

Abstract
Polynucleotide kinase/phosphatase (PNKP) has been implicated in non-homologous end joining (NHEJ) of DNA double-strand breaks (DSBs). To assess the consequences of PNKP deficiency for NHEJ of 3′-phosphate-ended DSBs, PNKP-deficient derivatives of HCT116 and of HeLa cells were generated using CRISPR/CAS9. For both cell lines, PNKP deficiency conferred sensitivity to ionizing radiation as well as to neocarzinostatin (NCS), which specifically induces DSBs bearing protruding 3′-phosphate termini. Moreover, NCS-induced DSBs, detected as 53BP1 foci, were more persistent in PNKP−/− HCT116 cells compared to their wild-type (WT) counterparts. Surprisingly, PNKP-deficient whole-cell and nuclear extracts were biochemically competent in removing both protruding and recessed 3′-phosphates from synthetic DSB substrates, albeit much less efficiently than WT extracts, suggesting an alternative 3′-phosphatase. Measurements by ligation-mediated PCR showed that PNKP-deficient HeLa cells contained significantly more 3′-phosphate-terminated and fewer 3′-hydroxyl-terminated DSBs than parental cells 5-15 min after NCS treatment, but this difference disappeared by 1 hour. These results suggest that, despite presence of an alternative 3′-phosphatase, loss of PNKP significantly sensitizes cells to 3′-phosphate-terminated DSBs, due to a 3′-dephosphorylation defect.
1. Introduction
Free radical-mediated DNA double-strand breaks (DSBs) are formed by fragmentation of deoxyribose, and typically bear 3′-phosphate or, less frequently, 3′-phosphoglycolate termini [1–3]. The bifunctional enzyme polynucleotide kinase/phosphatase (PNKP) specifically removes phosphate from 3′ ends of DSBs [4]. Diverse evidence indicates that this phosphatase activity is important for repair of radiation-induced DSBs by the nonhomologous end joining (NHEJ) pathway. PNKP is recruited to the NHEJ complex by interaction with XRCC4 [5], and its presence is essential for rejoining of DSBs bearing 5′-hydroxyl termini in human cell extracts [6]. Furthermore, knockdown of PNKP confers radiosensitivity in A549 lung cancer cells [7]. Consequently, PNKP has been proposed as a therapeutic target for radiosensitization in cancer therapy [8]. In order to determine the importance of PNKP in NHEJ, and to determine whether and how 3′-phosphate DSB termini are resolved when PNKP is absent, PNKP was disrupted in HeLa and HCT116 cells. DSB 3′-phosphate processing was examined in cells and cell extracts, and the response of PNKP-deficient cells to NCS, a radiomimetic antibiotic that specifically induces 3′-phosphate DSBs, was also assessed. The results indicate that loss of PNKP confers a severe deficiency in resolution of 3′-phosphate DSBs and increases their cytotoxicity, despite presence of an alternative but less efficient 3′-phosphatase.
2. Methods
2.1 Cell lines and CRISPR knockout of PNKP
HCT116 and HeLa cells were purchased from the American Type Culture Collection through Cedarlane Corporation (Burlington, ON). XRCC4−/− HCT116 cells [9], constructed by homologous recombination, were obtained from Dr. Eric A. Hendrickson, University of Minnesota.
The constructs for CRISPR knockout of PNKP in HCT116 cells were generated by incorporating the short guide sequences targeting exon three (all oligonucleotide sequences written 5′→3′), GCGGGTCTCTTCCCAGCGCA (guide sequence A) and TCCCAGCCAGATACTCCGCC (guide sequence B) in the pSpCas9n(BB)-2A-Puro (pX462) vector (Addgene, Cambridge, MA). For HeLa cells the construct was prepared by incorporating the guide sequence C targeting exon 3, GACTTCCGCATACGCTTCTT, in the pSpCas9(BB)-2A-GFP (pX458) vector (Addgene). The new constructs were confirmed by DNA sequencing.
HCT116 cells were co-transfected with 2.5 μg DNA (pX462 plasmid containing guide sequence A) and 2.5 μg DNA (pX462 plasmid containing guide sequence B) and the HeLa cells were transfected with 5 μg DNA (pX458 plasmid containing guide sequence C) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Puromycin, according to kill curve data, was added to the dishes the next day, and cells were cultured in puromycin-containing medium for another 48 h.
To ensure that the short guide RNAs have CRISPR activity, 72 h after transfection, aliquots of the cells were harvested and genomic DNA isolated using the KAPA Express Extract Kit (Kapa Biosystems, Roche, Laval, PQ) according to the manufacturer’s instructions. The primers used to amplify the genomic region of PNKP exon three were: forward, CTCCCTCTCTTTCTGCAGCT and reverse, TGAGAGCACGCAACAAACG. Surveyor nuclease mutation detection assay was performed using a Surveyor Mutation Detection Kit (IDT, Coralville, Iowa) according to manufacturer’s protocol.
For single clone selection and expansion, cells were sorted into 96-well plates 72 h after transfection by flow cytometry and single cells expanded to provide sufficient material for Western blot analysis and DNA sequencing confirmation. For DNA sequencing, cells were harvested and genomic DNA isolated. The primers used to amplify genomic region of PNKP exon three were: forward, GGAATTCCTCCCTCTCTTTCTGCAGCT and reverse, GGGGTACCTGAGAGCACGCAACAAACG. PCR products were subcloned into pEGFP-C2 vector (Clontech, Mountain View, CA) and extracted plasmid DNA from individual clones were subsequently sequenced.
2.2 Western blots
Subconfluent cells growing in a 10-cm dish were detached by scraping, pelleted, and suspended in 50 μL RIPA Lysis and Extraction Buffer (Thermo Fisher) containing fresh protease inhibitor cocktail (Biomake, Houston, TX). The cell lysates were incubated on ice for 30 min (vortexing every 10 min) and then centrifuged at 14000 ×g for 15 min. The supernatants were transferred to a new tube and the total protein was determined by Bradford assay (Bio-Rad). The cell lysates were aliquoted and stored at −80°C. For western blotting, Laemmli buffer (2X: 4% SDS, 20% glycerol, 5% β-mercaptoethanol and 120 mM Tris pH 6.8) was added to 50 μg of each lysate and they were heated for 10 min at 90°C. Proteins were resolved on either a BioRad 10% or Any-KD (gradient) SDS-Polyacrylamide gel. Pure human recombinant PNKP protein [10] was used as a positive control. Antibodies used for PNKP detection included a mouse monoclonal antibody (H101, Millipore) against an epitope in the aa 1-140 region, diluted 1:5000; a second mouse monoclonal antibody (B5, Santa Cruz) against an epitope in the aa 379-411 region, diluted 1:500; or an in-house rabbit polyclonal antibody (#122), raised against full-length protein and diluted 1:10,000. Other mouse monoclonal antibodies included anti-Ku80 from Abcam (#ab3107), anti-β-actin from Santa Cruz (#SC47778) and anti-XPF from ThermoFisher (#MA5-12060). Primary antibodies were diluted in 1% casein in PBS, added to the membranes and incubated for 16 hr at 4°C, with rocking. After washing the blots with 3 changes of Tris-buffered saline, 10 minutes each, the membranes were incubated in 10 ml of respective peroxidase conjugated (chemiluminiscent) secondary antibodies, recombinant anti-mouse antibody (Santa Cruz) at a concentration of 1:1000 or goat anti-rabbit (Cell Signaling) at a concentration of 1:3000, in 1% casein in PBS, for one hour at room temperature. After washing the membranes with 3 more changes of Tris-buffered saline, 10 ml of ECL substrate (ThermoFischer) was added to develop an image with detection either with X-ray film or a BioRad ChemiDoc imager. ThermoFischer Supersignaling western blot enhancer kit was used for the B5 antibody from Santa Cruz, to increase the sensitivity of detection. Image J was used for the quantification of the bands. For the blots in Fig. 1B, F and G, PBS containing 5% milk powder (Carnation) and 0.1% Tween 20 was used instead of casein for antibody adsorption and washing, with a final wash in PBS, and blots were developed with Super signal West Pico chemiluminescent substrate (Thermo Fisher).
Figure 1.
