

Abstract
Castration-resistant prostate cancer (CRPC) is characterized by reactivation of androgen receptor (AR) signaling in part by elevated expression of AR splice variants (ARv) including ARv7, a constitutively active, ligand binding domain (LBD)-deficient variant whose expression has been correlated with therapeutic resistance and poor prognosis. In a screen to identify small molecule dual inhibitors of both androgen-dependent and androgen-independent AR gene signatures, we identified the chalcone C86. Binding studies using purified proteins and CRPC cell lysates revealed C86 to interact with heat shock protein 40 (Hsp40). Pulldown studies using biotinylated-C86 found Hsp40 present in a multi-protein complex with full-length (FL-) AR, ARv7 and Hsp70 in CRPC cells. Treatment of CRPC cells with C86 or the allosteric heat shock protein 70 (Hsp70) inhibitor JG98 resulted in rapid protein destabilization of both FL-AR and ARv, including ARv7, concomitant with reduced FL-AR- and ARv7-mediated transcriptional activity. The glucocorticoid receptor (GR), whose elevated expression in a subset of CRPC also leads to androgen-independent AR target gene transcription, was also destabilized by inhibition of Hsp40 or Hsp70. In vivo, Hsp40 or Hsp70 inhibition demonstrated single agent and combinatorial activity in a 22Rv1 CRPC xenograft model. These data reveal that, in addition to recognized roles of Hsp40 and Hsp70 in FL-AR LBD remodeling, ARv lacking the LBD remain dependent on molecular chaperones for stability and function. Our findings highlight the feasibility and potential benefit of targeting the Hsp40/Hsp70 chaperone axis to treat prostate cancer that has become resistant to standard anti-androgen therapy.
Keywords: Castrate-resistant prostate cancer, androgen receptor, androgen receptor splice variants, molecular chaperones, drug targeting
Introduction
Surgical or chemical androgen deprivation therapy (ADT, encompassing both androgen depletion and anti-androgen strategies) is the first line treatment for locally advanced or metastatic prostate cancer (1). However, ADT efficacy is limited and despite systemic androgen depletion, the cancer often continues to progress to a terminal disease state known as castration resistant prostate cancer (CRPC) (1). In support of the continuing importance of androgen receptor (AR) signaling in CRPC, two potent second line agents, abiraterone and enzalutamide, have been approved by the FDA. Abiraterone is a CYP17A1 inhibitor (blocking both 17α-hydroxylase and 17,20-lyase, two enzymes important for androgen synthesis) (2), while enzalutamide is a potent AR ligand binding domain (LBD) competitive antagonist that blocks nuclear translocation and AR-dependent gene transcription (3). Although these agents extend overall survival, 20-40% of metastatic CRPC patients have no response to these drugs, while those that initially respond eventually develop secondary resistance (4).
CRPC therapeutic resistance is associated with reactivated and sustained AR signaling mediated by several mechanisms including AR overexpression and gene amplification, AR point mutations, enhanced AR coregulator activity or expression, and intratumoral androgen synthesis (5). Notably, CRPC is frequently associated with expression of a number of AR splice variants (ARv), including, but not limited to, ARv7 and ARv567es, which lack a complete carboxy-terminal LBD. Consequently, these ARv are insensitive to anti-androgens or androgen ablation, and are constitutively active (6). In particular, ARv7 has been shown to provide a growth advantage in androgen-depleted environments, leading to castration-resistant growth in vivo (7). Furthermore, ARv7 expression in CRPC correlates with poor prognosis, shorter progression-free and overall survival, and resistance to standard of care treatments (4, 8). In part, these characteristics are likely the outcome of a distinct transcriptional program mediated by ARv7 that is able to sustain cancer cell growth in androgen-depleted environments. For example, ligand-dependent full-length AR (FL-AR) regulates the expression of KLK3 (prostate-specific antigen, PSA), TMPRSS2, and NDRG1 (9–11), while ligand-independent ARv7 transcriptionally activates UBE2C and EDN2 (9–12).
In addition to increased expression of ARv7 and other ARv, upregulation of the glucocorticoid receptor (GR, like AR, is a member of the nuclear receptor family (13)) provides an alternative mechanism for generating resistance to AR LBD-targeted therapy (14–16). GR can occupy approximately one-half of the AR cistrome and can induce expression of approximately one-third of AR-regulated genes (17). While most prostate cancers express AR, approximately 30% of prostate cancers not exposed to LBD-targeted therapy also express GR (15, 16). Intriguingly, in the presence of androgen, AR-regulated transcription is frequently suppressed by the GR ligand dexamethasone. However, in the context of androgen deprivation, ligand-occupied GR drives the transcription of a host of AR target genes, many of which are overexpressed in prostate cancer. Thus, GR expression in CRPC also has been shown to promote clinical resistance to enzalutamide (14, 17). Due to the morbidity associated with late-stage disease, identification of novel strategies able to simultaneously disrupt the FL-AR/ARv7 and GR signaling axes in CRPC are of significant clinical importance and remain a critical unmet need.
Molecular chaperones (Heat shock proteins, Hsp), including Hsp40, Hsp70 and Hsp90, participate in protein folding, quality control of misfolded proteins, and the stability of metastable proteins (e.g., protein homeostasis) (18). Nuclear hormone receptors, including AR and GR, require the collaborative activity of the Hsp40 (DnaJ)/Hsp70/Hsp90 chaperone complex to maintain their LBD in an activation-competent, high-affinity ligand binding state (13, 19–23). Hsp90 has been shown to be important for androgen binding to the AR LBD in yeast (24) and the chaperone interacts directly with the human AR LBD (25). Further, Hsp90 inhibitors cause destabilization and degradation of FL-AR and GR (26, 27). However, recent studies have shown that ARv proteins lacking the LBD, including ARv7, are independent of both Hsp90 and androgen, and are insensitive to Hsp90 inhibitors (28, 29). It should be noted that two recent reports identify a role for Hsp90 in regulation of alternative splicing, including alternative splicing of AR (30, 31). Nevertheless, in contrast to the rapid FL-AR destabilizing effects of Hsp90 inhibitors, indirect effects on AR splicing require longer drug exposure and have general effects on alternative splicing beyond AR (31).
The present study is an outgrowth of a small molecule screen whose aim was to identify novel dual inhibitors of both androgen-dependent and androgen-independent transcriptional activity. As shown herein, a potent compound (C86) identified in this screen was found to be an Hsp40 interactor, suggesting a continued, Hsp90-independent role for Hsp40, and perhaps also Hsp70 (with which Hsp40 frequently associates), in functional regulation of ARv lacking an LBD. The current work uses C86 and the allosteric Hsp70 inhibitor JG98 to explore this possibility in detail, and our findings identify both Hsp40 and Hsp70 as potential therapeutic targets in CRPC that is resistant to abiraterone and enzalutamide.
Materials and Methods
Compounds/SAR/Synthesis
C86 (3-nitro-2’,4’,6’-trimethoxychalcone) was obtained from Indofine Chemical Company and the inactive analog (1,3,5-trimethoxybenzene) was obtained from Sigma. To synthesize biotin-C86 (b-C86) or biotin-inactive analog (b-IA), C86 or the inactive analog was conjugated with an iodinated biotin reagent in the presence of Cs2CO3 and dimethylformamide. JG98 (32) and JG231 were synthesized and provided by J. Gestwicki. Synthesis and screening strategy of chalcone compounds have been described previously (33).
Cell culture
LNCaP, 22Rv1, VCaP, HEK293, COS7, SkBr3, and A549 cells were obtained from ATCC and cultured in RPMI-1640 (Corning) or DMEM (Corning), each supplemented with 10% FBS. The LNCaPAR-V7/pHage line, which expresses AR-V7 in response to doxycycline, was generated by lentiviral infection of a pHage vector containing the AR-V7 sequence as previously described (12). All cell lines used in this study were tested for mycoplasma prior to use and were authenticated by ATCC using short tandem repeat analysis. For drug treatment, LNCaP cells were transferred to medium containing 10% charcoal stripped serum (CSS) and cultured overnight prior to the addition of 10 nM R1881 for 6 additional hours. LNCaP-ARv7 cells were transferred to medium containing 10% CSS and treated with drug 2-4 hours prior to the addition of 20 ng/ml doxycycline to induce ARv7. All cells were maintained in a 37°C and 5% CO2 humidified incubator, used for < 20 passages following thawing, and cultured for no longer than 3 months.
Immuno and chemical precipitation
FLAG- or V5-tagged proteins were immunoprecipitated from cell lysate by incubation with FLAG- or V5-conjugated agarose beads for 1 hour (Sigma). For chemical precipitation, b-C86, GA beads, or PU-H71 beads (synthesized as described previously (34, 35)) were incubated with recombinant Hsp40 (DnaJB1, Abcam), Hsp70 (provided by Sue Wickner, NCI), Hsp90 (R&D Systems) or cell lysate for 1 hour, followed by streptavidin beads (Sigma) for 1 hour. In each case, beads were then washed at least 3 times with TNES buffer and bound protein eluted with 3xFLAG peptide (Sigma), V5 peptide (Sigma), or 2X gel loading buffer. Samples were analyzed by SDS-PAGE and WB.
RT-qPCR
Total RNA was isolated from cultured cells using the TRIzol (Invitrogen) or the RNeasy RNA Isolation Kit (Qiagen) and reverse transcribed using the Maxima First Strand cDNA Synthesis Kit (Thermo). Transcript levels were detected by RT-qPCR using an ABI 7500 Real Time Detection System (Life Technologies) with experiments performed in triplicate. Primer sequences are listed in Supplemental Information.
Cell viability
Twenty-four hours following seeding (5,000 cells/well) in a 96-well plate, 22Rv1 cells (60-70% confluent) were treated with vehicle (DMSO), C86, MDV3100 (enzalutamide, Selleck Chemicals), bicalutamide (Selleck Chemicals), or JG98 at indicated concentrations and duration. Cell viability was then assessed by MTT ((3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium bromide, Sigma) or CellTiter-Glo (Promega) assay.
