

Abstract
Purpose
Despite initial benefit from tyrosine kinase inhibitors (TKI), advanced non-small cell lung cancer (NSCLC) patients harboring ALK (ALK+) and ROS1 (ROS1+) gene fusions ultimately progress. Here we report on the potential resistance mechanisms in a series of ALK+ and ROS1+ NSCLC patients progressing on different types and/or lines of ROS1/ALK targeted therapy.
Experimental Design
We used a combination of next generation sequencing (NGS), multiplex mutation assay, direct DNA sequencing, RT-PCR, and fluorescence in situ hybridization (FISH) to identify fusion variants/partners and copy number gain (CNG), kinase domain mutations (KDMs) and copy number variations (CNV) in other cancer-related genes. We performed testing on 12 ROS1+ and 43 ALK+ patients.
Results
One of 12 ROS1+ (8%) and 15/43 (35%) ALK+ patients harbored KDM. In the ROS1+ cohort, we identified KIT and β-catenin mutations and HER2-mediated bypass signaling as non-ROS1 dominant resistance mechanisms. In the ALK+ cohort we identified a novel NRG1 gene fusion, a RET fusion, 2 EGFR, and 3 KRAS mutations, as well as mutations in IDH1, RIT1, NOTCH and NF1. Additionally, we identified CNV in multiple proto-oncogenes genes including PDGFRA, KIT, KDR, GNAS, K/HRAS, RET, NTRK1, MAP2K1, and others.
Conclusions
We identified a putative TKI resistance mechanism in 6 of 12 (50%) ROS1+ patients and 37/43 (86%) ALK+ patients. Our data suggest that a focus on KDMs will miss most resistance mechanisms; broader gene testing strategies and functional validation is warranted to devise new therapeutic strategies for drug resistance.
Keywords: non-small cell lung cancer, ALK, ROS1, acquired resistance, tyrosine kinase inhibitor
INTRODUCTION
Large scale chromosomal alterations involving c-ros oncogene 1 (ROS1) or the anaplastic lymphoma kinase (ALK) exhibit oncogenic activity in non-small cell lung cancer (NSCLC) (1–3). ROS1 and ALK gene fusions result in the expression of chimeric proteins that constitutively activate downstream proliferation and survival pathways (2,4,5). These fusions can be detected by multiple methods including fluorescence in situ hybridization (FISH), RT-PCR with direct sequencing, next generation sequencing (NGS) and immunohistochemistry (IHC) (6–11).
Treatment of NSCLC patients whose tumors harbor ROS1 and ALK fusions using cognate tyrosine kinase inhibitors (TKIs) has allowed for dramatic improvements in response rates, progression free survival and overall survival compared to chemotherapy (12–16). Crizotinib, the first U.S. Food and Drug Administration (FDA)-approved TKI for ALK+ and the only FDA-approved TKI for ROS1+ patients, demonstrated a PFS of 9.7 months in ALK+ patients and 19.2 months in ROS1+ patients, both studies enrolled patients with and without prior lines of therapy (3,16). Ceritinib, alectinib, and brigatinib have also been approved for ALK+ patients who have progressed on crizotinib and these drugs demonstrated response rates in the second line setting of 56%, 50%, and 54% respectively (17–19). In ROS1+ patients who have progressed on crizotinib there are no FDA approved TKIs; however, multiple drugs including ceritinib, brigatinib, lorlatinib, and cabozantinib are being evaluated as post-crizotinib options (20–23). For ALK+ patients there are also multiple new ALK targeted TKIs that are currently being investigated including lorlatinib and ensartinib (24–28).
Our group and others have previously reported on the mechanisms of crizotinib resistance in ALK+ patients, which include somatic mutations in the kinase domain (KDM), gene copy number gains (CNG) and alternate oncogenic mutations (29,30). In addition, series describing the resistance mechanisms to next generation ALK TKIs have been recently published, however many ALK+ tumors did not have an identifiable mechanism of resistance (30–34). Currently little is known about resistance mechanisms in patients with ROS1+ NSCLC after treatment with crizotinib and/or other ROS1 TKIs. Here we report on the potential resistance mechanisms of cohorts of ROS1+ and ALK+ positive NSCLC patients treated across multiple lines of therapy and with different TKIs.
METHODS
Patient population
Patients with advanced ROS1+ or ALK+ NSCLC were considered for re-biopsy following progression on specific ROS1 or ALK TKI therapy. All patients gave informed consent for collection of clinical correlates, tissue collection, research testing, and cell line derivation under Institutional Review Board (IRB)-approved protocols. Formalin-fixed paraffin embedded (FFPE), frozen (placed in liquid nitrogen), and/or fresh tissue samples were obtained according to the safety standards of the interventional radiologist, pulmonologist, or surgeon. Prior therapies and days until progression for each patient were obtained from chart review. Days until progression were determined based on imaging studies which demonstrated definitive growth of a known tumor site or new extra-CNS metastatic deposits. If questionable, serial scans were evaluated to confirm continued growth. Patient studies were conducted according to the Declaration of Helsinki, the Belmont Report, and the U.S. Common Rule.
Next Generation Sequencing
We analyzed samples from 10 ROS1+ and 29 ALK+ patients with a custom capture-based NGS panel of 48 genes (NimbleGen SeqCap EZ Choice Library, Roche, Pleasanton, CA). See Supplementary Table 1 for gene list. Samples were run on a NextSeq sequencer (Illumina, San Diego, CA). We used ~100ng (range 35–150ng) of DNA for each NGS assay. Total genomic DNA were isolated from tissue using the QIAamp DNA Mini Kit (Qiagen) and processed accordingly to the manufacture’s protocol depending on whether the material was FFPE or frozen. The isolated genomic DNA were then sheered to 300bp using Covaris S220 Focused-ultrasonicator and libraries were made using Kapa Hyper Prep Kit (Kapa Biosystems, Wilmington, MA). The average read depth for each region was 2233 independent reads.
ROS1 exons 36–42 and ALK exons 21–25 were sequenced to detect kinase domain mutations and average read depth for each exon is shown in Supplementary Table 2. Coverage for ALK intron 19, the most common breakpoint region, is also shown in Supplementary Table 2.
Bioinformatics analysis
A bioinformatics pipeline was utilized in which sequence reads were analyzed using the Genomic Short Read Nucleotide Alignment Program (GSNAP) and the ‘Clipping REveals STructure’ (CREST) algorithm to identify structural rearrangements (35,36). We calculated copy number variation (CNV) for each gene locus by first calculating the number of unique reads for each gene in each tumor sample and taking the average of all 48 genes in that sample. The number of unique reads for each gene locus was then normalized to this 48-gene average to account for differences in depth of coverage between each sample. Non-ALK and –ROS1 cohort samples were utilized in this analysis to improve the reliability of the median unique reads at each locus. A value that was greater than 2.5 standard deviations from the median unique read count at each locus was chosen as the cut-off for a significant increase. Gitools version 2.3.1 (available at www.gitools.org) was used to generate heatmaps.
Direct Sequencing, SNaPshot, Anchored-multiplexed PCR
As described previously, genomic DNA was isolated from manually micro-dissected FFPE tumor samples using the QiaAmp FFPE DNA isolation kit from Qiagen (Valencia, CA) (29). Samples were PCR amplified using custom primer sets from exons 21–25 of ALK and exons 36–42 of ROS1 and directly sequenced using the ABI Big Dye Thermocycle Sequencing kit and analyzed on an ABI 3730 DNA Sequencer (37). Mutation analysis was performed with the Mutation Surveyor software v3.97-4.0.0 from Soft Genetic (State College, PA). The reference sequence used for ROS1 was NM 002944.2, for ALK NM_004304.4, and for EML4 NM_019063.3. Anchored-multiplex PCR (Archer FusionPlex assay, Boulder, CO) was utilized in three tumor samples to further characterize ALK fusions, but also detected other gene fusions.
The SNaPshot assay for evaluation of multiple oncogenic mutations in APC, AKT1, BRAF, CTNNB1, EGFR, FLT3, JAK2, KIT, KRAS, MAP2K1 (MEK1), NOTCH1, NRAS, PIK3CA, PTEN, and TP53 was performed by amplification using 13 multiplexed PCR reactions followed by single nucleotide base extension reactions. The products were separated by capillary electrophoresis and analyzed using GeneMapper 4.0(38).
Subcloning and sequencing of ROS1
RNA was isolated from frozen tissue sample via Rneasy Mini Kit (Qiagen) which then underwent 1st strand synthesis using SuperScript III Reverse Transcriptase (Invitrogen) according to manufacturer protocol. The resulting cDNA was subjected to 30 rounds of PCR amplification targeting exons 35 to exon 39 of ROS1. The 387bp PCR product was then inserted into a TA cloning vector (Invitrogen) and used to transform competent TOP10 bacteria (Invitrogen) and subsequently plated on LB amp plates. Bacterial colonies were miniprepped (Qiagen) and DNA sequenced using the T7 primer.
