 1998).
The white spot is also useful for studying the dynamics and regulation of meiotic spindles and polar body emission, a highly asymmetric form of cytokinesis. As the white spot forms before the spindle is fully assembled, it provides a clear landmark that allows the researcher to predict the site of both meiotic spindle assembly and polar body formation. Multifocal plane (4D) time-lapse imaging of the white spot revealed that polar body emission occurs via the activation of the small GTPase, Rho, in a ring-like zone at the animal pole (Bement, Benink, & Von Dassow, 2005; Fig. 2). The ring of Rho activity surrounds a disc-like patch of activity of another small GTPase, Cdc42 (Ma et al., 2006; Zhang et al., 2008), and the two proteins coordinate polar body emission not only in Xenopus oocytes, but in mammalian oocytes as well (e.g., Dehapiot, Carrière, Carroll, & Halet, 2013; Pomerantz et al., 2012; Zhang et al., 2014). This indicates that local, complementary Rho and Cdc42 activation is likely a common feature of polar body emission.
FIG. 2.

Polar body emission in a Xenopus egg expressing fluorescent tubulin (red) to label the meiotic spindle and GFP-rGBD (green) to label active Rho. Top row shows an en face view, revealing formation of a ring-like zone of Rho activity around the nascent polar body; bottom rows show z-views of the same process. Time in min:s. Scale bar is 25μm.
From Bement, W. M., Benink, H. A., & Von Dassow, G. (2005). A microtubule-dependent zone of active RhoA during cleavage plane specification. Journal of Cell Biology, 170(1), 91–101. https://doi.org/10.1083/jcb.200501131.
Liu and colleagues have used multifocal plane (4D) time-lapse imaging of meiosis in Xenopus eggs to reveal several surprising features of this process. First, they found that Aurora B kinase plays a role in the regulation of spindle bipolarity (Shao et al., 2012). Second, by combining 4D imaging with a novel, single-egg genotyping approach, they demonstrated that meiosis in this system lacks a spindle assembly checkpoint (Shao, Li, Ma, Chen, & Liu, 2013). Surprisingly, it is nonetheless quite accurate (Liu, Shao, Wang, & Liu, 2014). Most recently, they have provided evidence that meiotic progression may depend on nanodomain calcium increases in the region around the meiotic spindle (Li, Leblanc, He, & Liu, 2016).
1.3 MITOSIS
The activation and inactivation of cyclin-dependent kinase 1 (Cdk1), the so-called master regulator of the cell cycle, can be visualized in living Xenopus embryos by cycles of cortical contraction and relaxation, evident via time-lapse imaging using just a dissecting microscope. As with white spot formation, this feature of Xenopus embryos was used as a means to speed investigation of cell cycle control mechanisms in the 1970s and 1980s (e.g., Hara, Tydeman, & Kirschnert, 1980). In a much more recent study (Anderson, Gelens, Baker, & Ferrell, 2017), cycles of contraction and relaxation were used to investigate the relationship between cell–cell adhesion and the timing of Cdk1 activation and inactivation.
The spindle cannot be directly imaged for most of the early rounds of cell division, but by the time the early embryonic epithelium has formed (~6–12h postfertilization, depending on the temperature of culture), spindles can be imaged using time lapse or 4D microscopy in conjunction with the appropriate fluorescent probes (Woolner, O’Brien, Wiese, & Bement, 2008). Such approaches, combined with morpholino-mediated loss of function, revealed a role for myosin-10 (Myo10)—an actin-based motor protein that binds microtubules (Hirano et al., 2011; Weber, Sokac, Berg, Cheney, & Bement, 2004)—in mitotic spindle stability and dynamics (Woolner et al., 2008; Woolner & Papalopulu, 2012; Fig. 3). Remarkably, at least part of Myo10’s contribution to mitosis is somehow mediated through control of cell cycle progression, as the depletion of Myo10 (Woolner et al., 2008) or expression of its isolated “Myth4” domain (Sandquist, Larson, & Hine, 2016) greatly slows the metaphase–anaphase transition. While it is not yet known how widely required Myo10 is for cell division in other systems, Myo10 has been implicated in spindle formation in Xenopus and mammalian meiosis (Brieño-Enríquez et al., 2017; Weber et al., 2004), and in spindle stability and dynamics in several cultured cell types (Chan, Hsu, Liu, Lai, & Chen, 2014; Iwano et al., 2015; Kwon, Bagonis, Danuser, & Pellman, 2015; Kwon et al., 2008; Toyoshima & Nishida, 2007).
FIG. 3.

Time-lapse image series from confocal movie of a Xenopus embryonic epithelium expressing fluorescent tubulin (green) and fluorescent histone H2B (red). The top row shows a metaphase spindle in a control epithelial cell; the spindle undergoes a characteristic series of rotations and movements to and from the cortex before undergoing anaphase at 02:18. The bottom row shows a metaphase spindle in an epithelial cell depleted of myosin-10 via morpholino injection. The spindle is initially normal but becomes abnormally long and undergoes pole fragmentation. The metaphase–anaphase transition is greatly delayed, and when it happens (at 27:24), highly abnormal. Time in min:s.
Due to the accessibility and relative abundance of epithelial spindles in early Xenopus embryos, their dynamic behavior could be quantitatively analyzed using time-lapse imaging combined with a computational approach (Larson & Bement, 2017). This analysis revealed that the mitotic spindles undergo a stereotyped “dance” that commences after the achievement of metaphase and ceases upon the beginning of anaphase. The spindles first undergo a slow rotation that brings them approximately parallel to the long axis of the cells. Then, the spindles move rapidly toward and then rapidly away from the cortex. The close approach of spindle poles to the cortex was correlated with anaphase onset, suggesting a possible link between these two events. While most studies have focused on spindle movements occurring prior to metaphase, a growing body of evidence indicates that postmetaphase spindle movements are a common feature of intact epithelia (e.g., Adams, 1996; Peyre et al., 2011). However, because previous studies in such systems were conducted with much longer sampling intervals (30–300s vs 6s) and without the benefit of automated analysis, it is unknown if the dynamic features of Xenopus epithelial spindles are conserved in other systems.
1.4 CYTOKINESIS
The large size of Xenopus blastomeres, the easy accessibility of their cortices to time lapse and 4D imaging, and their high tolerance for laser light make them excellent models for studies of cytokinesis. These features were exploited to reveal that the spindle controls cytokinesis via generation of an equatorial stripe (zone) of Rho activity in Xenopus embryos as well as those of several echinoderms (Bement et al., 2005; Fig. 4). Subsequently, the same features figured prominently in the demonstration of the importance of Rho turnover (aka Rho GTPase “flux”) during cytokinesis, and the contributions of the cytokinetic regulator, MgcRacGAP, to Rho flux in the cytokinetic Rho zone (Miller & Bement, 2009). Live imaging of Xenopus gastrulae also revealed flux of both Rho and Rac (Breznau, Semack, Higashi, & Miller, 2015) in the cell–cell junctions. MgcRacGAP is known to localize to the plus ends of microtubules; live imaging studies from Xenopus were used to demonstrate that this localization is critically dependent on a putative EB1-binding motif within MgcRacGAP (Breznau, Murt, Blasius, Verhey, & Miller, 2017).