SDS- PAGE and Western blot (WB)
Cells were rinsed with ice-cold PBS, lysed with TNES buffer (50 mM Tris [pH 7.5], 2 mM EDTA, 100 mM NaCl, 1% NP-40) containing fresh protease and phosphatase inhibitor (Roche) and centrifuged at 4°C, 13,200 g for 30 minutes. Where applicable, soluble sample was removed and the insoluble pellet was rinsed with PBS following centrifugation. After pelleting and removal of PBS, TNES buffer was added and samples sonicated. All samples were quantified by BCA assay (Thermo). 10-20 μg of protein was separated by SDS-PAGE using Mini-PROTEAN TGX 4-20% precast gradient gels (BioRad), transferred to nitrocellulose membranes, blocked for 1 hour with TBS-T (50 mM Tris [pH 7.5], 300 mM NaCl, 0.5% Tween 20) containing 5% nonfat milk (blocking buffer), and incubated with primary antibody overnight at 4°C. The following day, membranes were rinsed in TBS-T and incubated with species-specific secondary antibody in blocking buffer for 1 hour at room temperature. Proteins were detected using the SuperSignal chemiluminescence detection system (Thermo) and films were scanned. The same blot was probed multiple times, and if necessary, was sliced horizontally for better exposure when the same antibody species was used. If brightness/contrast was adjusted, it was equivalently adjusted for the entire image. Antibodies used are listed in Supplemental Information.
Transfection
HEK293, COS7, or 22Rv1 cells were seeded at 1 × 106 cells per 10cm2 dish. The following day, X-tremeGENE 9 (Roche) was used to transfect cells with 2 μg of: FLAG-DnaJA1, -A4, -B2a, -B4, -B6b, -C5, -C7 (provided by Chad Dickey, USF); tetracycline-inducible pcDNA3-V5-DnaJB6b, -B6b H/Q, -B8, -B8 H/Q (provided by Harm Kampinga, University of Groningen); pCMV-3xFLAG-ARv7, -FL-AR; pEGFP-ARv7, -FL-AR (provided by Lisa Butler, University of Adelaide); -3xFLAG-Hsp70. Cells were treated or lysate was collected 24 hours after transfection for binding assays (IPs) and WB.
Animals
All animal experiments were approved by the NCI-Bethesda Animal Care and Use Committee (approved animal protocol number UOB-013-A). Four-week-old male athymic mice (Charles River) were allowed to mature and acclimate for 2 weeks. To establish CRPC xenografts, 1 × 106 low passage 22Rv1 cells in a 1:4 mixture of media/matrigel (v/v; BD Biosciences) were injected into the right flank of each mouse. Once tumors reached a volume of 75-125 mm3, mice were randomized and administered either vehicle (10% DMSO, 18% Cremophor RH 40 [Sigma], 3.6% dextrose, 68.4% 1 M HEPES) or 15 mg/kg C86 by tail vein (IV) injection every Monday, Wednesday, and Friday for 2 weeks. For experiments using JG231, tumor bearing mice were randomized and treated with either vehicle (as described above) or 8 mg/kg JG231 by intraperitoneal (IP) injection every other day for 3 weeks. Finally, for combination studies, tumor bearing mice were randomized and treated with vehicle or 4 mg/kg JG231 Monday, Wednesday, and Friday +/− 15 mg/kg C86 Tuesday, Thursday, and Saturday, with Sundays off, for 2.5 weeks. Tumor volumes and mouse body weights were measured 2-3 times per week. For pharmacodynamic analyses, tumors were excised from vehicle or drug treated mice after 10 days, homogenized, and processed for WB.
GR Activity Assay
The W303 yeast strain was utilized to measure GR activity and drug sensitivity. Cells were grown in YPD (1% Bacto yeast extract, 2% peptone, 2% dextrose) or defined synthetic media supplemented with 2% dextrose at 30°C unless otherwise specified. Yeast cells were transformed with the p/G795GR plasmid encoding the mammalian steroid hormone receptor GR in combination with either the β-galactosidase reporter pUCΔSS-26X GRE, pRS416GPD control vector, or the pLacZi reporter control (Addgene). Resultant colonies were grown in 50 mL of defined minimal media at 30°C overnight. Overnight culture density was measured and diluted to an O.D.600 of 0.08 followed by incubation at 30°C for 30-45 minutes, until reaching an O.D.600 of 0.1. Once cell density reached an O.D.600 of 0.1, 100 μL of each culture was aliquoted in triplicate into a white 96-well plate, followed by the immediate addition of DMSO or C86. After an incubation period of 30 minutes, dexamethasone (10 μM, Sigma) was added to each well and incubated for 2 hours at 30°C. Subsequently, 100 μL of Tropix Gal-Screen reagent (Applied Biosystems) was added to each well and incubated for 1.5 hours at room temperature. Luminescence was then measured using a microplate luminometer. To determine the effect of C86 on yeast growth, two isolated yeast colonies were inoculated in 50 mL of YPD media followed by an overnight incubation at 30°C. Inoculum densities were adjusted to an O.D.600 of 0.5, either DMSO or C86 were added to the flasks, and cell density was subsequently measured at O.D.600 every 2 hours for 8 hours.
Results
The small molecule C86 inhibits FL-AR/ARv7 signaling in CRPC cells
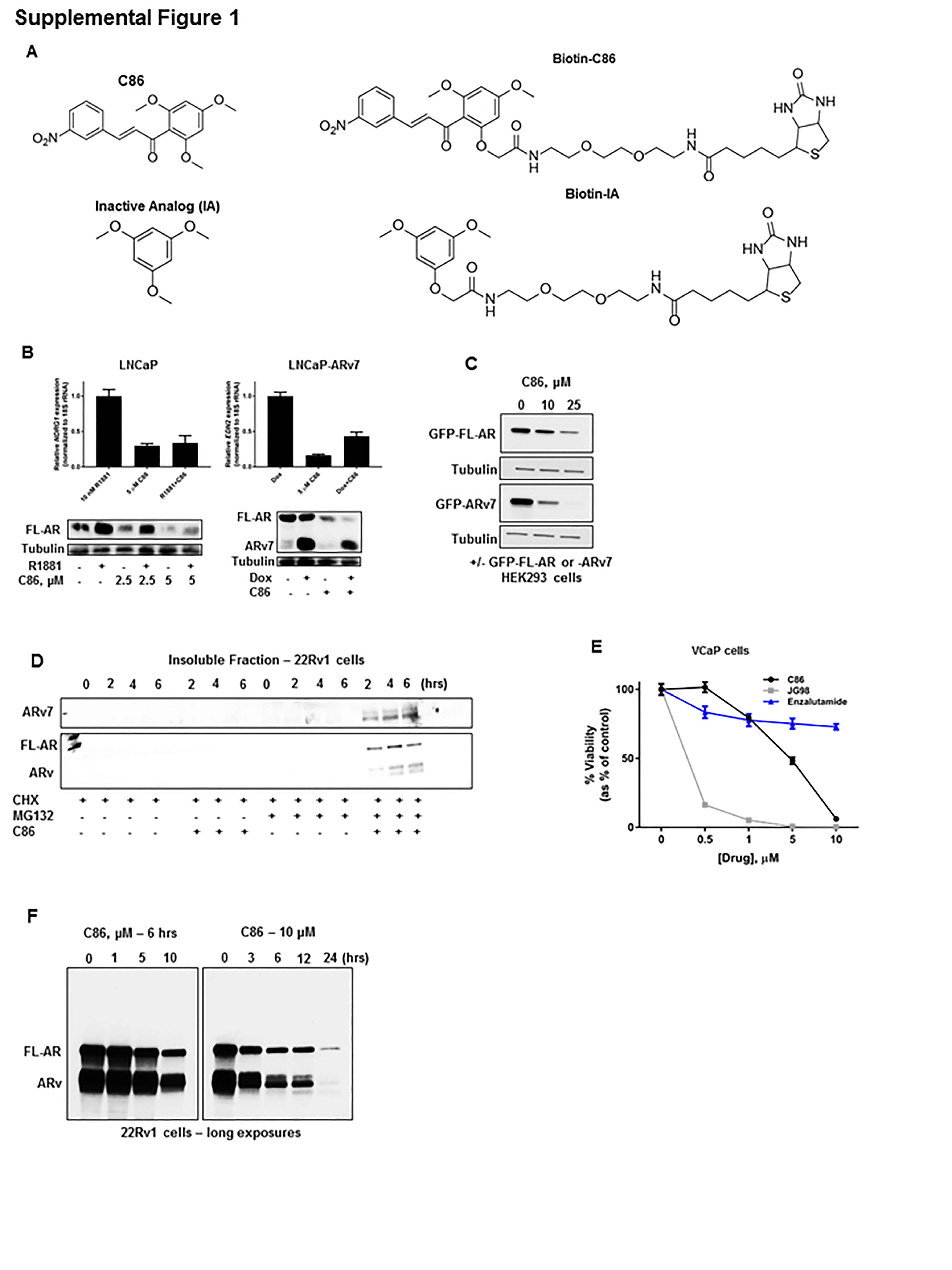
To identify compounds that perturb the FL-AR/ARv7 transcriptional program, a library of chalcones was screened for their ability to simultaneously inhibit transcription of the androgen-responsive genes KLK3 (PSA) and TMPRSS2, and the androgen-independent but AR-regulated gene UBE2C, that are endogenously expressed in 22Rv1 CRPC cells (which endogenously express both FL-AR and ARv7). Following several rounds of structure-activity relationship analyses, C86 (Supplemental Figure 1A) was identified as the most potent inhibitor of the expression of these genes (red arrow, Figure 1A), whereas bicalutamide, a clinically relevant FL-AR antagonist, was ineffective (green arrow, Figure 1A). To further verify these results, C86 was tested in parental LNCaP cells (expressing only FL-AR) and in LNCaP cells stably expressing exogenous, doxycycline-inducible ARv7 (12). In agreement with the data obtained in 22Rv1 cells, C86 reduced androgen-dependent transcription of NDRG1 (Supplemental Figure 1B, top left) (10), as well as transcription of the androgen-independent, ARv7-specific target gene EDN2 (12) (Supplemental Figure 1B, top right).