Fluorescence in situ hybridization (FISH)
ALK and ROS1 FISH positivity was determined using break-apart probes (Vysis LSI ALK 2p23) Dual Color, Break Apart Rearrangement Probe and Vysis LSI ROS1 (Cen) SpectrumGreen and Vysis LSI ROS1 (Tel) SpectrumOrange (Abbott Molecular, Abbott Park, IL). The FISH assays and analyses were performed as described previously with minor modifications (39). Specimens were considered positive for rearrangement when ≥15% of cells carried split 3′ and 5′ signals, that is physically separated by ≥2 signal diameters for ALK or ≥1 signal diameter for ROS1, or single 3′ signals. Copy number of the rearranged genes was based on determination of the mean of split and isolated red signals per tumor cell (40). At least 50 tumor cells were analyzed per specimen. Copy number gain was defined as a more than two-fold increase in the mean of the rearranged gene per cell in the post-treatment specimen compared with the pre-treatment specimen.
Cell line and reagents
Primary cell lines were derived from patients by placing fresh tumor tissue into sterile tissue culture dishes and culturing in RPMI supplemented with 10% FBS. Specifically, fresh core biopsies were minced with a razor blade in order to dislodge tumor clusters and subsequently plated on collagen coated plates to encourage adherence. When pleural effusion sample contained excessive red blood cell (RBC) an additional RBC lysis step was included using ACK Lysis Buffer (KD Medical, Columbia, MD). Cell cultures that had undergone 20 or more passage were deemed a cell line. Propagation of these lines were done in the absence of TKI in the culturing media. CUTO16 was an early live cell culture, but did not develop into an immortal cell line, whereas CUTO23, CUTO27, and CUTO28 were established as immortal cell lines. These were utilized in this study and were derived by our laboratory from ROS1+ patient tumor samples following written-informed consent approved by local IRB. Mycoplasma and STR testing is routinely performed on all human cell lines in our laboratory.
Immunoblotting
Immunoblotting was done as previously described with minor modifications (41). Briefly, cells were lysed in modified RIPA buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail from Thermo Scientific (Waltham, MA). Total protein was separated by SDS-PAGE, transferred to nitrocellulose and stained with the primary antibodies. Antibodies used in this study included pErbB2 (6B12), pErbB3 (21D3),total ErbB3 (D22C5), pROS1 (3078), total ROS1 (D4D6), pShp2 (3751), pAKT (D9E), total AKT (40D4), total ERK (L34F12) and pERK (clone D13.14.4E) from Cell Signaling Technologies (Danvers, MA), total ErbB2 (clone 42), total Shp2 (clone 79) from BD Biosciences (San Jose, CA), and GAPDH from Millipore (Burlington, MA). Protein detection was achieved by imaging with an Odyssey Imager and Image Studio 5.2 analysis software from LI-COR (Lincoln, NE).
Generation of the RALGAPA1-NRG1 gene fusion via CRISPR/Cas9
SgRNAs targeting introns of RALGAPA1 and NRG1 were designed using the Zhang lab CRISPR design tool (crispr.mit.edu) and synthesized by Integrated DNA Technologies. The sgRNAs were tandemly cloned into the pX333 CRISPR/Cas9 system (a gift from Andrea Ventura (Addgene plasmid # 64073)) using the BbsI and BsaI cloning sites. The H3122 cell line was transiently transfected using TansIT-X2 (Mirus) with this construct and then cultured in the presence of 1.5μM of crizotinib for 4 weeks. After 4 weeks, the surviving cells were assessed for the genomic rearrangement using primers that flank RALGAPA1 and NRG1 breakpoint.
Proximity Ligation Assay
This assay, performed on FFPE tumor samples, was described previously using the Duolink® In Situ PLA® (Sigma, St. Louis, MO) (42). Antibodies used for this assay included ALK (D5F3) and ErbB2 (29D5) from Cell Signaling Technology and GRB2 (clone 81) from BD Biosciences.
RESULTS
Demographics
Twelve ROS1+ and 43 ALK+ patients underwent biopsies after demonstrating radiographic progression while on treatment with a ROS1- or ALK-directed TKI. Three of 12 (25%) ROS1+ patients and 18 of the 43 (42%) ALK+ patients had more than one line of ROS1/ALK targeted therapy prior to the biopsy that was evaluated in this study. Five patients (all ALK+) had >1 repeat biopsy following progression on different treatments. Fifteen of the ALK+ patients were previously reported, with updated sequencing using NGS (Supplementary Table 3). (29,43). Table 1 shows the characteristics of both cohorts. The majority of ROS1+ and ALK+ patients were males with adenocarcinoma and were never or light smokers. The median ages were 47 and 53 for the ROS1+ and ALK+ patients, respectively. All patients received crizotinib as their initial TKI therapy against ROS1 or ALK with median time (range) on therapy before progression of 187 days (106–533) and 206 days (28–1035). In the ALK+ cohort 2 patients received >1 TKI and received crizotinib TKI rechallenge prior to biopsy, including those patients the median time on therapy before progression was 194 days (Table 2). The pre-progression biopsy treatments for the ROS1+ patients were crizotinib (9), ceritinib (2) and brigatinib (1) (Table 2, Supplementary Table 3). The pre-progression biopsy treatments for the ALK+ patients were crizotinib (30), ceritinib (6), brigatinib (7), alectinib (4), lorlatinib (2).
Table 1.
Patient Demographics
| ROS1 (n = 12) | ALK (n = 43) | Total (n = 55) | |
|---|---|---|---|
| Age | 47 (37–65) | 53 (21–78) | 50 (21–78) |
| Sex (%) | |||
| Male | 8 (67) | 28 (65) | 36 (65) |
| Female | 4 (33) | 15 (35) | 19 (35) |
| Histology (%) | |||
| Adenocarcinoma | 11 (92) | 42 (98) | 53 (9) |
| Large Cell | 1 (8) | 1 (2) | 2 (4) |
| Smoking Statusa (%) | |||
| Never/Light | 11 (92) | 37 (86) | 48 (87) |
| Current/Former | 1 (8) | 6 (14) | 7 (13) |
| Prior Treatmentb | |||
| Crizotinib | 12 (100) | 43 (100) | 55 (100) |
| Ceritinib | 2 (17) | 7 (16) | 9 (16) |
| Brigatinib | 1 (8) | 9 (21) | 10(18) |
| Alectinib | 0 | 4 (9) | 4 (7) |
| Lorlatinib | 0 | 1 (2) | 1 (2) |
| ≥ 2 ALK/ROS1 targeted tx pre bx | 3 (25) | 19 (44) | 22 (40) |
| Biopsy Sitec | |||
| Lymph Node | 1(8) | 22 (41) | 23 (35) |
| Lung Parenchyma | 3 (25) | 13 (24) | 16 (24) |
| Liver | 1 (8) | 6 (11) | 7 (11) |
| Other/unknown | 7 (58) | 13 (24) | 20 (30) |
Abbreviations: TKI –tyrosine kinase inhibitor, tx- treatment, bx, biopsy
Never/Light was considered to be = 10 pack years
All patients received crizotinib as the first ALK or ROS1 targeted therapy. Prior treatments are those targeted treatments the patient received prior to resistance biopsy
percentage based on total number of rebiopsies. 5 patients had >1 resistance biopsy.
Table 2.
Pre-biopsy treatment characteristics in ROS1+ and ALK+ cohorts.
| A. ROS1 | ||||
|---|---|---|---|---|
| Immediate Pre-biopsy treatment | crizotinib | ceritinib | brigatinib | >1 TKI |
| Number | 9 | 2 | 1 | 3 |
| Median days until progression or stopped due to SE | 187 | 41 | 57 | 51 |
| Range | 74–1461 | 30–51 | N/A | 30–57 |
| Chemotherapy prior n (%) | 3 (33%) | 1 (50%) | 1 (100%) | 2 (66%) |
| Median ROS1 targeted treatments | 1 | 2 | 2 | 2 |
| B. ALK | ||||||
|---|---|---|---|---|---|---|
| Immediate Pre-biopsy treatment | crizotinib | ceritinib | brigatinib | alectinib | lorlatinib | >1 TKI |
| Number | 30 | 6 | 7 | 4 | 2 | 21 |
| Median days until progression or stopped due to SE | 194 | 190 | 390 | 314 | 84 | 226 |
| Range | 28–1035 | 76–672 | 128–612 | 41–497 | 22–146 | 22–1546 |
| Chemotherapy prior n (%) | 25 (83%) | 5 (83%) | 4 (57%) | 2 (50%) | 1(50%) | 13 (62%) |
| Median ALK targeted treatments | 1 | 2 | 2 | 2 | 3 | 2 |
Fusion Partner and Variant Frequencies
Ten ROS1+ and 29 ALK+ samples underwent custom capture NGS. The coverage for ROS1 intron 31 was poor due to repetitive sequences. Coverage for introns 32 and 33 was sufficient for fusion detection (Supplementary Table 2). The fusion partner was identified in 8/10 ROS1+ tumors patients who were tested by NGS (Fig. 1A). Notably, one of the CD74-ROS1 fusions was only detected through the baiting of the CD74 gene and would not have been detected if only the ROS1 gene had been baited in the NGS assay (data not shown). In the remaining two patients, we were unable to identify the fusion partners using NGS, however, both patients were FISH positive and responded to ROS1 TKI therapy. The ROS1 fusion partners we identified in this cohort have all been described previously (1,10).