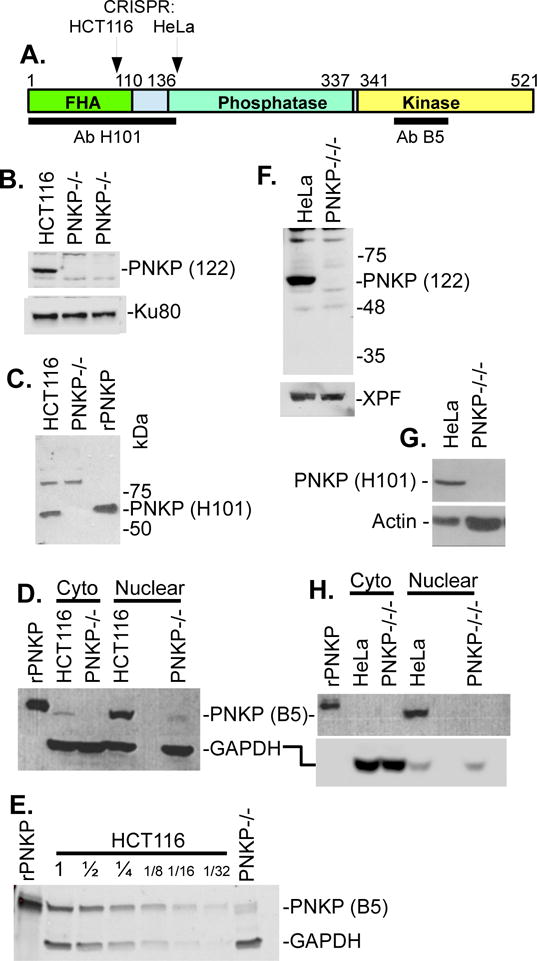
Analysis of PNKP knockout cell lines by western blot. A. Domain structure of PNKP showing location of epitopes recognized by the H101 and B5 anti-PNKP antibodies, and the locations of CRISPR/CAS9-mediated gene knockout. B. Lack of detectable PNKP in whole-cell lysates of PNKP−/− HCT116 cells, as probed with a polyclonal antibody (122) raised against full-length PNKP. Ku80 is a loading control. C. Lack of detectable PNKP in nuclear extracts of same cell line, using the N-terminal H101 antibody. A nonspecific band at ~90 kD serves as a loading control. rPNKP = 50 ng His-tagged recombinant human PNKP. D. Detection of a trace of an apparent truncated PNKP in PNKP−/− HCT116 nuclear extract, using the C-terminal B5 antibody. GAPDH is the loading control. E. Estimation of the level of truncated PNKP in PNKP−/− HCT116 nuclear extract, by comparison with serial dilutions of WT extract. F-H. Lack of detectable PNKP in whole-cell lysates (F, G) or nuclear extract (H) of PNKP−/−/−HeLa cells by any of the three antibodies, with XPF, Actin and GAPDH as loading controls. Nuclear and cytoplasmic fractions were separated using a fractionation kit (BioVision).
2.3 Clonogenic survival assay
Cells were seeded at densities ranging from 300 to 10,000 in 6 cm dishes and incubated for 12 hr to allow attachment. Cells were then treated with NCS (stock concentration 37 μM diluted to 2 μM in 20 mM sodium citrate buffer, pH 4.0) at concentrations ranging from 0.25 nM to 2 nM for HCT116 cells and 2 nM to 6 nM for HeLa cells for 6 hr. Following treatment, cells were incubated in fresh medium for 8-9 days to form colonies. Colonies were fixed with 100% methanol for 10 min, stained with 0.5% crystal violet in 20% methanol for 10 min, washed under tap water, air dried and counted manually. Plating efficiency (PE) was calculated as the number of colonies formed/number of cells seeded *100 for each dose. Surviving fraction (SF) was calculated as PE of treated/PE of control *100. Dose-modifying factor (DMF) was calculated as IC90 of control/IC90 of the mutant cell line. For experiments using ionizing radiation, cells were irradiated using a MDS Nordion Gammacell 40 research irradiator (ON, Canada), with a 137Cs source. For experiments with KU-60019, NU-7441, olaparib (AZD-2287) and veliparib (ABT-888), the respective inhibitor was added 1 hr prior to NCS treatment and left in the medium during and 24 hr after NCS treatment. Inhibitors were from Selleck Chemicals (Houston, TX).
2.4 Immunofluorescence
Twenty-five thousand cells were seeded in 4-well chamber slides (Nunc Lab Tek) and incubated overnight. Cells were then serum-starved by incubating in 0.5% FBS/RPMI for 72 hr. Cells were treated with 4 nM NCS for 1 hr and fixed at different time points using ice-cold 4% paraformaldehyde (PFA) in 1X PBS for 10 min. Cells were permeabilized in 0.5% Triton X-100/PBS for 10 min and blocked in 1X PBS 1% Casein blocker (Bio-Rad, 1610783) for 1 hr at 22°C. Primary mouse monoclonal anti 53BP1 antibody at 1:1000 (BD Pharmingen) was added and incubated overnight at 4°C. Slides were washed 4 times with PBS for 15 min each and incubated with secondary goat anti-mouse CFL594 antibody at 1:1000 (sc-362277) for 2 hr at 22°C. Slides were washed 4 times with PBS for 15 min each and post-fixed using ice-cold 4% PFA for 10 min. Nuclei were counterstained with Vectashield mounting medium containing 1.5 μg/mL 4′ -6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, H-1200). Confocal images were obtained with the Zeiss LSM700 Confocal Laser Scanning Microscope equipped with a 63X, 1.4 NA oil immersion objective, located in the Virginia Commonwealth University Microscopy Core Facility using a 405 nm laser (DAPI) and a 555 nm laser (CFL594).
2.5 Substrates
Linear plasmid substrates bearing 3′-phosphate termini with an internal radiolabel near one end were generated by ligation of appropriate 3′-phosphate oligonucleotides into 5′ overhangs of plasmid pRZ56, as described [11,12]. The oligonucleotides were purchased from IDT and in some cases 5′-32P labeled using a 3′-phosphatase-free polynucleotide kinase (NEB). To construct an internally labeled substrate bearing a recessed 5′-hydroxyl terminus, plasmid pUC19 was digested with XmaI, dephosphorylated with calf intestinal phosphatase (CIP), and 5′-32P-labeled with T4 polynucleotide kinase and [γ-32P]ATP. A 6-fold excess of an annealed oligomeric duplex with an Xma-compatible 5′-phosphorylated pCCGG-5′ overhang at one end and a 3-base-recessed 5′-hydroxyl terminus at the other end was ligated to the plasmid using T4 ligase, and unit-length plasmid ligation products were isolated from an agarose gel. The resulting substrate has an internal label 15 bases from the recessed 5′-hydroxyl end (see Fig.6), that can be released as a labeled oligonucleotide by cleavage with SmaI, to assess 5′-phosphorylation.
Figure 6.
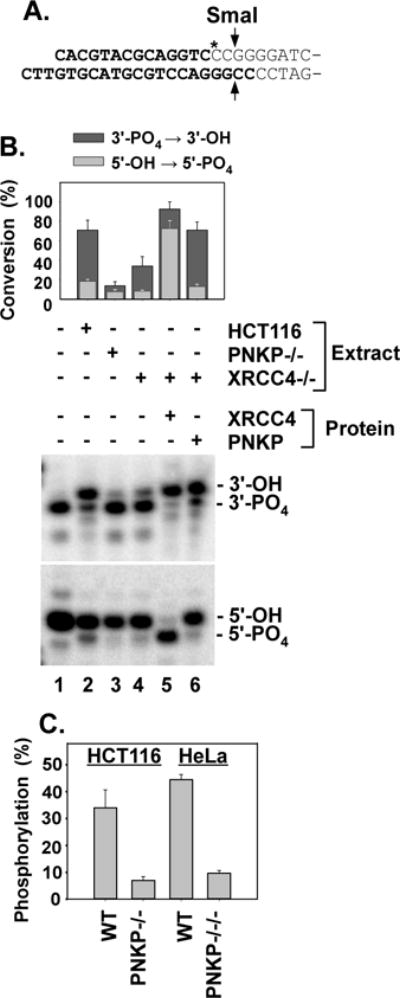
Dependence of 3′-phosphatase and 5′-kinase activities on XRCC4. A. Plasmid-length 5′-hydroxyl DSB substrate for 5′-phosphorylation. Bold lettering indicates oligomeric duplex that was ligated to pUC19. SmaI releases a labeled 16-base oligonucleotide. B. Upper gel: The 3′ overhang 3′-phosphate substrate (Fig. 4A) was incubated in HCT116 WT, XRCC4−/− or PNKP−/− extracts for 1 hr, cut with TaqαI and BstXI and 3′-dephosphorylation was assessed as in Fig. 3. Lower gel: The 5′-hydroxyl substrate shown in (A.) was incubated in the same extracts for 4 hr, cut with SmaI, and analyzed by gel electrophoresis to assess 5′-phosphorylation. C. The same substrate was incubated for 6 hr in whole-cell extracts of HCT116 and HeLa cells or their PNKP-deficient derivatives, and 5′-phosphorylation was similarly determined. Error bars show mean ± SEM for 3 experiments.
2.6 Extracts
HCT116 and HeLa cells were grown to ~70% confluence and then serum-starved in medium containing 0.5% fetal bovine serum for 3 days. Whole-cell extracts were prepared by Dounce homogenization followed by high-salt extraction and ultracentrifugation, as described [13,14]. Nuclear and cytoplasmic extracts were prepared using a BioVision cell fractionation kit. The protein concentration in the extracts was determined by BCA (BioRad).
2.7 DNA end processing in extracts
Reactions contained 50 mM triethanolammonium acetate pH 7.5, 1 mM ATP, 1 mM dithiothreitol, 50 μg/ml BSA, 1.3 mM magnesium acetate and dNTPs (or ddNTPs) at 100 μM each. Typically, a 16-μL reaction contained 10 μl of extract, resulting in a final concentration of 8 mg/ml protein (4 mg/ml for nuclear extracts), 66 mM potassium acetate and 16% glycerol, and an effective Mg++ concentration of 1 mM (taking into account ~0.3 mM EDTA from the extract). Buffer components were first mixed with cell extract at 22°C. In some cases, recombinant proteins and/or inhibitors (in 0.2 μL DMSO) were then added, and the solution mixed by pipetting. Finally, substrate (10 ng) was added 10 min after inhibitors, and the reaction again mixed by pipetting. Samples were incubated in a 37°C water bath for various times, and deproteinized as described (13). pRZ56-based substrates were then cut with BstXI and either TaqαI or AvaI, while the pUC19-based substrate was cut with SmaI, and fragments were analyzed on 20% polyacrylamide DNA sequencing gels. Storage phosphor screens were exposed to frozen polyacrylamide gels, and were quantitatively analyzed on a GE Typhoon imager using ImageQuant 5.1 software. All enzymes were from New England Biolabs (NEB). Recombinant XRCC4●DNA ligase IV (X4L4) complex was a gift from Dr. Dale Ramsden (University of North Carolina, Chapel Hill).
