

Summary
Through a gain-of-function kinome screen, MEX3B was identified as a mediator of resistance to T cell immunotherapy not previously identified using CRISPR based screens. MEX3B is a post-transcriptional regulator of HLA-A, validating the critical role of tumor-intrinsic antigen presentation in T cell immunotherapy and indicating a new putative molecular target.
In this issue of Clinical Cancer Research, Huang and colleagues used a kinome library screen and identified MEX3B as an important player in melanoma resistance to T cell immunotherapy(1). Low expression of MEX3B was strongly associated with response in a cohort of patients with melanoma treated with anti-PD1 checkpoint blockade. In functional studies using patient-derived melanoma cell lines and autologous TIL, MEX3B overexpression inhibited recognition and killing of melanoma by autologous TIL. Notably, this effect of MEX3B was dependent on endogenous expression of HLA-A and could be reversed by overexpression of exogenous HLA-A. In elegant studies using a dual luciferase reporter assay, the authors demonstrate that MEX3B disrupts HLA-A by binding the 3′UTR of its mRNA (Figure 1). Indeed, MEX3B expression was inversely correlated with HLA-A expression in both the anti-PD1 treated cohort and in the skin cutaneous melanoma TCGA cohort.
Figure 1. Molecular screens reveal regulators of antigen presentation as key factors in tumor sensitivity to immunotherapy.
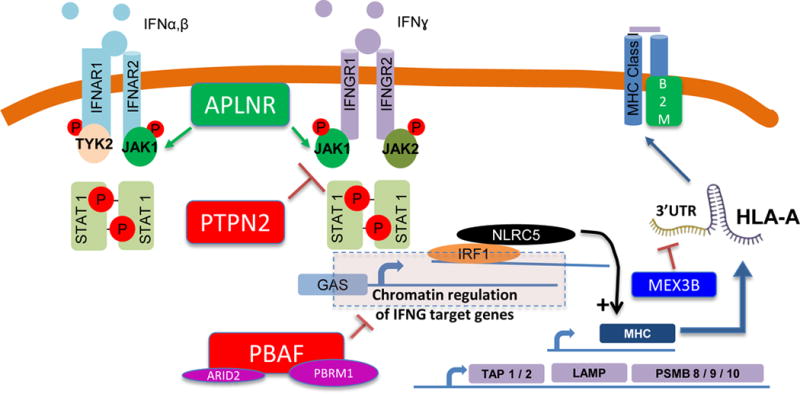
In the 1990s it was well documented that some patients who initially responded to cancer immunotherapies with interleukin-2 or tumor infiltrating lymphocyte adoptive cell transfer therapy may develop acquired resistance through loss of B2M, which leads to absence of surface expression of HLA class I. More recently, we identified defects in B2M from patients with acquired resistance to immune checkpoint blockade, along with defects in interferon signaling (JAK1, JAK2). Interferon signaling activates transcription of antigen processing machinery. Two in vitro and one in vivo CRISPR based screens have identified APLNR, PTPN2 and PBAF as immunotherapy targets, and each is a regulator of interferon signaling, and thereby each indirectly impacts downstream antigen presentation. The CRISPR screens also identified components of antigen processing machinery (e.g. TAP1, TAP2, and immunoproteasome subunits including PSMB9) as determinants of sensitivity to immunotherapy. NLRC5, a transcriptional regulator of MHC-I expression, has also been implicated in immunotherapy sensitivity in the CRISPR based screens. The work reported by Huang, et al. (1) in this issue of Clinical Cancer Research identifies MEX3B as a post-transcriptional regulator of HLA-A, via binding and disruption of the 3′UTR of the HLA-A mRNA.
The authors’ work follows in a line of recent studies using molecular screening methods to identify tumor-intrinsic mechanisms of resistance to T cell immunotherapy. Their approach was a gain-of-function screen to test the impact of a set of 384 genes from a kinome library on the sensitivity of melanoma to direct T cell killing in culture. In a similar approach, Patel, et al. used an in vitro assay to identify tumor-intrinsic genes important in regulating T cell anti-tumor efficacy(2). A CRISPR screen was performed on human melanoma cells in a 12 hour coculture with tumor-specific T cells. Of the genes most enriched in tumor cells surviving T cell killing, those involved in antigen presentation machinery were most prominent (in particular, HLA-A). The authors also identified APLNR as a critical gene for sensitivity of cancer cells to T cell killing through its association with JAK1 and interferon signaling (Figure 1). In a similar in vitro CRISPR screen approach, Pan et al. cocultured B16-F10 murine melanoma with tumor specific T cells and also identified key genes in antigen presentation (as well as interferon signaling and other pathways) as critical to T cell mediated anti-tumor immunity(3). However, in these CRISPR screens, MEX3B was not identified as a resistance mechanism, highlighting the importance of the approach by Huang, et al.