Figure 1.
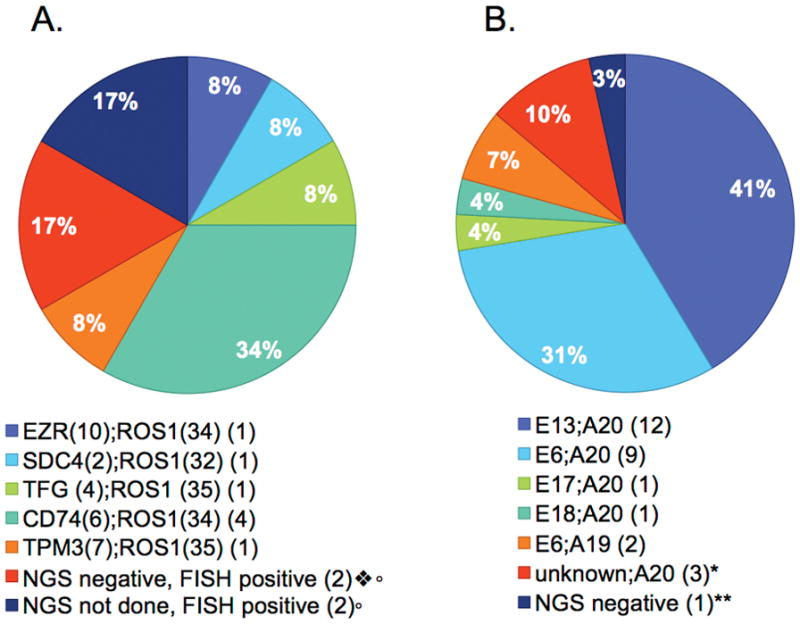
ROS1 and ALK fusion partner and variant frequencies. A, ROS1 fusion partner frequencies in the 10 patients whose tumor samples underwent NGS. Three patient’s tumor samples were positive by FISH but negative by NGS. B, ALK fusion variant frequencies in the 28 patients whose tumor samples underwent NGS.
○ All NGS negative and NGS not done patients responded to crizotinib
❖ Though fusion partner not identified, one patient found to have two ROS1 kinase domain mutations, illustrated in figure 2
* One patient whose fusion partner was not identifiable by NGS underwent additional testing with Archer DX and was found to have an E2;A20 fusion variant.
**One patient’s NGS and FISH was negative for ALK fusion however the patient’s pre-treatment tumor sample was positive for ALK fusion indicating loss of ALK during treatment.
Fig. 1B demonstrates the distribution of ALK fusion variants. All identified ALK gene partners were EML4. The most common variants identified were E13;A20 (41%) and E6;A20 (31%) (7,44). Using custom-capture NGS, we observed ALK gene rearrangements that required additional testing to confirm the presence of an in-frame gene fusion. In the first case, the genomic breakpoint was in intron 17 of EML4 and exon 20 of ALK (E17;A20). Inspection of the genomic sequence did not support an in-frame transcript between EML4 exon 17 and ALK exon 20. However, further testing using RT-PCR with direct sequencing and anchored-multiplexed PCR (AMP) revealed that the fusion transcript contains EML4 exon 17 and sequences derived from EML4 intron 17 and is lacking the first 79 nucleotides from exon 20 of ALK (Supplementary Fig. 1A). This patient demonstrated a tumor response to alectinib (Supplementary Fig. 1B–E). We identified three additional ALK fusions using the capture-based NGS assay in which there was evidence of a rearrangement within intron 19 of ALK, but in which the 5′ gene partner could not be positively identified; one of these was also further characterized by AMP and identified as E2;A20, whereas another had a complex rearrangement of E16;A20. In patients for whom we had biopsy samples at more than one treatment time point we did not see any evidence of change in the fusion variant. Finally, one acquired resistance sample was negative for an ALK fusion using both NGS and FISH, indicating likely loss of the ALK fusion gene, a finding we have previously reported in resistance samples (29).
ROS1 kinase domain mutations
Among our ROS1+ patients, exons 36–42 were sequenced by NGS and/or direct sequencing to evaluate for the presence of KDM. The average read-depth for these exons was greater than 3000X for each exon using NGS (Supplementary Table 2). We identified only one patient whose resistance biopsy demonstrated ROS1 KDMs (Fig. 2). The patient had been treated with crizotinib for ~17 months prior to the post-progression biopsy. The post-progression sample harbored dual ROS1 mutations generating a L2026M substitution, the gatekeeper position, and L1951R, which is located at the solvent front (Fig. 2A). The mutations occurred at different variant allele frequencies (VAF), 19% and 9%, respectively. We confirmed that these mutations were in trans by subcloning the ROS1 gene and demonstrating that no clones contained both mutations (data not shown). To further explore the clinical significance of these co-occurring mutations we tested sensitivity to treatment with crizotinib and ceritinib in an early live culture (CUTO16) derived from this patient sample by evaluating phosphorylation of ROS1, SHP2 and ERK1/2. Drug treatment only partially decreased ROS1 activation as well as partially decreased downstream signaling of SHP2 and ERK1/2 (Fig. 2B). The patient was treated with ceritinib 750mg daily by mouth given the lack of clinical trial options at the time of tumor progression. After 1 month of treatment there was minimal change and by 3 months the nodule demonstrated progression and the patient went on to receive chemotherapy (Fig. 2C).
Figure 2.
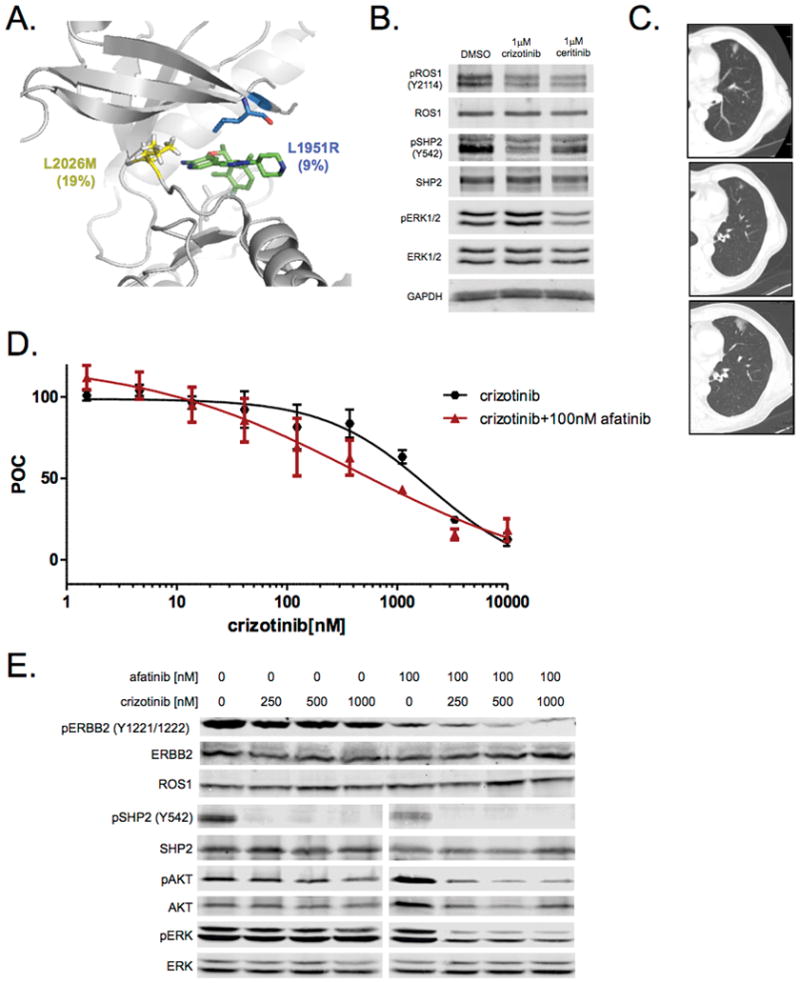
ROS1 resistance in human-derived cell lines. A, Crystal structure of ROS1 bound to crizotinib (PDB 3ZBF) highlighting the two mutations identified in this tumor sample showing the L2026M gatekeeper mutation (yellow) and the L1951R mutation (green). VAF is shown in parentheses for each mutation. B, Western blot using protein extract from cell line derived from patient’s crizotinib-resistant tumor sample (CUTO16) demonstrating only partial inhibition of pROS1, pSHP2 and pERK1/2 with 1 mm crizotinib or ceritinib following 2 hour drug treatment. C, CT scan of the ROS1+ patient after development of resistance to crizotinib (top) and the first interval scan at 1 month (middle) after starting ceritinib and the same nodule after 3 months on ceritinib with progression (bottom). D, Cell viability of CUTO23 treated with increasing doses of crizotinib alone (black) or in the presence of 100 nM afatinib (red) for 72 hours. The IC50 for CUTO23 cells in crizotinib alone was 1959 ± 31nM and for crizotinib with 100nM afatinib 438 ± 79nM. Error bars, mean ± SEM for three triplicate experiments (n = 9). E, Western blot analysis of CUTO23 cells treated with the indicated doses of crizotinib in the absence (left) or presence (right) of 100 nM afatinib for 2 hours demonstrating inhibition of pERBB2 with afatinib and concomitant rescue of inhibition of downstream signaling by AKT and ERK1/2 with the addition of afatinib to crizotinib.