2.8 Analysis of DSB termini in intact cells
NCS-induced DSBs are formed preferentially at AGT●ACT hotspots, and bear at one end a 5′-aldehyde and either a 3′-phosphate or a 3′-phosphoglycolate on a one-base 3′ overhang; the opposite end bears a 5′ phosphate and a 3′-phosphate on a 2-base 3′ overhang [15,16]. To detect 3′-phosphates and the removal thereof at the latter end of DNA in NCS-treated cells, a ligation-mediated PCR (LMPCR) method used previously to detect 3′-phosphoglycolate ends [17] was modified. Cells were grown to ~50% confluence, serum-starved (0.5%) for 3 days, trypsinized, suspended in PBS at a concentration of 2×106/mL, and then treated with the isolated nonprotein chromophore of NCS (NCS-C), prepared as described [18]. NCS-C, typically 5.5 μL of an 80 μM solution in 20 mM sodium citrate pH 4 and 80% methanol, was placed in a 0.5 mL microfuge tube on ice. Cells in ice-cold PBS, typically 75 μL, were added and quickly mixed by pipetting, yielding a final NCS-C concentration of 5 μM. The cells were kept on ice for 5 min and then incubated for an additional 5-180 min at either 22°C or 37°C as indicated. Aliquots of 17 μl were removed, frozen in liquid nitrogen and stored at −80°C for no more than 24 hr. For 37°C incubations longer than 5 min, 17 μL of treated cells were diluted into 1.3 ml complete medium in 12-well culture dishes, incubated, harvested by centrifugation at 15000 × g, frozen and stored as pellets at −80°C. DNA was isolated using Qiagen DNeasy spin columns as prescribed by the manufacturer, except that for all incubations at 22°C and for 5-min incubations at 37°C, PBS, lysis solution and proteinase K were premixed before addition to the cell suspension. Eluted DNA was ethanol-precipitated, treated with NaBH4 to stabilize abasic sites and reduce 5′-aldehydes to 5′-hydroxyls, then treated with putrescine to hydrolyze any remaining abasic sites, as described previously [17]. Following ethanol precipitation, DNA was dissolved in 27 μL of 10 mM Tris pH8/0.1 mM EDTA (TE) for 1 hr at 37°C. A 5.5-μL aliquot was removed and diluted to 20 μL, and the remainder was treated with terminal transferase (TdT) in the presence of dTTP/ddTTP (40:1) as described to cap any 3′-hydroxyl ends. Finally, one-half of each sample was dephosphorylated with CIP [17], phenol/chloroform extracted, again precipitated, and dissolved in 20 μL TE. The concentration of each untreated, TdT-treated or TdT/CIP-treated DNA sample was determined by Alu PCR [17], and then 20 ng of the DNA in 12 μL was ligated to a 3′-dideoxy-terminated anchor (1.5 μM), i.e., pAGATCGCGATTTGACGCAGAGTTGCATGATTCGGCATCA(InvT) annealed to TGATGCCGAATCATGCAACTCTGCGTCAAATCGCGATCTGddT with 180 units T4 DNA ligase (NEB) in the buffer provided (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM dithiothreitol, 1 mM ATP). The 5′-phosphorylated end of this anchor has a 2-base 3′ overhang complementary to the 2-base (-AC) overhang of many NCS-induced DSBs and so will in principle be ligated to any such DSBs that have been 3′-dephosphorylated, while its 3′-dideoxy terminus prevents ligation of the opposite strand. The inverted T (InvT) was attached to prevent ligation of the other end of the anchor. Each ligated sample was subjected to Taqman PCR using a probe (6-FAM-CCAGCACAGATCG-MGBNFQ, Applied Biosystems; MGBNFQ = minor-groove-binding non-fluorescent quencher) that spans the joint formed by ligation of the anchor to NCS-induced DSBs at bp 38 of the human consensus Alu sequence. The Alu primer for PCR was GGTGGCTCACGCCTGTAATC (bases 11-30 of the consensus Alu sequence [19]); the anchor primer was CCTATC-CCCTGTGTGC-CTTGGCAGTC-TCAGTGCAAC-TCTGCGTCAAATCG.
3. Results
3.1 Disruption of the PNKP gene with CRISPR/CAS9
To generate PNKP-deficient derivatives of HCT116 and HeLa cells, PNKP mutations were induced by CRISPR/CAS9. For HCT116, the forkhead-associated (FHA) domain was targeted (Fig. 1A). A clone, henceforth designated PNKP−/−, with no detectable PNKP expression as judged by western blotting with a polyclonal antibody raised against full-length PNKP, was selected (Fig. 1B). Sequencing showed that one allele had tandem 24- and 32-bp deletions separated by 10 bp, and was predicted to encode a 103-aa protein with phosphatase and kinase domains completely eliminated. The other allele had an 87-bp insertion predicted to encode an extended protein with a 29-aa insertion in the FHA domain (Supplemental Fig. 1). However, no such larger PNKP protein was detectable with any PNKP antibody. Blots probed with a monoclonal antibody (H101, Millipore) to the N-terminus showed complete elimination of the normal PNKP band, with no detectable new bands of any size (Fig. 1C). An antibody to the C-terminal kinase domain (aa 379-411) detected a very weak doublet band with an estimated size 4 kD smaller than PNKP (Fig. 1D). Comparison of its intensity with that of titrated extracts of WT cells indicated that it is present at ~1/13 the WT concentration (Fig 1E). Presumably it represents some form of alternatively spliced and/or proteolyzed PNKP expressed from one of the mutant PNKP alleles. It would necessarily have a disrupted FHA domain and therefore is unlikely to be efficiently recruited to single-strand break (SSB) or DSB repair complexes by XRCC1 or XRCC4, respectively, but the possibility that it harbors residual phosphatase and/or kinase activity cannot be discounted.
Mutagenesis in HeLa cells (which have three copies of the PNKP gene) was targeted to the N-terminal portion of the PNKP phosphatase domain (Fig. 1A), and clones were screened with the same full-length PNKP antibody. A HeLa clone with no detectable PNKP was isolated that harbored insertions of 1 bp, 171 bp and 205 bp at the target site (Supplemental Fig. 2). The 1-bp and 205-bp insertions are predicted to encode the same truncated protein of 137 aa, with both the phosphatase and kinase domains completely eliminated, while the 171-bp insertion would encode a protein ~7 kD larger than PNKP with a disrupted phosphatase domain. However, no PNKP protein of either normal or modified size could be detected on western blots with the rabbit polyclonal antibody (122), or either an N-terminal or a C-terminal mouse monoclonal antibody, under conditions where a band representing as little as 2% of the WT level of PNKP would have been visible Fig. 1F-H). Hence, this clone appears to lack expression of any form of PNKP and is referred to as PNKP−/−/−.
3.2 PNKP-deficient cells are sensitive to radiation and NCS
Previous work showed that shRNA-mediated knockdown of PNKP in A549 lung carcinoma cells conferred significant sensitivity to radiation as well as NCS, with a DMF of ~1.5 [7,20]. Similarly, the PNKP-deficient derivatives of HCT116 and HeLa cells showed enhanced sensitivity to both radiation and NCS, as determined by clonogenic survival assays (Fig. 2A-D). The PNKP−/− HCT116 cells were considerably more sensitive to NCS, which specifically induces DSBs bearing protruding 3′-phosphate-terminated DSBs [15,16] (DMF = 2.5) than to radiation (DMF = 1.5), suggesting that PNKP deficiency confers a specific sensitivity to 3′-phosphate-terminated DSBs, consistent with a possible deficiency in processing and repair of such lesions (Fig. 2A and 2B). However, PNKP−/−/−HeLa cells were equally as sensitive to NCS as to radiation (DMF = 1.37), suggesting the presence of more robust alternative PNKP-independent DSB repair pathway(s) in HeLa cells than in HCT116 (Fig. 2C and 2D).
Figure 2.
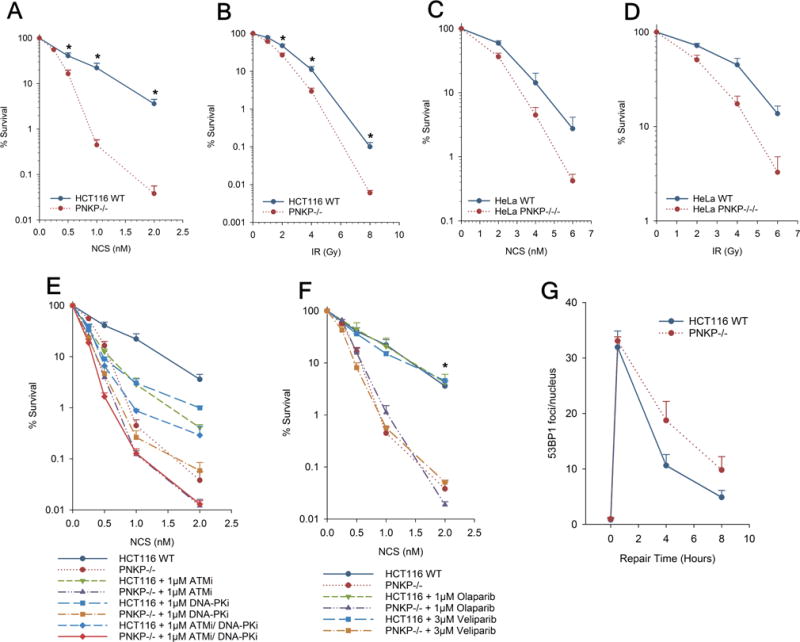
Sensitivity of PNKP-deficient cells to radiation and NCS. HCT116 WT or PNKP−/− cells (A, B, E, F), or HeLa WT or PNKP−/−/− cells (C, D) were seeded at low density, treated wIth NCS for 4 hr (A, C, E, F) or irradiated (B, D), and colonies were counted 7-9 days later. In (E) and (F), ATMi, DNA-PKi or olaparib were added 1 hr before NCS and were present for 24 hr after. Error bars show mean ± SEM from 3 independent experiments. In (G), serum-starved cells were treated with 4 nM NCS for 1 hr and then incubated for up to 8 hr., and then 53BP1 foci were assessed by confocal microscopy.