Figure 1.
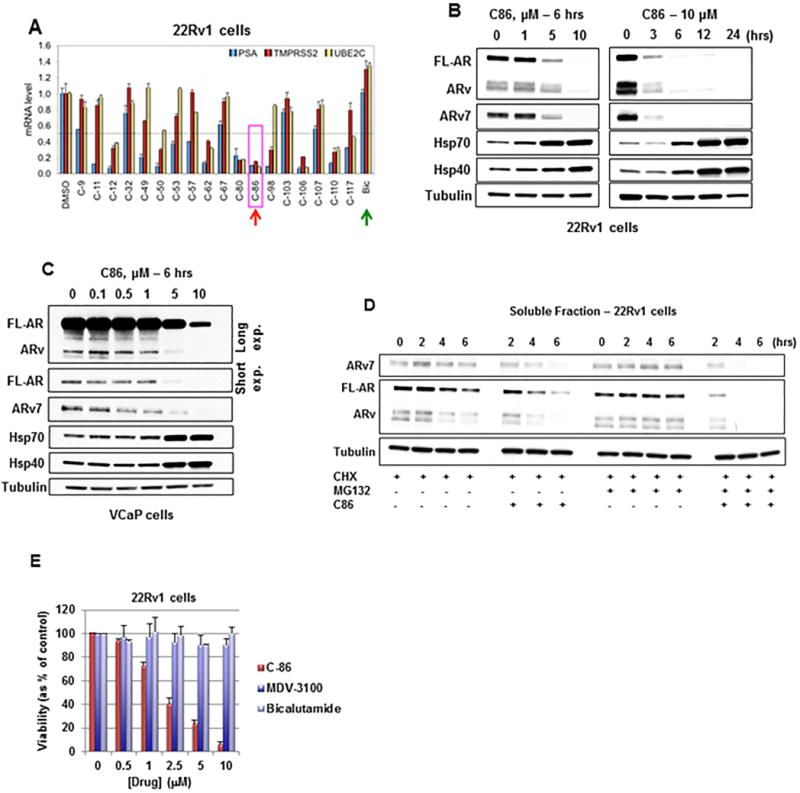
C86 is an inhibitor of the AR/ARv7 signaling axis in CRPC cells.
(A) RT-qPCR of AR/ARv7 target genes, normalized to 18S rRNA, from 22Rv1 CRPC cells treated with 5 μM of each compound or bicalutamide (Bic) for 20 hours. Data represent a mean of three replicates +/− S.D. PSA (prostate specific antigen) refers to gene designation KLK3.
(B) WB of cell lysate from 22Rv1 CRPC cells treated with increasing concentrations of C86 (left) or 10 μM C86 for up to 24 hours (right).
(C) WB of cell lysate from VCaP CRPC cells treated with increasing concentrations of C86 for 6 hours.
(D) WB of soluble AR, ARv, and ARv7 from 22Rv1 CRPC cells following pre-incubation with 10 μM MG132 and/or 10 μg/mL cycloheximide (CHX) for 1 hour, followed by +/− 10 μM C86 for 6 additional hours. Equal amounts of protein (10 μg) were loaded for WB of soluble and insoluble fractions (see also Supplemental Figure 1D).
(E) Viability of 22Rv1 CRPC cells exposed to increasing concentrations of C86 or anti-androgens (MDV3100 - enzalutamide, Bic) for 72 hours as determined by MTT assay. Data represent a mean of four to six replicates +/− S.D.
Experiments were repeated at least three times. AR antibody N-20 recognizes full-length AR (FL-AR) and likely also a number of similarly sized splice variants.
To obtain further mechanistic insight, we examined effects of C86 on FL-AR and ARv7 protein levels. Treatment of 22Rv1 cells with C86 resulted in a dose- and time-dependent loss of FL-AR and ARv (as detected with AR antibody N-20 and likely representing a number of similarly sized splice variants), including ARv7 (Figure 1B), with nearly complete loss of these proteins observed by 6 hours (10 μM C86). C86 exposure also caused a modest increase in Hsp40 and Hsp70 levels (Figure 1B), suggesting activation of the heat shock response. However, the fact that levels of FL-AR, ARv, and ARv7 proteins are substantially decreased after exposure to 10 μM C86 for 3 hours without a concomitant increase in Hsp40 or Hsp70 (Figure 1B, right), suggests it is possible to destabilize these nuclear receptors with pulsed/brief treatments that avoid potential compensatory induction of a heat shock response. Similar results were observed in VCaP CRPC cells (which also express FL-AR and ARv7) (Figure 1C), and in HEK293 cells transiently expressing exogenous GFP-FL-AR or GFP-ARv7 (Supplemental Figure 1C). Finally, consistent with its transcriptional inhibition of NDRG1 and EDN2, C86 destabilized FL-AR and ARv7 proteins in parental LNCaP and LNCaP-ARv7 cells (Supplemental Figure 1B, bottom).
To explore whether C86 effects on FL-AR/ARv7 protein levels may be due to modulation of AR/ARv7 gene expression, we evaluated effects on protein half-life. As shown in Figure 1D, loss of FL-AR, ARv, and ARv7 proteins occurred more rapidly in 22Rv1 cells treated with cycloheximide and 10 μM C86 compared to cells treated with cycloheximide alone. Further, treatment with the proteasome inhibitor MG132 enhanced the loss of FL-AR, ARv, and ARv7 (Figure 1D) from the soluble lysate fraction and caused their accumulation in a detergent-insoluble lysate fraction (Supplemental Figure 1D), suggesting C86 potentiates FL-AR/ARv7 aggregation and targets these nuclear receptors for degradation by the proteasome. Finally, while the clinically approved FL-AR antagonists bicalutamide and MDV3100 (enzalutamide) failed to display robust growth inhibitory activity in these CRPC cells, C86 treatment led to a significant, dose-dependent reduction in 22Rv1 (Figure 1E) and VCaP (Supplemental Figure 1E) viability by 72 hours at concentrations that cause loss of FL-AR/ARv7 protein expression and reduced target gene expression. Taken together, these data identify C86 as a novel small molecule that affects FL-AR/ARv7 signaling in vitro in CRPC-derived cell lines with activity that is superior to that of FDA-approved standard of care treatments targeting FL-AR.
Importance of the highly conserved J-domain for C86 binding to Hsp40 (DnaJ) family members
In some respects, the activity of C86 resembles that of Hsp90 inhibitors, since both classes of inhibitors cause client protein instability and degradation (18). Further, like many Hsp90 inhibitors (18), C86 appears to activate a heat shock response, as evident by induction of Hsp40 and Hsp70 (Figure 1B, C), and to promote accumulation of clients in a detergent-insoluble lysate fraction when combined with proteasome inhibitor (Figure 1D; Supplemental Figure 1D) (36). Thus, we first tested whether the mechanism of C86’s anti-CRPC activity was due to its ability to interact with Hsp90. As shown in Supplemental Figure 2A, biotinylayed-C86 (b-C86, Supplemental Figure 1A) was unable to bind recombinant Hsp90 protein (Supplemental Figure 2A, top left) or endogenous Hsp90 from SkBr3 (Supplemental Fig 2A, top right) and LNCaP (Supplemental Figure 2A, bottom left) cell lysate, whereas a significant fraction of this Hsp90 was captured with Hsp90-inhibitor-conjugated agarose beads (Supplemental Figure 2A, bottom left). The lack of direct Hsp90 interaction is also consistent with the recent observation that ARv lacking the LBD are resistant to Hsp90 inhibitors (28, 29), as Hsp90 is involved specifically in remodeling the LBD of nuclear receptors (23). Further, Hsp90 inhibitor promoted degradation of FL-AR protein in 22Rv1 cells but had no impact on ARv7 (Supplemental Figure 2B).
Due to the collaborative nature of the AR chaperone cycle, we next tested whether C86 could interact with other chaperone proteins known to function together with Hsp90 or Hsp70. We found that b-C86 was also unable to bind recombinant Hsp70 protein (Supplemental Figure 2A, bottom right). However, b-C86 bound a significant fraction of recombinant Hsp40 (DnaJB1), relative to a biotinylated-inactive control analog (Supplemental Figures 1A, 2C). Additionally, b-C86 captured endogenous Hsp40 (DnaJB1) from 22Rv1 and A549 lung cancer cell lysates (Supplemental Figure 2C), confirming that this interaction is not cell-type specific.
Figure 2.
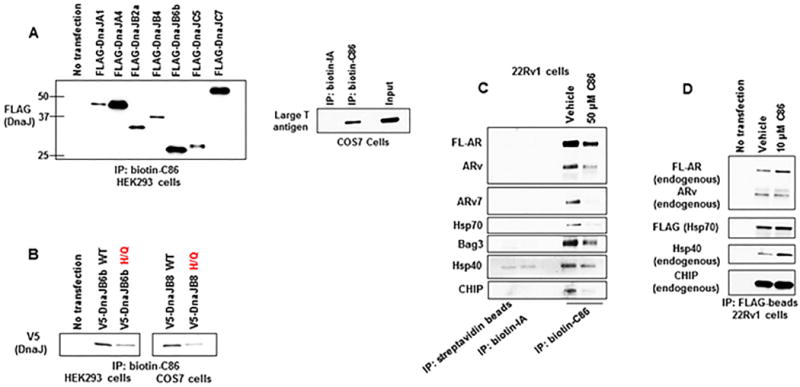
C86 binding to Hsp40/DnaJ family members likely involves the highly conserved J-domain.
(A) Biotin-C86 or -inactive analog (b-IA) binding to FLAG-DnaJ family members from transfected HEK293 cell lysates (left) or to endogenous Large T antigen from COS7 cell lysate (right) as assessed by streptavidin-bead IP and WB.
(B) Biotin-C86 binding to WT or H/Q mutant V5-DnaJB6b (left) or V5-DnaJB8 (right) as assessed by streptavidin-bead IP and WB.