Another of the ROS1 samples, (ROS1-8), harbored no KDM. A cell line, CUTO23, was derived from the malignant pleural effusion of this patient following progression on crizotinib. This cell line, confirmed to harbor CD74-ROS1 by NGS, showed resistance to crizotinib in cell proliferation assays (Fig. 2D). Although ROS1 protein was detectable at the predicted molecular weight for CD74-ROS1, pROS1 could not be detected (Fig. 2E and Supplementary Fig. 2). Crizotinib failed to inhibit pERK1/2, even at high concentrations, but did inhibit pSHP2, a known ROS1 signaling adaptor, suggesting that a bypass signaling pathway that did not utilize SHP2 was the cause of resistance (Fig. 2E). Given prior evidence by our group that HER family RTK members can mediate resistance to ROS1 inhibitors, we queried whether a pan-HER inhibitor, afatinib, might overcome the ROS1 resistance in this cell line.(45,46) Addition of afatinib partially restored crizotinib sensitivity in cell proliferation assays (Fig. 2D). Furthermore, afatinib inhibited critical downstream signaling via AKT and ERK1/2 (Fig. 2E). We were unable to detect EGFR or pEGFR in these samples (data not shown), but HER2 (ERBB2) was expressed and phosphorylated in this cell line suggesting it was mediating resistance. No evidence of EGFR or ERBB2 mutations or CNV were identified in this sample using NGS. Although we did not have a pre-crizotinib tumor sample or cell line for ROS1-8/CUTO23, we evaluated total and phosphorylated levels of HER2 in CUTO23 compared to the ROS1 cell lines CUTO27 (derived from sample ROS1-9) and CUTO28 (derived from sample ROS1-11), which are sensitive to ROS1 inhibitors (data not shown). CUTO23 displayed higher levels of total HER2 protein and showed markedly increased pHER2 compared to CUTO27 and CUTO28 (Supplementary Fig. 2).
ALK kinase domain mutations
ALK exons 21–25 were sequenced by NGS and/or direct sequencing to evaluate for the presence of KDM. The average read-depth was greater than 2000x for each of these exons using NGS (Supplementary Table 2). Of the 49 samples evaluated, immediate prior therapy was first line crizotinib (n=28), ceritinib (n=6), brigatinib (n=7), alectinib (n=4), lorlatinib (n=2), and 2 patients were treated with third-line crizotinib after crizotinib and ceritinib (Supplementary Table 3). Eight of 28 patients (25%) demonstrated an ALK KDM after treatment with crizotinib as the initial ALK TKI. Four different mutations were noted, the most common being L1196M (n=3) (Fig. 3). In the 21 patients who had a post-progression biopsy after treatment with more than one ALK-targeted TKI, 9 (45%) displayed a KDM.
Figure 3.
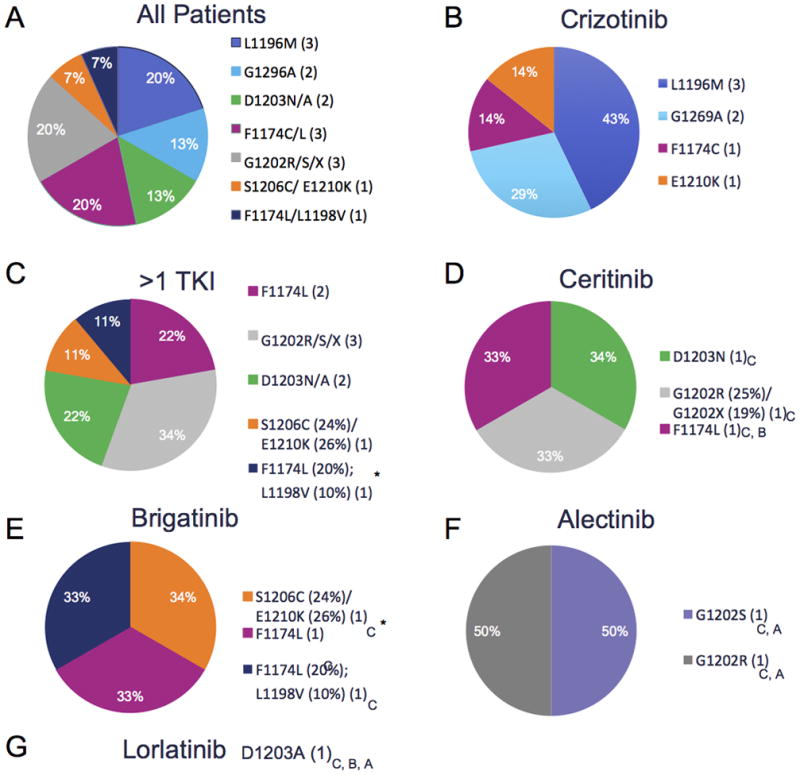
Kinase Domain Mutation Distribution in ALK+ Resistance samples.
A, Distribution of kinase domain mutations across ALK+ samples. One sample with G1202* (VAF 19%) in combination with G1202R (VAF 25%). 3 patients demonstrated compound mutation after multiple ALK-targeted TKIs. B, Distribution of kinase domain mutations in those patient who had a post progression biopsy after crizotinib, firstline therapy for all patients. C, Distribution of kinase domain mutations in patients who had post progression biopsy after >1 TKI (D) Distribution of kinase domain mutations in patients who had post progression biopsy after ceritinib. E, Distribution of kinase domain mutations in patients who had post progression biopsy after brigatinib. F, Distribution of kinase domain mutations in patients who had post progression biopsy after alectinib. G, One patient had a post progression biopsy after treatment with lorlatinib. Prior treatments for each patient listed in subscript. Patients with serial biopsies on different treatments indicated with Asterisk. If multiple biopsies KDM listed for biopsy on which it appeared. Abbreviations: C- crizotinib, B- brigatinib, A-Alectinib, TKI – tyrosine kinase inhibitor, X- stop codon.
In the ALK rebiopsy cohort, five patients were biopsied after multiple different targeted treatments (patients 17, 22, 24, 29, and 44). Two of these patients, 17 and 29, had KDM on post-progression biopsy after crizotinib. Patient 17 maintained an F1174C mutation on three different post-treatment evaluations (crizotinib, ceritinib and crizotinib rechallenge). Direct sequencing of the post-crizotinib tumor in patient 29 revealed the presence of an E1210K mutation (Supplementary Fig. 3A) and inspection of the post-brigatinib tumor specimen revealed that the majority of the sequencing reads harbored both an E1210K and an S1206C mutation in cis with VAFs of 26% (E1210K) and 24% (S1206C) (Supplementary Fig. 3B). The crystal structure of the ALK kinase domain with the position of these two mutations is illustrated in Supplementary Fig. 3C. S1206 and E1210 are in the solvent exposed region, adjacent to the adenosine triphosphate-binding pocket (47,48). Thus, co-occurring mutations in this region may interfere with TKI activity.
ALK-47 harbored two ALK KDM, F1174L (VAF 20%) and L1198V (VAF 10%) on a post-brigatinib tumor sample derived from a brain tumor specimen (Supplementary Fig. 3D). The L1198V mutation occurred in cis with F1174 as it was always identified on the same sequencing reads using NGS (Supplementary Fig. 3E) and suggests that it occurred after acquisition of the F1174L mutation given its lower VAF. The distribution of KDMs is illustrated by pre-progression TKI in Fig. 3 and Supplementary Table 3.