PNKP is phosphorylated at S114 and S126 by ATM and DNA-PK in response to DSB production and S114A/S126A phosphomutant PNKP proteins show decreased 3′-phosphatase activity [20].
To examine the interplay between PNKP and the DNA damage response to NCS, survival of HCT116 WT and PNKP−/− cells was assessed following treatment with NCS in the presence of an inhibitor of DNA-PK, ATM kinase, or both (Fig. 2E). DNA-PK is a critical factor in NHEJ [21], while ATM is important for DSB repair by homologous recombination [22,23] as well as for repair of a subset of DSBs by NHEJ [24]. NU7441 (DNA-PKi) and KU60019 (ATMi) equally sensitized WT HCT116 cells, although at the highest NCS concentration, ATMi was more potent. Intriguingly, at low NCS concentrations, each inhibitor was more deleterious to survival than was PNKP deficiency, but at the highest concentration, PNKP deficiency was more deleterious than the inhibitors, either alone or in combination. Furthermore, at high levels of damage, DNA-PKi had almost no effect on survival of PNKP−/− cells. These results are consistent with the presence of some alternative process or enzyme that can substitute for PNKP in NHEJ, but that saturates at very low levels of damage. Thus, in severely damaged cells, repair of NCS-induced DSBs by NHEJ becomes almost completely dependent on PNKP, so that DNA-PKi has little effect on PNKP−/− survival. In the presence of low-level damage, DSBs would be repaired by NHEJ despite PNKP deficiency, so that DNA-PKi markedly reduces survival of PNKP−/− cells. The additive effects of ATMi and PNKP deficiency on survival are likely to result from ATM’s role in DSB repair by homologous recombination, which does not require PNKP [25].
Poly(ADP-ribose) polymerase (PARP) adds poly(ADP-ribose) chains to a variety of nuclear proteins when DNA is damaged [26], and is essential for efficient SSB repair, as well as for an alternative Ku-independent end joining pathway involving XRCC1, DNA ligase III and DNA polymerase θ [27–29]. In most genetic backgrounds, PARP inhibitors confer a replication-dependent radiosensitivity by inhibiting SSB repair and by inappropriately channeling the resulting single-ended DSBs into NHEJ [30,31]. However, radiosensitivity can be replication-independent when Ku-dependent NHEJ is compromised [32]. Previous work showed that HCT116 cells were radiosensitized by the PARP inhibitor olaparib [33,34]. However, olaparib did not sensitize HCT116 WT cells to NCS (Fig. 2F), suggesting that the relatively small number of NCS-induced SSBs do not contribute significantly to cytotoxicity even when PARP is inhibited. Furthermore, neither olaparib nor veliparib further sensitized PNKP−/− cells. This result is consistent with reports that PNKP is involved in end joining by both Ku-dependent NHEJ [25] and PARP-dependent Alt-NHEJ [35]. Thus, both repair systems are already compromised in PNKP−/− cells, so that blocking Alt-NHEJ with a PARP inhibitor would be expected to have little if any effect.
To directly investigate whether the hypersensitivity shown by the mutants to NCS was attributable to a defect in repairing 3′-phosphate-ended DSBs, measurements of NCS-induced DSBs in cells, detected as 53BP1 foci, were performed. Serum-starved G1/G0 cells were used to specifically analyze DSB repair by NHEJ while avoiding spontaneous focus formation at stalled replication forks. As shown in Fig. 2G, there were about twice as many residual DSBs in PNKP-deficient as in WT HCT116 cells at 4 and 8 hr after NCS treatment. This result is consistent with a model wherein increased persistence of 3′-phosphate termini due to lack of PNKP interferes with efficient DSB rejoining.
3.3 Extracts of PNKP-deficient cells have residual but greatly reduced 3′-phosphatase activity toward DSB ends
To more directly assess the effect of PNKP deficiency on 3′-phosphate removal during NHEJ, internally labeled DSB substrates bearing either recessed or protruding 3′-phosphates were constructed (Figs. 3A and 4A, respectively) and incubated in whole-cell extracts of WT parental and PNKP-deficient cells. Previous work has shown that end joining of such substrates in these extracts is strictly dependent on DNA-PK and XLF [12,14], suggesting that it is carried out exclusively by classical NHEJ. The recessed-end substrate has complementary 5′ overhangs and so following dephosphorylation can be joined by simple ligation (Fig. 3A), while the 3′ overhang is partially complementary and requires fill-in of one nucleotide in each strand prior to ligation (Fig 4A). Following incubation of these plasmid-length substrates in extracts, 3′-dephosphorylation and end joining can be assessed by cutting off both ends of the substrate with restriction enzymes to yield labeled oligomers, which are analyzed on denaturing sequencing gels.
Figure 3.
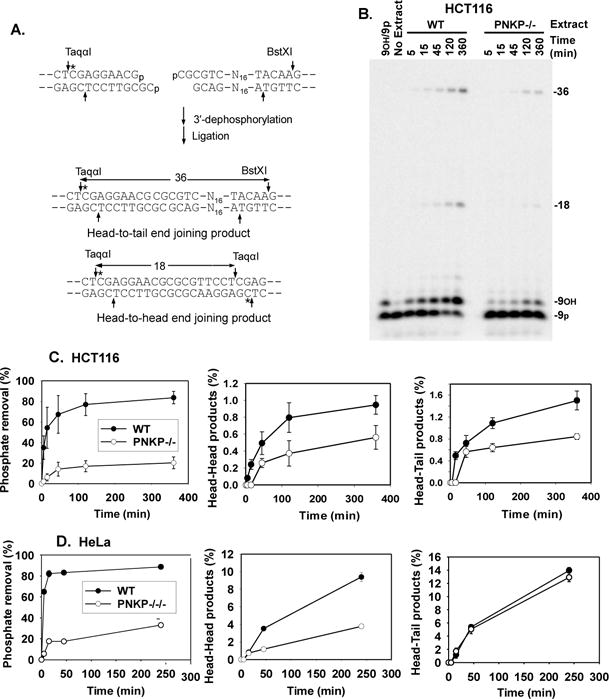
End processing and end joining of a DSB bearing a recessed 3′-phosphate. Following incubation in cell extract and digestion with TaqαI and BstXI cleavage, unprocessed substrate is detected as a 3′-phosphate 9-mer; 3′-dephosphorylation and ligation yields labeled 18-base head-to-head and 36-base head-to-tail end joining products. A. Recessed 3′-phosphate substrate with complementary ends. All substrates are drawn 5′→3′ in the top strand and 3′→5′ in the bottom strand. B. Representative gel from an experiment with extracts of HCT116 cells. C. Quantitation of results in HCT116 cells. D. Quantitation of results from HeLa cells. Error bars show mean ± SEM from 3 independent experiments.
Figure 4.
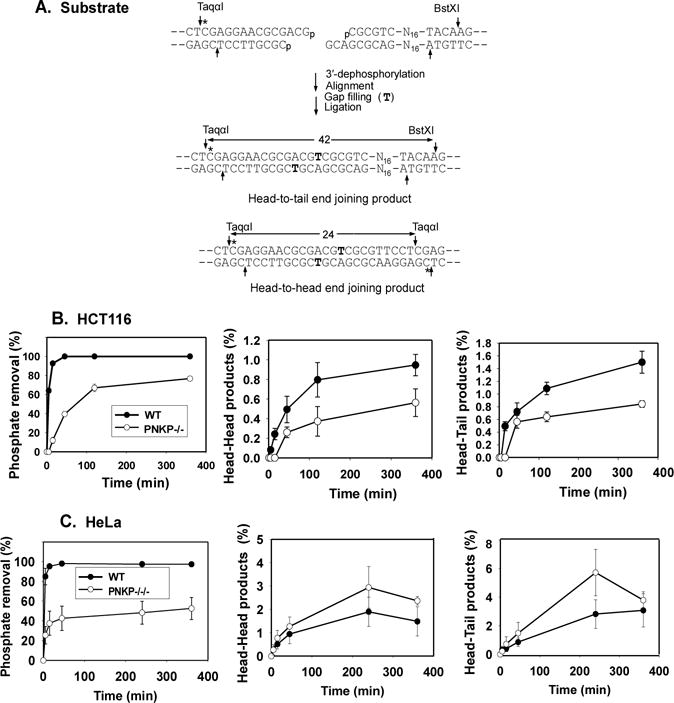
End processing and end joining of a DSB bearing a 3′-phosphate on a 3′ overhang. A. Overhang 3′-phosphate substrate with partially complementary ends. Following incubation in cell extract for the indicated times and digestion with TaqαI and BstXI, unprocessed substrate is detected as a 3′-phosphate 14-mer; 3′-dephosphorylation, single-base gap filling (T) and ligation yields labeled 24-base head-to-head and 42-base head-to-tail end joining products. B. Quantitation of results from HCT116 cells. C. Quantitation of results from HeLa cells. Error bars show mean ± SEM from 3 independent experiments. Representative gels are shown in Supplemental Fig. 4.