(C) Biotin-C86 binding profile from 22Rv1 CRPC cells incubated with excess unlabeled vehicle (DMSO) or 50 μM C86 for 1 hour as assessed by streptavidin-bead IP and WB.
(D) 22Rv1 CRPC cells were transfected with FLAG-Hsp70 and treated with vehicle or 10 μM C86 for 1 hour. Lysate was then probed with FLAG-beads and bound proteins were detected by WB.
Experiments were repeated at least three times.
The mammalian Hsp40 (DnaJ) chaperone family consists of more than 40 members each with different functions and client binding profiles (37). Thus, it is possible that C86 may interact with some Hsp40 proteins and not with others. To determine if C86 acts as a pan-DnaJ/Hsp40 inhibitor, b-C86 was incubated with cell lysates from HEK293 cells transfected with various FLAG-tagged DnaJ proteins. Our data revealed that C86 interacts with DnaJ/Hsp40 proteins representative of the three DnaJ families A, B, and C (37) (Figure 2A, left; Supplemental Figure 2D, left). Since the presence of a J-domain (the site which interacts with Hsp70 for client protein transfer and to promote Hsp70 ATPase activity) (37) is the only common structural motif shared by all DnaJs, our findings implicate the J-domain in C86 binding. This hypothesis is consistent with the observation that b-C86 recognizes the Simian virus 40 (SV40) large T antigen in COS7 cells (Figure 2A, right), which is not a member of the Hsp40 family but also contains a J-domain (38).
To explore the possibility that J-domain structure is important for C86 binding, we transfected HEK293 or COS7 cells with DnaJs containing a known mutation in the highly conserved J-domain HPD motif (a three-amino acid motif comprised of histidine, proline and aspartic acid). This mutation (H/Q) perturbs the interaction of Hsp40 with Hsp70 by disrupting J-domain architecture (37). As shown in Figure 2B, b-C86 was able to bind both wild-type (WT) V5-tagged DnaJB6b (confirming the interaction in Figure 2A, left) and V5-DnaJB8 (Figure 2B; Supplemental Figure 2E). However, binding of V5-DnaJB6b-H/Q and V5-DnaJB8-H/Q to b-C86 was markedly decreased (Figure 2B; Supplemental Figure 2E), lending support to the importance of J-domain structural integrity for optimal recognition of Hsp40 by C86.
Next, we expanded our b-C86 pulldown experiments in 22Rv1 cells to determine whether C86 can pull down Hsp40 proteins bound to FL-AR/ARv7. Indeed, probing cell lysate with b-C86 identified endogenous Hsp40 (DnaJB1) in a complex with FL-AR, ARv, and ARv7 (Figure 2C). Compared to vehicle treatment, excess free C86 competed away Hsp40, AR, and ARv7, indicative of a specific interaction (Figure 2C). Hsp70, and its cochaperones the nucleotide-exchange factor Bag3 and the E3 ubiquitin ligase CHIP, were also present in this complex and were similarly affected by addition of unlabeled C86, suggesting that b-C86, in addition to interacting with free Hsp40, also recognizes an Hsp40/Hsp70/Bag3/CHIP complex that associates with FL-AR and/or ARv7 (Figure 2C). To further validate the interaction of C86 with an Hsp40/Hsp70 chaperone complex, we transfected 22Rv1 cells with FLAG-tagged Hsp70 and treated with 10 μM C86 (a concentration that causes AR/ARv7 destabilization at later time points, Figure 1B) for 1 hour. Under these conditions, C86 exposure resulted in increased FL-AR, ARv, Hsp40, and CHIP association with Hsp70 (assessed following anti-FLAG immunoprecipitation, see Figure 2D), while total FL-AR/ARv expression was diminished relative to vehicle treated cells (Supplemental Figure 2F). Taken together, these data are consistent with interaction of C86 with an Hsp40/Hsp70 multi-protein complex.
Multiple DnaJ families interact with AR and ARv7
To explore whether FL-AR and ARv7 preferentially interact with distinct Hsp40 proteins, first we transiently transfected HEK293 cells with either FLAG-tagged FL-AR or ARv7 and monitored interaction with endogenous Hsp40 (DnaJB1) by WB after immunoprecipitation with FLAG-beads. As shown in Figure 3A (see also Supplemental Figure 3A), endogenous Hsp40 coimmunoprecipitated with both FLAG-tagged FL-AR and ARv7. Endogenous Hsp70 was also present in these pulldowns (Figure 3A; Supplemental Figure 3A). We next examined whether various FLAG-DnaJ proteins shown to bind b-C86 may preferentially interact with either FL-AR or ARv7. As demonstrated in Figure 3B (see also Supplemental Figure 3B), and similar to our previous b-C86 binding data (Figure 2C), all FLAG-DnaJs tested interacted to some extent with FL-AR and ARv7. For FLAG-DnaJA1, -A4, -B2a, -B6b, -C5, and -C7, interaction with FL-AR was stronger relative to interaction with ARv7, while binding of FLAG-DnaJB1 and -B4 to FL-AR and ARv7 was relatively equal (Figure 3B; Supplemental Figure 3B). From 22Rv1 cells, FLAG-DnaJB2a, -B6b, -C5, and -C7 each coimmunoprecipitated with endogenous FL-AR and ARv (in this experiment, ARv was detected with FL-AR antibody) (Figure 3C; Supplemental Figure 3C). Endogenous Hsp70 was also present in these complexes (Figure 3C; Supplemental Figure 3C). Taken together, these data confirm that loss of the AR LBD does not confer loss of Hsp40/Hsp70 interaction.
Figure 3.
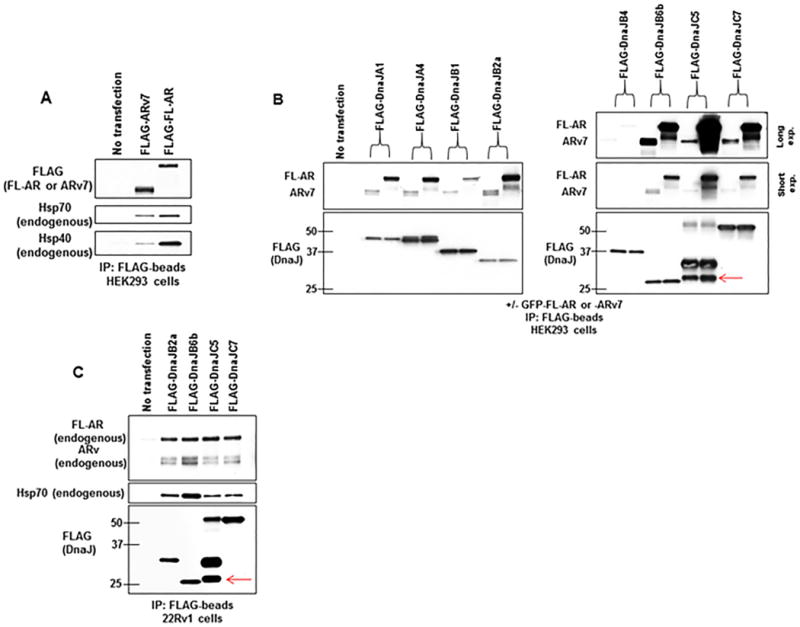
Multiple DnaJ isoforms interact with AR and ARv7.
(A) Lysate from HEK293 cells transfected with FLAG-ARv7 or FLAG-AR was probed with FLAG-beads. Bound proteins were detected by WB.
(B) Lysate from HEK293 cells cotransfected with indicated FLAG-DnaJ family members and GFP-AR or GFP-ARv7 was probed with FLAG-beads. Bound proteins were detected by WB. Red arrow notes detection of native FLAG-DnaJC5, which tends to oligomerize. AR antibody N-20 was used to detect FL-AR and ARv7.
(C) Lysate from 22Rv1 CRPC cells transfected with indicated FLAG-DnaJ family members was incubated with FLAG-beads. Bound proteins were detected by WB. Red arrow notes detection of native FLAG-DnaJC5, which tends to oligomerize.
Experiments were repeated at least three times.