FIG. 4.

Blastomeres from a Xenopus embryo expressing GFP-rGBD to detect active Rho. Cytokinetic Rho zones indicated by arrowheads. Time in min:s. Scale bar is 25μm.
From Bement, W. M., Benink, H. A., & Von Dassow, G. (2005). A microtubule-dependent zone of active RhoA during cleavage plane specification. Journal of Cell Biology, 170(1), 91–101. https://doi.org/10.1083/jcb.200501131.
Using a high-contrast marker for microtubules, it was found that the Xenopus Rho zone forms in regions of low cortical microtubule density, as do the Rho zones in embryos of echinoderms (Von Dassow, Verbrugghe, Miller, Sider, & Bement, 2009; Fig. 5). Using faster and higher resolution imaging, it was found that the Xenopus cortex continuously generates waves of Rho activity that stimulate subsequent waves of actin assembly and disassembly (Bement et al., 2015; Fig. 6; see also below). These studies were performed in parallel with work on starfish eggs and embryos, which also generate Rho activity and F-actin waves. While there are differences between the two systems, in both starfish and Xenopus, cytokinesis ensues as the waves become focused at the cell equator. It remains to be seen if such Rho and F-actin waves are conserved features of cytokinesis, but recent work has revealed cortical Rho activity waves in cultured mammalian cells (Graessl et al., 2017).
FIG. 5.

Blastomeres from a Xenopus embryo expressing GFP-rGBD to detect active Rho (red) and 3xmch-EMTB to detect microtubules (blue). Cytokinetic Rho zones form in regions of low cortical microtubule density (arrowheads). Time in min:s.
From Von Dassow, G., Verbrugghe, K. J. C., Miller, A. L., Sider, J. R., & Bement, W. M. (2009). Action at a distance during cytokinesis. Journal of Cell Biology, 187(6), 831–845. https://doi.org/10.1083/jcb.200907090.
FIG. 6.

Blastomeres from a Xenopus embryo expressing GFP-UtrCh to detect actin filaments. Top left panel shows a blastomere with the area used for the kymograph indicated by a thin rectangle. Bottom left panel shows resultant kymograph which reveals F-actin waves as jagged lines. White lines on left show relationship of kymograph to images above and to the right of the kymograph. Montage shows cortex of blastomere at different times. Cytokinesis ensues at 11:29; this corresponds to fourth white line on kymograph. Time in min:s.
From Bement, W. M., Leda, M., Moe, A. M., Kita, A. M., Larson, M. E., Golding, A. E., et al. (2015). Activator–inhibitor coupling between Rho signalling and actin assembly makes the cell cortex an excitable medium. Nature Cell Biology, 17(11), 1471–1483. https://doi.org/10.1038/ncb3251.
Live imaging of Xenopus embryos has also revealed other unexpected features of cytokinesis. For example, cytokinesis differs morphologically in different cell types, apparently as a function of differences in the degree of development of the spindle midzone, which, in turn, appears to vary as a function of expression of the microtubule bundling protein PRC (Kieserman, Glotzer, & Wallingford, 2008). In addition, the first demonstration of a role for the serine–threonine kinase MELK in cytokinesis was obtained by a combination of live imaging and morpholino-based depletion in Xenopus embryos (Le Page, Chartrain, Badouel, & Tassan, 2011).
2 METHODS
2.1 GENERAL CONSIDERATIONS
2.1.1 Microscopes and imaging
As implied earlier, we have had far more success with confocal imaging than other approaches, and in particular, laser scanning confocals work well. On the few occasions we have tried spinning disc confocals, we had difficulty obtaining sufficient signal. With respect to different microscope brands, we currently use the Bruker Prairie Laser Scanning confocal microscope for most of our imaging, which collects high-quality images with remarkable stability and ease of use. However, in the past, we have also successfully used the Fluoview FV1000 and the Zeiss 1024, so we suspect that most laser scanning confocal microscopes will work.
Recently, we have started using the Bruker Opterra swept-field confocal microscope (Davenport, Sonnemann, Eliceiri, & Bement, 2016). Like the Prairie, it is also stable and simple to use. However, its great advantage is speed—it permits collection of 5–15 512 × 512 focal planes in 1–2 s. Its major disadvantage for use with Xenopus is that it is more prone to suffer from image degradation at deeper focal planes than the laser scanning confocal microscopes. For many processes, this is not an issue (see above).
High quality 4D movies can be generated using a 60 ×, 1.4 NA oil objective with an electronic zoom of 2 (note that swept-field confocal microscopy does not permit an electronic zoom; so, to achieve higher magnification, we use the 100 × objective with this instrument). One simple trick we use if we wish to increase imaging speed while maintaining high resolution with the laser scanning confocal microscope is to image using a 1024 × 100- or 1024 × 200-pixel box. This works simply because the laser moves much more quickly across the sample than down the sample.
2.1.2 Wild-type vs albino eggs and embryos
Wild-type Xenopus are pigmented, as are their oocytes, eggs, and embryos. This is often a very useful feature, for example, when judging meiotic maturation (see above) or when the samples need to be mounted with a particular orientation. For imaging cortical cytoskeletal dynamics (e.g., waves of Rho activity and F-actin assembly and disassembly, cortical microtubules), perfectly acceptable movies can be obtained with wild-type eggs and embryos. However, when imaging spindles, which are below the cortex, albinos provide a clear advantage in terms of signal intensity and clarity.
2.1.3 Simple, useful image display and processing tricks
Most of our image analysis and processing are performed using FIJI, which can be obtained free of charge at https://imagej.net/Fiji/Downloads. One of its most useful features is the Bio-Formats importer (Plugins→Bio-Formats→Bio-Formats importer), which permits the user to open an enormous variety of different file types, useful for users of microscopes from more than one company.
2.1.3.1 Kymographs (aka space-time plots)
Kymographs are extraordinarily useful for extracting dynamic spatial information t from time-lapse movies. These can be made by importing an image stack into FIJI, drawing a line over the desired area of the stack, and then reslicing the image (Image→Stacks→Reslice) to generate a kymograph of the spacing and orientation chosen by the user.
2.1.3.2 Montages
The montage tool is not only useful for preparing figures for display and publication but also for revealing unexpected features of time-lapse movies. For example, the montage can reveal repetitive patterns in cortical waves of Rho activity and actin assembly–disassembly that might be overlooked when viewing the original movies (Bement et al., 2015). To make a montage, import the image stack and crop the area of interest by drawing a box and selecting “Image→Crop.” Then, create the montage by selecting “Image→Stacks→Make Montage” and specify the layout, scale, intervals, and increment.