As shown in Fig. 3B and 3C, dephosphorylation of a recessed 3′-phosphate terminus in extracts of HCT116 cells was nearly complete within 2 hr, while in PNKP−/− extracts, most 3′-phosphate termini remained intact and unprocessed even after 6 hr. This reduced dephosphorylation was reflected in a marked decrease in head-to-head products that result from joining of two ends each bearing 3′-phosphate termini (18-base fragment in Fig. 3B). Similar deficiencies in both dephosphorylation and head-to-head end joining were seen in PNKP−/−/− HeLa cells (Fig. 3D and Supplemental Fig. 3). As might be expected, there was in each case less effect on head-to-tail products, which derive from joining of one 3′-phosphate and one 3′-hydroxyl DSB end. Overall, the results indicate that PNKP is the main 3′-phosphatase for DSB ends during NHEJ, and that recessed 3′-phosphates can be quite persistent when PNKP is absent. Nevertheless, extracts of PNKP-deficient cells still retain some 3′-phosphatase activity. Although PNKP−/− HCT116 cells appear to express a trace of a truncated PNKP that could account for residual 3′-dephosphorylation (Fig. 1D), PNKP−/−/−HeLa cells have no detectable PNKP, implying that they must contain some less efficient alternative 3′-phosphatase.
In extracts of both HeLa and HCT116 WT cells, 3′-phosphate DSB termini on a 3-base 3′-overhang were dephosphorylated much faster than 3′-recessed ends, and much faster than the subsequent gap filling and ligation that continued for several hours (Fig. 4 and Supplemental Fig. 4). PNKP deficiency decreased the initial rate of dephosphorylation by approximately tenfold, and in HCT116 PNKP−/− extracts (although not HeLa extracts), the decreased 3′-dephosphorylation was reflected in reduced production of ligated repair products. Nevertheless, by 6 hr the majority of 3′ DSB termini were dephosphorylated in extracts of both PNKP-deficient cell lines. Essentially identical results were obtained for a 3′-phosphate on a 1-base 3′ overhang; moreover, addition of recombinant PNKP to PNKP−/− HCT116 and PNKP−/−/− HeLa extracts restored 3′-dephosphorylation to the level seen in parental cells (Supplemental Fig. 5), confirming that loss of PNKP was responsible for the deficiency in 3′-dephosphorylation. Again, however, the residual 3′-dephosphorylation seen in PNKP−/−/− extracts suggests that there must be an alternative 3′-phosphatase that acts on protruding 3′-phosphate termini.
Tyrosyl-DNA phosphodiesterase 1 (TDP1) catalyzes resolution of 3′-phosphotyrosyl ends left by abortive topoisomerase 1 reactions, leaving a 3′-phosphate that is subsequently removed by PNKP [36]. When a DSB substrate with a 3′-phosphotyrosine terminus on a 3-base overhang was incubated in parental HCT116 extracts, 3′-hydroxyl ends were rapidly formed, but no 3′-phosphate intermediates could be detected, presumably because dephosphorylation by PNKP was very efficient (Fig. 5). However, in PNKP-deficient cells a band with mobility of the 3′-phosphate intermediate was readily detected, again suggesting less efficient 3′-dephosphorylation.
Figure 5.
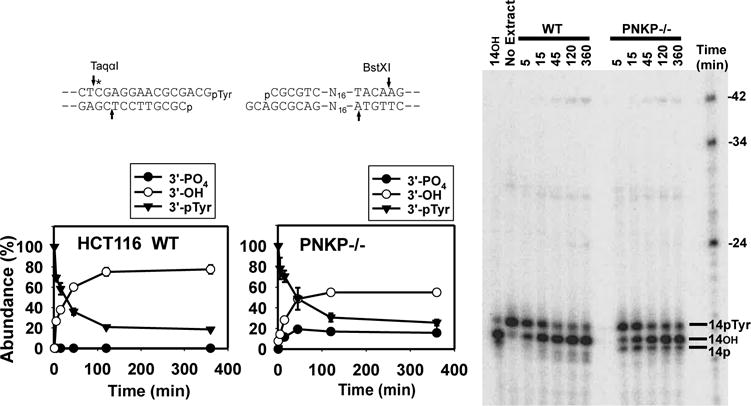
Processing and end joining of a 3′-pTyr DSB substrate. The substrate shown, with a 3′-pTyr terminus on a 3-base 3′ overhang is otherwise identical to the 3′-phosphate substrate in Fig. 4A, and was incubated in HCT116 or PNKP−/− extracts and products analyzed as in Fig. 3. The 3′-pTyr 14-mer represents unprocessed substrate. Removal of 3′-pTyr yields a 3′-hydroxyl (14OH). A 3′-phosphate intermediate (14p) is apparent in PNKP−/− but not WT extracts. The substrate yields the same 24-base head-head and 42-base head-tail end joining products as the 3′-phosphate substrate (see Fig. 4A). Graphs show the fraction of each type of terminus as a function of time, derived from quantitation of gels from 3 experiments (mean ± SEM).
3.4 Dephosphorylation occurs in context of NHEJ in both WT and PNKP-deficient extracts
During NHEJ, PNKP is recruited to DSB ends by the XRCC4●DNA ligase IV (X4L4) complex [5]. To assess the X4L4 dependence of PNKP-mediated end processing, 3′-dephosphorylation of the 3′ overhang substrate, as well as 5′-phosphorylation of a substrate with a recessed 5′-hydroxyl terminus (Fig. 6A), were examined in extracts of XRCC4 knockout cells. As shown in Fig. 6B, there was greatly reduced 3′-dephosphorylation and almost no 5′-phosphorylation of DSB ends in XRCC4−/− extracts (compare lanes 2 and 4), emphasizing the importance of X4L4 in recruiting PNKP for both these functions. Addition of recombinant X4L4 restored efficient 3′-dephosphorylation and promoted 5′-phosphorylation to an extent even greater than that seen in WT extracts (lane 5), suggesting that the level of X4L4 in WT extracts was a limiting factor. As expected, both PNKP−/− HCT116 cells and PNKP−/−/− HeLa cells showed greatly reduced DSB 5′-phosphorylation. Nevertheless, there was still detectable 5′-kinase activity in extracts of both PNKP-deficient cell lines (Fig. 6C).
To determine whether the residual 3′-dephosphorylation in PNKP-deficient cells occurred in the context of NHEJ, incubations were performed in the presence of NU7441 (DNA-PKi), a specific DNA-PK inhibitor [37]. As expected, DNA-PKi eliminated end joining completely (Supplemental Figs. 3B and 4B). In addition, DNA-PKi blocked most but not all dephosphorylation of both 3′-recessed and 3′ overhang substrates by endogenous 3′-phosphatases in whole-cell extracts of HCT116 and HeLa cells as well as their PNKP-deficient derivatives (Fig. 7A-F and Supplemental Figs. 3B and 4B). These results suggest that both PNKP and the residual 3′- in PNKP-deficient extracts function in the context of NHEJ through interactions with NHEJ proteins.
Figure 7.
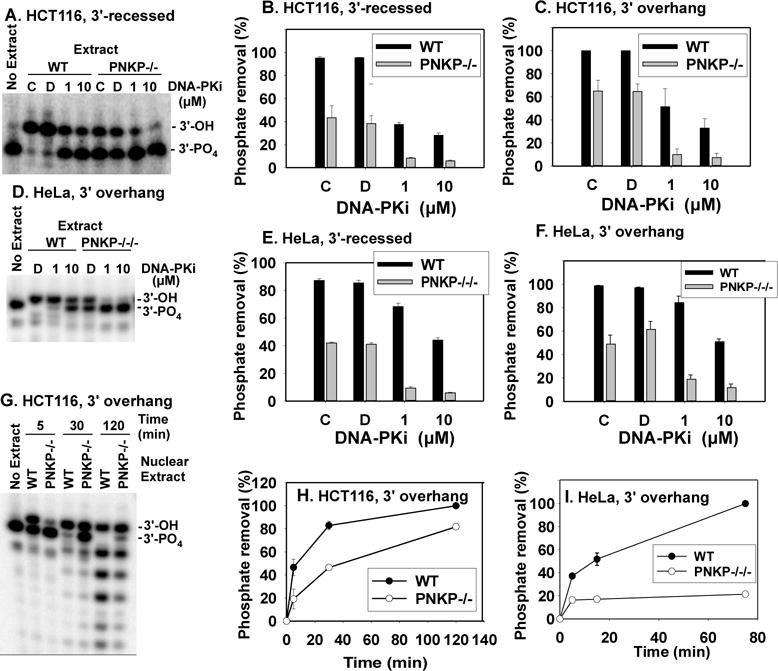
Effect of DNA-PK inhibitor NU7441 (DNA-PKi) on dephosphorylation in whole-cell extracts and detection of 3′-phosphatase in nuclear extracts. A. Gel showing 3′-dephosphorylation of the 3′-recessed DSB substrate in HCT116 WT and PNKP−/− extracts. No-inhibitor control reactions were performed both without (“C”) and with (“D”) DMSO, which is the solvent for DNA-PKi. B. and C. Quantitation of 3′-dephosphorylation for recessed and overhang substrates in HCT116 WT and PNKP−/− extracts. D. Gel showing 3′-dephosphorylation of the 3′ overhang substrate in HeLa WT and PNKP−/−/− extracts. E. and F. Quantitation of 3′-dephosphorylation for recessed and overhang substrates in HeLa WT and PNKP−/−/− extracts. G. Gel showing 3′-dephosphorylation of the 3′ overhang substrate in nuclear extracts of HCT116 WT and PNKP−/− cells. H. Quantitation of 3′-dephosphorylation in nuclear extracts of HCT116 WT and PNKP−/− cells. I. Quantitation of 3′-dephosphorylation in nuclear extracts of HeLa WT and PNKP−/−/− cells. Shorter labeled products resulting from subsequent 3′→5′ exonucleolytic resection were included in calculation of dephosphorylation. Error bars = mean ± SEM for 3 experiments, except H. and I., 2 experiments.