FISH identified copy number gain in ALK+ but not ROS1+ resistance samples
ALK fusion copy number gain has been previously implicated in TKI resistance (29,47,49,50). We further characterized a subset of patients with available matched biopsies before and after treatment with TKIs to identify those with evidence of copy number change by FISH. Biopsy specimens were available for comparative FISH for two ROS1 samples, but no CNG was identified. Of the 28 ALK+ patients in whom biopsy specimens were available for comparative FISH we identified CNG in 6 patients (21%) (Table 3). Patients 7 and 8 were reported previously (29). Overall, all biopsies with ALK CNG demonstrated both an increase in the number of rearranged copies per cell and in the number of cells with detectable ALK rearrangement (51). Among the 6 patients with biopsies with CNG, patient 7 also had a KDM (G1269A, VAF 29%) and patient 31 was found to have a mutation in an alternate cancer-related gene (EGFR exon 19 deletion). Additionally, patient 24 acquired a CNG after treatment with ceritinib when compared with a prior, post-crizotinib sample.
Table 3.
FISH analysis to evaluate for copy number alterations.
| Pre-treatment | Post-treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient ID | ALK FISH % cells positive | ALK FISH pattern | Abnormal ALK copy number/cell | ALK FISH | ALK FISH % cells positive | ALK FISH pattern | Abnormal ALK copy number/cell | ALK FISH |
| 7 | 28% | split | 0.3 sR, sG | Positive | 82% | split | 1.5 sR, sG | Positive |
| 8 | 48% | split | 0.5 sR,sG | Positive | 66% | split | 2.2 sR, sG | Positive |
| 20 | 76% | sR | 0.78 sR | Positive | 90% | sR | 1.92 sR | Positive |
| 22 | 76% | sR | 1.32 sR | Positive | 100% | sR | 3.08 sR | Positive |
| 24 | 72% | split | 1.66 sR, 1.68 sG | Positive | 98% | split | 3.68 sR, 3.92 sG | Positive |
| 31 | 46% | sR | 0.62 sR | Positive | 80% | sR | 1.84 sR | Positive |
Abbreviations – PT ID- patient ID, sR = single red, sG = single green, split = split red/green, mix = split red/green and single red
Rounded to the nearest tenth decimal
Identification of potential ROS1- and ALK-independent mechanisms of resistance
We and others have previously identified bypass signaling pathways as a mechanism of drug resistance that obviates the need for the original dominant oncogene such as ROS1 and ALK (29,45). The NGS panel was thus designed to evaluate alterations in additional oncogenes beyond ALK and ROS1.
Among the 12 ROS1 patients, we identified a mutation in the kit proto-oncogene tyrosine kinase (KIT) (D816G) that we have previously characterized in vitro as an acquired resistance mechanism to crizotinib (52). We also found a mutation in β-catenin (CTNNB1 S45F), that has been identified as a potential oncogenic driver in lung cancer, although we were not able to demonstrate its absence prior to therapy due to lack of available tissue (53).
Among the 43 ALK+ patients we identified 13 oncogenic alterations in 6 different genes that may have resulted in new oncogenic activity or resistance to treatment (two fusions, one incidence of loss of ALK fusion gene, and 10 oncogenic mutations) (Supplementary Table 3).
Gene fusions identified as ALK resistance mechanisms
Patient ALK-22 demonstrated a RALGAPA1–NRG1 fusion on the post-alectinib tumor sample (Supplementary Fig. 4A,B). Although NRG1 fusions have been described, RALGAPA1 has not been described as a partner with NRG1. To demonstrate this fusion was both functional and could induce resistance to an ALK Inhibitor we used CRISPR to engineer this fusion into the H3122 cell line to (H3122-NRG1). The presence of the NRG1 fusion in H3122 was confirmed by genomic sequencing (not shown). H3122-NRG1 cells demonstrated marked resistance to crizotinib and sensitivity was restored by the pan-HER inhibitor afatinib (Fig. 4A). Western blot analysis demonstrated increased pHER3 (ERBB3), the receptor for the ligand neuregulin 1 (Fig. 4B). H3122-NRG1 cells showed persistent pAKT and pERK1/2 in the presence of crizotinib. Afatinib inhibited pHER3 and inhibited pAKT and pERK1/2 and the addition of afatinib to crizotinib similarly inhibited AKT and ERK1/2 signaling. Notably the induction of the NRG1 fusion in H3122 cells led to the loss of phosphorylation of SHP2, a known signaling adaptor for ALK. Analysis of the ALK-22_2 sample by proximity ligation assay demonstrated the presence of both ALK-GRB2 and HER2 (ERBB2)-GRB2 complexes consistent with functional signaling by both of the ALK and NRG1 gene fusions (Fig. 4C,D). HER2 is known to heterodimerize with HER3 upon ligand binding to HER3.
Figure 4.
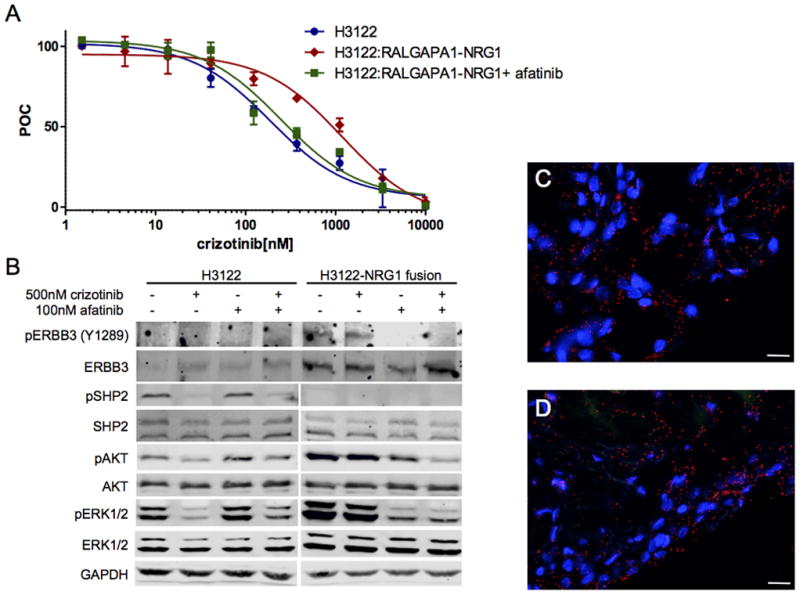
RALGAPA1-NRG1 fusion induces drug resistance in ALK+ cancer.
A, Cellular proliferation of the EML4-ALK cell lines, H3122 (blue)or its derivative H3122-NRG1 (red) which harbors a CRISPR induced RALGAPA1-NRG1 fusion using increasing doses of crizotinib demonstrating marked resistance crizotinib resistance. Addition of afatinib (100nM) resensitized the H3122-NRG1 (black) to crizotinib. IC50 for crizotinib was H3122 cells was 187 ± 3.7nM, for H3122-NRG1 was 1182 ± 5.3nM and for H3122-NRG1 with afatinib was 200 ± 10.5nM. Error bars, mean ± SEM for three triplicate experiments (n = 9). B, Western blot analysis of H3122 or H3122-NRG1 cells treated with crizotinib 500 nM and/or afatinib 100nM as indicated for 2 hours. H3122-NRG1 cells demonstrated increased levels of phosphorylated and total ERBB3, pAKT and pERK1/2 compared to H3122 cell lines, but lack of pSHP2. Crizotinib inhibits SHP2, AKT and ERK1/2 in H3122 cells. AKT and ERK are not inhibited by crizotinib alone in H3122-NRG1, but are inhibited by afatinib as is ERBB3 indicating oncogene switch from ALK to HER3, the receptor for neuregulin. C, Proximity ligation assay (PLA) of ALK and GRB2 from ALK-22_2 FFPE tumor sample showing functional ALK signaling. D, PLA of ERBB2 and GRB2 from ALK-22_2 sample demonstrating functional ERBB2 signaling.
Notably, when we examined earlier tumor specimens from patient ALK-22, including the patient’s pre-crizotinib specimen, we identified the presence of the RALGAPA1-NRG1 fusion suggesting that this alteration co-existed with the ALK fusion and was therefore not a de novo mechanism of resistance. The patient’s very short-lived response to crizotinib (4.5 months) was consistent with this representing an intrinsic mechanism of resistance.
The tumor sample ALK-44_2 was found to have a CCDC6-RET gene fusion on the post-brigatinib biopsy in addition to the known EML4-ALK fusion (not shown). This gene fusion, which fuses exon 1 of CCDC6 to exon 12 of RET has been described as a primary oncogenic driver in NSCLC and other cancers (10). This RET gene fusion was not identified on the prior biopsy, which occurred post-alectinib (ALK-44_1).