2.1.3.3 Registration
In some samples, the entire field of view moves as the egg or embryo contracts. If the movement is not too fast, nor too extensive, it can be corrected by using registration. StackReg is a useful registration plugin that can be downloaded from Imagej.net. After installing StackReg in your plugins, open the image stack and select “Plugins→StackReg.” Then select the “rigid body” option.
2.1.3.4 Subtraction movies
Subtraction movies allow the viewer to selectively visualize changes in a dynamic sample by dropping out unchanging features of the data. This can be useful in samples where relatively stable, bright background structures or signal hampers visualization of relatively dynamic but dimmer signal of interest. For example, cortical Rho activity waves in control egg or embryo samples can be hard to detect due to the relatively high background of the unbound Rho activity probe (namely, GFP-rGBD; see Benink & Bement, 2005) and due to stable foci of Rho activity in the cortex. A subtraction movie reduces the contribution of both the unbound probe and the stable foci, providing more contrast to the Rho activity waves (Fig. 7).
FIG. 7.
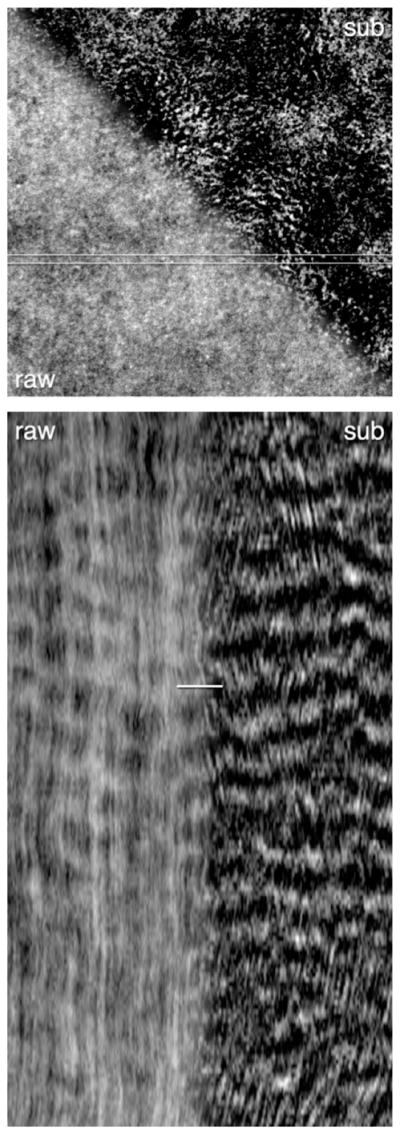
Activated Xenopus egg expressing 3 × GFP-rGBD to label active Rho: single frame (top) and kymograph (bottom), raw (left), and subtracted data (right, t0 − t−3), highlighting rising Rho activity. The kymograph demonstrates Rho activity waves without processing (left) and with subtraction processing (right). Arrowheads indicate waves.
From Bement, W. M., Leda, M., Moe, A. M., Kita, A. M., Larson, M. E., Golding, A. E., et al. (2015). Activator–inhibitor coupling between Rho signalling and actin assembly makes the cell cortex an excitable medium. Nature Cell Biology, 17(11), 1471–1483. https://doi.org/10.1038/ncb3251.
Subtraction movies are made by opening an image stack, duplicating it (Image→Duplicate), and then removing a set number of frames from the beginning of one copy of the movie and the same number of frames from the end of the other copy using the “delete slice” command, which is found in the “Stk” menu on the toolbar. (The number of frames removed depends on the dynamic feature being emphasized and is best determined empirically. For the Rho activity waves in frogs, 30 s worth of frames works well.) After shortening the two copies of the movies, the second copy is subtracted from the first using the Image Calculator (Process→Image Calculator).
3 CONSTRUCT PREPARATION
3.1 PLASMID DNA PREPARATION
Clone constructs of interest into a vector containing promoters amenable to in vitro transcription, and an SV40 polyadenylation site. We used the pCS2+ vector. The following methods assume the use of the SP6 promoter, but other promoters that allow for in vitro transcription, such as T3 or T7, may be used. The coding sequence of the protein of interest should be downstream in the sense direction of the promoter. It is important to include a start codon prior to the start of the coding sequence, and we have found it beneficial to use a partial Kozak start sequence of GCCACCATG.
3.2 LINEARIZATION OF DNA
Before mRNA can be made, plasmid DNA must be linearized. Choose a restriction enzyme with a single-cut downstream of the coding sequence for the DNA digest. For pCS2+, we use the Not1 site and prepare the following reaction in a 1.5-mL microtube:
| Plasmid DNA | 5μg |
| 10 × Buffer | 10μL (1×) |
| Not1-HF | 2μL (20,000UmL−1) |
| Nuclease-free water | To 100μL |
Incubate the reaction for a minimum of 2h at 37°C, and then clean up the linearized DNA using the Promega Wizard® SV Gel and PCR Clean-Up System (Promega, #A9282). Perform the elution step using 30μL of nuclease-free water. Concentrate the sample to 10μL using a SpeedVac (Savant, DNA100). It is strongly recommended to confirm the quality and size of linear DNA by running 0.5μL of the sample on a DNA gel. Store linearized DNA at −20°C.
3.3 IN VITRO TRANSCRIPTION OF CAPPED RNA
To avoid sample contamination and degradation, it is important to carry out any RNA work in an RNase-free area and use sterile technique. Use RNaseZap™ (Sigma-Aldrich, #R2020-250ML) to clean benches and labware prior to making RNA, and always use nuclease-free water (ThermoFisher, #AM9937). A dedicated workstation for RNA handling is ideal.
Perform in vitro transcription using the mMESSAGE mMACHINE® SP6 Transcription Kit (Ambion, #AM1340). Prepare a 20 μL reaction as follows and incubate for 2–4h at 37°C:
| 2× NTP/CAP | 10μL |
| 10× Reaction buffer | 2μL |
| Linear DNA | 1μg |
| Enzyme mix | 2μL |
| Nuclease-free water | Up to 20μL |
3.4 POLYADENYLATION OF RNA
Addition of a 3′ poly(A) tail will result in faster expression and longer lifetime of the mRNA, but it is not required for all mRNAs. If polyadenylation is desired, set up the following reaction (ThermoFisher, #AM1350):
| RNA reaction | 20μL |
| Nuclease-free water | 36μL |
| 5 × E-PAP buffer | 20μL |
| 25mM MgCl2 | 10μL |
| 10mM ATP | 10μL |
Aliquot 2.5μL of the reaction to use as a control for the RNA confirmation gel and place on ice or store at −80°C. Add 4μL of E-PAP polymerase to the reaction tube and mix gently. Incubate the reaction for an additional 45min to 1h.