Because whole-cell extracts were used for 3′-dephosphorylation and end-joining experiments, residual 3′-dephosphorylation in PNKP-deficient extracts could reflect action of cytoplasmic phosphatase(s) not normally involved in DNA metabolism. However, similar to whole-cell extracts, nuclear extracts of both of the PNKP-deficient cell lines harbored substantial 3′-phosphatase activity, although less than did WT extracts (Fig. 7G-I). Absence of vinculin in these nuclear extracts (Supplemental Fig. 6) indicates that they were not contaminated with cytoplasmic proteins. Thus, the putative alternate 3′-phosphatase in PNKP-deficient HCT116 and HeLa cells is a nuclear enzyme, as would be required for participation in NHEJ.
3.5 Terminal 3′-phosphates of DSBs are removed more slowly in PNKP-deficient cells
Because reactions in cell extracts may not accurately reflect those in intact cells, 3′-dephosphorylation of DSBs was also examined in cells. Previous work showed that sequence-specific DSBs induced by treatment of cells with the nonprotein chromophore of NCS (NCS-C), which is its active component, can be detected by LMPCR, and that various enzymatic manipulations can be used to specifically detect DSBs bearing 3′-hydroxyl, 3′-phosphate or 3′-phosphoglycolate termini [17]. Treatment with NCS-C, without the protective NCS carrier protein, facilitates very rapid production of extensive damage to cellular DNA [38]. NCS-induced DSBs are formed preferentially at AGT●ACT sequences [39], with one end of the break having a mixture of 3′-phosphate, 3′-phosphoglycolate and possibly, cleaved abasic sites [15,16]. The opposite end has almost exclusively 3′-phosphate termini on 2-base overhangs, formed by free radical attack on the 5′ carbon of the adjacent nucleotide (Fig. 8A). Removal of the terminal phosphate would yield a ligatable 3′ terminus, and an anchor was designed to link to these DSB ends, thus allowing their amplification and detection by LMPCR. The anchor has a 2-base unligatable (-GddT) 3′ overhang and a recessed ligatable 5′-phosphate terminus that can be directly ligated to the 3′ terminus of any NCS-induced DSBs in cellular DNA formed at AGT●ACT hotspots and 3′-dephosphorylated in the cell. A Taqman probe was designed to specifically detect NCS-induced DSBs at the AGT●ACT hotspot at bp 38-40 of the highly repetitive human Alu sequence [19]. This probe spans the ligation joint between cellular Alu sequences and the anchor, and thus permits quantitation of 3′-hydroxyl DSBs formed at this site, by Taqman PCR (Fig. 8B). DSBs that retain 3′-phosphates can also be detected, by first capping the existing 3′-hydroxyl ends with terminal transferase and dTTP/ddTTP, then dephosphorylating the remaining 3′-phosphate DSB ends with CIP [17]. The resulting 3′-hydroxyl DSB ends can then be similarly quantitated by LMPCR.
Figure 8.
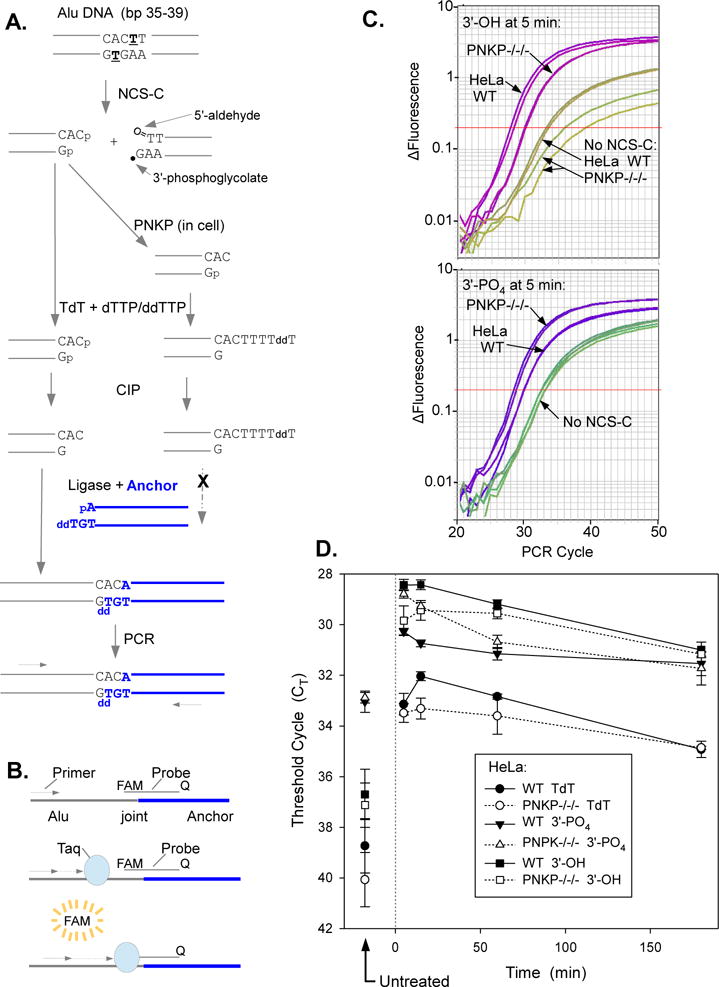
Deficiency in 3′-dephosphorylation of NCS-C-induced DSBs in PNKP−/−/− HeLa cells as determined by LMPCR. Cells were treated with 5 μM NCS-C and incubated at 37°C for 5-180 min. DNA was isolated and 3′ termini of DSBs were assessed by ligation-mediated PCR. A. Experimental scheme. NCS-C induces DSBs preferentially at AGT●ACT sequences, including at bp 36-38 of the human Alu repeat. The left-hand DSB end has an overhanging 3′-phosphate. To quantify these phosphate ends, any existing 3′-hydroxyl ends are first blocked by tailing with TdT and dTTP/ddTTP. The 3′-phosphates are then removed with CIP and the resulting 3′-hydroxyl DSB ends are ligated to an anchor and quantitated by ligation-mediated real-time PCR. To quantitate 3′-hydroxyl DBSs formed in the cell by PNKP, DNA from NCS-C-treated cells is subjected to LMPCR without any prior enzyme treatments, B. Principle of Taqman PCR. As PCR product accumulates, the fluorescent probe binds to the Alu/anchor junction after each denaturation cycle and the fluorophore is released by the 5′→3′ exonuclease of Taq polymerase as it replicates the product, resulting in an exponential increase in fluorescence as a function of cycle number. C. LMPCR amplification profiles from a representative experiment, showing more 3′-phosphate DSBs and fewer 3′-hydroxyl DSBs in PNKP−/−/− than in WT HeLa cells. Amplification profiles from duplicate PCR reactions are shown for each condition. D. Time course for formation and disappearance of 3′-phosphate and 3′-hydroxyl NCS-induced DSBs, as determined by LMPCR of cell DNA treated with TdT plus CIP or neither enzyme, respectively. CT values for DNA treated with TdT only are also shown. Error bars show the mean ± SEM from 4 independent experiments.
When either parental or PNKP-deficient HeLa cells were treated with NCS-C, both 3′-hydroxyl- and 3′-phosphate-terminated DSBs were readily detected in cellular DNA using the LMPCR method (Fig. 8C and 8D). As expected, capping the existing 3′-hydroxyl ends with terminal transferase reduced the LMPCR signal, while subsequent treatment with CIP increased it. Further, since no 3′-hydroxyl-terminated DSBs were detected when isolated human DNA was treated with NCS-C in vitro (Supplemental Fig. 7), the 3′-hydroxyl termini must have been formed by enzymatic processing of NCS-C-induced DSBs in the cell. For both parental and PNKP-deficient cells, both 3′-hydroxyl- and 3′-phosphate-terminated DSBs were formed within 5 min, reached a maximum within 15 min, and then decreased slowly upon extended incubation (Fig. 8D). However, at 5 and 15 minutes after treatment, there were about 3 times more 3′-phosphate-terminated DSBs in PNKP-deficient cells than in parental cells, as indicated by a difference of 1.5 PCR cycles in the amplification profiles (p<0.001 at both 5 and 15 min). It is unlikely that this difference reflects greater initial NCS-C-induced damage in PNKP-deficient cells, because PNKP−/−/− cells contained about twofold fewer 3′-hydroxyl-terminated DSBs than WT cells (p<0.05). The difference between the two cell lines was most striking at 15 min, by which time WT cells had 5 times more 3′-hydroxyl termini than 3′-phosphate termini, while PNKP−/−/− cells had equal numbers of the two species. Even at 22°C, 3′-hydroxyl termini were rapidly generated following NCS-C treatment, emphasizing the efficiency of 3′-phosphate removal from DSBs in the cell, but again, PNKP-deficient cells accumulated more 3′-phosphate and fewer 3′-hydroxyl termini than did WT cells (Supplemental Fig. 8). These results indicate that 3′-phosphate termini of at least some NCS-C-induced DSBs were more persistent in PNKP-deficient than in WT HeLa cells.