Pharmacologic targeting of Hsp70 destabilizes AR and ARv7 in CRPC cells
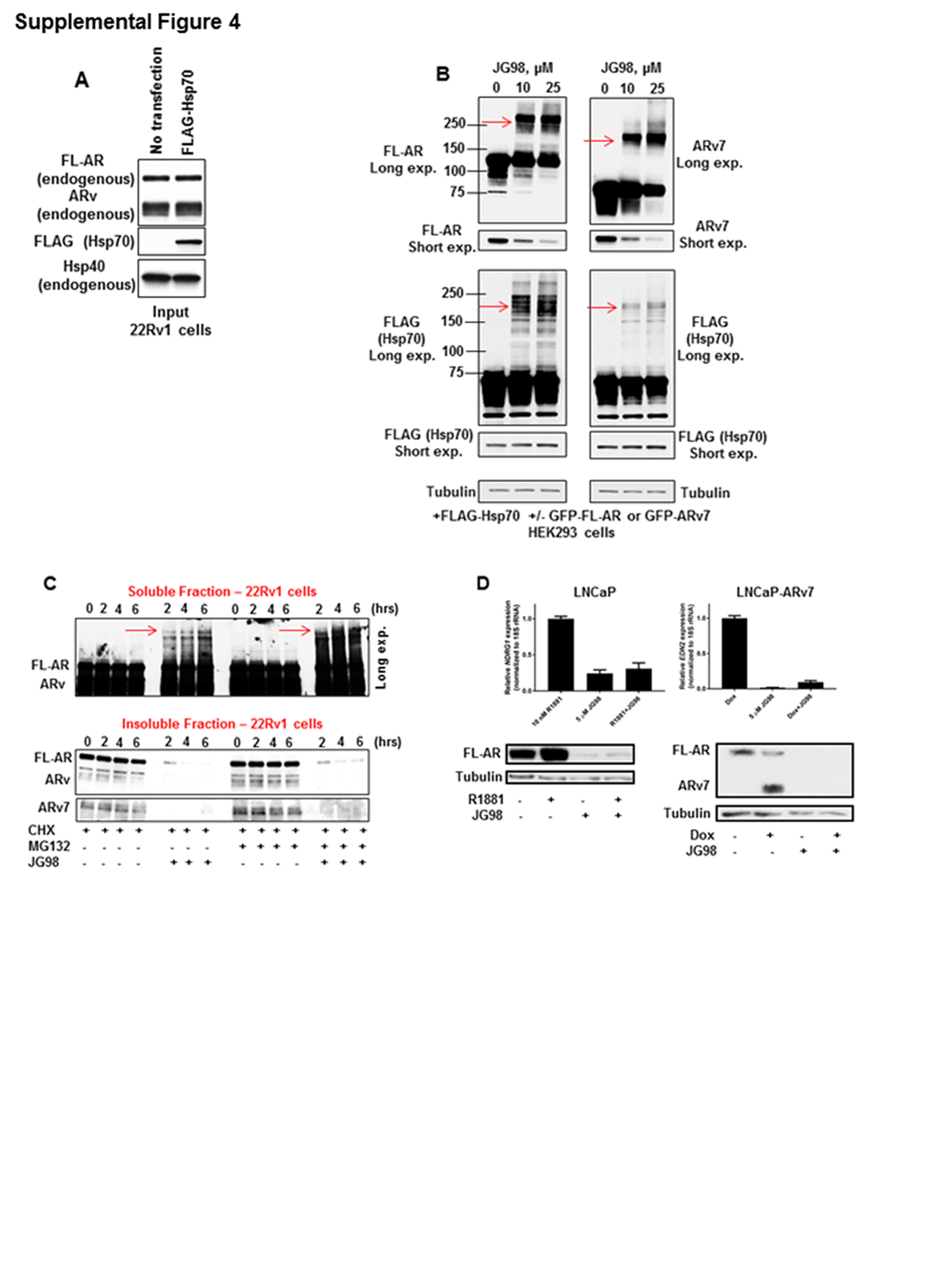
The fact that Hsp70 appears to be present in complexes with FL-AR and ARv7, and that Hsp70 is found in endogenous complexes of Hsp40 and AR/ARv7 pulled down with either b-C86, FLAG-AR, or FLAG-DnaJ (Figures 2C, 3A, 3C), suggests that Hsp70 contributes to the stability and/or function of AR and ARv7 and provides a rationale for targeting Hsp70 in addition to Hsp40 in CRPC. JG98 is a potent allosteric inhibitor of Hsp70 derived from the rhodacyanine MKT-077 (32) that inhibits Hsp70 by preventing nucleotide exchange and thus stabilizing a high-affinity ADP-bound Hsp70/client interaction (32, 39). Preventing release of clients from Hsp70 eventually leads to degradation in proteasomes (20) and studies of JG98 in MCF7 breast cancer cells have demonstrated robust anti-cancer activity (39). To begin to explore the impact of JG98 on FL-AR and ARv in CRPC, we first assessed whether Hsp70 directly interacts with endogenous FL-AR and/or ARv7 in 22Rv1 cells, knowing that it coimmunoprecipitates with transiently expressed FLAG-FL-AR and FLAG-ARv7 in HEK293 cells (Figure 3A). As shown in Fig 4A (see also Supplemental Figure 4A), endogenous FL-AR and ARv coimmunoprecipitated with FLAG-Hsp70 in 22Rv1 cells. Concurrently, and in agreement with our previous data (Figure 3A), endogenous Hsp40 was also present in these FLAG pulldowns (Figure 4A).
Figure 4.
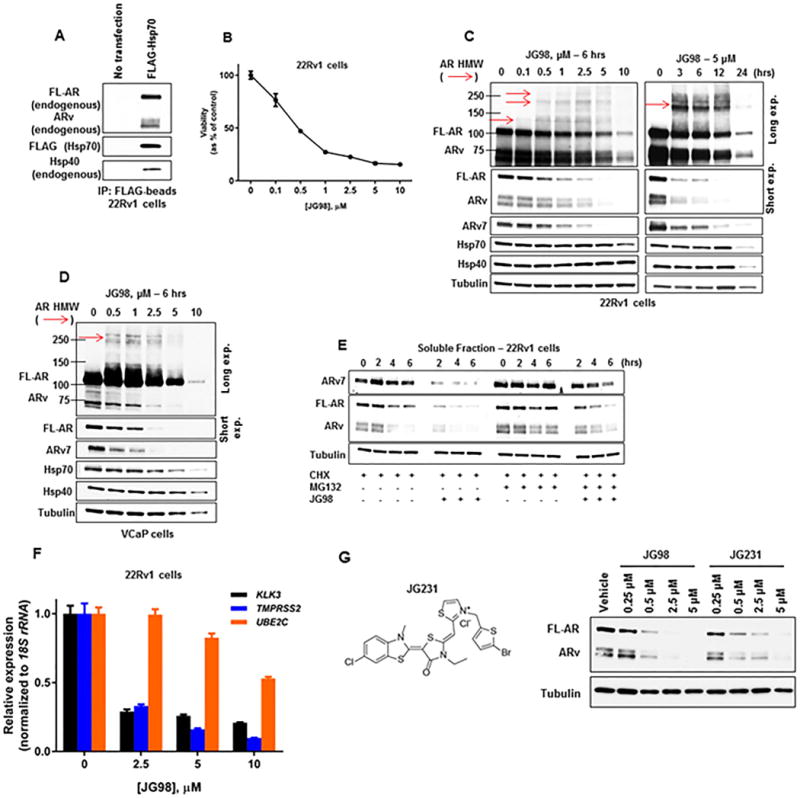
Pharmacologic targeting of Hsp70 destabilizes the AR/ARv7 signaling axis in CRPC cells.
(A) Lysate from 22Rv1 CRPC cells transfected with FLAG-Hsp70 was probed with FLAG-beads. Bound protein was detected by WB.
(B) Viability of 22Rv1 CRPC cells treated with increasing concentrations of JG98 for 72 hours as determined by MTT assay. Data represent a mean of four to six replicates +/− S.D.
(C) WB of cell lysate from 22Rv1 CRPC cells treated with increasing concentrations of JG98 for 6 hours (left) or 5 μM JG98 for up to 24 hours (right). Red arrows note detection of HMW AR species detected with AR antibody N-20.
(D) WB of cell lysate from VCaP CRPC cells treated with increasing concentrations of JG98 for 6 hours. Red arrow notes detection of HMW AR species detected with AR antibody N-20.
(E) WB of the stability of soluble AR, ARv, and ARv7 from 22Rv1 CRPC cells following pre-incubation with 10 μM MG132 and/or 10 μg/mL cycloheximide (CHX) for 1 hour, followed by +/− 1 μM JG98 for 6 additional hours. Equal amounts of protein (10 μg) were loaded for WB of soluble and insoluble fractions (see also Supplemental Figure 4C).
(F) RT-qPCR of AR/ARv7 target genes, normalized to 18S rRNA, from 22Rv1 CRPC cells treated with increasing concentrations of JG98 for 16 hours. Data represent a mean of three replicates +/− S.D.
(G) Structure of JG231 (left). WB of cell lysate from 22Rv1 CRPC cells treated with increasing concentrations of JG98 or JG231 for 6 hours (right).
Experiments were repeated at least three times.
With evidence that endogenous FL-AR and ARv engage with Hsp70 as well as with Hsp40, we next characterized the consequences of Hsp70 inhibition with JG98 in CRPC cells. A significant, dose-dependent decrease in cell viability was observed, with JG98 displaying an IC50 between 400-500 nM in 22Rv1 (Figure 4B) and VCaP (Supplemental Figure 1E) cells at 72 hours. Importantly, like 22Rv1, VCaP cells were highly resistant to enzalutamide (Supplemental Figure 1E). Not surprisingly, we observed destabilization of FL-AR, ARv, and ARv7 proteins with increasing JG98 dose and incubation time in both 22Rv1 and VCaP cells (Figures 4C, 4D). Notably, coincident with destabilization of FL-AR/ARv7 proteins, JG98 also led to a concomitant increase in the formation of soluble high molecular weight (HMW) AR species (Figure 4C, 4D, red arrows, detected with AR antibody N-20) in both cell types, which were not detected in vehicle control samples or C86 treated cells (Supplemental Figure 1F). Unlike C86, JG98 did not induce a compensatory upregulation in either Hsp40 (DnaJB1) or Hsp70 in vitro. In fact, levels of these chaperones decreased in 22Rv1 cells after treatment with 10 μM JG98 for 6 hours (Figure 4C, left) or after longer treatment (12-24 hours) with 5 μM JG98 (Figure 4C, right). JG98 also caused a reduction in Hsp70 expression in VCaP cells after 6 hours (Figure 4D).
To determine whether Hsp70 is present in these HMW AR species, we transiently co-expressed FLAG-Hsp70 with GFP-AR or GFP-ARv7 in HEK293 cells. JG98 caused a dose-dependent loss of FL-AR (Supplemental Figure 4B, left) and ARv7 (Supplemental Figure 4B, right) in these cells, accompanied by formation of HMW soluble FL-AR and ARv7 species. JG98 treatment also led to formation of HMW FLAG-Hsp70 species (detected with FLAG-tag antibody), which appear to colocalize with HMW AR species (red arrows, Supplemental Figure 4B).
When FL-AR/ARv7 protein half-life was examined, we found that contemporaneous treatment with both cycloheximide and JG98 caused a greater loss (compared to cycloheximide alone) of “native” (e.g., with predicted mobility in SDS gels) AR, ARv, and ARv7, and increased accumulation of HMW AR species (Figure 4E; Supplemental Figure 4C). Blocking proteasome activity with MG132 somewhat reversed Hsp70-inhibitor mediated destabilization of native FL-AR/ARv7 and the appearance of HMW AR species (Figure 4E; Supplemental Figure 4C). However, in contrast to C86 (Supplemental Figure 1D), JG98 did not promote accumulation of these nuclear receptors in a detergent-insoluble protein lysate fraction (Supplemental Figure 4C). Consistent with these data, and similar to inhibition of Hsp40 with C86, Hsp70 inhibition affected expression of FL-AR and ARv7 target genes, as treatment of 22Rv1 cells with JG98 led to a dose-dependent decrease in KLK3, TMPRSS2, and UBE2C gene expression (Figure 4F). Additionally, JG98 inhibited androgen-dependent NDRG1 expression (Supplemental Figure 4D, top left) and ARv7-dependent EDN2 expression (Supplemental Figure 4D, top right), while also destabilizing AR and ARv7 proteins (Supplemental Figure 4D, bottom) in LNCaP and LNCaP-ARv7 cells.