3.5 CLEANUP AND CONFIRMATION OF RNA
Cleanup can be accomplished using an RNA cleanup kit, such as the RNeasy MinE-lute Cleanup Kit (Qiagen, #74204). After cleanup, determine the RNA concentration and 260/280 ratio (we use a NanoDrop, ThermoFisher). It is also important to confirm the size and quality of the RNA by running a sample on a denaturing gel alongside a molecular size standard (Ambion, #AM7150).
Prepare a 10 × MOPS stock: Dissolve 41.8g MOPS in 700mL DEPC water. Adjust the pH to 7.0 with NaOH. Add 20mL of 1M sodium acetate (made with DEPC water). Add 20mL of 0.5M EDTA (pH 8.0, made with DEPC water). Bring the final volume to 1L with DEPC water. Protect the solution from light.
Make a 1 × MOPS running buffer using DEPC-treated water (0.05% (v/v)) and the 10 × MOPS stock. Then, prepare a 0.5-cm gel using a 1% agarose solution in 1 × MOPS. Transfer agarose solution to a 50-mL conical tube. When agarose is cool to the touch, add 37% formaldehyde such that the final concentration becomes 2%–3% in solution and pour the gel. Make loading dye with 50μL of loading dye (Invitrogen, #AM8546G) and 1μL of ethidium bromide. Then, add 4μL of loading dye to 1μL of sample or standard. Boil samples for 2min, at 100°C. Load 5μL of each sample into gel and run at 50V until samples travel at least halfway down the gel (around 2–3h). Once confirmed, it is recommended to dilute the remaining RNA stocks to 1mgmL−1 and aliquot 1μL samples into 1.5-mL microtubes. Seal the lids with parafilm and store at −80°C.
4 ACTIVATED EGG EXPERIMENTS
This section describes an experimental setup for working with activated eggs. Activated eggs are oocytes that have been matured via progesterone and are primed for fertilization. Eggs can be “tricked” into activating without fertilization through pricking with a glass needle or incubation with a calcium ionophore. Activated eggs are advantageous for studying cytoskeletal dynamics because they are easily acquired from oocyte surgeries, they provide a one-cell, large field of view for imaging, and they allow for more flexibility in terms of experimental timing when compared to embryos.
4.1 SURGERY
Prepare 1L of 5 × Barth’s concentrated stock (store at 16–18°C).
| NaCl | 440mM |
| KCl | 5mM |
| NaHCO3 | 12mM |
| MgSO4 | 41mM |
| Ca(NO3)2 | 16.5mM |
| CaCl2 | 34mM |
| HEPES | 50mM |
pH solution to 7.4 with NaOH.
Prepare 1L of 1 × Barth’s from the concentrated stock and readjust the pH to 7.4. Supplement the 1 × Barth’s with the following antibiotics:
| Ampicillin | 1mL (25mgmL−1 stock) |
| Tetracycline | 0.5mL (12mgmL−1 stock) |
| Gentamicin | 0.1g (Sigma, #G3632) |
Store the 1 × Barth’s solution at 16–18°C. Extract oocytes from a female frog by removing pieces of ovary tissue according to the surgical procedure detailed in Kay and Peng (1991). Place the ovary tissue in a Petri dish containing 1 × Barth’s solution and snip the tissue into small bunches of ~40 oocytes with surgical scissors. Transfer the oocytes to a 50-mL conical tube and wash with 1 ×Barth’s until the solution is clear. Then, pour off any liquid and transfer the oocytes to Petri dishes, taking care to not overcrowd the dish. High densities result in premature death and maturation. Let the oocytes sit for at least 30min at 16°C, and then proceed with collagenase treatment.
4.2 COLLAGENASE TREATMENT
Collagenase treatment allows for faster isolation of oocytes and prevents needle clogging during injection. Transfer oocytes to 50-mL conical tubes and pour off Barth’s solution. Add collagenase type 1 powder (ThermoFisher, #17100017) to create an 8mgmL−1 solution. Gently rock the conical to mix in the collagenase, and then set on a shaker in a slightly elevated position with the cap off. Rock gently at ~50 RPM for 1h at 16°C. After the collagenase treatment, rinse three times in 1 × Barth’s, five times with deionized water, and four more times with 1 × Barth’s. Separate into clean Petri dishes and incubate in 1 × Barth’s at 16°C until use. Oocytes can remain healthy for up to 4–5 days with daily media changes.
4.3 DEFOLLICULATION
Oocytes must be defolliculated prior to injection. The follicle cells appear as a shimmery translucent layer surrounding the oocyte. Defolliculation is difficult immediately following collagenase treatment, so it is advisable to wait at least 1–2h before attempting. First, isolate stage VI oocytes in a separate Petri dish filled with fresh 1 × Barth’s solution. Refer to Kay and Peng (1991) for help with staging oocytes. Stage VI oocytes should be large, turgid and have even pigmentation in the animal hemisphere. If time permits, it can be helpful to let the sorted oocytes sit for 20min prior to defolliculation. This allows the follicle cells to stick to the bottom of the Petri dish. Using forceps, gently grasp the oocyte. With a second pair of forceps, kept closed, lightly roll across the top of the oocyte in the direction of the first set of forceps, gently pushing down and rolling the oocyte away. This motion, if done correctly, will cause the follicle cells to stick to the bottom of the dish and the oocyte to roll out of them. Do not try to pinch the follicle cells or use excessive force while defolliculating, as this can damage the oocyte. Transfer defolliculated oocytes into a small Petri dish with fresh 1 × Barth’s solution. It is advisable to have at least 25 oocytes per injection condition.
4.4 MICROINJECTION OF OOCYTES
Microinjection should be performed as detailed in Kay and Peng (1991). We use the following equipment:
Microinjector: Picoinjector, Harvard Apparatus, #PLI-100, with foot pedal
Needle puller: Flaming/Brown Micropipette Puller, Sutter Instrument CO, #P-97
Needle glass: Micro Capillary Tubes, Disposable, Kimble Chase, #71900-10
Dissecting microscope: 0.7–4.2 × dissecting microscope, American Optical, #570
Micromanipulator: Narishige, #MN-153
Needles are pulled using the following settings on the micropipette puller:
Pressure: 500
Heat: 520
Pull: 80
Velocity: 60
Time: 70
Needles are then broken with forceps to the desired width (see below for detailed instructions).
Microinjections should typically be performed around 24h prior to imaging, depending on the constructs used and incubation temperature. Our lab typically injects 1 day before imaging and incubates at 16°C overnight. Times and expression levels can be modulated by incubating at higher temperatures (such as 18°C) or by leaving oocytes at room temperature for 2–5h after injection prior to 16–18°C overnight incubation. Calibrate the needle with nuclease-free water (Fisher, #AM9937) to accommodate 40nL injections. To do this, first place a droplet of oil on a glass micrometer slide with 0.01mm increments (Ted Pella, #2280-13). Then, inject nuclease-free water into the oil droplet, measure the diameter of the droplet, and calculate the total volume. A droplet diameter of ~0.42mm gives an injection volume of roughly 40nL. For our equipment, the pressure is usually set between 6.0 and 8.0psi, and the injection time is at 1s. A 40 nL injection volume can be used to inject ~ 25 oocytes with 1μL of RNA.