In HCT116 cells, 3′-phosphate and 3′-hydroxyl ends were likewise formed very rapidly upon NCS-C treatment. However, at 37°C, PNKP deficiency did not result in a measurable difference in the abundance of either 3′-phosphate or 3′-hydroxyl DSB ends, as nearly all 3′-phosphate DSB ends appeared to be dephosphorylated within 5 min of treatment even in PNKP−/− cells (data not shown). Nevertheless, when the incubation temperature was reduced to 22°C, there was a consistent delay in generation of 3′-hydroxyl termini in PNKP−/− cells, suggesting a deficiency in 3′-phosphate removal (Fig. 9).
Figure 9.
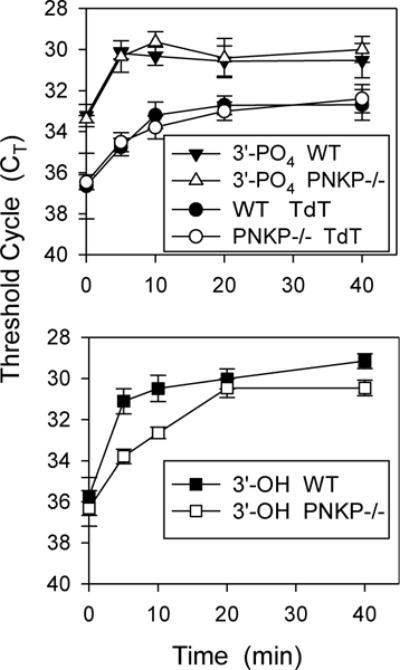
Delay in 3′-dephosphorylation of NCS-C-induced DSBs in PNKP−/− HCT116 cells. Cells were treated with NCS-C, incubated, and 3′-termini were quantitated as in Fig. 8, except incubation was at 22°C for the indicated times. Error bars show the mean ± SEM from 3 independent experiments.
4. Discussion
Since PNKP is capable of dephosphorylating a variety of single- and double-strand substrates including DSB ends [4], and is recruited to DSB ends by the core NHEJ protein XRCC4 [5,6], a role for PNKP in resolving 3′-phosphate termini of free radical-mediated DSBs is highly likely, but has never been explicitly demonstrated. In order to assess the importance of this function for DSB repair by NHEJ and to examine the fate of 3′-phosphate-terminated DSBs in the absence of PNKP, the PNKP gene was disrupted in HCT116 and in HeLa cells using CRISPR/CAS9. As expected from PNKP’s putative role in removing 3′-phosphates from DSB ends, PNKP-deficient HCT116 and HeLa cells were hypersensitive to both NCS and radiation. However, PNKP deficiency conferred much greater hypersensitivity to HCT116 cells than to HeLa cells. Since HCT116 cells have reduced expression of Mre11 [40], these results could potentially be explained by the existence of two parallel pathways for repair of NCS-induced DSBs, one requiring PNKP and another requiring Mre11, as has been proposed for repair of topoisomerase I-mediated strand breaks [36,41]. Thus, PNKP−/− HCT116 cells, being deficient in both pathways, would be highly NCS-sensitive.
Extracts from both HCT116 and HeLa cells with disrupted PNKP showed a dramatic reduction in 3′-phosphatase as well as 5′-kinase activity at DNA ends, suggesting that PNKP is largely responsible for such dephosphorylation and phosphorylation. Nevertheless, extracts of PNKP-deficient cells still contained substantial 3′-phosphatase activity, despite the absence of detectable PNKP protein in PNKP−/−/− HeLa cells. This residual 3′-dephosphorylation was largely blocked by a DNA-PK inhibitor, suggesting that it occurs in the context of classical NHEJ. Moreover, DNA-PK inhibition in PNKP−/− HCT116 cells caused increased cytotoxicity to NCS at low concentrations, suggesting a DNA-PK-dependent alternative enzyme capable of resolving the 3′-phosphate DSB termini in the absence of PNKP. All these results suggest that loss of PNKP confers a significant, but only partial, deficiency in resolution of 3′-phosphate DSB ends for NHEJ.
Similarly, analysis of termini of NCS-induced DSBs in cellular Alu DNA by LMPCR confirms a significant but only partial deficit in 3′-dephosphorylation in PNKP-deficient cells. Particularly in HeLa cells, PNKP disruption results in increased 3′-phosphate termini and decreased 3′-hydroxyl termini at 5-15 min after treatment with NCS-C. Nevertheless, substantial numbers of 3′-hydroxyl DSBs are rapidly formed in NCS-C-treated PNKP-deficient cells, indicating residual 3′-dephosphorylation. Such dephosphorylation must occur via an alternative 3′-phosphatase rather than a nuclease, as removal of even a single nucleotide from the 3′ terminus would alter the sequence of the ligation joint with the anchor and thus prevent detection by Taqman LMPCR. After 1 hr of incubation, no difference between PNKP-deficient and parental cells is seen, and there appears to be a surprisingly large number of persistent unrepaired 3′-hydroxyl DSBs 3 hr after treatment. This may be a consequence of the high level of damage induced by NCS-C, which is necessary to allow direct detection of these lesions in cells by LMPCR. Nevertheless, even a short delay in 3′-dephosphorylation of DSBs could enhance their lethality, for example by increasing the probability that the two DSB ends become physically separated and then either remain unrejoined or are misjoined to a DSB on another chromosome.
In HCT116 cells, the effect of PNKP disruption was more subtle, a brief delay in generation of 3′-hydroxyl ends only, and only at 22°C. The reason for this difference between cell lines, and whether it is related to the apparent presence of residual modified PNKP protein in HCT116 cells, is not known.
Despite the quantitative differences between cell lines, overall the data suggest an association between PNKP deficiency, delayed DSB 3′-dephosphorylation, and increased cytotoxicity of 3′-phosphate-terminated DSBs. Although involvement of PNKP in NHEJ [6,25], as well as radiosensitivity of PNKP knockdown cells [7], had been demonstrated previously, the heterogeneous nature of radiation damage, the high ratio of SSBs to DSBs, and the involvement of PNKP in multiple repair pathways including SSB repair, complicates interpretation of radiosensitivity data. Because NCS induces a much higher proportion of DSBs (SSB/DSB ratio of approximately 5 [42,43]), and all NCS-induced DSBs have at least one 3′-phosphate end, the finding that PNKP-deficient HCT116 cells are more sensitive to NCS than to radiation strengthens the implication of PNKP, and its 3′-dephosphorylation activity, in DSB repair by NHEJ. It is unlikely that NCS sensitivity is due to loss of PNKP-mediated 5′-phosphorylation, as NCS does not produce 5′-hydroxyl DSBs [15,16].
The identity of the apparent alternative 3′-phosphatase activity in PNKP-deficient cells is not known. Although one disrupted PNKP allele in both HCT116 and HeLa cells is a large in-frame insertion, no PNKP protein of increased size is detectable in either cell line, and no PNKP protein at all can be detected in the HeLa cell derivative by antibodies to either the N- or C-terminal portions of the protein. Hence, it is unlikely that this activity is some alternatively spliced or otherwise altered form of PNKP. Although APE1 has weak 3′-phosphatase activity [44], it requires a base-paired 3′-terminal nucleotide and does not act on 3′ overhangs [45]. Another possibility is aprataxin, as purified recombinant aprataxin has been reported to remove both 3′-phosphates and 3′-phosphoglycolates from DNA ends. However, a similar study [46] as well as our own efforts (unpublished) failed to detect any 3′-phosphatase or 3′-phosphodiesterase activity in aprataxin. On the other hand, it seems unlikely that the residual 3′-dephosphorylation is the result of general broad-specificity phosphatases, since it is regulated by DNA-PK. Furthermore, it appears to act specifically on 3′- and not 5′-phosphates, as indicated by the retention of 5′-32P label on DSB substrates even after incubation in whole-cell extracts for many hours [14,47]. Rather, overall the data suggest an enzyme that acts specifically in NHEJ, in a manner similar to PNKP but less efficiently. In principle, it should be possible to purify this activity from PNKP-deficient cells and identify it.
Supplementary Material
Acknowledgments
We thank Eric Hendrickson for providing XRCC4−/− cells and Dale Ramsden for providing recombinant X4L4. This work was supported by grants CA40615 and CA166264 from the National Cancer Institute, USDHHS, grant MOP 15385 from the Canadian Institutes of Health Research and grant 26603 from the Alberta Cancer Foundation Transformative Program Project.