Two patients were found to have EGFR mutations (L858R and exon 19 deletion). Three KRAS mutations were also observed (G12C, G12V, and G13D). The clinical details of the patients with G12C and G12V have been previously reported (29); however, we were previously unable to validate the presence of a functional ALK fusion (other than ALK FISH positivity) in the sample also harboring KRAS G12C. Using NGS, we demonstrated both an EML4-ALK (E6;A20) fusion as well as a KRAS G12C mutation in the same biopsy specimen. Notably, KRAS G12C was present in the tumor sample prior to crizotinib therapy and heralded primary progression as best response to therapy. This is in contrast to the patient with KRAS G13D who did not demonstrate this mutation in the pre-TKI clinical sample and had a much longer response to crizotinib.
We also identified mutations in isocitrate dehydrogenase 1 (IDH1) (R132C) and neurofibromatosis 1 (NF1) (Q642X) in different patients. Interestingly, the NF1 mutation was found in a patient (ALK-28) with a clinical syndrome consistent with neurofibromatosis and this patient demonstrated short-lived responses to both crizotinib (139 days) and alectinib (110 days). In another patient (ALK-46), we identified a previously unreported RIT1 mutation, K139N, though the significance of this mutation is unknown. Mutations in RIT1 have been associated with lung adenocarcinoma via activation of the RAS/RAF/MEK and PI3K pathways (54). Finally, we identified mutations in notch homolog 1 (NOTCH1 D1533 and D1538) in patients ALK-8 and ALK-42. Mutations in NOTCH1 have been identified in hematologic, head and neck and lung squamous cell carcinomas (55,56) though their clinical significance in this setting is unknown.
We performed analysis of CNV using our target capture NGS data (57). (Fig. 5 and Supplementary Fig. 5). In ROS1+ cohort, three patient samples demonstrated evidence of CNV of cancer-related genes by NGS ROS1-1 (SRC, ERBB2, STK11, and NOTCH1), ROS1-9 (PDGFRA, KIT and KDR) and ROS1-11 (FGFR3, RET, and ERBB2).
Figure 5.

Copy Number Variation Distribution. Median read depths across the 48 gene NGS panel illustrating outliers in our cohort. Genes are arranged by chromosome. Patients who have a gene with a read depth > 2.5 SD above the median are annotated. ROS1 samples (red), ALK samples (blue), and other NSCLC samples (open circles).
We also observed increases in CNV in several proto-oncogenes genes within our ALK dataset including KRAS (ALK-10): EGFR (ALK-21 and -27_2), FGFR1 (ALK-35 and -47) GNAS (ALK-40), DDR2 (ALK-10 and -41), HRAS (ALK-25 and -37) and NTRK1 and RIT1 in sample ALK-25. These genes may have a role in bypass pathway activation and resistance to targeted therapy. Notably, patient ALK-10 with evidence of KRAS CNV also harbored the pre-existing KRAS G12C mutation which is notable given that mutant KRAS alleles frequently demonstrate CNG (58).
DISCUSSION
In this study, we evaluated potential mechanisms of resistance at the time of progression on targeted therapy in 12 ROS1+ patients and 43 ALK+ patients across multiple different TKIs and lines of therapy.
Within the ROS1 cohort of 12 patients, a possible mechanism of resistance was identified in 50% of ROS1+ patients (summarized in Fig. 6A). Together with the relatively small number of patients in the ROS1+ cohort, the confidence intervals around the exact frequency of any mechanism must be considered broad. Despite this we found very few KDM in ROS1 patients compared with the frequency we and others have identified in ALK positive patients (29,30). Previously, KDMs have been identified in patient tumor samples include G2032R, D2033N and S1986Y/F (59–61). Although the KDM mutations we identified (L2026M and L1951R) have been previously reported in mutagenesis screens they have not been reported in patient samples (21,61,62).
Figure 6.

Resistance Mechanisms after TKI in ROS1+ ALK+ patients. Venn Diagrams demonstrating the distribution of resistance mechanisms identified in each cohort. A, ROS1 cohort (n = 12). B, ALK 1 TKI cohort (n = 28). C, ALK >1 TKI cohort (n = 21).
Within the subcategories of ALK+ patients, a mechanism of resistance was identified in 71% of 28 patients evaluated after first line crizotinib (Fig. 6B). In the 21 patients who received more than one ALK-targeted therapy, 95% of patients demonstrated a possible mechanism of resistance (Fig. 6C). Thus, similar to prior reports, we found that patients treated with >1 TKI were more likely to have developed a KDM compared with patients who had received crizotinib as a first line of targeted therapy (30). While it is possible that this represents a difference in resistance mechanisms to crizotinib vs. next generation ALK inhibitors, an alternative explanation may be patient selection bias. Whereas most patients who progressed on crizotinib as the first ALK TKI were offered biopsy at progression, patients on 2nd or 3rd line ALK TKIs who experienced immediate progression were not generally offered re-biopsy at our institution as they had typically undergone biopsy 6–8 week prior and it was deemed unlikely that new information would be identified from these tumor samples. Patients with primary progression to an appropriate ALK TKI are the least likely to have an ALK KDM, thus skewing the denominator for this cohort of patients. In the ALK+ cohort who received >1 ALK targeted TKI the small numbers of patients within each treatment group and frequent lack of sufficient pretreatment tissue for comparison makes it difficult to draw significant conclusions about the relationship between different KDM and prior TKI therapy. For example, one of the two patients who received treatment with lorlatinib had a post-treatment biopsy that demonstrated a D1203A mutation. This may be a mutation that evolved in response to lorlatinib; however, the patient has received several prior TKIs without pre-treatment biopsy so it is difficult to ascribe causality. Despite the small numbers, it is notable that 2/7 brigatinib samples harbored compound mutations in cis.
ALK + patient 17 illustrates the challenges of focusing on KDM characterization as the main source of resistance. This patient had an F1174C mutation in a post-first-line crizotinib biopsy sample. Despite this mutation, which has documented resistance to ceritinib (63), the patient responded to ceritinib for nearly one year. After re-challenges with crizotinib then ceritinib, rebiopsy again demonstrated the F1174C mutation. This case illustrates several important points; first, though resistance mechanisms can be inferred on progression after a period of response and known sensitivity patterns, definitive determination of an acquired resistance mechanism requires pre/post treatment testing demonstrating that the mechanism was acquired during the most recent treatment and/or demonstration of resolution of the resistant clone on new effective treatment. Consideration of alternative oncogenic drivers and evaluation of bypass pathway activation may identify clinically informative information. Future efforts may need to rely on multiple genomic and proteomic evaluations to determine the full extent of resistance mechanisms.
With regard to KDM in ROS1+ patients, it is notable that other small clinical series have suggested a much higher rate of ROS1 KDM (45%), with almost all KDM (4 of 5, 80%) being G2032R(64). We did not observe this in our cohort, raising questions about the true frequency of ROS1 KDM. The two KDM in our cohort, L2026M and L1951R, co-occurred in one patient.
Use of early passage live cell culture from this patient’s tumor allowed us to identify a drug with potential clinical activity that was initially stable on serial CT imaging. The rapid progression observed in this patient was consistent with the lack of in vitro activity of ceritinib against the L1951R subclone, inadequate ceritinib exposures to achieve the potential inhibition of L2026M observed in vitro, or the activity of a bypass signaling pathway not detected by our testing. In tumor sample ROS1-8 we were unable to identify any acquired resistance alterations using NGS; however, we demonstrated the role of the HER2 pathway in mediating bypass of a ROS1 TKI using CUTO23, a patient-derived cell line from this tumor sample. This confirms previous preclinical work by our lab demonstrating the critical role for the HER family in ROS1+ NSCLC and highlights the difficulty in uncovering mechanisms of resistance not driven by mutations (45,46). In the ALK cohort, we identified an NRG1 fusion further highlighting the role of the HER family RTK signaling in drug resistance.
Dardei et al. recently demonstrated that a SHP2 inhibitor could overcome resistance in ALK+ NSCLC models (65). Importantly, two resistance models generated in this work, H3122-NRG1 (ALK) or CUTO23 (ROS1), did not show significant SHP2 modulation by inhibition of the bypass resistance mechanisms, nor did our prior ROS1 resistance models of HCC78- and CUTO2-KITD816G, suggesting that SHP2 inhibition might not be able to overcome some modes of bypass-mediated resistance (66).
We also identified a RET fusion as a mechanism of acquired resistance in ALK+ NSCLC. Recently ALK, RET, FGFR3, and NTRK1 fusions have been described as resistance mechanisms to the EGFR TKIs (67–70); therefore, our data demonstrate yet another shared TKI resistance mechanism between EGFR and ALK+ NSCLC. Additionally, we found EGFR mutations and KRAS mutations, both of which have been reported previously (29,47,71). We also identified mutations in IDH1, NF1 and RIT. IDH1 mutations encoding substitutions at R132 have been implicated as oncogenic drivers in hematologic malignancies, CNS malignancies and cholangiocarcinoma (29,72,73). Mutation in the tumor suppressor gene NF1 results in increased cell proliferation via activation of p21, RAS, and the mammalian target of rapamycin protein (mTOR) pathway (74,75). This case was particularly compelling as the NF1 mutation was likely a germline mutation for the patient with a VAF 50% and a prior clinical diagnosis of neurofibromatosis. Notably, this patient progressed systemically after ~4 months on crizotinib and ~3.5 months on alectinib suggesting that this germline mutation may have played a role in intrinsic drug resistance.