To load the RNA, place a square of parafilm on the stage and add the RNA in a droplet. It is important to work quickly in order to minimize evaporation while loading RNA into the needle. Be sure not to suck up air, as this can prevent liquid from being ejected. Once the RNA has been loaded, test the injection setup by pressing the foot pedal and watching for a droplet at the end of the needle. Then, proceed to inject oocytes through the animal hemisphere and into the cytoplasm. The animal hemisphere is the upper half of the cell, above the cell equator. In wild-type oocytes, the animal hemisphere is a dark brown color. Periodically check that the needle has not clogged by injecting into the air. Rinse the needle in between RNA samples by loading and clearing nuclease-free water three times prior to loading the next RNA type. After the injections are completed, the samples can be incubated at 16–18°C until progesterone treatment (which is performed at the end of the day). Alternatively, oocytes can be left at room temperature until progesterone treatment if quicker expression of the injected RNA is desired.
If one wishes to study polar body emission via egg activation, oocytes can be microinjected with a caged form of inositol-1,4,5-trisphosphate (InsP3) (Sokac & Bement, 2006). When uncaged, InsP3 increases internal calcium levels and thus, stimulates activation. After microinjection, induce maturation in oocytes via progesterone incubation overnight (see below). Once mature, uncaging and activation can be achieved by briefly exposing the cells to UV light from an arc lamp or laser through the microscope objective.
4.5 MATURATION WITH PROGESTERONE
To induce maturation, treat oocytes with 5μgmL−1 progesterone (Fisher Scientific, #C225650250) for 15min, then place them in dishes with fresh 1 × Barth’s solution and incubate overnight at 16–18°C.
4.6 ACTIVATION OF MATURE EGGS
Assess maturationthe following morning by looking for the appearance of a white spot at the top of the animal hemisphere. It is best to let the eggs sit at room temperature for at least 30min–1h to ensure that eggs are fully matured. Discard any immature oocytes from the dish. Activation can be achieved by pricking the oocytes with a glass injection needle or by a calcium ionophore, such as ionomycin (Sigma-Aldrich, #I0634-1MG). We have found that eggs survive longer after activation in MMR solutions than in Barth’s, therefore, prior to activation, switch matured eggs in to a solution of 0.1 × MMR.
-
10 × MMR stock:
1M NaCl 20mM KCl
20mM CaCl2
10mM MgCl2
50mM HEPES
Mix together in 800mL of deionized water. Adjust the pH to 7.4 and then bring volume to 1L. Store at 4°C.
For ionophore activation, make a solution of 10μgmL−1 ionomycin in 0.1 × MMR and fill a separate dish. Activate eggs by placing them in ionophore solution for 2–4min. Activation can be assessed within 2–3min by upward pigment contraction in the animal hemisphere. Eggs should then be rinsed three times in 0.1 × MMR and then maintained at 16°C and checked periodically for expression. Depending on the construct, expression starts around 2h after activation.
5 EMBRYO EXPERIMENTS
5.1 TESTES PREP MEDIA
Isolated male testes are stored in testes prep media. To make, assemble:
| 1 × MMR | 20mL (see Section 4.6 for preparation of 10 × stock) |
| FBS | 5mL, heat inactivated |
| Gentamycin | 25μL, (50mgmL−1 stock) |
| Penicillin/streptomycin | 125μL, (10UμL−1 stock) |
Mix together all materials and sterile filter with 0.22-μm syringe filter into 5 × 5mL aliquots. Store at 4°C.
5.2 TESTES ISOLATION
Prior to collecting embryos, isolate the testes of a male frog according to the surgical procedure detailed in Kay and Peng (1991). Place testes in a small dish of 1 × MMR and use forceps to remove blood vessels and fat. Take care to avoid nicking or damaging the tissue. Place the cleaned testes in a small Petri dish of testes prep media and seal with parafilm. Keep on ice or store at 4°C. Testes tissue will remain viable for 2 weeks after surgery at 4°C.
5.3 PRIMING FEMALES (OPTIONAL)
Female frogs can be primed with a low dose of human chorionic gonadotropin (hCG) (Merk Animal Health, Chorulon®) 3–5 days before ovulation to increase production and the chances of successful egg laying. Inject 50U of hCG (1UμL−1 stock) into the dorsal lymph sac of a female frog. Keep the needle angle shallow and you should see the dorsal lymph sac swell as it fills. Place the frog in a separate tank from the rest of the colony and perform full ovulation injections the night before intended egg collection.
5.4 FULL OVULATION
Place ovulation frogs in separate tanks with system water supplemented with 20mM NaCl. Inject 700U hCG into the dorsal lymph sac. Keep the needle angle shallow. You should see the dorsal lymph sac swell as it fills. Place the frogs back in tanks and incubate at 17–18.5°C overnight. Frogs should lay eggs within 12–14h after injection (see below).
5.5 HARVESTING AND FERTILIZING EMBRYOS
Fill a small Petri dish (60 × 15 mm) with 1 × MMR until the liquid is slightly raised above the rim of the dish. Pick up the female frog such that the hind legs are separated, and hold the frog over the Petri dish. The natural movements of the female will usually result in egg release, but gentle lateral strokes along the side of the frog can also help stimulate egg laying. Be careful not to touch the cloaca, and do not rub or “squeeze” the back of the frog. There is very little subcutaneous fat, and these actions can lead to bruising. Collect for 2–3 min at a time, dipping the cloaca into the 1 × MMR periodically to catch the eggs. To fertilize, make sure the eggs are dispersed in the dish and not overcrowded or clumped together. Remove excess liquid until eggs are barely covered and do not float. Move a whole testis into dish and cut off a small piece with forceps. Normally, ¼ of a testis is sufficient; depending on the age of the tissue, more or less can be used. Place the remaining testes back in the testes prep media. Shred the testis to release the sperm and drag it around the dish, touching all of the eggs. Incubate for 30–60 s without shaking. Then, gently fill the dish with 0.1 × MMR to mobilize the sperm. Mark the time of fertilization, which is time zero for all experimental timing. Incubate the fertilized eggs at room temperature for 30 min then check for fertilization and rotation using a dissecting microscope. The fertilization envelope should rise off the eggs and become visible as a second layer inside the outer jelly coat. The animal hemisphere will be facing upward for all of the embryos at this stage. The first cleavage furrow will form around 2 h postfertilization.
5.6 DeJELLYING EMBRYOS
The embryo jelly coat is made up of polysaccharides, and treatment with cysteine disrupts the disulfide bonds. Wait at least 20min after fertilization before dejellying, but after that dejellying can be done any time prior to injection. Be sure to dejelly the fertilized eggs from different frogs separately as the thickness of the jelly coat varies from frog to frog. First, make up a 2% cysteine (w/v) solution in 0.1 × MMR and pH to 7.8 with 10N NaOH. Seal the solution and mark the time; it is good for 1 day.