It is worth noting that mechanisms of resistance to direct T cell cytotoxicity in short-term in vitro coculture may not capture the complexity of resistance mechanisms to immune checkpoint blockade in vivo. As such, Manguso, et al. used an in vivo CRISPR screen to identify genes involved in sensitivity or resistance to immune checkpoint blockade(4). Their CRISPR screen was tested in a murine melanoma model treated with an irradiated tumor vaccine with or without anti-PD1 therapy, and identified interferon signaling as a critical pathway in sensitivity to therapy. They also identified PTPN2 as a negative regulator of antigen presentation (and therapeutic response) through decreased interferon gamma signaling sensitivity (Figure 1). That antigen presentation is identified as a critical pathway in the Huang, et al.’s kinome library screen, the CRISPR screens noted above, as well as the mutations identified in patients who have primary or acquired resistance to immune checkpoint blockade, is highly reassuring and biologically consistent(5). In each model – whether it is in vitro coculture, in vivo model of anti-PD1 therapy, or even an in vivo model of adoptive cell therapy – the end effector of anti-tumor function is the tumor-specific T cell. Thus, any deficiency in antigen presentation would render the T cells (and the immunotherapy) ineffective.
In conjunction with antigen presentation machinery, tumor interferon signaling has been well-defined as a critical component of response to immune checkpoint blockade. Disruptions in both type I and II interferon signaling, in the form of mutations in JAK1, JAK2, APLNR, STAT1, among others, have been described as mediators of resistance to immune checkpoint blockade. This is at least in part mediated by the indirect role of interferon signaling as an upstream promoter of antigen presentation machinery (Figure 1). Thus, better understanding of regulators of antigen presentation machinery downstream of interferon signaling could lead to approaches to overcome immunotherapy resistance in interferon signaling defective tumors. For that reason MEX3B is unique from the targets identified by the two CRISPR based screens described above. Unlike PTPN2 and APLNR, which regulate antigen presentation by impacting interferon sensing, MEX3B regulates antigen presentation at the level of HLA-A mRNA. These and other studies that provide insight into mechanisms of interferon-independent regulation of antigen presentation, shed light on approaches which may restore antigen presentation in tumors with interferon signaling defects.
More immediately, the relevance of these studies in the clinic relates to patient selection. Across different malignancies, T cell infiltration, PD-L1 expression, and mutational burden have been used as biomarkers for response to immune checkpoint blockade. However, there are an increasing number of studies describing defined molecular and genetic changes that impact antigen presentation, interferon signaling and ultimately, response to immune checkpoint blockade. It is plausible that a defined set of these molecular and genetic changes could serve as a biomarker to exclude non-responding and enrich for responding patients.
For defined molecules that negatively impact antigen presentation like MEX3B, development of targeted therapeutic approaches should be considered. The melanoma cell lines used by Huang, et al. had high basal expression of HLA-A, and thus disruption of MEX3B was unlikely to have a phenotypic impact. There are subsets of human melanoma with low basal expression of MHC-I molecules, and it would be intriguing to examine whether MEX3B inhibition or deletion could induce MHC-I overexpression. This would be particularly valuable for tumors lacking interferon signaling. Needless to say, Huang and colleagues have identified a regulator of antigen presentation downstream of interferon signaling that is associated with resistance to immune checkpoint blockade and in so doing, piqued interest in this gene as a combinatorial target.
Acknowledgments
The Parker Institute for Cancer Immunotherapy (A. Ribas), NIH grant R35 CA197633 (A. Ribas), RSNA Research Scholar Grant (A. Kalbasi), and the Ressler Family Fund (A. Ribas).
Footnotes
Conflict of Interest Statement: Dr. Kalbasi and Dr. Ribas have no competing financial or other conflicts of interest.
References
- 1.Huang L, Malu S, McKenzie JA, Andrews MC, Talukder AH, Tieu T, et al. The RNA-binding protein MEX3B mediates resistance to cancer immunotherapy by downregulating HLA-A expression. Clin Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-17-2483. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29496759. [DOI] [PMC free article] [PubMed]
- 2.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–42. doi: 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359:770–5. doi: 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–8. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]