In our ROS1+ cohort we identified 3 alternative pathway mutations: KIT, β-catenin, and GNA11. The activating mutation in KIT has been described previously by our lab and is notable for being resistant to most KIT-specific TKIs (66). One patient had a mutation in the gene encoding β-catenin (S45F, VAF 22%). Beta-catenin is involved in cell-cell adhesion and is thought to be involved in the wingless/WNT signaling pathway. This mutation has been previously reported in SCLC lung cancer and implicated in its development, and was reported in another cohort of ROS1 patients (53,64). GNA11 is known to be involved in development of uveal melanoma, although the functional significance of this mutation is unknown (76,77). In this study, we did not assay for RAS CNG by FISH or WT EGFR activation, which are known mechanisms of resistance in ROS1+ NSCLC, this may have decreased our detection of resistance mechanisms in the ROS1+ cohort (45,78).
Copy number gain of the dominant oncogene provide another source of resistance that have been described previously (29,47,49,50). We did not identify evidence of ROS1 CNG in the patients evaluated in our ROS1+ cohort, however ROS1 FISH was performed in a small number of these samples. However, in our ALK+ patient cohort, we report six of 28 evaluated patients (21%) demonstrated ALK fusion CNG. These may be underestimates of incidence, as evaluation of CNG requires pre- and post- treatment samples, which were not always available in our series.
CNV evaluation by NGS allows the monitoring of multiple genes simultaneously, where FISH can typically only test one gene at a time. Using CNV by NGS, we identified several putative resistance mechanisms not identified by other analyses. In the ROS1 and ALK cohorts treated with crizotinib, none of the 9 cases with CNV identified had evidence of a KDM, suggestive of alterations that contribute to resistance. Finally, samples ROS1-9 and ALK-33 showed evidence of CNV of genes located at the chromosomal locus of 4q12, which is frequently amplified in lung cancer and may generate drug resistance via bypass signaling mediated by these RTKs (79).
This analysis was subject to several limitations. First, it was a retrospective analysis and thus some cases relied on chart review and the presence of archival tissue, restricting us from performing every analysis on every patient. When available we have included in descriptions of pre-existing mutations, but to comprehensively determine and attribute resistance mechanisms requires testing of clinical samples before and after each treatment.
In this report, we describe the patterns of resistance mutations in biopsies from a cohort of patients with ROS1 or ALK rearrangements. As larger datasets are accumulated and we have increased experience with patients on multiple different TKIs, we may be able to identify unique patterns of resistance that develop with individual TKIs. Additionally, although KDMs remain an important mechanism of resistance, our data clearly shows that bypass pathway signaling through both genetic alterations or non-mutation driven pathways are at least equally important and we are likely to benefit from inquiry into these mechanisms for patient care.
Supplementary Material
Statement of Translational Relevance.
ROS1 and ALK positive non-small cell lung cancer patients treated with oncogene-targeted therapy will inevitably develop drug resistance and disease progression. Two general biological mechanisms of resistance are described: alterations that restore signaling through the original oncogene driver in the presence of the drug or alterations that switch dependence to other signaling pathways. Currently, little is known about ROS1 resistance and the spectrum of ALK resistance mechanisms may be changing with the introduction of new inhibitors. As specific resistance mechanisms could influence subsequent drug treatment choices and future drug combination strategies, data on the type and frequency of different resistance mechanisms for ROS1 and ALK+ cancers are likely to become increasingly important. Our data suggest comprehensive methods, beyond the identification of kinase domain mutations, are needed to identify the full spectrum of drug resistance in ROS1 and ALK+ cancer.
Acknowledgments
Funding
This work was made possible by generous support from the Christine J. Burge Endowment for Lung Cancer Research at the University of Colorado Cancer Center, the Burge family, and the Miramont Cares Foundation. This work was also supported by funding from the SWOG/HOPE Foundation, and the NIH/NCI P50 CA058187 (University of Colorado Lung SPORE in Lung Cancer) and R01 CA193935 grants to R.C. Doebele.
We like to acknowledge the University of Colorado Molecular Pathology Shared Resource (P30 CA46934) for their assistance. We would like to thank the ROS1ders patient advocacy group (ros1cancer.com) for their support of ROS1 research and their donation of tissue samples for the creation of novel ROS1 cell lines.
Footnotes
Conflicts of Interest.
CEM- honoraria for Takeda, Guardant Health, DRC- honoraria from Takeda; RD – Pfizer speaker’s bureau and advisory board, Novartis, Roche, Ignyta – advisory board; RCD – AstraZeneca – advisory board, Guardant Health – honoraria, Pfizer – honoraria, Ignyta – advisory board and sponsored research grant, Takeda – advisory board, Spectrum Pharmaceuticals – advisory board, Abbott Molecular – remuneration for patent license, Rain Therapeutics – stock owner; PAB – consultant for Genentech and Pfizer; ATL – Abbott Molecular – remuneration for patent license; DTM, ATL, KG, KJ, LS, AD, AEB, KDD, MV-G, WTP, DLA - declare no potential conflicts of interest;
References
- 1.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 2.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014;371(21):1963–71. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 5.Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(24):9270–4. doi: 10.1073/pnas.84.24.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inamura K, Takeuchi K, Togashi Y, Nomura K, Ninomiya H, Okui M, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2008;3(1):13–7. doi: 10.1097/JTO.0b013e31815e8b60. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(20):6618–24. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 8.Shaw AT, Solomon B, Kenudson MM. Crizotinib and testing for ALK. Journal of the National Comprehensive Cancer Network : JNCCN. 2011;9(12):1335–41. doi: 10.6004/jnccn.2011.0115. [DOI] [PubMed] [Google Scholar]
- 9.Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(17):4570–9. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nature medicine. 2012;18(3):378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 11.Pekar-Zlotin M, Hirsch FR, Soussan-Gutman L, Ilouze M, Dvir A, Boyle T, et al. Fluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancer. The oncologist. 2015;20(3):316–22. doi: 10.1634/theoncologist.2014-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. The Lancet Oncology. 2011;12(11):1004–12. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013;368(25):2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 14.Shaw AT, Camidge DR, Engelman JA, Solomon BJ, Kwak EL, Clark JW, et al. Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. J Clin Oncol. 2012;30(15S) abstr 7508. [Google Scholar]
- 15.Ou SH, Bang YJ, Camidge DR, Riely GJ, Salgia R, Shapiro G, et al. Efficacy and safety of crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer (NSCLC) J Clin Oncol. 2013;31(Supple) Abstr 8032. [Google Scholar]
- 16.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. The Lancet Oncology. 2012;13(10):1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014;370(13):1189–97. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ou SH, Ahn JS, De Petris L, Govindan R, Yang JC, Hughes B, et al. Alectinib in Crizotinib-Refractory ALK-Rearranged Non-Small-Cell Lung Cancer: A Phase II Global Study. J Clin Oncol. 2016;34(7):661–8. doi: 10.1200/JCO.2015.63.9443. [DOI] [PubMed] [Google Scholar]
- 19.Kim DW, Tiseo M, Ahn MJ, Reckamp KL, Hansen KH, Kim SW, et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J Clin Oncol. 2017 doi: 10.1200/JCO.2016.71.5904. JCO2016715904. [DOI] [PubMed] [Google Scholar]
- 20.De Braud FG, Pilla L, Niger M, Damian S, Bardazza B, Martinetti A, et al. Phase 1 open label, dose escalation study of RXDX101, an oral pan-trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol. 2014;32(5s)(suppl) abstr 2502. [Google Scholar]
- 21.Katayama R, Kobayashi Y, Friboulet L, Lockerman EL, Koike S, Shaw AT, et al. Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(1):166–74. doi: 10.1158/1078-0432.CCR-14-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Squillace R, Anjum R, Miller D, Vodala S, Moran L, Wang F, et al. AP26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions; 2013 Apr 6–10; Washington, DC. AACR; [Google Scholar]
- 23.Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(11):3493–8. doi: 10.1073/pnas.1420785112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gettinger SN, Bazhenova LA, Langer CJ, Salgia R, Gold KA, Rosell R, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. The Lancet Oncology. 2016;17(12):1683–96. doi: 10.1016/S1470-2045(16)30392-8. [DOI] [PubMed] [Google Scholar]
- 25.Horn L, Wakelee H, Reckamp KL, Blumenschein G, Jr, Infante JR, Carter CA, et al. MINI01. 02: Response and Plasma Genotyping from Phase I/II Trial of Ensartinib (X-396) in Patients (pts) with ALK+ NSCLC: Topic: Medical Oncology. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2016;11(11S):S256–S7. doi: 10.1016/j.jtho.2016.09.017. [DOI] [Google Scholar]