Inhibition of Hsp40 and/or Hsp70 demonstrates in vivo efficacy in CRPC-derived 22Rv1 xenografts
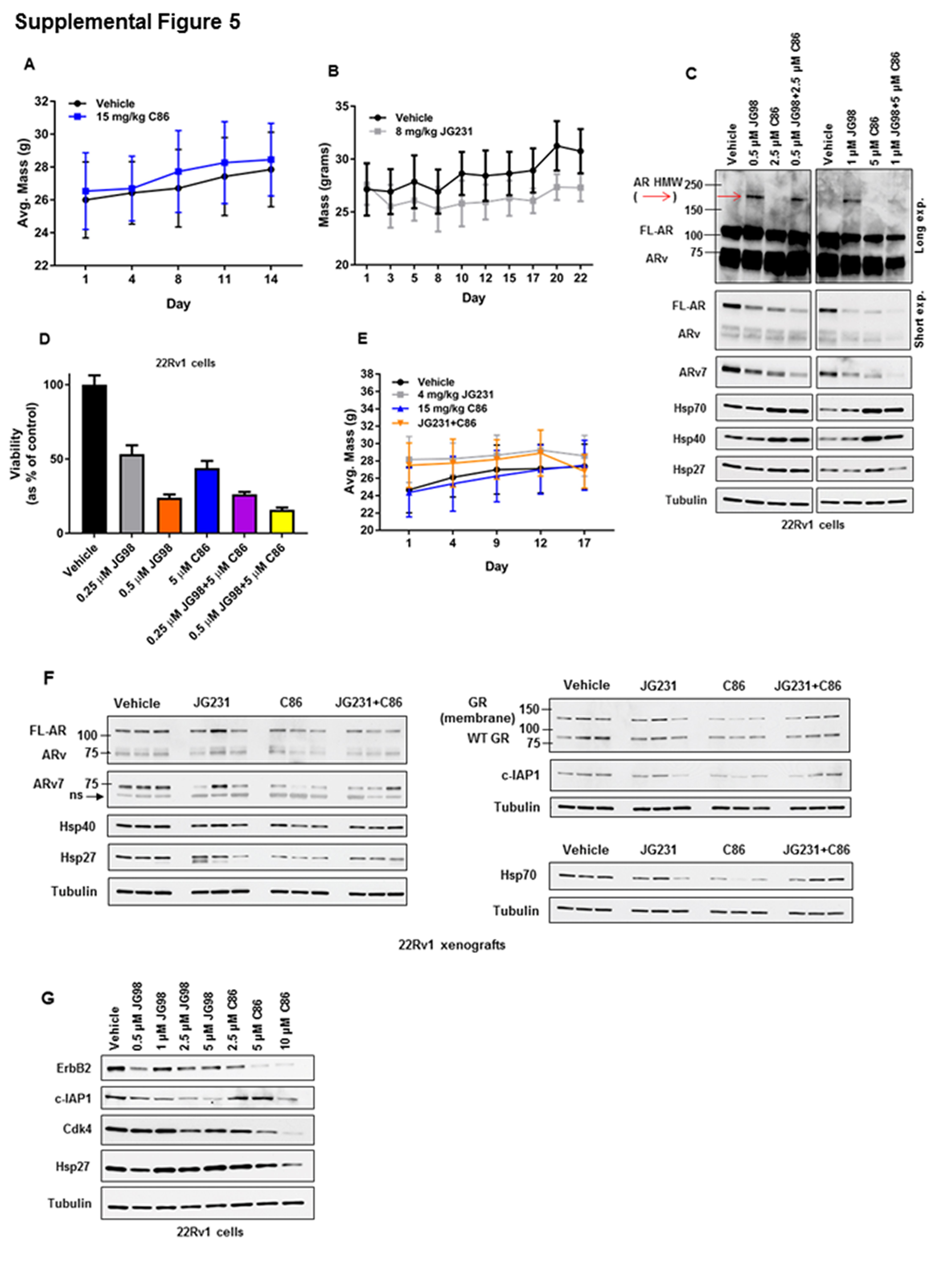
With sufficient evidence in hand that AR/ARv7 rely on both Hsp40 and Hsp70 for protein stability, and that C86 and JG98 individually destabilize these nuclear receptors and inhibit their transcriptional activity in vitro, we next explored the efficacy of Hsp40- and/or Hsp70-inhibitor mediated anti-CRPC activity in vivo (either single agent or in combination). After confirming the in vitro equivalence of JG98 and JG231 (a JG98 analog with improved in vivo characteristics) in promoting AR/ARv destabilization (Figure 4G), we used JG231 for Hsp70 inhibition in these in vivo studies. For single agent treatments, once palpable tumors were evident (tumor volume of 75-125 mm3), mice bearing 22Rv1 xenografts were treated by tail vein injection every Monday, Wednesday, and Friday for 2 weeks with vehicle or C86 at 15 mg/kg, or by intraperitoneal injection every other day for three weeks with vehicle or 8 mg/kg JG231. Relative to vehicle control animals, tumor growth was significantly inhibited in mice treated with either C86 (Figure 5A) or JG231 (Figure 5B) and both agents were well-tolerated, as no overt signs of toxicity were observed with either treatment (Supplemental Figures 5A, 5B).
Figure 5.
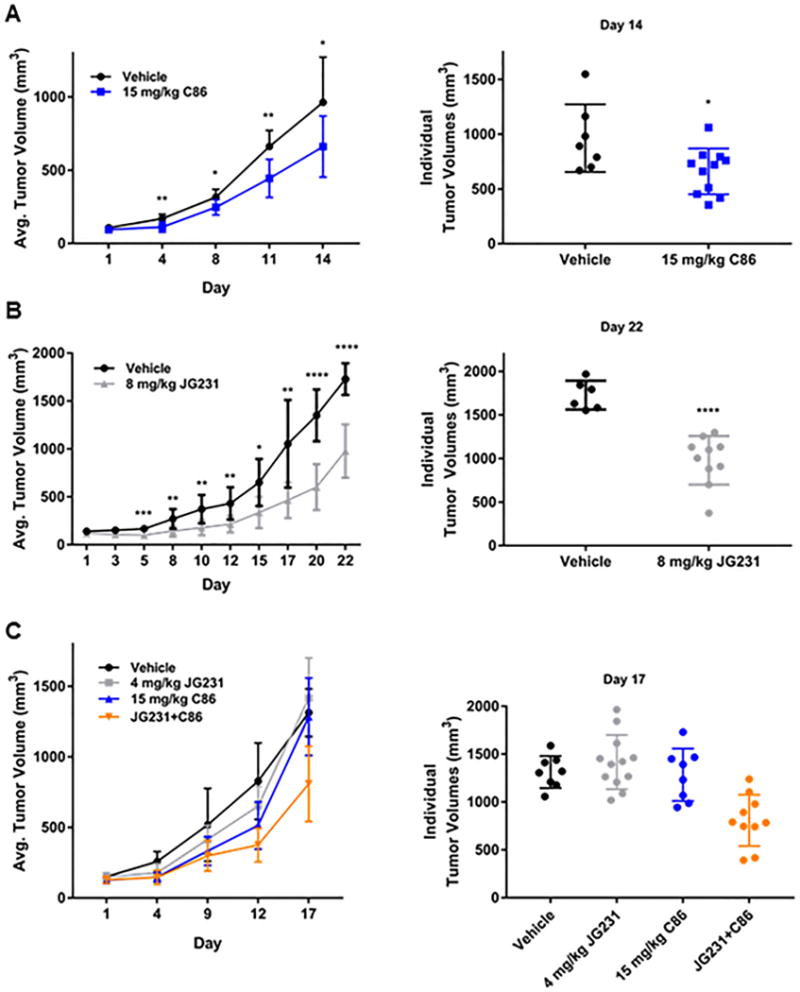
C86 and the pharmacokinetically improved JG231 display anti-CRPC activity in vivo.
(A) Average tumor volume (+/− S.D.) of 22Rv1 CRPC xenografts in mice treated with vehicle (n=7) or C86 (n=11) every Monday, Wednesday, and Friday for 2 weeks (left). Individual tumor volume measurements for vehicle and C86 treated mice after 14 days (right).
(B) Average tumor volume (+/− S.D.) of 22Rv1 CRPC xenografts in mice treated with vehicle (n=6) or JG231 (n=10) every other day for 3 weeks (left). Individual tumor volume measurements for vehicle and JG231 treated mice after 22 days (right).
(C) Average tumor volume (+/− S.D.) of 22Rv1 CRPC xenografts in mice treated with vehicle (n=8), JG231 (n=12), C86 (n=8), or JG231+C86 (n=10) for 2.5 weeks (see methods for schedule) (left). Individual tumor volume measurements for vehicle, JG231, C86, and JG231+C86 treated mice after 17 days (right). Statistics at day 17 – Vehicle vs. JG231 – NS; Vehicle vs. C86 – NS; Vehicle vs. JG231+C86***. JG231+C86 vs. Vehicle***; JG231+C86 vs. JG231****; JG231+C86 vs. C86**.
Experiments were repeated at least two times. NS – not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by unpaired t-test or one-way ANOVA with multiple comparisons.