Gather the following materials:
2% Cysteine solution
50-mL conical
1 × MMR
0.1 × MMR
Petri dish (60 × 15mm)
Transfer the embryos to a 50-mL conical tube and replace the 0.1 × MMR solution with 2% cysteine. Bring the volume to 25mL with the cysteine solution. Gently swirl the fertilized eggs until they pack with no space between them. Quickly wash with 1 × MMR (3 × 25mL), pouring off the buffer after each wash. Then, wash with 0.1 × MMR (3 × 25mL), pouring off the buffer after each wash. Transfer washed embryos to a new Petri dish at low density and remove damaged, nonrotated, or otherwise unwanted fertilized eggs. Store in 0.1 × MMR at 16°C until injection.
5.7 MICROINJECTION OF EMBRYOS
Equipment and general setup have been described under Section 4.4. Calibrate the needle to inject 5nL for embryo injections (~0.25mm droplet diameter). Try to keep the pressure between 15 and 22psi and the injection time between 0.5 and 1s. The ideal stage to inject embryos is at the two-cell stage, but injections at the four-cell stage are also possible. If injecting at the four-cell stage, a 2.5-nL injection volume should be used. Fill the injection dish with injection buffer (0.1 × MMR+5% Ficol (Sigma, #F4375-100G)) and transfer embryos to the dish. Load the needle with RNA and inject each cell at the two-cell stage. Periodically check that the needle is not clogged by injecting into the air. Due to the small size of the needle tip, separate needles should be used for different RNA samples. After the injections are complete, place the embryos at room temperature (if imaging on the same day), or at 16°C for next day imaging.
5.8 MICROINJECTION OF MORPHOLINOS
X. laevis is tetraploid, which makes genetic modification difficult. Morpholinos are a useful tool for knocking down expression. We order morpholinos from Gene Tools, LLC (http://www.gene-tools.com), who provide an oligo design service for free. Oligos are shipped as lyophilized stocks, which can be made into solution with distilled water and stored at room temperature. Although it is recommended to maintain stocks at or below 1mM to avoid solubility issues, we have often found that needle concentrations of up to 4–5mM can be used. Typically, both blastomeres at the two-cell stage are injected with morpholino to ensure that the knockdown occurs throughout the entire embryo. A second injection of RNA or protein can be done at the four-cell stage. Morpholinos targeting the 5′UTR of endogenous RNA can be used in tandem with RNA for the same protein, but lacking a 5′UTR, in order to achieve gene replacement.
5.9 RECOVERY OF OVULATION FEMALES
Septicemia is most often induced after ovulation, either by stress or infection caused by handling. It is important to allow the ovulation frogs to recover before reintroduction to the main colony. This reduces the risk of septicemia and avoids hormones released by the ovulated females getting introduced into the system water. Females should be kept in a 20-mM NaCl solution of system water during egg laying and for at least 12 h after. Frogs typically recover after 6–7 days and may then be reintroduced to the main colony. Change the water every day during the recovery period.
6 MOUNTING SAMPLES FOR LIVE IMAGING
There are two common ways to mount samples for live imaging. Detailed instructions for using each method are given below.
6.1 MOUNTING ON GLASS SLIDES
The simplest method is to mount samples on plain, glass slides using a ring of vacuum grease and a coverslip. This approach has the virtue of compensating for the slight differences in size that may occur in different batches of eggs and embryos, because the coverslip is pushed down as much or as little as needed. It has the disadvantage that only one side of the egg or embryo can be imaged (the side facing the coverslip) unless the sample is remounted.
First, fill a 10-mL syringe with vacuum grease (Dow Corning) to serve as an applicator. Next, make a ring of vacuum grease on a plain glass slide (Fisher, #12-549-3). The ring should be approximately the height of the sample and relatively even so that the coverslip lies flat when applied. After the ring is made, mount a sample in the center in a small droplet of media. Avoid using too much liquid, as overflow can reduce the grip of the vacuum grease on the coverslip, once applied. Orient the embryo or activated egg such that the animal hemisphere is facing directly up. Then, under a dissecting microscope, gently lower the coverslip on top of the sample and grease ring, pressing evenly on all four corners at once. Maneuver the coverslip and depress the grease enough to gently flatten the sample, taking care not to lyse the sample or introduce air bubbles. If the sample has shifted in orientation, use the coverslip to reorient the animal hemisphere facing up (Fig. 8).
FIG. 8.

(A) Schematic showing how to mount a sample on a glass slide. First, a ring of vacuum grease is applied to the slide. Then, a sample is mounted in a droplet of media to the center of the ring and oriented such that the animal hemisphere is facing up. Lastly, a coverslip is lowered on top of the sample and grease ring. (B) Schematic showing how to mount a sample on a metal slide. First, a ring of vacuum grease is applied around the center hole. Then, a coverslip is pressed into the grease ring, and the slide is flipped over. A second ring of vacuum grease is applied, and the sample is then mounted in the center in a droplet of media, animal hemisphere facing up. Lastly, a coverslip is lowered on top of the sample and grease ring.
6.2 MOUNTING ON METAL SLIDES
Metal slide mounting devices are made from 0.5 to 0.9mm thick metal rectangles of the same dimensions as glass microscopy slides, with a 3–6mm hole punched in the middle. Essentially, a coverslip is attached to each side, with the sample pressed between them. This approach has two advantages. First, it doubles the amount of surface available for imaging, because the sample can be imaged through either coverslip. Second, it tends to hold the samples more firmly in place, because the coverslips cannot flex as much as they can when mounted onto glass slides as above. However, it has the disadvantage that it is harder to compensate for variations in sample thickness.
To prepare metal slides, create a ring of vacuum grease around the center hole on one side of the slide. The height of the grease ring should be thinner than with glass slides, as the thickness of the metal and the second grease ring will add to the overall chamber depth. Attach a coverslip on top of the grease ring and press down to seal it in place. Flip the slide over and create a second grease ring on the other side. Mount the sample in a small droplet of media inside the imaging chamber. Take care to reduce the amount of media to avoid overwetting the grease ring. Orient the sample so that the animal hemisphere is facing directly upward. Then, under a dissecting microscope, gently lower a coverslip on top of the sample and press evenly on all four corners to fuse it to the grease ring. Maneuver the coverslip and depress the grease enough to gently flatten the sample, taking care not to lyse the sample or introduce air bubbles. If the sample has shifted in orientation, use the coverslip to reorient the animal hemisphere facing up (Fig. 8).