References
- 1.Ward JF. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 2.Hutchinson F. Chemical changes induced in DNA by ionizing radiation. Prog Nucleic Acids Res Mol Biol. 1985;32:115–154. doi: 10.1016/s0079-6603(08)60347-5. [DOI] [PubMed] [Google Scholar]
- 3.Henner WD, Rodriguez LO, Hecht SM, Haseltine WA. Gamma-ray induced deoxyribonucleic acid strand breaks. 3′ glycolate termini. J Biol Chem. 1983;258:711–713. [PubMed] [Google Scholar]
- 4.Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. Tidying up loose ends: The role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci. 2011;36:262–271. doi: 10.1016/j.tibs.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koch CA, Agyei R, Galicia S, Metalnikov P, O’Donnell P, Starostine A, Weinfeld M, Durocher D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. Embo j. 2004;23:3874–3885. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. Embo j. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasouli-Nia A, Karimi-Busheri F, Weinfeld M. Stable down-regulation of human polynucleotide kinase enhances spontaneous mutation frequency and sensitizes cells to genotoxic agents. Proc Natl Acad Sci U S A. 2004;101:6905–6910. doi: 10.1073/pnas.0400099101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein NK, Karimi-Busheri F, Rasouli-Nia A, Mani R, Dianov G, Glover JN, Weinfeld M. Polynucleotide kinase as a potential target for enhancing cytotoxicity by ionizing radiation and topoisomerase I inhibitors. Anticancer Agents Med Chem. 2008;8:358–367. doi: 10.2174/187152008784220311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy S, de Melo AJ, Xu Y, Tadi SK, Negrel A, Hendrickson E, Modesti M, Meek K. XRCC4/XLF interaction is variably required for DNA repair and is not required for ligase IV stimulation. Mol Cell Biol. 2015;35:3017–3028. doi: 10.1128/MCB.01503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fanta M, Zhang H, Bernstein N, Glover M, Karimi-Busheri F, Weinfeld M. Production, characterization, and epitope mapping of monoclonal antibodies against human polydeoxyribonucleotide kinase. Hybridoma. 2001;20:237–242. doi: 10.1089/027245701753179811. [DOI] [PubMed] [Google Scholar]
- 11.Bennett RAO, Gu XY, Povirk LF. Construction of a vector containing a site-specific DNA double-strand break with 3′-phosphoglycolate termini and analysis of the products of end-joining in CV-1 cells. Intl J Radiat Biol. 1996;70:623–636. doi: 10.1080/095530096144509. [DOI] [PubMed] [Google Scholar]
- 12.Povirk LF, Zhou RZ, Ramsden DA, Lees-Miller SP, Valerie K. Phosphorylation in the serine/threonine 2609-2647 cluster promotes but is not essential for DNA-dependent protein kinase-mediated nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2007;5:3869–3878. doi: 10.1093/nar/gkm339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumann P, West SC. DNA end-joining catalyzed by human cell-free extracts. Proc Natl Acad Sci U S A. 1998;95:14066–14070. doi: 10.1073/pnas.95.24.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akopiants K, Zhou RZ, Mohapatra S, Valerie K, Lees-Miller SP, Lee KJ, Chen DJ, Revy P, de Villartay JP, Povirk LF. Requirement for XLF/Cernunnos in alignment-based gap filling by DNA polymerases λ and μ for nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2009;37:4055–4062. doi: 10.1093/nar/gkp283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Povirk LF. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: Bleomycin, neocarzinostatin and other enediynes. Mutat Res. 1996;355:71–89. doi: 10.1016/0027-5107(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 16.Dedon PC, Goldberg IH. Free-radical mechanisms involved in the formation of sequence-dependent bistranded DNA lesions by the antitumor antibiotics bleomycin, neocarzinostatin, and calicheamicin. Chem Res Toxicol. 1992;5:311–332. doi: 10.1021/tx00027a001. [DOI] [PubMed] [Google Scholar]
- 17.Akopiants K, Mohapatra S, Menon V, Zhou T, Valerie K, Povirk LF. Tracking the processing of damaged DNA double-strand break ends by ligation-mediated PCR: Increased persistence of 3′-phosphoglycolate termini in SCAN1 cells. Nucleic Acids Res. 2014;42:3125–3137. doi: 10.1093/nar/gkt1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Povirk LF, Goldberg IH. Binding of the nonprotein chromophore of neocarzinostatin to DNA. Biochemistry (N Y) 1980;19:4773–4780. doi: 10.1021/bi00562a009. [DOI] [PubMed] [Google Scholar]
- 19.Kariya Y, Kato K, Hayashizaki Y, Himeno S, Tarui S, Matsubara K. Revision of consensus sequence of human alu repeats–a review. Gene. 1987;53:1–10. doi: 10.1016/0378-1119(87)90087-4. [DOI] [PubMed] [Google Scholar]
- 20.Segal-Raz H, Mass G, Baranes-Bachar K, Lerenthal Y, Wang SY, Chung YM, Ziv-Lehrman S, Strom CE, Helleday T, Hu MC, Chen DJ, Shiloh Y. ATM-mediated phosphorylation of polynucleotide kinase/phosphatase is required for effective DNA double-strand break repair. EMBO Rep. 2011;12:713–719. doi: 10.1038/embor.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jette N, Lees-Miller SP. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol. 2015;117:194–205. doi: 10.1016/j.pbiomolbio.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kocher S, Rieckmann T, Rohaly G, Mansour WY, Dikomey E, Dornreiter I, Dahm-Daphi J. Radiation-induced double-strand breaks require ATM but not artemis for homologous recombination during S-phase. Nucleic Acids Res. 2012;40:8336–8347. doi: 10.1093/nar/gks604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Löbrich M. ATM and artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. Embo j. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Löbrich M. A pathway of double-strand break rejoining dependent upon ATM, artemis, and proteins locating to γ-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Karimi-Busheri F, Rasouli-Nia A, Allalunis-Turner J, Weinfeld M. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer Res. 2007;67:6619–6625. doi: 10.1158/0008-5472.CAN-07-0480. [DOI] [PubMed] [Google Scholar]
- 26.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 28.Black SJ, Kashkina E, Kent T, Pomerantz RT. DNA polymerase theta: A unique multifunctional end-joining machine. Genes (Basel) 2016;7 doi: 10.3390/genes7090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood RD, Doublie S. DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst) 2016;44:22–32. doi: 10.1016/j.dnarep.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, Koyama H, de Murcia G, Takeda S. Parp-1 protects homologous recombination from interference by ku and ligase IV in vertebrate cells. Embo j. 2006;25:1305–1314. doi: 10.1038/sj.emboj.7601015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9:1775–1787. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alotaibi M, Sharma K, Saleh T, Povirk LF, Hendrickson EA, Gewirtz DA. Radiosensitization by PARP inhibition in DNA repair proficient and deficient tumor cells: Proliferative recovery in senescent cells. Radiat Res. 2016;185:229–245. doi: 10.1667/RR14202.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, Wang L, El-Rayes B, Shu HK. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys. 2013;86:469–476. doi: 10.1016/j.ijrobp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 35.Audebert M, Salles B, Weinfeld M, Calsou P. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J Mol Biol. 2006;356:257–265. doi: 10.1016/j.jmb.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 36.Kawale AS, Povirk LF. Tyrosyl-DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 2018;46:520–537. doi: 10.1093/nar/gkx1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14:6083–6087. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 38.Kappen LS, Ellenberger TE, Goldberg IH. Mechanism and base specificity of DNA breakage in intact cells by neocarzinostatin. Biochemistry. 1987;26:384–390. doi: 10.1021/bi00376a008. [DOI] [PubMed] [Google Scholar]
- 39.Dedon PC, Goldberg IH. Sequence-specific double-strand breakage of DNA by neocarzinostatin involves different chemical mechanisms with a staggered cleavage site. J Biol Chem. 1990;265:14713–14716. [PubMed] [Google Scholar]
- 40.Takemura H, Rao VA, Sordet O, Furuta T, Miao ZH, Meng L, Zhang H, Pommier Y. Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J Biol Chem. 2006;281:30814–30823. doi: 10.1074/jbc.M603747200. [DOI] [PubMed] [Google Scholar]
- 41.Sacho EJ, Maizels N. DNA repair factor MRE11/RAD50 cleaves 3′-phosphotyrosyl bonds and resects DNA to repair damage caused by topoisomerase 1 poisons. J Biol Chem. 2011;286:44945–44951. doi: 10.1074/jbc.M111.299347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Povirk LF, Houlgrave CW. Effect of apurinic/apyrimidinic endonucleases and polyamines on DNA treated with bleomycin and neocarzinostatin: Specific formation and cleavage of closely opposed lesions in complementary strands. Biochemistry (N Y) 1988;27:3850–3857. doi: 10.1021/bi00410a049. [DOI] [PubMed] [Google Scholar]
- 43.Beerman TA, Goldberg IH. DNA strand scission by the antitumor protein neocarzinostatin. Biochem Biophys Res Commun. 1974;59:1254–1261. doi: 10.1016/0006-291x(74)90449-5. [DOI] [PubMed] [Google Scholar]
- 44.Demple B, Harrison L. Repair of oxidative damage to DNA: Enzymology and biology. Ann Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 45.Suh D, Wilson DM, III, Povirk LF. 3′-phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Res. 1997;25:2495–2500. doi: 10.1093/nar/25.12.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris JL, Jakob B, Taucher-Scholz G, Dianov GL, Becherel OJ, Lavin MF. Aprataxin, poly-ADP ribose polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function together to protect the genome against oxidative damage. Hum Mol Genet. 2009;18:4102–4117. doi: 10.1093/hmg/ddp359. [DOI] [PubMed] [Google Scholar]
- 47.Almohaini M, Chalasani SL, Bafail D, Akopiants K, Zhou T, Yannone SM, Ramsden DA, Hartman MC, Povirk LF. Nonhomologous end joining of complex DNA double-strand breaks with proximal thymine glycol and interplay with base excision repair. DNA Repair (Amst) 2016;41:16–26. doi: 10.1016/j.dnarep.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.