- 26.Facchinetti F, Tiseo M, Di Maio M, Graziano P, Bria E, Rossi G, et al. Tackling ALK in non-small cell lung cancer: the role of novel inhibitors. Transl Lung Cancer Res. 2016;5(3):301–21. doi: 10.21037/tlcr.2016.06.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. Journal of medicinal chemistry. 2014;57(11):4720–44. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- 28.Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer cell. 2015;28(1):70–81. doi: 10.1016/j.ccell.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(5):1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ou SH, Greenbowe J, Khan ZU, Azada MC, Ross JS, Stevens PJ, et al. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung cancer. 2015;88(2):231–4. doi: 10.1016/j.lungcan.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Ou SH, Milliken JC, Azada MC, Miller VA, Ali SM, Klempner SJ. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression Lung cancer. 2016;91:70–2. doi: 10.1016/j.lungcan.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Tchekmedyian N, Ali SM, Miller VA, Haura EB. Acquired ALK L1152R Mutation Confers Resistance to Ceritinib and Predicts Response to Alectinib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2016;11(7):e87–8. doi: 10.1016/j.jtho.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. The New England journal of medicine. 2016;374(1):54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26(7):873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature methods. 2011;8(8):652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971–4. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 38.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO molecular medicine. 2010;2(5):146–58. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667–74. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camidge DR, Theodoro M, Maxson DA, Skokan M, O’Brien T, Lu X, et al. Correlations between the percentage of tumor cells showing an anaplastic lymphoma kinase (ALK) gene rearrangement, ALK signal copy number, and response to crizotinib therapy in ALK fluorescence in situ hybridization-positive nonsmall cell lung cancer. Cancer. 2012;118(18):4486–94. doi: 10.1002/cncr.27411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong J, Doebele RC, Lingen MW, Quilliam LA, Tang WJ, Rosner MR. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J Biol Chem. 2007;282(27):19781–7. doi: 10.1074/jbc.M700128200. [DOI] [PubMed] [Google Scholar]
- 42.Davies KD, Ng TL, Estrada-Bernal A, Le AT, Ennever PR, Camidge DR, et al. Dramatic Response to Crizotinib in a Patient With Lung Cancer Positive for an HLA-DRB1-MET Gene Fusion. JCO Precision Oncology. 2017;(1):1–6. doi: 10.1200/PO.17.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doebele RC, Aisner DL, Le AT, Berge EA, Pilling AB, Kutateladze TG, et al. Analysis of resistance mechanisms to ALK kinase inhibitors in ALK+ NSCLC patients. J Clin Oncol. 2012;30(suppl):2012. abstr 7504. [Google Scholar]
- 44.Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer research. 2008;68(13):4971–6. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 45.Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PloS one. 2013;8(12):e82236. doi: 10.1371/journal.pone.0082236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, et al. EGFR Mediates Responses to Small-Molecule Drugs Targeting Oncogenic Fusion Kinases. Cancer research. 2017;77(13):3551–63. doi: 10.1158/0008-5472.CAN-17-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine. 2012;4(120):120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78(6):999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim S, Kim TM, Kim DW, Go H, Keam B, Lee SH, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2013;8(4):415–22. doi: 10.1097/JTO.0b013e318283dcc0. [DOI] [PubMed] [Google Scholar]
- 50.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(18):7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weickhardt AJ, Aisner DL, Franklin WA, Varella-Garcia M, Doebele RC, Camidge DR. Diagnostic assays for identification of anaplastic lymphoma kinase-positive non-small cell lung cancer. Cancer. 2013;119(8):1467–77. doi: 10.1002/cncr.27913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella-Garcia M, et al. An Activating KIT Mutation Induces Crizotinib Resistance in ROS1-Positive Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2016 doi: 10.1016/j.jtho.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shigemitsu K, Sekido Y, Usami N, Mori S, Sato M, Horio Y, et al. Genetic alteration of the beta-catenin gene (CTNNB1) in human lung cancer and malignant mesothelioma and identification of a new 3p21. 3 homozygous deletion. Oncogene. 2001;20(31):4249–57. doi: 10.1038/sj.onc.1204557. [DOI] [PubMed] [Google Scholar]
- 54.Berger AH, Imielinski M, Duke F, Wala J, Kaplan N, Shi GX, et al. Oncogenic RIT1 mutations in lung adenocarcinoma. Oncogene. 2014;33(35):4418–23. doi: 10.1038/onc.2013.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(43):17761–6. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Zhao L, Wang Y, Cao M, Gelowani V, Xu M, et al. SeqCNV: a novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinformatics. 2017;18(1):147. doi: 10.1186/s12859-017-1566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sasaki H, Hikosaka Y, Kawano O, Moriyama S, Yano M, Fujii Y. Evaluation of Kras gene mutation and copy number gain in non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6(1):15–20. doi: 10.1097/JTO.0b013e31820594f0. [DOI] [PubMed] [Google Scholar]
- 59.Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. The New England journal of medicine. 2013;368(25):2395–401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS, et al. A Novel Crizotinib-Resistant Solvent-Front Mutation Responsive to Cabozantinib Therapy in a Patient with ROS1-Rearranged Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(10):2351–8. doi: 10.1158/1078-0432.CCR-15-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Facchinetti F, Loriot Y, Cassin-Kuo MS, Mahjoubi L, Lacroix L, Planchard D, et al. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0917. [DOI] [PubMed] [Google Scholar]
- 62.Davare MA, Vellore NA, Wagner JP, Eide CA, Goodman JR, Drilon A, et al. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(39):E5381–90. doi: 10.1073/pnas.1515281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer discovery. 2014;4(6):662–73. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gainor J, Friboulet L, Yoda S, Alghalandis L, Farago A, JL, et al. Frequency and spectrum of ROS1 resistance mutations in ROS1-positive lung cancer patients progressing on crizotinib. J Clin Oncol. 2016;34(suppl) abstr 9072. [Google Scholar]
- 65.Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nature medicine. 2018 doi: 10.1038/nm.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella-Garcia M, et al. An Activating KIT Mutation Induces Crizotinib Resistance in ROS1-Positive Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2016;11(8):1273–81. doi: 10.1016/j.jtho.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piotrowska Z, Nagy R, Fairclough S, Lanman R, Marcoux N, Gettinger S, et al. OA 09.01 Characterizing the Genomic Landscape of EGFR C797S in Lung Cancer Using ctDNA Next-Generation Sequencing. Journal of Thoracic Oncology. 12(11):S1767. doi: 10.1016/j.jtho.2017.09.375. [DOI] [Google Scholar]
- 68.Ou SI, Horn L, Cruz M, Vafai D, Lovly CM, Spradlin A, et al. Emergence of FGFR3-TACC3 fusions as a potential by-pass resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR mutated NSCLC patients. Lung cancer. 2017;111:61–4. doi: 10.1016/j.lungcan.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakanishi Y, Masuda S, Iida Y, Takahashi N, Hashimoto S. Case Report of Non-Small Cell Lung Cancer with STRN-ALK Translocation: A Nonresponder to Alectinib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2017;12(12):e202–e4. doi: 10.1016/j.jtho.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 70.Klempner SJ, Bazhenova LA, Braiteh FS, Nikolinakos PG, Gowen K, Cervantes CM, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung cancer. 2015;89(3):357–9. doi: 10.1016/j.lungcan.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 71.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer research. 2011;71(18):6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32(25):3091–100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia. 2010;24(6):1146–51. doi: 10.1038/leu.2010.77. [DOI] [PubMed] [Google Scholar]
- 74.Sandsmark DK, Zhang H, Hegedus B, Pelletier CL, Weber JD, Gutmann DH. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer research. 2007;67(10):4790–9. doi: 10.1158/0008-5472.CAN-06-4470. [DOI] [PubMed] [Google Scholar]
- 75.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356(6371):713–5. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 76.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. The New England journal of medicine. 2010;363(23):2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Lange MJ, Razzaq L, Versluis M, Verlinde S, Dogrusoz M, Bohringer S, et al. Distribution of GNAQ and GNA11 Mutation Signatures in Uveal Melanoma Points to a Light Dependent Mutation Mechanism. PloS one. 2015;10(9):e0138002. doi: 10.1371/journal.pone.0138002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cargnelutti M, Corso S, Pergolizzi M, Mevellec L, Aisner DL, Dziadziuszko R, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015;6(7):5182–94. doi: 10.18632/oncotarget.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramos AH, Dutt A, Mermel C, Perner S, Cho J, Lafargue CJ, et al. Amplification of chromosomal segment 4q12 in non-small cell lung cancer. Cancer Biol Ther. 2009;8(21):2042–50. doi: 10.4161/cbt.8.21.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.