References
- Abreu-Blanco MT, Verboon JM, Parkhurst SM. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. Journal of Cell Biology. 2011;193(3):455–464. doi: 10.1083/jcb.201011018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu-Blanco MT, Verboon JM, Parkhurst SM. Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Current Biology. 2014;24(2):144–155. doi: 10.1016/j.cub.2013.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RJ. Metaphase spindles rotate in the neuroepithelium of rat cerebral cortex. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1996;16(23):7610–7618. doi: 10.1523/JNEUROSCI.16-23-07610.1996. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8922417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GA, Gelens L, Baker JC, Ferrell JE. Desynchronizing embryonic cell division waves reveals the robustness of Xenopus laevis development. Cell Reports. 2017;21(1):37–46. doi: 10.1016/j.celrep.2017.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Benink HA, Von Dassow G. A microtubule-dependent zone of active RhoA during cleavage plane specification. Journal of Cell Biology. 2005;170(1):91–101. doi: 10.1083/jcb.200501131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Leda M, Moe AM, Kita AM, Larson ME, Golding AE, et al. Activator–inhibitor coupling between Rho signalling and actin assembly makes the cell cortex an excitable medium. Nature Cell Biology. 2015;17(11):1471–1483. doi: 10.1038/ncb3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. Journal of Cell Biology. 2005;168(3):429–439. doi: 10.1083/jcb.200411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breznau EB, Murt M, Blasius TL, Verhey KJ, Miller AL. The MgcRac-GAP SxIP motif tethers Centralspindlin to microtubule plus ends in Xenopus laevis. Journal of Cell Science. 2017;130(10):1809–1821. doi: 10.1242/jcs.195891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breznau EB, Semack AC, Higashi T, Miller AL. MgcRacGAP restricts active RhoA at the cytokinetic furrow and both RhoA and Rac1 at cell-cell junctions in epithelial cells. Molecular Biology of the Cell. 2015;26(13):2439–2455. doi: 10.1091/mbc.E14-11-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieño-Enríquez MA, Moak SL, Holloway JK, Cohen PE, Hayes M, Faragher A. NIMA-related kinase 1 (NEK1) regulates meiosis I spindle assembly by altering the balance between α-Adducin and Myosin X. PLoS One. 2017;12(10):e0185780. doi: 10.1371/journal.pone.0185780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PC, Hsu RYC, Liu CW, Lai CC, Chen HC. Adducin-1 is essential for mitotic spindle assembly through its interaction with myosin-X. Journal of Cell Biology. 2014;204(1):19–28. doi: 10.1083/jcb.201306083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MI, Kwon T, Tu F, Brooks ER, Gupta R, Meyer M, et al. Coordinated genomic control of ciliogenesis and cell movement by RFX2. eLife. 2014;3:e01439. doi: 10.7554/eLife.01439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Miller AL, Vaughan E, Yu HYE, Penkert R, Bement WM. Integration of single and multicellular wound responses. Current Biology. 2009;19(16):1389–1395. doi: 10.1016/j.cub.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport NR, Sonnemann KJ, Eliceiri KW, Bement WM. Membrane dynamics during cellular wound repair. Molecular Biology of the Cell. 2016;27(14):2272–2285. doi: 10.1091/mbc.E16-04-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehapiot B, Carrière V, Carroll J, Halet G. Polarized Cdc42 activation promotes polar body protrusion and asymmetric division in mouse oocytes. Developmental Biology. 2013;377(1):202–212. doi: 10.1016/j.ydbio.2013.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE, Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280(5365):895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- Graessl M, Koch J, Calderon A, Kamps D, Banerjee S, Mazel T, et al. An excitable Rho GTPase signaling network generates dynamic subcellular contraction patterns. The Journal of Cell Biology. 2017;216(12):4271–4285. doi: 10.1083/jcb.201706052. jcb.201706052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Tydeman P, Kirschnert M. A cytoplasmic clock with the same period as the division cycle in Xenopus eggs (surface contraction waves/cell cycle/cleavage/ time-lapse cinematography/biological oscillations) Cell Biology. 1980;77(1):462–466. doi: 10.1073/pnas.77.1.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Arnold TR, Stephenson RE, Dinshaw KM, Miller AL. Maintenance of the epithelial barrier and remodeling of cell-cell junctions during cytokinesis. Current Biology. 2016;26(14):1829–1842. doi: 10.1016/j.cub.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Hatano T, Takahashi A, Toriyama M, Inagaki N, Hakoshima T, et al. Structural basis of cargo recognition by the myosin-X MyTH4-FERM domain. The EMBO Journal. 2011;30(13):2734–2747. doi: 10.1038/emboj.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano S, Satou A, Matsumura S, Sugiyama N, Ishihama Y, Toyoshima F. PCTK1 regulates integrin-dependent spindle orientation via protein kinase a regulatory subunit KAP0 and myosin X. Molecular and Cellular Biology. 2015;35(7):1197–1208. doi: 10.1128/MCB.01017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay BK, Peng HB. Xenopus laevis: Practical uses in cell and molecular biology. San Diego: Academic Press, Inc; 1991. [PubMed] [Google Scholar]
- Kieserman EK, Glotzer M, Wallingford JB. Developmental regulation of central spindle assembly and cytokinesis during vertebrate embryogenesis. Current Biology. 2008;18(2):116–123. doi: 10.1016/j.cub.2007.12.028. [DOI] [PubMed] [Google Scholar]
- Kim HY, Davidson LA. Microsurgical approaches to isolate tissues from Xenopus embryos for imaging morphogenesis. Cold Spring Harbor Protocols. 2013;8(4):362–365. doi: 10.1101/pdb.prot073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010;329(5997):1337–1340. doi: 10.1126/science.1191184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono K, Saeki Y, Yoshida S, Tanaka K, Pellman D. Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell. 2012;150(1):151–164. doi: 10.1016/j.cell.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Kwon M, Bagonis M, Danuser G, Pellman D. Direct microtubule-binding by myosin-10 orients centrosomes toward retraction fibers and subcortical actin clouds. Developmental Cell. 2015;34(3):323–337. doi: 10.1016/j.devcel.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes and Development. 2008;22(16):2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M, Bement W. Automated mitotic spindle tracking suggests a link between spindle dynamics, spindle orientation, and anaphase onset in epithelial cells. Molecular Biology of the Cell. 2017;28(6):746–759. doi: 10.1091/mbc.E16-06-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page Y, Chartrain I, Badouel C, Tassan JP. A functional analysis of MELK in cell division reveals a transition in the mode of cytokinesis during Xenopus development. Journal of Cell Science. 2011;124(6):958–968. doi: 10.1242/jcs.069567. [DOI] [PubMed] [Google Scholar]