Since Hsp40 frequently interacts with Hsp70, it is possible that combined treatment with both JG231/JG98 and C86 would additively promote AR/ARv7 protein instability in vitro and would display enhanced anti-tumor activity in vivo. Due to the observable increase in Hsp40/Hsp70 expression upon C86 exposure in vitro (Figure 1B), we pretreated 22Rv1 cells with JG98 to attempt to counter any Hsp70-mediated compensatory effects caused by C86 treatment, followed by the addition of C86. A low dose of this combination (0.5 μM JG98+2.5 μM C86) more potently destabilized FL-AR, ARv, and ARv7 in vitro, relative to vehicle and single agent treatments, but was unable to block the increase in Hsp40/Hsp70 expression caused by C86 (Supplemental Figure 5C). Additive destabilization of AR/ARv7 was also observed at higher doses of JG98+C86 (1 μM JG98+5 μM C86) relative to vehicle and JG98 or C86 alone. Furthermore, this concentration of JG98 was also able to partially blunt the C86-induced increase in Hsp40/Hsp70 expression, suggesting it is possible to inhibit any potential chaperone-dependent compensatory effects caused by C86 treatment (Supplemental Figure 5C). The combinatorial activity of JG98+C86 is also evident in the observed effect on viability in vitro, where a significant decrease was found in 22Rv1 cells treated with JG98+C86 compared to JG98, C86, or vehicle single agent treatments (Supplemental Figure 5D).
When tested against 22Rv1 xenografts in vivo, a significant inhibition of tumor growth was detected in JG231+C86 treated mice relative to vehicle and both monotherapies (Figure 5C) with no overt toxicity observed (Supplemental Figure 5E). Tumor homogenates from JG231 or C86 treated mice showed decreased FL-AR, ARv, and ARv7 protein levels compared to vehicle treated mice (Supplemental Figure 5F), thereby recapitulating our in vitro data (Figures 1B, 4C). In addition, levels of c-IAP1, an Hsp70 client and biomarker of Hsp70 inhibition (40) were decreased in tumors from JG231 treated mice, confirming selective, on-target engagement with Hsp70. Combination treatment of JG231 with C86 also decreased these client proteins (Supplemental Figure 5F), but the effects were not additive as in our in vitro data (Supplemental Figure 5C). It is possible this combination impacts a number of other Hsp70 and Hsp90 client proteins, as several additional chaperone clients are affected by Hsp40 and Hsp70 inhibition in vitro (Supplemental Figure 5G). Finally, it should be noted that no induction of the heat shock response by C86 was found in vivo since Hsp27/Hsp40/Hsp70 protein levels were not induced by JG231, C86, or JG231+C86 (Supplemental Figure 5F). In fact, expression of these chaperone proteins was decreased relative to vehicle treated mice, which may also play a role in the in vivo efficacy observed (Figure 5). Collectively, these data suggest multiple dynamic client interactions with the Hsp40/Hsp70/Hsp90 chaperone machinery in CRPC cells, consistent with the pleiotypic nature of molecular chaperones. Importantly, these data also support the feasibility and need for further exploration of targeting the Hsp40/Hsp70 chaperone axis as an alternative, multifactorial therapeutic strategy to treat CRPC.
Hsp40 and Hsp70 inhibition destabilizes GR in CRPC cells
Increased GR expression in a significant percentage of CRPC and in a drug-resistant CRPC xenograft model, coupled with the ability of GR to occupy and regulate a portion of the AR cistrome, supports GR upregulation as an alternative mechanism of resistance to enzalutamide and other AR-targeted therapies in a subset of CRPC patients (14–17). Like AR, GR is a nuclear receptor that requires interaction with the Hsp40/Hsp70/Hsp90 chaperone axis for stability and function (41). Thus, we tested the ability of C86 and JG98 to affect GR protein levels. Using the D8H2 GR antibody (Cell Signaling), we detected two distinct GR bands in an SDS gel: one pertaining to “native” (e.g., having the predicted mobility) GR and the other consistent with a lesser studied membrane-associated form of GR which has also been reported to interact with Hsp70 and Hsp90 (Hsp40 was not tested) (42). In response to either C86 or JG98, levels of both forms of GR protein in 22Rv1 and VCaP cells were decreased (Figure 6A, B). These effects were confirmed in vivo, where a reduction in membrane-associated GR and native GR was found in 22Rv1 tumor lysate from JG231, C86, or JG231+C86 treated mice (Supplemental Figure 5F). Similar to its impact on AR, JG98 induced the formation of soluble HMW GR species (Figure 6B, red arrows). We also found both forms of GR interacted with b-C86, were pulled down in a complex containing Hsp40/Hsp70, and that b-C86-mediated pulldown of these proteins was competed by excess unlabeled C86 (Figure 6C).
Figure 6.
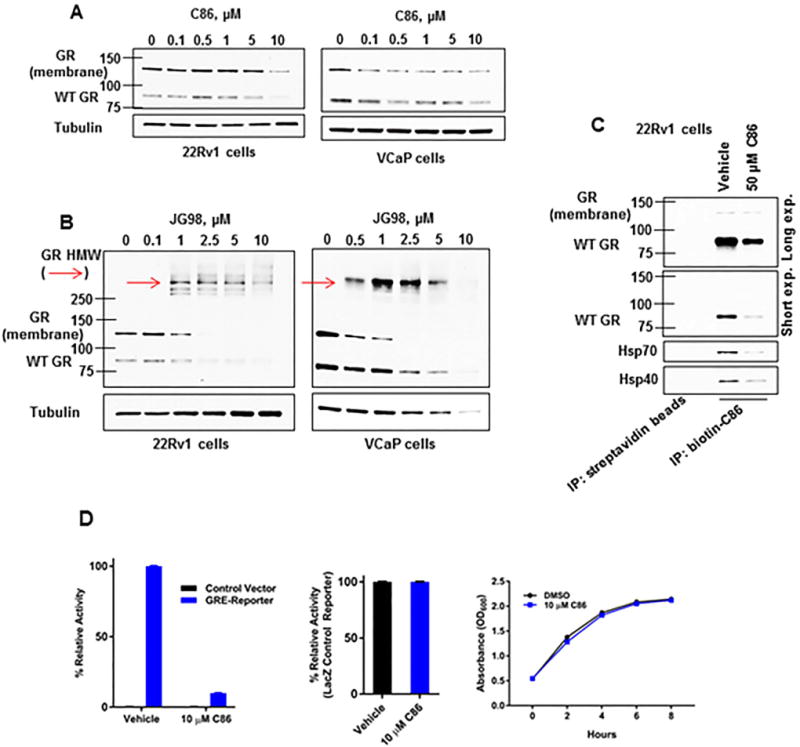
Hsp40 and Hsp70 inhibition destabilizes GR in CRPC cells.
(A, B) WB of cell lysate from 22Rv1 or VCaP CRPC cells treated with increasing concentrations of C86 (A, left, right, respectively) or JG98 (B, left, right, respectively) for 6 hours. Red arrows note detection of HMW GR species.
(C) Biotin-C86 binding profile from 22Rv1 CRPC cells incubated with excess unlabeled vehicle (DMSO) or 50 μM C86 for 1 hour as assessed by streptavidin-bead IP and WB.
(D) GRE-(left) or LacZ Control-Reporter (middle) assay of yeast cells treated with vehicle or C86 as detected by chemiluminescence. Absorbance at O.D. 600 was utilized to measure yeast viability +/− C86 (right).
Experiments were repeated at least three times.
Finally, we co-expressed human GR and a GR reporter in yeast to examine the influence of C86 on GR transcriptional activity (JG98 was not used in this experiment as it directly affected the LacZ reporter). Consistent with the effects of C86 on GR protein levels (Figure 6A), C86 markedly reduced GR transcriptional activity in this yeast model system (Figure 6D, left), while negligibly affecting the LacZ control reporter (Figure 6D, middle) or yeast viability (Figure 6D, right). Together, these data provide evidence of the reliance of GR on the Hsp40/Hsp70 chaperone axis and they further emphasize the potential benefit of developing inhibitors of these two chaperones to combat therapeutic resistance in CRPC.
Discussion
Current standard of care therapies for the treatment of CRPC, including enzalutamide and abiraterone, disrupt AR signaling by targeting the AR LBD (directly or indirectly, respectively). Although these agents prolong patient survival, they are not curative, and alternative splicing of the AR to a constitutively active, LBD-deficient form (e.g., ARv7) represents one mechanism of resistance (6). Furthermore, expression of the nuclear receptor GR is significantly increased in an enzalutamide-resistant CRPC model and in a portion of human CRPC patient tumors, and GR has been shown to regulate a subset of AR-dependent genes in CRPC (14, 17). Thus, identification of agents able to simultaneously target FL-AR and GR, as well as ARv lacking the LBD, may provide a novel strategy to treat several mechanisms of ADT resistance. In this study, we have identified Hsp40 and Hsp70 chaperones as critical regulators of these nuclear receptors, including ARv lacking the LBD.
These observations stem from our initial identification of C86 as a small molecule able to inhibit transcription of both androgen-dependent and androgen-independent AR target genes, and by our subsequent characterization of C86 as an inhibitor of Hsp40. Further, our data showing that b-C86 recognizes a number of structurally diverse Hsp40 proteins (and even SV40 large T antigen), all of which have in common a J domain which allows Hsp40 to interact with Hsp70, are key observations suggesting that C86 is a modulator of pan-Hsp40-dependent proteostasis. In 22Rv1 and VCaP CRPC cells, C86 was able to simultaneously affect FL-AR, ARv7, and GR protein stability and transcriptional activity.
Using b-C86 to pull down Hsp40-containing complexes, we not only identified FL-AR, ARv, including ARv7, and GR, but also Hsp70 and its cochaperones Bag3 and CHIP. Thus, we explored whether targeting Hsp70 may also impact FL-AR/ARv/GR protein stability and transcriptional activity. For that purpose, we used JG98, a potent allosteric inhibitor of Hsp70 (32, 39). We found that, similar to Hsp40 inhibition with C86, Hsp70 inhibition with JG98 also destabilized these nuclear receptor proteins and inhibited their transcriptional activity. However, unlike C86, which promoted accumulation of the nuclear receptors in a detergent-insoluble fraction upon proteasome inhibition, JG98 promoted the formation of detergent-soluble HMW nuclear receptor species in which Hsp70 is present. This is a likely consequence of this inhibitor’s mechanism of action, which involves stabilization of a high affinity client-ADP-bound Hsp70 complex (32, 39).