- Li R, Leblanc J, He K, Liu XJ. Spindle function in Xenopus oocytes involves possible nanodomain calcium signaling. Molecular Biology of the Cell. 2016;27:3273–3283. doi: 10.1091/mbc.E16-05-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Zhu H, Cai C, Wang X, Cao C, Xiao R, et al. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. The FASEB Journal. 2012;26(5):1875–1883. doi: 10.1096/fj.11-188599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Shao H, Wang H, Liu XJ. Meiosis I in Xenopus oocytes is not error-prone despite lacking spindle assembly checkpoint. Cell Cycle. 2014;13(10):1602–1606. doi: 10.4161/cc.28562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Benink HA, Cheng D, Montplaisir V, Wang L, Xi Y, et al. Cdc42 activation couples spindle positioning to first polar body formation in oocyte maturation. Current Biology. 2006;16(2):214–220. doi: 10.1016/j.cub.2005.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller JL. Xenopus oocytes and the biochemistry of cell division. Biochemistry. 1990;29(13):3157–3166. doi: 10.1021/bi00465a001. [DOI] [PubMed] [Google Scholar]
- Mandato CA, Bement WM. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. Journal of Cell Biology. 2001;154(4):785–797. doi: 10.1083/jcb.200103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandato CA, Bement WM. Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Current Biology. 2003;13(13):1096–1105. doi: 10.1016/S0960-9822(03)00420-2. [DOI] [PubMed] [Google Scholar]
- McDade JR, Archambeau A, Michele DE. Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB Journal. 2014;28(8):3660–3670. doi: 10.1096/fj.14-250191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklavc P, Hecht E, Hobi N, Wittekindt OH, Dietl P, Kranz C, et al. Actin coating and compression of fused secretory vesicles are essential for surfactant secretion—A role for Rho, formins and myosin II. Journal of Cell Science. 2012;125(Pt. 11):2765–2774. doi: 10.1242/jcs.105262. [DOI] [PubMed] [Google Scholar]
- Milberg O, Shitara A, Ebrahim S, Masedunskas A, Tora M, Tran DT, et al. Concerted actions of distinct nonmuscle myosin II isoforms drive intracellular membrane remodeling in live animals. Journal of Cell Biology. 2017;216(7):1925–1936. doi: 10.1083/jcb.201612126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AL, Bement WM. Regulation of cytokinesis by Rho GTPase flux. Nature Cell Biology. 2009;11(1):71–77. doi: 10.1038/ncb1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Verboon JM, Parkhurst SM. Prepatterning by RhoGEFs governs Rho GTPase spatiotemporal dynamics during wound repair. The Journal of Cell Biology. 2017;216(12):3959–3969. doi: 10.1083/jcb.201704145. jcb.201704145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyre E, Jaouen F, Saadaoui M, Haren L, Merdes A, Durbec P, et al. A lateral belt of cortical LGN and NuMA guides mitotic spindle movements and planar division in neuroepithelial cells. Journal of Cell Biology. 2011;193(1):141–154. doi: 10.1083/jcb.201101039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz Y, Elbaz J, Ben-Eliezer I, Reizel Y, David Y, Galiani D, et al. From ubiquitin-proteasomal degradation to CDK1 inactivation: Requirements for the first polar body extrusion in mouse oocytes. FASEB Journal. 2012;26(11):4495–4505. doi: 10.1096/fj.12-209866. [DOI] [PubMed] [Google Scholar]
- Reyes CC, Jin M, Breznau EB, Espino R, Delgado-Gonzalo R, Goryachev AB, et al. Anillin regulates cell-cell junction integrity by organizing junctional accumulation of Rho-GTP and actomyosin. Current Biology. 2014;24(11):1263–1270. doi: 10.1016/j.cub.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousso T, Schejter ED, Shilo BZ. Orchestrated content release from Drosophila glue-protein vesicles by a contractile actomyosin network. Nature Cell Biology. 2016;18(2):181–190. doi: 10.1038/ncb3288. [DOI] [PubMed] [Google Scholar]
- Sandquist JC, Larson ME, Hine KJ. Myosin-10 independently influences mitotic spindle structure and mitotic progression. Cytoskeleton. 2016;73(7):351–364. doi: 10.1002/cm.21311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedzinski J, Hannezo E, Tu F, Biro M, Wallingford JB. Emergence of an apical epithelial cell surface in vivo. Developmental Cell. 2016;36(1):24–35. doi: 10.1016/j.devcel.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Li R, Ma C, Chen E, Liu XJ. Xenopus oocyte meiosis lacks spindle assembly checkpoint control. Journal of Cell Biology. 2013;201(2):191–200. doi: 10.1083/jcb.201211041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Ma C, Zhang X, Li R, Miller AL, Bement WM, et al. Aurora B regulates spindle bipolarity in meiosis in vertebrate oocytes. Cell Cycle. 2012;11(14):2672–2680. doi: 10.4161/cc.21016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokac AM, Bement W. Kiss-and-coat and compartment mixing: Coupling exocytosis to signal generation and local actin assembly. Molecular Biology of the Cell. 2006;17(4):1495–1502. doi: 10.1091/mbc.E05-10-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokac AM, Co C, Taunton J, Bement W. Cdc42-dependent actin polymerization during compensatory endocytosis in Xenopus eggs. Nature Cell Biology. 2003;5(8):727–732. doi: 10.1038/ncb1025. [DOI] [PubMed] [Google Scholar]
- Toyoshima F, Nishida E. Integrin-mediated adhesion orients the spindle parallel to the substratum in an EB1- and myosin X-dependent manner. The EMBO Journal. 2007;26(6):1487–1498. doi: 10.1038/sj.emboj.7601599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dassow G, Verbrugghe KJC, Miller AL, Sider JR, Bement WM. Action at a distance during cytokinesis. Journal of Cell Biology. 2009;187(6):831–845. doi: 10.1083/jcb.200907090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KL, Sokac AM, Berg JS, Cheney RE, Bement WM. A microtubule-binding myosin required for nuclear anchoring and spindle assembly. Nature. 2004;431(7006):325–329. doi: 10.1038/nature02834. [DOI] [PubMed] [Google Scholar]
- Woolner S, Miller AL, Bement WM. Imaging the cytoskeleton in live Xenopus laevis embryos. Methods in Molecular Biology (Clifton, NJ) 2009;586:23–39. doi: 10.1007/978-1-60761-376-3_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolner S, O’Brien LL, Wiese C, Bement WM. Myosin-10 and actin filaments are essential for mitotic spindle function. Journal of Cell Biology. 2008;182(1):77–88. doi: 10.1083/jcb.200804062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolner S, Papalopulu N. Spindle position in symmetric cell divisions during epiboly is controlled by opposing and dynamic Apicobasal forces. Developmental Cell. 2012;22(4):775–787. doi: 10.1016/j.devcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HYE, Bement WM. Control of local actin assembly by membrane fusion-dependent compartment mixing. Nature Cell Biology. 2007;9(2):149–159. doi: 10.1038/ncb1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Duan X, Cao R, Liu HL, Cui XS, Kim NH, et al. Small GTPase RhoA regulates cytoskeleton dynamics during porcine oocyte maturation and early embryo development. Cell Cycle. 2014;13(21):3390–3403. doi: 10.4161/15384101.2014.952967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ma C, Miller AL, Katbi HA, Bement WM, Liu XJ. Polar body emission requires a RhoA contractile ring and Cdc42-mediated membrane protrusion. Developmental Cell. 2008;15(3):386–400. doi: 10.1016/j.devcel.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]