Given the ability of C86 and JG98 to simultaneously target FL-AR, ARv, and GR, Hsp40/Hsp70 inhibition has the ability to affect multiple mechanisms of CRPC therapeutic resistance. For example, ADT can lead to reactivated AR signaling due to AR gene amplification, which sensitizes prostate cancer cells to low androgen levels, or to point mutations in the AR, which promote promiscuous/non-androgenic ligand binding capable of upregulating AR transcriptional activity (5). Our data using VCaP cells (which overexpress wild type FL-AR due to gene amplification, while also expressing a number of ARv), show that both C86 and JG98 promote the degradation of overexpressed FL-AR as well as ARv. Further, our data show that the endogenous FL-AR H874Y mutant expressed in 22Rv1 cells, which can be activated by the anti-androgen flutamide (43), remains sensitive to inhibition of Hsp40 and Hsp70. FL-AR mutations in response to selective pressure from enzalutamide have been described in an in vitro random mutagenesis screen (44) and have also been detected in human CRPC patients (45). Based on our data, it is likely that these resistance-mediating AR mutants will also retain sensitivity to C86 and JG98.
There are a number of ARv whose contributions to CRPC and development of therapeutic resistance are not completely understood. For example, in addition to ARv7, both 22Rv1 and VCaP cells, and some human CRPC patient tumors, express ARv567es, another constitutively active AR splice variant (6). Expression of ARv567es promotes tumor growth in castrated mice (46), its transcript level has been shown to be increased in CRPC bone metastases, and its expression correlates with reduced survival (8). Like ARv7, ARv567es is also resistant to Hsp90 inhibitors (28). Although we did not specifically examine the effect of C86 or JG98 on ARv567es expression, it would be worthwhile to investigate if it and other ARv also require the Hsp40/Hsp70 chaperone machinery for stability and transcriptional activity. This is likely to be the case, since the AR antibody N-20 (used to detect FL-AR) appears to recognize additional splice variants besides ARv7 that are similarly sensitive to both C86 and JG98.
Although there is strong evidence to implicate the Hsp40/Hsp70/Hsp90 chaperone complex in FL-AR/GR LBD refolding and stabilization (20, 23), other observations support an LBD-independent role for Hsp40/Hsp70 chaperones in regulating nuclear receptor stability. As general modulators of protein homeostasis, Hsp40 and Hsp70 are known to interact with and stabilize hydrophobic patches of nascent proteins (41), and mutations in Hsp40 result in AR aggregation (47). Further, exogenous expression of the Hsp40 family members DnaJB6b and DnaJB8 reverses the aggregation propensity of poly-glutamine-expanded AR (48). Taken together, these observations suggest that ARv7 (and other ARv) likely require Hsp40 and Hsp70 for proper folding and prevention of aggregation. Additionally, each of the currently identified ARv share a common N-terminal transactivation domain, which is required for interactions with cofactors to activate transcription (6). Because Hsp40/Hsp70 may reassociate with the FL-AR after ligand binding and assist with its nuclear translocation (41), and given that our data confirm LBD-deficient ARv7 interacts with Hsp40 and Hsp70, it is likely that one or both of these chaperones may play a role in stabilizing FL-AR and ARv7 for dimerization events (including heterodimerization) (49, 50), assistance in nuclear translocation, and interactions with cofactors and/or DNA to stimulate transcriptional activity.
No matter the ultimate mechanism or mechanisms by which C86 and JG98 affect the stability and function of these overexpressed, mutated or aberrantly spliced nuclear receptors driving CRPC, this study provides an in vitro and in vivo proof-of-concept of the potential benefit of inhibiting the Hsp40/Hsp70 chaperone axis in ADT-resistant CRPC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the investigators listed in Methods for kindly providing reagents used in this study. Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. In conducting research using animals, the investigators have adhered to the laws of the United States and regulations of the Department of Agriculture. In the conduct of research utilizing recombinant DNA, the investigators have adhered to NIH Guidelines for research involving recombinant DNA molecules. The authors declare no conflicts of interest. This study was supported by funds from the Intramural Research Program, National Cancer Institute, Center for Cancer Research (L.M.N. and J.B.T.), NIH grant NS059690 (J.E.G), the Cancer Prevention & Research Institute of Texas (RP150648, N.L.W.), and the Office of the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program under Award No. W81XWH-16-1-0562 (L.M.N. and J.E.G.).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 3.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boudadi K, Antonarakis ES. Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin Med Insights Oncol. 2016;10:1–9. doi: 10.4137/CMO.Ss34534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu J, Van der Steen T, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nat Rev Urol. 2015;12:137–144. doi: 10.1038/nrurol.2015.13. [DOI] [PubMed] [Google Scholar]
- 7.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 11.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krause WC, Shafi AA, Nakka M, Weigel NL. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol. 2014;54:49–59. doi: 10.1016/j.biocel.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Echeverria PC, Picard D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim Biophys Acta. 2010;1803:641–649. doi: 10.1016/j.bbamcr.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szmulewitz RZ, Chung E, Al-Ahmadie H, Daniel S, Kocherginsky M, Razmaria A, et al. Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate. 2012;72:157–164. doi: 10.1002/pros.21416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yemelyanov A, Bhalla P, Yang X, Ugolkov A, Iwadate K, Karseladze A, et al. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle. 2012;11:395–406. doi: 10.4161/cc.11.2.18945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73:1570–1580. doi: 10.1158/0008-5472.CAN-12-2350. [DOI] [PubMed] [Google Scholar]
- 18.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, et al. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999;69:307–313. doi: 10.1016/s0960-0760(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 20.Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2015;55:353–371. doi: 10.1146/annurev-pharmtox-010814-124332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 22.Pratt WB, Morishima Y, Osawa Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J Biol Chem. 2008;283:22885–22889. doi: 10.1074/jbc.R800023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014;157:1685–1697. doi: 10.1016/j.cell.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang Y, Fliss AE, Robins DM, Caplan AJ. Hsp90 regulates androgen receptor hormone binding affinity in vivo. J Biol Chem. 1996;271:28697–28702. doi: 10.1074/jbc.271.45.28697. [DOI] [PubMed] [Google Scholar]
- 25.Marivoet S, Van Dijck P, Verhoeven G, Heyns W. Interaction of the 90-kDa heat shock protein with native and in vitro translated androgen receptor and receptor fragments. Mol Cell Endocrinol. 1992;88:165–174. doi: 10.1016/0303-7207(92)90021-w. [DOI] [PubMed] [Google Scholar]
- 26.He S, Zhang C, Shafi AA, Sequeira M, Acquaviva J, Friedland JC, et al. Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. Int J Oncol. 2013;42:35–43. doi: 10.3892/ijo.2012.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kakar M, Kanwal C, Davis JR, Li H, Lim CS. Geldanamycin, an inhibitor of Hsp90, blocks cytoplasmic retention of progesterone receptors and glucocorticoid receptors via their respective ligand binding domains. AAPS J. 2006;8:E718–728. doi: 10.1208/aapsj080481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gillis JL, Selth LA, Centenera MM, Townley SL, Sun S, Plymate SR, et al. Constitutively-active androgen receptor variants function independently of the HSP90 chaperone but do not confer resistance to HSP90 inhibitors. Oncotarget. 2013;4:691–704. doi: 10.18632/oncotarget.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shafi AA, Cox MB, Weigel NL. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids. 2013;78:548–554. doi: 10.1016/j.steroids.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferraldeschi R, Welti J, Powers MV, Yuan W, Smyth T, Seed G, et al. Second-Generation HSP90 Inhibitor Onalespib Blocks mRNA Splicing of Androgen Receptor Variant 7 in Prostate Cancer Cells. Cancer Res. 2016;76:2731–2742. doi: 10.1158/0008-5472.CAN-15-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y, Xu W, Ji J, Feng D, Sourbier C, Yang Y, et al. Alternative splicing of the cell fate determinant Numb in hepatocellular carcinoma. Hepatology. 2015;62:1122–1131. doi: 10.1002/hep.27923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, et al. Analogs of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, as Anti-Cancer Agents. ACS Med Chem Lett. 2013;4 doi: 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pepe A, Pamment M, Kim YS, Lee S, Lee MJ, Beebe K, et al. Synthesis and structure-activity relationship studies of novel dihydropyridones as androgen receptor modulators. J Med Chem. 2013;56:8280–8297. doi: 10.1021/jm301714s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106:8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther. 2004;3:551–566. [PubMed] [Google Scholar]
- 37.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelley WL, Landry SJ. Chaperone power in a virus? Trends Biochem Sci. 1994;19:277–278. doi: 10.1016/0968-0004(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Colvin T, Rauch JN, Acosta-Alvear D, Kampmann M, Dunyak B, et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol Cancer Ther. 2015;14:642–648. doi: 10.1158/1535-7163.MCT-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cesa LC, Shao H, Srinivasan SR, Tse E, Jain C, Zuiderweg ERP, et al. X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition. J Biol Chem. 2018;293:2370–2380. doi: 10.1074/jbc.RA117.000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prescott J, Coetzee GA. Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 2006;231:12–19. doi: 10.1016/j.canlet.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 42.Powell CE, Watson CS, Gametchu B. Immunoaffinity isolation of native membrane glucocorticoid receptor from S-49++ lymphoma cells: biochemical characterization and interaction with Hsp 70 and Hsp 90. Endocrine. 1999;10:271–280. doi: 10.1007/BF02738626. [DOI] [PubMed] [Google Scholar]
- 43.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–153. [PubMed] [Google Scholar]
- 44.Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2015;21:2315–2324. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 46.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan CY, Ren HY, Lee P, Caplan AJ, Cyr DM. The type I Hsp40 zinc finger-like region is required for Hsp70 to capture non-native polypeptides from Ydj1. J Biol Chem. 2005;280:695–702. doi: 10.1074/jbc.M410645200. [DOI] [PubMed] [Google Scholar]
- 48.Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, et al. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell. 2010;37:355–369. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res. 2015;75:3663–3671. doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646–1656. doi: 10.18632/oncotarget.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.